

Abstract
The arms race among microbes is a key driver in the evolution of not only the weapons but also defence mechanisms. Many gram-negative bacteria use the type six secretion system (T6SS) to deliver toxic effectors directly into neighbouring cells. Defence against effectors requires cognate immunity proteins. However, here we show immunity-independent protection mediated by envelope stress responses in Escherichia coli and Vibrio cholerae against a V. cholerae T6SS effector, TseH. We demonstrate that TseH is a PAAR-dependent species-specific effector highly potent against Aeromonas species but not against its V. cholerae immunity mutant or E. coli. Structural analysis reveals TseH is likely a NlpC/P60 family cysteine endopeptidase. We determine that two envelope stress response pathways, Rcs and BaeSR, protect E. coli from TseH toxicity by mechanisms including capsule synthesis. The two-component system WigKR (VxrAB) is critical for protecting V. cholerae from its own T6SS despite expressing immunity genes. WigR also regulates T6SS expression, suggesting a dual role in attack and defence. This deepens our understanding of how bacteria survive T6SS attacks and suggests that defending against the T6SS represents a major selective pressure driving the evolution of species-specific effectors and protective mechanisms mediated by envelope stress responses and capsule synthesis.
Introduction
In every environment where life exists, competition is a critical evolutionary driving force of all organisms from Darwin’s finches to simple microbes1. Microbes employ offensive weapons, such as producing antibiotics that inhibit neighbouring bacteria and, in response, bacteria have evolved defensive mechanisms to repair injuries and survive in hostile environments. These include specific resistance genes that inhibit antibiotic activity or nonspecific resistance pathways, such as producing exopolysaccharides (EPS) and biofilms or using envelope stress responses (ESRs) to recognize cellular damage and coordinate repair regulons. These systems have demonstrated protection against antibiotics and other cellular stressors, yet in the natural environment – where encountering an antibiotic might be rare – the evolutionary pressures selecting for these defences can be unclear.
The type six secretion system (T6SS) is a molecular weapon that injects toxic effector proteins into neighbouring cells to kill and out-compete them2,3. Resembling a molecular spear, a contractile outer sheath thrusts a long tube forward and out of the attacking ‘killer’ cell. Tipped by a VgrG protein trimer and further sharpened by a PAAR-domain containing protein, this tube physically punctures ‘prey’ cells to deliver the toxic cargo, which are often attached or covalently fused to the VgrG or PAAR proteins. These effectors have a variety of targets in both eukaryotic and prokaryotic prey and most bacteria with a T6SS produce several effectors with multiple molecular targets. V. cholerae O37 strain V52 constitutively expresses its T6SS and encodes five effectors2,4–6. The most recently identified, TseH, is a peptidoglycan hydrolase encoded outside of the main T6SS operons and likely acquired by horizontal gene transfer4.
The best understood defence against T6SS attacks is self-protection conferred by immunity proteins, which neutralize specific effector toxicities3,7,8. Usually encoded directly adjacent to their cognate effector, immunity genes are considered the gold standard of defence and in previous works their expression has consistently proven sufficient to survive a T6SS attack by their associated bacteria-targeting effector. Accordingly, deletion of an immunity gene renders the cell susceptible to killing by the effector; for instance in V52 where deletion of tsiV1, tsiV2, or tsiV3 cause the bacteria to be killed by TseL, VasX, or VgrG3 (respectively)5. An immunity gene (tsiH) is encoded downstream of tseH but its deletion did not reduce survival when attacked by wild-type V52, nor did deletion of tseH affect killing of E. coli prey that lack T6SS immunity genes entirely4. This raises the question of why do bacteria maintain cryptic effectors in their genome if they do not show activity when delivered by the T6SS?
Here we demonstrate that V. cholerae deploys TseH to outcompete specific species sharing common ecological niches. We report that TseH is a PAAR-dependent chaperone-independent effector highly toxic to Aeromonas and Edwardsiella species. We determine that E. coli is protected against TseH activity by two ESR pathways, BaeSR and Rcs. Expressing a BaeSR-regulated chaperone, Spy, also protects otherwise susceptible A. dhakensis. Surprisingly, the ESR in V. cholerae, WigKR (also known as VxrAB), is critical to survive self-killing by its own T6SS despite expressing a full complement of immunity genes. Considering that the T6SS can greatly influence survival of bacteria in mixed populations, including a significant role in host colonization by pathogens such as V. cholerae9–12, and that the T6SS is encoded by about 25% of gram-negative bacteria13, including co-inhabitants in various niches, our results impact a wide range of bacteria and highlight the striking microbial arms-race selecting for acquisition of effectors and immunity-independent envelope stress protection mechanisms.
Results
TseH is a PAAR-dependent effector displaying species-specific killing
The V. cholerae T6SS effector, TseH, is encoded adjacent to a PAAR protein (PAAR2) but isolated from other known T6SS components. In previous T6SS competition assays, TseH did not show any killing activity of V. cholerae lacking the immunity gene (tsiH), nor did it influence killing of E. coli4. Contrastingly, high concentrations of TseH during overexpression with a periplasmic localization (TAT) tag was able to kill E. coli, suggesting that it is an active effector but E. coli and V. cholerae are able to survive its damage at T6SS-delivered concentrations4. To isolate the activity of TseH we employed a V52 strain with catalytic mutations in all other antibacterial effectors: TseL, VasX and VgrG314. This strain (called TseHWT in this work) retains TseH activity and T6SS function but does not kill E. coli or V. cholerae ΔtsiH prey (Fig. 1a).
Figure 1: TseH is an NlpC/P60 peptidase delivered by PAAR2 that kills A. dhakensis but not E. coli or V. cholerae.

a. T6SS competition assay wherein E. coli, A. dhakensis , or V. cholerae ΔtsiH prey were subjected to killing by wild-type (WT) or catalytically inactive (H64A) TseH delivered from V52 with all other anti-bacterial effectors inactivated. One-way ANOVA with Sidak’s multiple comparisons test comparing each sample to the same prey killed by the TseHH64A strain; ***, p < 0.001; ns, not significant. DL, detection limit. b. Survival of A. dhakensis prey after killing by ΔPAAR2 (in TseHWT background) strain expressing plasmid-borne TseH and/or PAAR2. V, vector control; One-way ANOVA with Dunnett’s multiple comparisons test comparing to the sample expressing TseH and Vector control of PAAR2; ***, p < 0.001; ns, not significant. Graphs show the mean of at least three independent replicates and error bars show one standard deviation. Dots show individual replicates. WT, wild-type V52; TseHWT, V52 with all anti-bacterial effectors inactivated except TseH; TseHH64A, V52 with all anti-bacterial effectors inactivated including TseH. c. Crystal structure of TseH. Brown highlights conserved regions identified previously4. C172 was modified by a cacodylate ion in the crystal structure but is illustrated in stick format without the modification for illustration purpose. The area cleaved by thermolysin is depicted as a dotted line. d. Putative substrate binding pocket of TseH. The catalytic triad is shown as a stick model. The loop covering the active site (pink) is stabilized by the hydrogen bonding interaction (yellow dotted line) between Arg84 side chain and backbone carbonyls of Val39 and Ala40. Val39 side chain is not shown for illustration purpose.
We considered that perhaps TseH could show species-specific killing against bacteria that V. cholerae encounters in natural environments or common hosts. We tested waterborne bacteria prey, including Edwardsiella and Aeromonas species (the latter shares a marine host with V. cholerae15), and several were highly susceptible to TseH-mediated toxicity (Extended Data Fig. 1a). Active TseH killed 100-fold more A. dhakensis SSU than the catalytically inactive mutant (TseHH64A), which showed as little killing as a strain lacking T6SS activity (Fig. 1a). Killing was due to TseH activity, as expression of the immunity gene protected A. dhakensis (Extended Data Fig. 1b), and complementation with exogenous wild-type TseH restored killing to the TseHH64A strain (Extended Data Fig. 1c).
