

Abstract
Protein trans-splicing catalyzed by split inteins has increasingly become useful as a protein engineering tool. We solved the 1.0 Å-resolution crystal structure of a fused variant from the naturally split gp41–1 intein, previously identified from environmental metagenomic sequence data. The structure of the 125-residue gp41–1 intein revealed a compact pseudo-C2-symmetry commonly found in the Hedgehog/Intein (HINT) superfamily with extensive charge-charge interactions between the split N- and C-terminal intein fragments that are common among naturally occurring split inteins. We successfully created orthogonal split inteins by engineering a similar charge network into the same region of a cis-splicing intein. This strategy could be applicable for creating novel natural-like split inteins from other, more prevalent cis-splicing inteins.
Keywords: protein splicing, gp41–1 intein, orthogonal split intein, crystal structure, protein engineering
Graphical Abstract
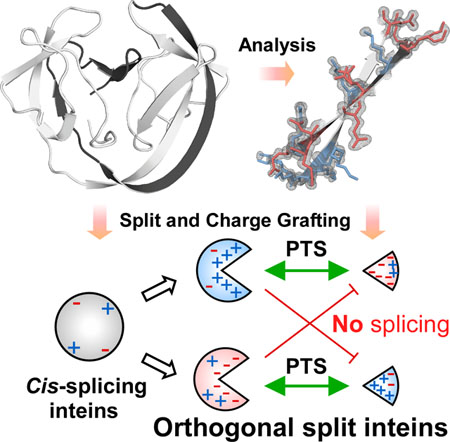
The high-resolution crystal structure of the naturally split gp41-1 intein revealed an intense charge network between the intein split fragments. We grafted a similar charge network onto the cis-splicing NpuDnaB mini-intein to design artificial natural-like split intein variants with orthogonal character.
Introduction
Protein splicing is a posttranslational modification where an intervening protein (intein) residing within an unrelated host protein excises itself, while covalently ligating the N- and C-terminally flanking sequences (exteins) with a standard peptide bond [1,2,3,4]. As a result, the ligated product is scar-less and devoid of any indication of its previous merged existence, while the function of the host protein is generally restored (Fig. 1). Inteins are commonly regarded as selfish parasitic elements, albeit some evidence attributes a regulatory role in controlling the activity of host proteins in response to environmental cues triggering the splicing reaction [3,5].
Fig. 1.

Schematic representation of protein splicing in cis and trans. (A) Cis-splicing inteins excise themselves from a precursor where the N- and C-exteins flank the intein on the same polypeptide. (B) Protein trans-splicing (PTS) ligates N- and C-exteins, each originating from an independent polypeptide, with a covalent peptide bond resulting in a trans-spliced product. Interaction of the N- (IntN) and C-terminal (IntC) split intein halves initiates the protein-splicing reaction. The N-terminal junction sequence at the front of an intein is termed as the −1 position. The +1 position after the intein sequence usually has a Cys, Ser, or Thr residue. The second residue after an intein is numbered as the +2 position.
During recent years, inteins have become increasingly popular for diverse applications in biotechnology, chemical biology, and synthetic biology due to the following properties. First, intein-mediated protein splicing tolerates the deliberate exchange of extein sequences [3,6]. Second, the existence of naturally occurring split inteins reconstituting a functional protein from two polypeptide chains, as well as the possibility of splitting cis-splicing inteins, generates ample possibilities for applications with protein trans-splicing (PTS) [3,7,8,9] (Fig. 1B). Ever since the discovery of protein splicing by inteins, engineering of inteins toward high performance, high tolerance of junction sequences, and smaller variants have been an ongoing quest [9,10]. Successfully engineered inteins arose from the accumulation of beneficial mutations upon directed evolution [11,12,13], propagation of consensus sequence [14], and as a result of rational design [15,16].