Due to their genetic linkage, TseH has been proposed to require PAAR2 for secretion4. We demonstrate that TseH requires PAAR2 for its delivery, as deletion or truncation of PAAR2 prevented TseH-mediated killing (Fig. 1b, Extended Data Fig. 1d). Notably, despite using the TseHWT background, deletion of PAAR2 likely influenced TseH expression, as both PAAR2 and TseH plasmids were required for complementation. Interestingly, the tseH operon does not include a T6SS chaperone protein and pull-down assays in E. coli suggest that the interaction between PAAR2 and TseH can occur in the absence of other T6SS proteins (Extended Data Fig. 1e). This demonstrates chaperone-independent delivery by a PAAR protein.
Crystal structure of TseH
We determined the crystal structure of TseH and identified random mutations reducing its toxicity that arose during cloning (see Methods) (Fig. 1c-d, Extended Data Fig. 2). TseH exhibits the bi-lobal architecture of papain-like NlpC/P60 enzymes with the catalytic center and substrate binding pocket at the interface of N- and C-terminal lobes (Extended Data Fig. 1f). According to the structural similarity Dali server16 the closest structural homologues of TseH among NlpC/P60 enzymes are representatives of YaeF/Poxvirus G6R subfamily, specifically E. coli metalloprotein YiiX (PDB: 2IF6, Z score of 9.3), B. cereus cysteine protease (PDB: 3KW0, Z score of 7.8) and human hydrolase PPPDE1 (PDB: 3EBQ, Z score of 7.7). The activity of these enzymes has not been characterized17. There is also structural similarity between TseH and Tse1, another T6SS effector that belongs to NlpC/P60 and was shown to degrade peptidoglycan18. TseH and Tse1 structures superimpose with RMSD of 4.11 Å2 over 71 Cα atoms yet the N- and C-terminal lobes appear swapped18,19. Several NlpC/P60 superfamily members are similarly swapped at the primary sequence while retaining relative tertiary positions20.
TseH residues Glu81 and His64 (N-terminal lobe) and Cys172 (C-terminal lobe) correspond to the catalytic triad in the conserved papain-like protease fold in NlpC/P60 superfamily20. Notably, while TseH Cys172 is solvent accessible in accordance with its proposed nucleophile role, the active site opening appears very narrow due to a long loop connecting β1 and β2 strands directly above the catalytic cysteine16. This configuration would preclude the access to large substrate molecules such as polypeptide. It is possible that the observed configuration is part of a regulatory “safety switch” mechanism, which have been reported for other NlpC/P60 superfamily proteins such as the peptidoglycan endopeptidase SaCwlT21,22. The active site of SaCwlT in apo form is occluded (Extended Data Fig. 1g) and it has been suggested that a change in rotomer conformations controls substrate access21,22. The only apparent interaction stabilizing TseH loop conformation is hydrogen bonding through Arg84, suggesting significant flexibility of this element.
Envelope stress responses protect E. coli against T6SS-delivered TseH
Since TseH was able to kill Aeromonas and Edwardsiella species, it is remarkable that the TseHWT strain showed no activity against other bacteria including E. coli and ΔtsiH V. cholerae. We confirmed that overexpression of periplasm-directed TseH caused lysis of A. dhakensis or E. coli, indicating that it is active even in the resistant species (Extended Data Fig. 3a-b) and led to apparent peptidoglycan degradation (Video 1)4. We therefore hypothesized that E. coli survives TseH attacks at T6SS-delivered concentrations by encoding defences capable of mitigating the damage. E. coli encodes five ESRs including the Rcs phosphorelay and the BaeSR two-component system, which can respond to cell wall damage23,24 potentially caused by TseH. Using E. coli ESR mutants25 as prey, we found that deletion of the response regulators rcsB or baeR rendered E. coli sensitive to killing by V. cholerae TseH, significantly reducing survival (Fig. 2a). Complementation by exogenous expression of the deleted genes restored survival (Fig. 2b). Impairing other ESRs (cpxR and pspF), the general stress response (rpoS), or the soxRS reactive oxygen defence, did not reduce survival (Fig. 2a, Extended Data Fig. 3c). These data indicate that the Rcs and BaeSR ESRs are critical for complete survival of E. coli against T6SS-delivered TseH.
Figure 2: Envelope stress responses protect E. coli against T6SS-mediated killing.

a. Relative survival (killing by TseHWT / TseHH64A) of E. coli wild-type or knockout mutants. One-way ANOVA with Dunnett’s multiple comparisons test comparing each sample to WT; ***, p < 0.001; ns, not significant. b. Relative survival of E. coli ΔrcsB or ΔbaeR mutants with empty vector (Vec) or complementation plasmids. One-way ANOVA with Sidak’s multiple comparisons test comparing samples to their respective empty vector controls; *, p < 0.05. c. Relative survival of select E. coli mutants involved in envelope integrity (Env.) or the RcsB and BaeR regulons. One-way ANOVA with Dunnett’s multiple comparisons test comparing each sample to WT; **, p < 0.01; ***, p < 0.001; ns, not significant. WT (E. coli) data is the same as in a. and is shown again for comparison. d. RT-qPCR comparing expression of the Rcs-regulated wza gene in wild-type E. coli prey after T6SS competition assay with TseHWT or TseHH64A killer strains. Value with TseHH64A killer was set to 1 to show fold induction in response to TseH activity upon delivery by the T6SS. One-way ANOVA (with samples shown in Extended Data Fig. 3d) with Sidak’s multiple comparisons test; ***, p < 0.001. e. Relative survival (killing by V52 wild-type / ΔT6SS) of E. coli wild-type or ΔwcaJ prey overexpressing empty vector or RcsA to induce colanic acid synthesis. Killer and prey strains were mixed at a 1:1 ratio. One-way ANOVA with Tukey’s multiple comparisons test; **, p < 0.01. DL, detection limit. f. Relative survival (killing by TseHWT / TseHH64A) of E. coli mutants with empty vector or expression plasmids. One-way ANOVA with Sidak’s multiple comparisons test comparing samples to their respective empty vector controls; **, p < 0.01; ***, p < 0.001; ns, not significant. Graphs show the mean of at least three independent replicates and error bars show one standard deviation. Dots show results from individual replicates. TseHWT, V52 with all anti-bacterial effectors inactivated except TseH; TseHH64A, V52 with all anti-bacterial effectors inactivated including TseH.
Rcs protection against TseH involves osmotic response genes and colanic acid capsule synthesis
To identify how RcsB and BaeR support E. coli survival against TseH, we examined additional knockout strains25. Multiple genes regulated by RcsB26 played a role in protecting against TseH-mediated killing, as survival of the mutant strains was significantly reduced (Fig. 2c, Extended Data Fig. 3c). These included osmB and bdm, which respond to osmotic stress, a potential consequence of peptidoglycan damage27,28. Moreover, deletion of genes involved in colanic acid synthesis (wza and wcaA) reduced survival, suggesting that this Rcs-induced EPS protects E. coli against T6SS attacks. Deletion of the wcaJ gene, resulting in loss of colanic acid while also abolishing potentially toxic intermediates during its synthesis29, also showed reduced survival compared to wild-type with a nearly significant p-value of 0.0527 (Extended Data Fig. 3c); however this difference in severity suggests that accumulating intermediates may further sensitize the wza and wcaA mutants to TseH. Importantly, the colanic acid synthesis operon was induced in E. coli prey when catalytically active TseH (compared to TseHH64A) was delivered by the T6SS (Fig. 2d). While other genes did not show significant induction (Extended Data Fig. 3d), this suggests that at least part of the defence against T6SS attacks is mediated by an active response to TseH damage.
To further demonstrate the role of a colanic acid capsule in protecting against T6SS attacks, we overexpressed the Rcs auxiliary component, RcsA, which has been shown to induce colanic acid synthesis resulting in capsule formation and mucoid colonies30,31. We examined the survival of rcsA-overexpressing E. coli against the T6SS of wild-type V. cholerae V52 with all four antibacterial effectors intact and active. Remarkably, overexpressing rcsA protected E. coli up to 10,000-fold compared to the vector only control (Fig. 2e). This protection did not occur in the ΔwcaJ background, demonstrating that expressing a colanic acid capsule can significantly defend E. coli against T6SS attacks despite lacking immunity genes. Interestingly, the defensive mechanism of the colanic acid capsule is likely via deflecting incoming attacks, as it could not protect against endogenously expressed TseH (Extended Data Fig. 4a).
The BaeSR-regulated periplasmic chaperone, Spy, provides cross-species protection against TseH
In contrast to RcsB, we did not identify any individual knockouts from the BaeR regulon26,32 that showed impaired survival against TseH, suggesting that the key BaeR-regulated defensive gene was not included in our targeted screen or that the regulon as a whole was able to compensate for the loss of a single member (Fig. 2c, Extended Data Fig. 3c). However, we observed that maintaining envelope integrity was important for surviving TseH attacks, as deletion of several genes involved in this process showed impaired survival when challenged with the TseHWT strain. These included the periplasmic protease and chaperone, DegP33, the most sensitive E. coli mutant identified.
Since a prominent member of the BaeSR regulon is the periplasmic chaperone, Spy26,32,34,35, we hypothesized that it may play a role in protecting against TseH. Though deletion of spy alone did not reduce E. coli recovery (Fig. 2c), we considered that expression of Spy may support overall envelope integrity to improve survival against TseH. We found that plasmid-borne exogenous BaeR fully protected sensitive E. coli mutants even with basal levels of expression (Fig. 2f). Moreover, while other members of the BaeR regulon did not improve survival of the highly sensitive ΔdegP mutant (Extended Data Fig. 4b), overexpression of Spy protected the ΔdegP strain and other sensitive E. coli mutants against TseH (Fig. 2f). While it is important to consider that this only occurred with overexpressed levels of Spy, we found that exogenous overexpression of E. coli Spy (but not BaeR) was also able to show cross-species protection in A. dhakensis, which lacks an obvious Spy homolog (Fig. 2f). How Spy overexpression conveys this protection remains unclear, but Spy did not interact with TseH in a pull-down assay (Extended Data Fig. 1e), and appeared to slightly – though not significantly (p = 0.0513) – improve cell permeability measured using propidium iodide (Extended Data Fig. 4c-d).
The WigKR ESR protects V. cholerae against TseH
TseH was also unable to kill V. cholerae lacking the immunity gene, tsiH (Fig. 1a and Altindis et al.4). Since V. cholerae lack the Rcs phosphorelay and BaeSR, we predicted that another ESR might similarly protect against TseH. Vibrio species encode a two-component system called WigKR (VxrAB) that can respond to cell wall damage and induce peptidoglycan repair genes36,37.
To investigate the role of WigKR in V. cholerae’s survival against TseH, we first employed periplasm-directed (TAT-tagged) TseH expressed from an arabinose-inducible plasmid. Interestingly, while wild-type V52 was not inhibited, overexpression of TseH in the wigR knockout strain yielded a distinct zone of clearance (Fig. 3a). Importantly, the ΔtsiH mutant showed no zone of clearing, suggesting that the protective effect of WigR is independent of TsiH. We next tested whether WigKR was responsible for V. cholerae surviving T6SS-mediated delivery of TseH. Individually, neither the ΔwigR nor the ΔtsiH mutants were sensitive to killing by T6SS-delivered TseH but the double mutant lacking both defences showed significantly reduced survival (Fig. 3b). This sensitivity could be complemented by expression of exogenous wigR, confirming that either the immunity protein or the WigKR ESR can provide sufficient protection to prevent killing by T6SS-delivered TseH.
Figure 3: V. cholerae WigR is necessary to survive T6SS self-killing, independent of immunity genes.

a. Induction of TAT-tagged TseH in V52 with intact or deleted tsiH and wigR. Representative of three independent replicates. b. Relative survival (V52 TseHWT / TseHH64A killer strain) of V. cholerae V52 prey with intact or deleted tsiH and wigR. Welch’s one-way ANOVA with Games-Howell test; *, p < 0.05; ns, not significant. For ΔtsiHΔwigR compared to tsiH+wigR+ and tsiH+ΔwigR prey, p < 0.05. c. Relative survival (wild-type V52 / TseHH64A killer strain) of stationary (Stat.) and log phase V52 prey with intact (+), deleted (Δ), or plasmid complemented (p) wigR. One-way ANOVA with Sidak’s test; ***, p < 0.001. Comparing stationary and log phase, p < 0.001 for wigR+ and ΔwigR, not significant for complemented strain. d. Relative survival (wild-type V52 / ΔT6SS killer strain) of V. cholerae V52 prey lacking the tsiV1, tsiV2 and tsiV3 immunity genes (Δimm background) with intact (+), deleted (Δ), or complemented (p) wigR. Stationary (Stat.) or log phase prey were used at a 1:5 killer:prey ratio. One-way ANOVA with Sidak’s multiple comparisons test; ***, p < 0.001; **, p < 0.01. e. Relative survival (killing by A. dhakensis SSU wild-type / ΔT6SS) of V. cholerae V52 prey (ΔT6SS background) with intact (+), deleted (Δ), or complemented (p) wigR. Log phase prey were used at a 1:1 killer:prey ratio. One-way ANOVA with Sidak’s multiple comparisons test; ***, p < 0.001; **, p < 0.01. Graphs show the mean of at least three independent replicates, error bars show standard deviation. Dots show individual replicates. tsiH+ prey in a and b, and all prey in c and e are in ΔT6SS V52 background. DL, detection limit.
WigKR is necessary for V. cholerae survival against its own wild-type T6SS, despite the presence of immunity genes
Since deletion of wigR rendered V52 sensitive to endogenous overexpression of TseH despite TsiH, we wondered if it plays a necessary role in protecting V. cholerae against self-killing by its own effectors even in the presence of immunity genes. Strikingly, we found that WigKR was crucial for protecting V. cholerae against its own effectors, as deletion of wigR significantly reduced survival during competition with wild-type V52, and exogenous WigR expression complemented this sensitivity (Fig. 3c). We observed sensitivity of wigR mutants to T6SS-mediated killing with ΔT6SS (Fig. 3c) or wild-type V52 prey (Extended Data Fig. 5a), and with pandemic V. cholerae O1 El Tor C6706 prey (Extended Data Fig. 5b). Importantly, this kin-killing of wigR mutants occurred despite the prey strain encoding a full array of immunity genes. While expression of immunity genes was slightly lower in the wigR mutant (Extended Data Fig. 5c), the ΔwigR strain also showed significantly reduced survival in a background lacking the immunity genes tsiV1-3 (Fig. 3d) and when attacked by the T6SS of A. dhakensis (Fig. 3e), against which V. cholerae has no immunity. Thus the protective role of WigR is independent of immunity genes.
The protective role of WigKR supersedes VPS expression
Similar to the Rcs pathway in E. coli, WigKR has been shown to positively regulate biofilm formation and synthesis of Vibrio polysaccharide (VPS)38.A recent study showed that loss of VPS renders V. cholerae immunity mutants further sensitive to T6SS activities39. Thus, in addition to its role in peptidoglycan repair, WigKR may also mediate protection against T6SS attacks via a VPS capsule. To examine this, we induced VPS synthesis in V. cholerae V52 by growing this strain at 30 °C, yielding cellular aggregates in a vpsA (a gene essential for VPS biosynthesis)-dependent manner (Extended Data Fig. 5d)40. Indeed, V52 lacking immunity genes or facing a T6SS attack by A. dhakensis (which is mutually destructive with V. cholerae when both encode an active T6SS41) were highly sensitive to killing when pre-grown at 37 °C but were impressively protected after growth at 30 °C (Fig. 4a, b). This protection was largely dependent on VPS, as the equivalent ΔvpsA mutants showed significantly reduced survival. However, even after growth at 30 °C, the wigR mutant remained sensitive to its own T6SS regardless of vpsA, and deletion of vpsA had no effect on V52 survival against its own T6SS when all immunity genes remained intact (Fig. 4c). This demonstrates that the protective ability of WigKR is not solely due to VPS regulation, but rather supersedes it.