Whereas more than 1500 putative inteins have been identified, only a few naturally occurring split inteins, such as the cyanobacterial DnaE inteins, have been reported [18]. The naturally fragmented gp41–1 intein was found as a result of metagenomic sequencing [19]. It is one of the smallest reported split inteins with very fast trans-splicing activity [20], consisting of 88-residue N-terminal (IntN) and 37-residue C-terminal (IntC) fragments. Its small size and robust protein splicing activity make it an attractive template for protein engineering [20]. Also, gp41–1 intein makes use of Ser as the catalytic residue at the +1 position, the first extein residue following the intein sequence. Given the much higher frequency of Ser over Cys within pro- and eukaryotic proteins, inteins with +1Ser allow a broader spectrum of possible insertion sites for scar-less protein ligation than naturally split inteins with Cys at the +1 position, thereby expanding potential applications.
Despite increasing interest in the utilization of various split inteins for protein engineering purposes, the repertoire of split inteins with both robust protein splicing activity and high sequence tolerance at the splice junctions is still very small. Particularly, pairs of orthogonal split inteins are desirable for one-pot multiple- fragment protein ligation and biorthogonal conjugation by PTS requiring two or more orthogonal split inteins [21,22,23,24]. Previous engineering attempts to derive novel split inteins from naturally occurring cis-splicing inteins did not result in highly robust split inteins, indicating that cis-splicing inteins are not optimized for trans-splicing, unlike naturally occurring split inteins [15,26,28].
Here we report the 1.0 Å-resolution crystal structure of a variant of the naturally split gp41–1 intein. Based on the crystal structure, we grafted the structural features of naturally split inteins onto a cis-splicing intein to develop novel natural-like split inteins and demonstrate the engineering of orthogonal split intein fragments from a cis-splicing intein.
Results
Crystal structure of the gp41–1 intein
As the first step to engineer inteins based on the gp41–1 intein, we created a cis-splicing gp41–1 intein variant by genetically fusing the gp41–1N and gp41–1C split fragments. We found that the cis-splicing gp41–1 intein retained high protein splicing activity when the three native extein residues were kept (Fig. 2). Next, for structure determination, we crystallized an inactive mutant of the cis-splicing gp41–1 intein bearing an alanine mutation as the first residue (C1A), to prevent N-cleavage due to the N-S acyl shift. Because the covalent fusion of gp41–1N and gp41–1C forces the association between the split intein fragments, we could solve the crystal structure of that inactive variant of the gp41–1 intein at the resolution of 1.0 Å. We used the crystal structure of the NpuDnaE intein as a search model for molecular replacement (Table 1). The structure of gp41–1 has the canonical intein horseshoe shape, termed HINT (Hedgehog/INTein) fold (Fig. 2A). The structure confirmed that the loop-engineering of naturally split gp41–1 intein fragments by introducing a peptide bond did not induce domain-swapping as it previously occurred with a short connection linker [55]. The active site region surrounding the terminal residues C1A and Asn125 is depicted in Fig. 2B and involves His63, Asp107, Thr123, and His124. The side-chain of Ala1 is pointing away from the last Asn125 residue, similar to other intein structures with “open” rather than “closed” conformation [15,16,17]. A Dali server search identified the engineered DnaB mini-intein (PDB ID: 4or1) from Nostoc punctiforme (NpuDnaBΔ290) as the closest structure to the gp41–1 intein, with a Z-score of 20.1 and an r.m.s.d. of 1.4 Å for Cα atoms of 127 residues [29]. NpuDnaBΔ290 intein is composed of 139 residues, which is 14 residues larger than the gp41–1 intein [15]. The main differences in the length between the two structures are found in two distinct regions (Fig. 2C and 2D). One is in the split fragment-connecting loop where canonical inteins harbor a homing endonuclease domain insertion (C35 site) [28], while the other is a loop at the pseudo-C2-symmetry related site (N35 site) [28]. These regions account for 11 residues of the size difference.
Fig. 2.