Figure 4: The protective role of WigKR supersedes VPS expression.

a. Relative survival (killing by V52 wild-type / ΔT6SS) of V. cholerae V52 immunity gene mutant prey encoding or lacking vpsA (and therefore VPS). VPS expression was induced by growth of prey strains at 30 °C. One-way ANOVA with Sidak’s multiple comparisons test; ***, p < 0.001. b. Relative survival (killing by A. dhakensis SSU wild-type / ΔT6SS) of V. cholerae V52 (ΔT6SS background) encoding or lacking vpsA. VPS expression was induced by pre-growth of prey strains at 30 °C. One-way ANOVA with Sidak’s multiple comparisons test; ***, p < 0.001. c. Relative survival (killing by wild-type V52 / ΔT6SS) of log phase V. cholerae V52 prey with intact (+) or deleted (Δ) wigR and vpsA genes. As indicated, prey were grown at 37 °C or at 30 °C to induce VPS expression. One-way ANOVA with Sidak’s multiple comparisons test; **, p < 0.01; ns, not significant. DL, detection limit. Graphs show the mean of at least three independent replicates and error bars show one standard deviation. Dots show results from individual replicates.
WigKR is required for T6SS expression in V. cholerae V52
In addition to its role in repairing peptidoglycan36,37, mediating VPS and biofilm formation38, and now defending against T6SS attacks, the WigKR system has also been shown to influence T6SS expression in V. cholerae El Tor strains that do not constitutively express their T6SS42. We examined if it is also involved in T6SS expression in V52 and indeed found that ΔwigR V52 was greatly impaired in its ability to kill E. coli prey (Fig. 5a). This reduced killing activity was reflected at both protein and mRNA levels as ΔwigR V52 produced fewer T6SS needles (Fig. 5b) and had reduced expression of T6SS genes, hcp and vipA, compared to wild-type (Fig. 5c). This demonstrates that even the constitutive T6SS expression in V52 relies substantially on WigKR.
Figure 5: WigR regulates the T6SS in V52.

a. Relative survival of E. coli prey after killing by V52 with intact (WT), deleted (Δ), or plasmid complemented (Δ,p) wigR. One-way ANOVA with Tukey’s test; ***, p < 0.001. DL, detection limit. b. Phase contrast (right) and VipA-GFP T6SS assembly (left) in V52 with intact (WT), deleted (Δ), or complemented (Δ,p) wigR. Representative of three independent replicates. Scale bar, 2 μm. c. Expression of main (vipA) and auxiliary (hcp) T6SS operons in wild-type (wigR+) or ΔwigR V52. One-way ANOVA with Sidak’s test; **, p < 0.01. Graphs show the mean of at least three independent replicates, error bars show standard deviation. Dots show individual replicates.
Discussion
In this work we demonstrate that TseH is a PAAR-dependent species-specific effector, and establish that ESRs play a significant role in protecting bacteria against T6SS attacks. Protection occurred both in the presence and absence of immunity genes, demonstrating that immunity genes are not always necessary or sufficient to survive incoming T6SS attacks, even from neighbouring kin bacteria. We highlighted specific ESRs (Rcs and BaeSR in E. coli, and WigKR in V. cholerae) that protect against a T6SS effector, TseH, and discovered that WigKR (VxrAB) is necessary for V. cholerae to survive attacks from its own T6SS despite expressing a full complement of immunity genes. Exemplified here, similar systems likely exist in many species encoding or encountering attacks from a T6SS.
We also identified protective mechanisms mediated by these ESRs, including by maintaining envelope integrity, osmotic response genes, and inducing EPS capsule synthesis, potentially improving the cell’s ability to resist lysis and deflect further incoming attacks. These ESRs likely also regulate genes that can repair effector-mediated damage, as WigKR has been shown to induce peptidoglycan synthesis36,37. Full protection is likely a combination of active response to damage and preparing the cell before encountering attacks. The importance of maintaining cell envelope integrity in surviving T6SS attacks was highlighted by the pronounced susceptibility of ΔdegP E. coli to TseH, and potentially by the ability of Spy to protect various sensitive E. coli mutants and A. dhakensis from TseH. While the exact mechanisms underlying these protections require further investigation, they might involve a healthy envelope providing a buffer of time to repair effector-mediated damage before the envelope fails and the cell lyses.
Since TseH showed killing activity against most Aeromonas strains tested, it may grant a competitive advantage to V. cholerae in aquatic and marine host environments shared with Aeromonas species15,41. While the basis of Aeromonas’ sensitivity to TseH was not fully elucidated, they notably do not encode Spy or any of the three defensive ESRs recognized in this work (Extended Data Fig. 5e), suggesting that Aeromonas may simply lack defences capable of adequately mitigating TseH-inflicted damage. The finding that E. coli Spy can protect A. dhakensis also supports the concept that they may have simply not yet evolved a coordinated defence against TseH. Cryptic T6SS effectors from other bacteria may show similar species-specific killing when targeting prey that lack defences.
We also noted that wild-type V. cholerae (encoding all immunity genes and WigKR) showed impaired survival against the V52 T6SS when the prey were in stationary phase (Fig. 3c, Extended Data Fig. 5a-b), suggesting that T6SS attacks against less metabolically active cells can overwhelm their defences. This sensitivity also existed in our prey strain lacking the immunity genes tsiV1–3 (Fig. 3d), suggesting it was not due to reduced immunity gene expression as previously implicated for Myxococcus xanthus43. Therefore, nonspecific defences may be impaired by nutrient limitation, especially energy-costly defences such as synthesizing EPS.
WigKR also regulates T6SS expression as shown previously for El Tor biotype V. cholerae42 and in this work for V52. This proposes the possibility that V. cholerae may use WigKR to sense incoming attacks (or environments where attacks are likely) and induce T6SS expression as a potential counterattack. This type of T6SS counterattack has precedent in Serratia marcescens, wherein reactive T6SS induction was mediated by the Rcs phosphorelay44. However, unlike WigKR, the Rcs pathway was not required for survival of S. marcescens during intraspecies competition and its role for interspecies competition remains unclear. Importantly, WigKR is also important during V. cholerae host colonization42, where its role in T6SS regulation and defence could tilt the balance against the host microbiome to help the pathogen establish infection.
Cumulatively, our findings highlight that beyond immunity genes nonspecific resistance to T6SS attacks exists in both T6SS-encoding and T6SS-lacking bacteria. This implies an evolutionary rationale for attacking cells to encode multiple T6SS effectors with divergent molecular targets to not only synergize with each other45 but also to overwhelm nonspecific defences. The T6SS is widespread – suggesting that bacteria are often subjected to T6SS attacks – and such a common selective pressure is expected to drive the evolution of defence mechanisms against it. Indeed, a few recent works have shown that forming kin-only clusters can prevent intoxication41,46, oxidative stress responses can limit prey mortality47, and V. cholerae VPS mutants are more susceptible to incoming T6SS attacks39. Though knowledge of non-immunity defence mechanisms is just scratching the surface they are likely as commonplace as the molecular weapon itself, fundamentally impacting bacterial T6SS interactions involving a wide variety of bacteria in the environment, amongst healthy microbiota, and during pathogen host colonization. ESRs and EPS are well known to protect against antibiotics and other cellular stressors, and here we propose that the T6SS also had a major influence on their evolution. This has important implications on how bacterial ecosystems have evolved, how stress responses and capsules impact survival in multispecies communities (including microbiomes resisting pathogen colonization), and reveals the possibility of targeting these defence pathways with next generation antibiotics to sensitize pathogenic bacteria not only to antibiotics but also to the T6SS of their neighbouring kin and competitors.
Methods
Bacterial strains and plasmids
Strains and plasmids used in this study are listed in Supplementary Information Table 1. Unless otherwise indicated, cells were grown shaking at 37 °C in LB media (0.5% NaCl) or on LB agar plates. Antibiotics were added for prey cell recovery or plasmid maintenance where required to final concentrations of 25 μg/mL chloramphenicol (for E. coli), 2.5 μg/mL chloramphenicol (for V. cholerae and A. dhakensis), 100 μg/mL carbenicillin, 50 μg/mL kanamycin, 5 μg/mL erythromycin. Plasmids and suicide vectors for chromosomal recombination were generated using standard molecular biology techniques and verified by Sanger sequencing. The V. cholerae ΔwigR (vxrB) mutant was also verified by whole genome Illumina sequencing to confirm that there were no secondary mutations. Random mutations in plasmid-borne TAT-TseH that likely reduce toxicity were identified during cloning in the absence of glucose repression. These were sequenced and point mutations are listed in Extended Data Fig. 2.
T6SS competition assay
Assessment of T6SS activity was conducted as described previously with minor modifications48. In brief, ‘killer’ strains and log phase ‘prey’ strains were subcultured 1/100 from overnight cultures for 3 hours (to an OD600nm ~ 1.0 – 1.5). Unless indicated, prey strains were from overnight cultures. Where indicated, V52 prey were grown at 30 °C to induce VPS expression. Killer and prey were mixed at a 10:1 ratio (unless otherwise stated in the figure legend), spotted on LB plates and incubated for 3 hours at 37 °C. VPS-induced cultures were pipetted and vortexed thoroughly to break up cellular aggregates prior to mixing with killer cells. Agar plugs containing the mixed bacteria were removed using wide-bore pipette tips, resuspended in PBS, serially diluted and plated for colony forming units (CFU). Data is reported as either a log CFU recovered or as a ratio of log CFU recovered relative to that with the TseHH64A or ΔT6SS control killer strains. For plasmid expression in prey cells, 0.1% arabinose or 1mM IPTG was included in the co-incubation plate. For plasmid expression in killer cells, 0.4% arabinose was included in the subculture and 0.1% arabinose was included in the co-incubation plate. For selective plating of prey CFU: plasmids for complementation were used as selective markers, E. coli wild-type, V. cholerae and A. dhakensis prey include pBAD33k as a selective marker, E. coli mutants encode chromosomal kanamycin resistance, Aeromonas species were selected by natural ampicillin resistance, Edwardsiella strains were selected by natural erythromycin resistance. Importantly, strains used as consistent controls across many experimental days have additional replicates and all are displayed in the data presented.
Disk diffusion assay
Plasmid encoding arabinose inducible TAT-tagged (periplasm-directed) TseH was freshly electroporated into strains of interest and maintained on 0.4% glucose to repress expression. Fresh colonies were resuspended in LB media and used to plate a bacterial lawn using a cotton swab. Sterile 16mm disks were placed on top of the inoculated agar and 50 μl of 40% arabinose or glucose (as indicated) was spotted onto the centre of the disk. Plates were imaged after overnight growth at 37 °C.
Microscopy
Microscopy was conducted as described previously49. In brief, overnight cultures were diluted 1/100 in fresh LB, grown to an OD600nm of approximately 1.0, resuspended to an OD600nm of 10 and spotted onto a 1% agar pad in 0.5 × PBS. Imaging was conducted on a Nikon Ti-E inverted microscope using NIS-Elements AR 4.40 software, and Fiji was used for image analysis. For WigR complementation, 0.4% arabinose was added to the subculture. For peptidoglycan visualization in Video 1, cells were concentrated to OD600 of 20 and stained with 100 μg/mL Wheat Germ Agglutinin (WGA) Alexa Fluor™ 488 Conjugate. Cells were spotted onto 1% agarose pads supplemented with diluted LB (1:5), PBS buffer (1:2), and 1 mM IPTG (for TseL and VgrG3 induction) or 0.2% L-arabinose (for TseH induction). Time lapse images were taken every 20 seconds for 25 minutes. For examining propidium iodide permeability, cells were resuspended in 0.5X PBS to OD600 of 15, stained with 0.5 μg/ml propidium iodide (PI) stain and spotted on 1% agarose-0.5X PBS pads for imaging.
RT-qPCR
For measuring gene expression in V. cholerae, strains were grown to an OD600 of approximately 1.1. For measuring gene expression in E. coli in response to V. cholerae-delivered TseH, a T6SS competition assay was conducted at a 2:1 killer:prey ratio; after 3 hours samples were scraped off the competition plate with a sterile loop, resuspended in LB, and prepped for RT-qPCR. Samples were prepped for RT-qPCR by lysing with hot SDS (1% SDS, 2 mM EDTA) and mixing with an equal volume of hot acidic phenol (pH 4.5). Phenol mixtures were incubated at 65 °C for 5 minutes followed by 5 minutes on ice before the phases were separated by centrifugation. RNA was purified from the aqueous phase with a Direct-zol RNA MiniPrep kit (Zymo Research). Reverse transcription was performed with SuperScript IV Reverse Transcriptase (Invitrogen) and qPCR was performed with Fast SYBR Green Master Mix (Applied Biosystems) in technical triplicate. Relative quantification was performed using a standard curve and the 16s rRNA gene, rrsA, was used as an internal standard. For E. coli response to V. cholerae-delivered TseH, E. coli-specific primers (including for 16s rRNA) were used.
Crystallography
E. coli BL21 (DE3) gold cells containing pET-SUMO-TseH were grown in 1 L LB media supplemented with ampicillin at 37 °C. Cytoplasmic TseH showed no toxicity to E. coli, enabling TseH overexpression and purification, followed by crystallization by a vapor diffusion approach in the presence of thermolysin as described previously50. Expression was induced by addition of 1 mM IPTG at OD600~0.5–0.7. The culture was continuously shaken overnight at 20 °C. Cells were collected by centrifugation and pellets resuspended in PBS buffer supplemented with 30 mM imidazole, DNaseI, PMSF, benzamidine and 0.5 mM TCEP. Subsequently the cells were lysed by Avastin (15,000psi). Debris were centrifuged and the supernatant was passed through Ni-NTA (ThermoFisher) resin by gravity flow. The resin was washed with 20 CV of wash buffer (50 mM sodium phosphate buffer pH 7.6, 300 mM NaCl, 50 mM imidazole, 0.5mM TCEP). The protein was eluted with 2 CV of elution buffer (wash buffer supplemented with 300 mM imidazole). The SUMO tag was cleaved by SUMO protease while dialyzing out imidazole overnight (Dialysis buffer: 10 mM HEPES pH 7.4, 300 mM NaCl, 0.5 mM TCEP). The cleaved TseH was separated from SUMO-tag, SUMO protease and uncleaved TseH by applying back to Ni-NTA resin. The flow through was collected and concentrated to 27 mg/ml prior to crystallization. Se-Met derivatized TseH was purified using the same protocol as above except that the protein was expressed using M9 high-yield growth procedure according to the manufacturer’s instructions (Shanghai Medicilon).
Native and Se-Met derivatized TseH were crystallized by using in situ proteolysis using Thermolysin50. TseH was incubated with thermolysin in a ratio of 1:1000 (wt) on ice for 30 min prior to setting up crystallization. The crystals were obtained by mixing TseH and mother liquor (18% w/v PEG8000, 0.2 M calcium acetate and 0.1 M sodium cacodylate pH 6.5) in 1:1 ratio at 22 °C. The crystal appeared overnight and matured in 1–2 weeks. The crystals were briefly transferred to cryo protectant containing the mother liquor and 30% glycerol, then flash frozen in liquid nitrogen. Native and Se-Met X-ray diffraction data were collected at Canadian Light Source (CMCF-08B1).