Crystal structure of the C1A variant of the engineered cis-splicing gp41–1 intein. (A) Ribbon representations of the gp41–1 intein structure. The region corresponding to the C-terminal split fragment (IntC) is colored in dark grey. N and C indicate N- and C-termini, respectively. (B) Stereoview of the active site residues of the gp41–1 intein structure. The distance between the Cβ atom of Ala1 and the carbonyl carbon of Asn125 is shown. (C) Stereo-view of an overlay of the crystal structures of the gp41–1 intein (red) and the closest related structure, the NpuDnaB mini-intein (blue) (PDB: 4o1r). A circle indicates the C35 and C2-symmetry-related N35 sites where additional residues are inserted in NpuDnaB mini-intein. (A-C) Structures were drawn using PyMOL. (D) Sequence alignment between the cis-splicing gp41–1 intein and NpuDnaB mini-intein (NpuDnaBΔ290). Underlined and italicized letters indicate sequence corresponding to the N- and C-terminal split fragments, respectively. (E) Engineering of the gp41–1 intein in the loop and splicing junction regions and their effects on protein splicing in cis. HB021, ADH21, HB182, BH37, BH321, HB088, and HB087 indicate the short names for different constructs with the sequence variations shown in the sequence alignment. (D-E) Sequence alignments were generated using Clustal Omega. (F) SDS-PAGE analysis of the cis-splicing activity of the engineered gp41–1 intein variants. M stands for molecular weight markers. H6-GB1-Int-GB1 indicates unspliced precursor proteins. H6-GB1-GB1 indicates cis-spliced products with various junction sequences causing minor variations in the migration profile. H6-GB1-Int indicates an off-pathway C-cleavage product. The splicing efficiency in percent quantified from the gel is shown at the bottom. The gel summarizes representative results of one to four individually performed experiments per construct.
Table 1.
Data collection and refinement statistics.
| Data collection | Diamond i02 |
|---|---|
| Wavelength | 0.9795 |
| Space group | I222 |
| Molecules/a.u. | 1 |
| Unit cell a, b, c (Å); | 48.81, 69.99, 71.24 |
| α=β=γ (°) | 90, 90, 90 |
| Resolution (Å)* | 49.92–1.02 (1.04–1.02) |
| Rmerge (%)† | 3.8 (28.8) |
| Rpim (%)& | 1.9 (23.1) |
| No. of reflections (measured/unique) |
242564/55708 |
| <I /σI> | 28.3 (2.8) |
| Completeness (%) | 89.4 (37) |
| Redundancy | 4.35 |
| Refinement | |
| Resolution (Å) | 49.92–1.02 |
| No. of reflections | 53080/2642 |
| (refinement/ Rfree) | |
| R / Rfree‡ | 12.10/15.00 |
| No. atoms | |
| Protein | 1093 |
| Ligand/ion | 51 |
| Water | 220 |
| R.m.s. deviations from ideal | |
| Bond lengths (Å) | 0.020 |
| Bond angles (°) | 1.98 |
| Ramachandran plot | |
| Favored (%) | 98.4 |
| Allowed (%) | 1.6 |
| PDB code | 6qaz |
The highest resolution shell is shown in parentheses.
Rmerge = ∑h∑i | Ii–〈I〉 | / ∑h∑I Ii, where Ii is the observed intensity of the i-th measurement of reflection h, and 〈I〉 is the average intensity of that reflection obtained from multiple observations.
Defined in Weisse et al., 2001 [59].
R = ∑ ||Fo|–|Fc|| / ∑|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively, calculated for all data. Rfree was defined in Brünger, 1992 [60].
The smaller size of the gp41–1 intein led to a compact pseudo-C2-symmetric structure found in the HINT fold with shorter loops due to the shortened insertions [30] (Fig. 2). The two C2 symmetry-related regions (residues 3–52 and residues 60–110) can be well superimposed [30,28] (Fig. 3A). The gp41–1 intein structure can thus be dissected into four distinct units: the first C2-symmetry related unit, β-strand 4 (β4), the second C2-symmetry related unit, and two β-strands (β8, β9) (Fig. 3B). The C2-symmetry related unit can further be divided into a globular region and two β-strands (β2 and β3, or β6 and β7). The natural split site of the gp41–1 intein is located within the second C2-symmetry related unit, separating the C2-symmetry unit into a globular region and two β-strands (Fig. 3B).