Diffraction data were processed using HKL3000 suite, XDS suite, Aimless51–53. Only the data up to 4.0 Å were used for obtaining crystallographic phase information using multiple anomalous dispersion method (MAD). Initial phase and atomic model were calculated using AutoSHARP54. Final figure of merit was 0.72563 (acentric)/049611 (Centric). The high resolution native structure was determined using Phenix.phaser55 with the partially built model from AutoSHARP. Model refinement was completed using Phenix.refine55 and Coot56. All B-factors were refined as isotropic with TLS parameterization. All geometry was verified using Phenix validation tools (Ramachandran statistics: Favoured [97.2%], additionally allowed [2.8%], disallowed [0.0%]) and the wwPDB server. Further information and statistics about the structure are listed in Extended Data Fig. 6.
Pull-down assay
Pull-down assays were conducted as described previously57. In brief, E. coli BL21 DE3 with plasmids encoding TseH-Flag and either PAAR2-His or Spy-His were grown to mid-log phase, induced for expression with 1 mM IPTG and 0.4% arabinose and grown for another 3 h at 37 °C prior to harvesting and sonication. Lysates were incubated with cobalt-NTA resin, washed, eluted in 500 mM imidazole, and analyzed by western blot.
KEGG ortholog analysis
The KEGG database Module Ortholog Table was used to identify species encoding orthologs of RcsCDB and BaeSR58. For WigK and WigR, KEGG’s SSDB Ortholog function was used to generate alignment SW-scores and those with over 50% of maximum (SW-score alignment against self) were considered orthologs. Only strains encoding both genes of the two-component system (or all three for the RcsCDB Module) were considered to have the orthologous pathway.
Statistics and Reproducibility
Statistics were computed using GraphPad Prism software. Exact n, p, and F values for all statistical analyses are provided in linked source data files. For all instances where data pertaining to the same experimental hypothesis are shown in multiple (main and extended data) figures, ANOVA analyses included all comparison groups across all figures to ensure conservative estimates of significance.
Data availability
Source data linked to figures is provided on the journal webpage. Final structural model was deposited to PDB (6V98). Further information is available from the corresponding author upon reasonable request.
Supplementary Material
Extended Data
Extended Data Fig. 1. Further analyses of TseH killing, delivery, and structure.
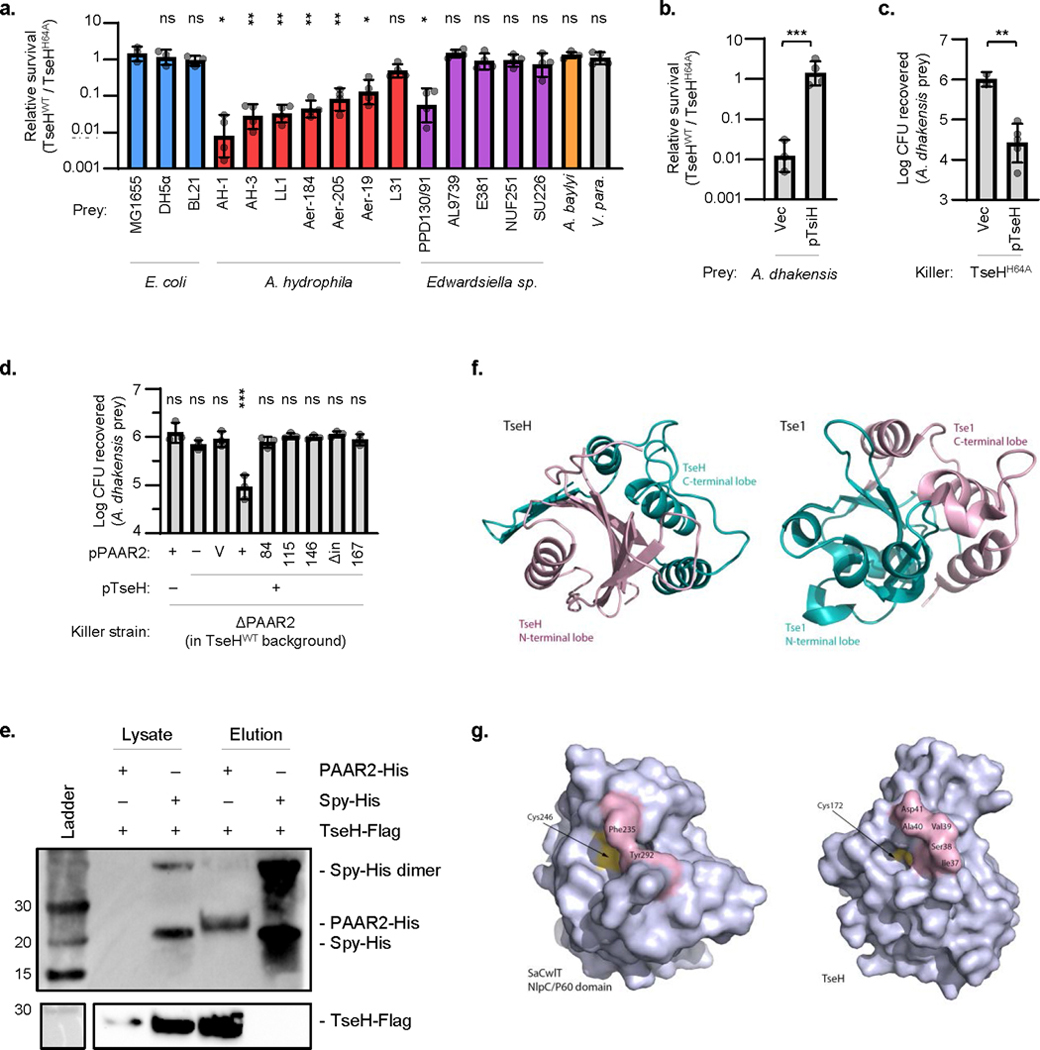
a. Relative survival (killing by V. cholerae V52 TseHWT / TseHH64A) of E. coli, A. hydrophila, Edwardsiella sp., A. baylyi ADP1 (ΔT6SS), or V. parahaemolyticus RIMD2210633 strains. Welch’s one-way ANOVA with Dunnett’s multiple comparisons test comparing each sample to E. coli MG1655; *, p < 0.05; **, p < 0.01; ns, not significant. b. Relative survival (killing by TseHWT strain relative to killing by TseHH64A strain) of A. dhakensis expressing TsiH or vector only control. Unpaired 2-tailed t-test; ***, p < 0.001. c. Survival of A. dhakensis after killing by TseHH64A strain expressing TseH or vector only. Vector control data resembles no-plasmid data shown in Fig. 1a and was only repeated in duplicate. Unpaired 2-tailed t-test with Welch’s correction; **, p < 0.01. d. Extension of Figure 1b in the main text. Survival of A. dhakensis prey after killing by ΔPAAR2 strain expressing plasmid-borne TseH and full-length or C-terminal truncations of PAAR2. For PAAR2: V, vector; numbers, PAAR2 truncated to indicated amino acid length; Δin, codons 146-156 removed leaving downstream residues intact. One-way ANOVA with Dunnett’s multiple comparisons test comparing to the sample expressing TseH and Vector control of PAAR2; ***, p < 0.001; ns, not significant. For all graphs, the mean and standard deviation are shown. Dots show individual replicates. e. Western blot showing whole cell lysate or eluted fraction after His-tag pull-down. BL21 DE3 E. coli expressed Flag-tagged TseH in combination with either His-tagged PAAR2 or Spy as a non-binding control. Number at left indicated ladder band size in kDa. Data is representative of two independent replicates. f. Circular permutation of catalytic residues causes swapping of N- and C-terminal lobes in the structure of TseH compared to Tse1 (PDB: 4EOB18), the other T6SS effector that belongs to NlpC/P60 family has reversed N- and C-lobe structure in comparison to TseH. g. Restricted access to the catalytic cysteine in certain peptidoglycan endopeptidases. The surface representations of SaCwlT and TseH show that the conformational changes in residues surrounding the active site are necessary for the substrate binding21,22. For all graphs, the mean and standard deviation are shown. Dots show individual replicates. TseHWT, V. cholerae V52 with all anti-bacterial effectors inactivated except TseH; TseHH64A, V52 with all anti-bacterial effectors inactivated including TseH.
Extended Data Fig. 2. Identified mutations that reduce TseH toxicity.

Extended Data Fig. 3. Additional E. coli mutants susceptible to T6SS-delivered TseH.

a. Disk diffusion induction of plasmid-borne TAT-tagged TseH expression in bacterial lawns of E. coli MG1655. Wild-type (top) or H64A mutant (bottom) TseH are compared. Right disks contain the inducer of expression, arabinose (Ara), and left disks contain the repressor, glucose (Glu). Representative of three independent replicates. b. As in a, comparing induction of plasmid-borne TAT-tagged TseHWT expression in E. coli MG1655 or A. dhakensis (ΔT6SS). Similar data for TseHWT in E. coli has been shown previously and is shown again here for comparison4. c. Relative survival (killing by TseHWT / TseHH64A) of E. coli wild-type or knockout mutants from indicated categories. One-way ANOVA with Dunnett’s multiple comparisons test comparing each sample to WT; **, p < 0.01; ***, p < 0.001; ns, not significant. Data for WT (E. coli) is shown in Figure 2 and is shown again here for comparison. Aux., auxotrophs. d. RT-qPCR comparing expression of genes in wild-type E. coli prey after T6SS competition assay with TseHWT or TseHH64A killer strains. Value with TseHH64A killer was set to 1 to show fold induction in response to TseH activity upon delivery by the T6SS. One-way ANOVA (with samples shown in Figure 2d) with Sidak’s multiple comparisons test; ns, not significant. For both graphs, the mean and standard deviation are shown. Dots show individual replicates. TseHWT, V. cholerae V52 with all anti-bacterial effectors inactivated except TseH; TseHH64A, V52 with all anti-bacterial effectors inactivated including TseH.
Extended Data Fig. 4. Additional data for RcsA and BaeR regulon overexpression, and effect of Spy overexpression on permeability.

a. Disk diffusion induction of plasmid-borne TAT-tagged TseH in bacterial lawns of E. coli BW25113 containing RcsA or vector only plasmids. IPTG was added throughout the lawn to induce RcsA. Disks contain arabinose to induce TAT-TseH expression. Representative of three independent replicates. b. Relative survival (killing by TseHWT / TseHH64A) of E. coli ΔdegP mutant or A. dhakensis with vector (Vec) or overexpressing genes from the BaeR regulon. One-way ANOVA with Sidak’s multiple comparisons test comparing samples to their respective empty vector controls; *, p < 0.05; **, p < 0.01; ns, not significant. Vector controls are shown in Figure 2 and are shown again here for comparison. Mean and standard deviations are shown. Dots show individual replicates. pSpyΔSec, plasmid expressing Spy with its Sec secretion tag deleted. c. Images of ΔbaeR E. coli overexpressing plasmid-borne TAT-TseH and either Spy or vector-only controls. The phase-contrast and propidium iodide(PI) channels are merged. Scale bar shows 5 μm. Representative of three independent replicates. d. Quantification showing the percent of total cells showing PI+ fluorescence. N number at top indicates total cells counted. Dots show mean results from three independent experiments (used to calculate statistics) and error bars show one standard deviation. Unpaired 2-tailed t-test, p = 0.0513.
Extended Data Fig. 5. Additional data for ΔwigR strain T6SS susceptibility, immunity gene expression, VPS-mediated aggregation, and Aeromonas species do not encode RcsCDB, BaeSR, or WigKR.

Relative survival (wild-type V52 / TseHH64A killer strain) of V. cholerae V52 (T6SS+ background) prey (a.) or V. cholerae C6706 prey (b.) with intact (+), deleted (Δ), or complemented (Δ,p) wigR. Stationary (Stat.) and log phase prey are shown. One-way ANOVA with Sidak’s multiple comparisons test; ***, p < 0.001; **, p < 0.01; *, p < 0.05; ns, not significant. c. RT-qPCR measuring expression of T6SS immunity genes in ΔwigR V. cholerae V52 relative to in the wigR+ strain (both in the ΔT6SS background). For all graphs, the mean and standard deviation are shown. Dots show individual replicates. d. Image showing settling of V. cholerae V52 grown at 37 °C or at 30 °C to induce VPS synthesis. Image after vortexing to disrupt aggregates is also shown for comparison (lower panel). Representative of three independent replicates. e. KEGG ortholog analysis across Gammaproteobacteria. KEGG Modules were compared for RcsCDB and BaeSR. WigK and WigR orthologs were assessed by sequence homology to VCA0565 and VCA0566 using an SW-score cutoff of 50% of maximum. Data is shown as the percentage of strains within each taxonomic group that contain an ortholog of all genes in the loci. Number in brackets indicates the number of strains in the taxonomic group in the KEGG database.
Extended Data Fig. 6. Table of data collection, phasing and refinement statistics for MAD (SeMet) structures.

Figure 6: Envelope stress responses defend against T6SS system attack.

Model depicting an attacking T6SS delivering toxic effectors such as TseH. The Rcs phosphorelay and BaeSR (in E. coli) or WigKR (in V. cholerae) two-component systems induce their respective regulons. The induced genes play various roles in protection, including inducing EPS to deflect further T6SS attacks, and mitigating effector-mediated damage. WigR also induces T6SS expression in V. cholerae, potentially instigating counter attacks. EPS, exopolysaccharide; PG, peptidoglycan.
Acknowledgements
This work was supported by grants from Canadian Institutes of Health Research (CIHR) and Canadian Natural Sciences and Engineering Research Council (NSERC) to T.G.D., and in part by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract nos. HHSN272201200026C and HHSN272201700060C to A.S. T.G.D. is also supported by a Government of Canada Research Chair award, and a Canadian Foundation for Innovation grant (CFI-JELF). A.P. was funded by the Markin Undergraduate Student Research Program in Health & Wellness. L.L. is supported by an Alberta Innovates Health Solutions (AIHS) Graduate Student Scholarship, K.M. by an Alberta Queen Elizabeth II Graduate Scholarship. B.B. was supported by AIHS and National Sciences and Engineering Research Council of Canada (NSERC) postdoctoral fellowships. S.J.H. holds a CIHR Postdoctoral Fellowship. We thank Z. Eltsova for assistance obtaining Keio and ASKA collection strains as well as W. Navarre for helpful discussions.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Lewin R. Finches show competition in ecology. Science. 219, 1411–1412 (1983). [DOI] [PubMed] [Google Scholar]
- 2.Pukatzki S. et al. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc. Natl. Acad. Sci. U. S. A. 103, 1528–1533 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hood RD et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7, 25–37 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altindis E, Dong T, Catalano C. & Mekalanos J. Secretome analysis of Vibrio cholerae type VI secretion system reveals a new effector-immunity pair. MBio 6, e00075–e00075 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong TG, Ho BT, Yoder-Himes DR & Mekalanos JJ Identification of T6SS-dependent effector and immunity proteins by Tn-seq in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 110, 2623–2628 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyata ST, Kitaoka M, Brooks TM, McAuley SB & Pukatzki S. Vibrio cholerae requires the type VI secretion system virulence factor VasX to kill Dictyostelium discoideum. Infect. Immun. 79, 2941–2949 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell AB et al. Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475, 343–347 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyata ST, Unterweger D, Rudko SP & Pukatzki S. Dual Expression Profile of Type VI Secretion System Immunity Genes Protects Pandemic Vibrio cholerae. PLOS Pathog. 9, e1003752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Logan SL et al. The Vibrio cholerae type VI secretion system can modulate host intestinal mechanics to displace gut bacterial symbionts. Proc. Natl. Acad. Sci. 115, E3779–E3787 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fast D, Kostiuk B, Foley E. & Pukatzki S. Commensal pathogen competition impacts host viability. Proc. Natl. Acad. Sci. 115, 7099–7104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu Y, Ho BT & Mekalanos JJ Tracking Vibrio cholerae Cell-Cell Interactions during Infection Reveals Bacterial Population Dynamics within Intestinal Microenvironments. Cell Host Microbe 23, 274–281.e2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Speare L. et al. Bacterial symbionts use a type VI secretion system to eliminate competitors in their natural host. Proc. Natl. Acad. Sci. 115, E8528–E8537 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bingle LE, Bailey CM & Pallen MJ Type VI secretion: a beginner’s guide. Curr. Opin. Microbiol. 11, 3–8 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Liang X. et al. An onboard checking mechanism ensures effector delivery of the type VI secretion system in Vibrio cholerae. Proc. Natl. Acad. Sci. 116, 23292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Senderovich Y, Gershtein Y, Halewa E. & Halpern M. Vibrio cholerae and Aeromonas: do they share a mutual host? Isme J. 2, 276 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Holm L. Benchmarking fold detection by DaliLite v.5. Bioinformatics 35, 5326–5327 (2019) [DOI] [PubMed] [Google Scholar]