Fig. 3.

The modular architecture of the gp41–1 intein and the charge network. (A) An overlay of the backbone atoms of the two C2-symmetry-related units (residues 3–52 and 59–110) observed in the gp41–1 intein structure. (B) The arrangement of the C2-symmetry-related units and connections of the secondary structures. The natural split site of the gp41–1 intein locates within the second C2-symmetry-related part and at the front of strand β6. (C) The charged network found in the gp41–1 intein structure. The side-chains of the charged residues in the β3 and β6 strands are shown together with the electron density map. Residues with negative and positive charges are highlighted in red and blue, respectively. A filled triangle indicates the natural split site. N and C indicate the termini. (D) Comparison of the charged residues in the β3 and β6 strands between the gp41–1, NpuDnaE, and NpuDnaB inteins. Thick lines indicate possible favored charge interactions. An asterisk indicates Glu112 modeled as Val112 in the coordinate of the NpuDnaB mini-intein structure (PDB: 4o1r). (A, C) Figures were produced by PyMOL.
Minimizing gp41–1 intein
The first question we asked was whether it is possible to minimize the cis-splicing gp41–1 intein to a size even smaller than 125 residues, which is already one of the very small functional inteins. The gp41–1 intein is among the smallest inteins identified to date, indicating that it underwent a natural selection for the size reduction. Because many intein-based applications would favor minimal interference, one would generally wish to use small rather than large inteins. The conserved insertion site for a homing endonuclease found in canonical inteins also overlaps with the split sites most commonly found for many split inteins [28]. We removed two residues from the linker where IntC and IntN were connected, i.e., at the natural split site of the gp41–1 intein. This deletion reduced the protein-splicing efficiency by about 40% (Fig. 2D–E), indicating that the naturally reduced size of the gp41–1 intein has presumably reached a functional minimum. Our attempt at optimizing the linker sequence to rescue the robust splicing activity of the gp41–1 intein did not succeed (Fig. 2D–E). Whereas NpuDnaBΔ290 intein, the closest crystal structure, shows higher B-factors for the backbone atoms of the corresponding linker region (43.4 ± 2.7 Å2), the B-factors for the corresponding regions in the gp41–1 intein crystal structure are much lower (22.8 ± 3.1 Å2), even though the linkers are not involved in crystal contacts. The lower B-factors thus suggest that this region might eventually be less flexible exhibiting a higher degree of order in the gp41–1 intein. This region also contains an unusual cis peptide bond between Lys87 and Glu88, presumably caused by the covalent bond to fuse the gp41–1 intein split fragments without any linker insertion, corroborating that further linker shortening likely affects the activity of the intein. The presence of the cis peptide bond is unambiguously supported by the excellent electron density, although part of the side chain of Glu88 appears to be disordered. Because the gp41–1 intein does not seem to easily tolerate any deletion, the linker length at this site could play an essential role during productive folding of some HINT superfamily members [31]. Moreover, we observed deteriorating effects on the protein splicing activity when deviating the extein sequences from the native sequences (“Y” as N-extein and “SSS” as C-extein) (Fig. 2D–E). Thus, gp41–1 intein might not be suitable for further engineering compared with the more robust DnaE intein from Nostoc punctiforme (NpuDnaE), the latter tolerating various amino-acid types at the splicing junctions [35].
The charge network in the gp41–1 intein
Previously, it has been suggested that local charge distributions between naturally split intein halves are important for their association [19,24,32]. We observed extensive charge-charge interactions in the crystal structure of the gp41–1 intein, as also identified among other naturally split inteins [32] (Fig. 3C and 3D). Particularly, they are located in the interacting regions within the β-strands of the two C2-symmetry-related units. Recently, a “capture and collapse” model has been proposed as a folding mechanism for the naturally split NpuDnaE intein, in which the first step of the interaction between the split fragments is initiated by electrostatic interactions on the extended β-strands [33]. A dissociation constant of 1.2 nM reported for the split NpuDnaE intein suggests strong binding between naturally split intein fragments although the binding constant could be extein-dependent [33,36]. We identified similar electrostatic networks between β6 at the beginning of IntC and β3 at the C-end region of IntN in the structure of the gp41–1 intein, but with inverted charges compared to the NpuDnaE intein (Fig. 3C and 3D). These two anti-parallel β-strands appeared to form a charge zipper, reminiscent of leucine zipper structures but embedded in extended strands rather than helices [34]. We also compared the charge patterns in the same regions with the naturally split NpuDnaE intein and its closest structural homolog, the cis-splicing NpuDnaB mini-intein (Fig. 3D). Whereas the NpuDnaB mini-intein does not contain such an extensive charge network in the corresponding region, the gp41–1 intein encompasses more prominent charge interactions than the NpuDnaE intein (Fig. 3D). This observation might support the notion that the “capture and collapse” model suggested for the naturally split NpuDnaE intein might also be valid for the naturally split gp41–1 intein [33].
Interestingly, the IntC region of the gp41–1 intein is dominated by negative charges, as opposed to more positive charges found in the same region of the NpuDnaE intein (Fig. 3D). The charge distributions in β3 and β6 are thus opposite between the naturally split NpuDnaE and gp41–1 inteins (Fig. 3D). Therefore, we decided to test if it is possible to swap the charge distribution in β3 and β6 by mimicking the charge pattern of the gp41–1 intein onto the NpuDnaE intein. We introduced three lysines in IntN and three glutamates in the IntC of the NpuDnaE intein (Fig. 4A). This charge swapped NpuDnaE intein (CS-NpuDnaE) could efficiently splice in cis (96% splicing efficiency), confirming that swapping these charges does not influence protein splicing in cis (Fig. 4B). This result is in line with the previous report in which the charge-swapping of NpuDnaE intein was successfully introduced into the entire NpuDnaEN and NpuDnaEC fragments to suppress the cross-reactivity [24].
Fig. 4.

Charge engineering of the NpuDnaE intein. (A) Charge distributions of the NpuDnaE and the Charge-Swapped NpuDnaE (CS-NpuDnaE) inteins corresponding to strands β3 and β6 in the gp41–1 intein structure. Color-coded arrows indicate the pairs of split inteins tested for trans-splicing in panel C with lines. (B) Cis-splicing analysis of the CS-NpuDnaE intein by SDS-PAGE. M, 0h, 3h, and E stand for molecular markers, 0 hours, 3 hours after induction, and elution from Ni-NTA spin columns. A red arrow indicates the band corresponding to the cis-spliced H6-GB1-GB1 product. A repesentative gel of three individual experiments is shown. (C) Cross-activity between split NpuDnaE and CS-NpuDnaE inteins. SDS-PAGE analysis of trans-splicing for three different pairs of split inteins, CS-NpuDnaEN/CS-NpuDnaEC (left), CS-NpuDnaEN/NpuDnaEC (middle), and NpuDnaEN/CS-NpuDnaEC (right), which are illustrated by arrows and lines in panel A. N, L, and C denote the N-terminal fragment with IntN, the ligated product, and the C-terminal fragment with IntC, respectively. I, A, I+A, and E stand for IPTG induction, arabinose induction, both IPTG and arabinose induction, and elution from Ni-NTA spin columns. IPTG induction produces the N-terminal precursor fragment, N. Arabinose induces the protein expression of the C-terminal precursor, C. Only dual induction by IPTG and arabinose (I+A) is expected to produce the ligated product, L by protein trans-splicing. A repesentative gel of two individual experiments for each combination is shown.