- 17.Xu Q. et al. Structural Analysis of Papain-Like NlpC/P60 Superfamily Enzymes with a Circularly Permuted Topology Reveals Potential Lipid Binding Sites. PLoS One 6, e22013- (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou S. et al. Structure of a peptidoglycan amidase effector targeted to Gram-negative bacteria by the type VI secretion system. Cell Rep. 1, 656–664 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y. & Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anantharaman V. & Aravind L. Evolutionary history, structural features and biochemical diversity of the NlpC/P60 superfamily of enzymes. Genome Biol. 4, R11–R11 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Q. et al. Structures of a bifunctional cell wall hydrolase CwlT containing a novel bacterial lysozyme and an NlpC/P60 DL-endopeptidase. J. Mol. Biol. 426, 169–184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Q. et al. Structural basis of murein peptide specificity of a gamma-D-glutamyl-l-diamino acid endopeptidase. Structure 17, 303–313 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laubacher ME & Ades SE The Rcs phosphorelay is a cell envelope stress response activated by peptidoglycan stress and contributes to intrinsic antibiotic resistance. J. Bacteriol. 190, 2065–2074 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raffa RG & Raivio TL A third envelope stress signal transduction pathway in Escherichia coli. Mol. Microbiol. 45, 1599–1611 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Baba T. et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bury-Moné S. et al. Global analysis of extracytoplasmic stress signaling in Escherichia coli. PLoS Genet. 5, e1000651–e1000651 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boulanger A. et al. Multistress regulation in Escherichia coli: expression of osmB involves two independent promoters responding either to sigmaS or to the RcsCDB His-Asp phosphorelay. J. Bacteriol. 187, 3282–3286 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Francez-Charlot A, Castanié-Cornet M-P, Gutierrez C. & Cam K. Osmotic regulation of the Escherichia coli bdm (biofilm-dependent modulation) gene by the RcsCDB His-Asp phosphorelay. J. Bacteriol. 187, 3873–3877 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ranjit DK & Young KD Colanic Acid Intermediates Prevent De Novo Shape Recovery of Escherichia coli Spheroplasts, Calling into Question Biological Roles Previously Attributed to Colanic Acid. J. Bacteriol. 198, 1230–1240 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gottesman S, Trisler P. & Torres-Cabassa A. Regulation of capsular polysaccharide synthesis in Escherichia coli K-12: characterization of three regulatory genes. J. Bacteriol. 162, 1111–1119 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stout V, Torres-Cabassa A, Maurizi MR, Gutnick D. & Gottesman S. RcsA, an unstable positive regulator of capsular polysaccharide synthesis. J. Bacteriol. 173, 1738–1747 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishino K, Honda T. & Yamaguchi A. Genome-wide analyses of Escherichia coli gene expression responsive to the BaeSR two-component regulatory system. J. Bacteriol. 187, 1763–1772 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strauch KL & Beckwith J. An Escherichia coli mutation preventing degradation of abnormal periplasmic proteins. Proc. Natl. Acad. Sci. U. S. A. 85, 1576–1580 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hagenmaier S, Stierhof YD & Henning U. A new periplasmic protein of Escherichia coli which is synthesized in spheroplasts but not in intact cells. J. Bacteriol. 179, 2073–2076 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quan S. et al. Genetic selection designed to stabilize proteins uncovers a chaperone called Spy. Nat. Struct. Mol. Biol. 18, 262 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weaver AI et al. Genetic Determinants of Penicillin Tolerance in Vibrio cholerae. Antimicrob. Agents Chemother. 62, e01326–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dörr T. et al. A cell wall damage response mediated by a sensor kinase/response regulator pair enables beta-lactam tolerance. Proc. Natl. Acad. Sci. U. S. A. 113, 404–409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teschler JK, Cheng AT & Yildiz FH The Two-Component Signal Transduction System VxrAB Positively Regulates Vibrio cholerae Biofilm Formation. J. Bacteriol. 199, e00139–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toska J, Ho BT & Mekalanos JJ Exopolysaccharide protects Vibrio cholerae from exogenous attacks by the type 6 secretion system. Proc. Natl. Acad. Sci. U. S. A. 115, 7997–8002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fong JCN, Syed KA, Klose KE & Yildiz FH Role of Vibrio polysaccharide (vps) genes in VPS production, biofilm formation and Vibrio cholerae pathogenesis. Microbiology 156, 2757–2769 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong M. et al. Microbial herd protection mediated by antagonistic interaction in polymicrobial communities. Appl. Environ. Microbiol. 82, 6881–6888 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng AT, Ottemann KM & Yildiz FH Vibrio cholerae Response Regulator VxrB Controls Colonization and Regulates the Type VI Secretion System. PLoS Pathog. 11, e1004933–e1004933 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Troselj V, Treuner-Lange A, Søgaard-Andersen L. & Wall D. Physiological Heterogeneity Triggers Sibling Conflict Mediated by the Type VI Secretion System in an Aggregative Multicellular Bacterium. MBio 9, e01645–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lazzaro M, Feldman MF & García Véscovi E. A Transcriptional Regulatory Mechanism Finely Tunes the Firing of Type VI Secretion System in Response to Bacterial Enemies. MBio 8, e00559–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.LaCourse KD et al. Conditional toxicity and synergy drive diversity among antibacterial effectors. Nat. Microbiol. 3, 440–446 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borenstein DB, Ringel P, Basler M. & Wingreen NS Established Microbial Colonies Can Survive Type VI Secretion Assault. PLOS Comput. Biol. 11, e1004520- (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong TG et al. Generation of reactive oxygen species by lethal attacks from competing microbes. Proc. Natl. Acad. Sci. U. S. A. 112, 2181–2186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacIntyre DL, Miyata ST, Kitaoka M. & Pukatzki S. The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc. Natl. Acad. Sci. U. S. A. 107, 19520–19524 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stietz MS, Liang X, Wong M, Hersch S. & Dong TG Double Tubular Contractile Structure of the Type VI Secretion System Displays Striking Flexibility and Elasticity. J. Bacteriol. 202, e00425–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong A. et al. In situ proteolysis for protein crystallization and structure determination. Nat. Methods 4, 1019–1021 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minor W, Cymborowski M, Otwinowski Z. & Chruszcz M. HKL-3000: the integration of data reduction and structure solution – from diffraction images to an initial model in minutes. Acta Crystallogr. Sect. D 62, 859–866 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Kabsch W. XDS. Acta Crystallogr. D. Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vonrhein C, Blanc E, Roversi P. & Bricogne G. Automated Structure Solution With autoSHARP in Macromolecular Crystallography Protocols: Volume 2: Structure Determination (ed. Doublié S) 215–230 (Humana Press, 2007). doi: 10.1385/1-59745-266-1:215 [DOI] [PubMed] [Google Scholar]
- 55.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emsley P. & Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Burkinshaw BJ et al. A type VI secretion system effector delivery mechanism dependent on PAAR and a chaperone–co-chaperone complex. Nat. Microbiol. 3, 632–640 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Kanehisa M, Sato Y, Furumichi M, Morishima K. & Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 47, D590–D595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data linked to figures is provided on the journal webpage. Final structural model was deposited to PDB (6V98). Further information is available from the corresponding author upon reasonable request.