Orthogonality of charge swapped split inteins
The naturally split gp41–1 intein seems to be more sensitive to sequence changes at the splice and loop junctions than we anticipated (Fig. 2E). This pooer tolerance could constrain its practical applications by PTS In contrast, the naturally split NpuDnaE intein and its homologs are more tolerant of amino-acid changes at the splice junctions, making them better suited for protein engineering applications than the gp41–1 intein [14,35,37,38,39]. However, naturally split DnaE inteins from cyanobacteria are cross-reactive to each other [19,35]. This cross-activity limits their application for, e.g., one-pot three-fragment ligation by PTS requiring two split inteins. For such multi-fragment applications, two or more non-cross active (orthogonal) split inteins are needed to suppress undesired cross-activity. Several approaches have been used to circumvent the cross-reactivity, such as utilizing different split sites of the NpuDnaE intein, or kinetic control of two split inteins [22,23,24]. The three-dimensional structure of the gp41–1 intein revealed charge distributions different from the NpuDnaE intein in the corresponding β3 and β6 strands. Next, we asked if the charge network found in β3 and β6 strands of naturally split inteins can be responsible for the orthogonality of split inteins. We created a split intein from the Charge-Swapped NpuDnaE intein (CS-NpuDnaE) and tested the cross-activity of the split fragments. CS-NpuDnaEN and CS-NpuDnaEC could still efficiently splice with the wild-type IntC (NpuDnaEC, 54% yield) and IntN (NpuDnaEN, 92% yield) fragments of the NpuDnaE intein, respectively, compared to the reference sample where both fragments were charge-swapped (100% yield). This high cross-activity of CS-NpuDnaE suggests that the charge network in the region of β3 and β6 alone cannot account for the cross-activity among the naturally split DnaE inteins (Fig.4C). Nevertheless, the charges in this region may play an important role, e.g., for keeping split intein fragments soluble. This observation is consistent with the previous report that the C-terminal 16-residue fragment of NpuDnaE intein is sufficient for efficient trans-splicing of the NpuDnaE intein [36,40,41].
Engineering of orthogonal split inteins
In contrast to naturally split inteins, cis-splicing inteins generally possess a less pronounced charge network within the regions corresponding to β3 and β6 in the gp41–1 structure [32]. Artificially split inteins derived from cis-splicing inteins are often poorly soluble and might not be suitable for protein ligation because they would require unfolding/refolding processes to initiate protein trans-splicing [44,45]. Hence, it would be of particular interest if one could convert cis-splicing inteins into natural-like split inteins by grafting the charge network similar to the one observed in naturally split inteins. We chose the cis-splicing NpuDnaB mini-intein for the grafting approach. This intein is the closest structural homolog to the gp41–1 intein and is superior in tolerating sequence alterations at the splice junctions while retaining high splicing activity [15,42,43]. We introduced five lysine residues (three in β3 and two in β6) (Fig. 5A). The Charge-Introduced NpuDnaB mini-intein (CI-NpuDnaB) was still able to splice efficiently in cis (Fig. 5B, 98% splicing efficiency). As the introduced charged residues did not impair cis-splicing, we derived a split intein pair from the CI-NpuDnaB mini-intein (CI-NpuDnaBN/CI-NpuDnaBC) by splitting at the conserved insertion site of the homing endonuclease domain [15,28]. Trans-splicing of the split CI-NpuDnaB mini-intein became less efficient than that of the split intein derived from the NpuDnaB mini-intein, as increased amounts of unreacted precursors appeared (Fig. 5C, yield 25% vs. 77%). This observation suggests that the charge interactions in the β3 and β6 could play a critical role in the association of the two split intein fragments derived from the NpuDnaB mini-intein. To confirm this hypothesis, we introduced unfavorable interactions by mutating Lys58 to Glu in β3 and Glu116 to Lys in β6. This orthogonal design of NpuDnaB mini-intein (Oth-NpuDnaB) was still able to efficiently splice in cis as no precursor had been left due to spontaneous splicing when expressed in E. coli (Fig. 5B, 97% splicing efficiency). The efficient cis-splicing of Oth-NpuDnaB intein verifies that the introduced mutations were not detrimental to protein splicing reaction, which is an important prerequisite for engineering split inteins. We thus split Oth-NpuDnaB intein into a pair of two fragments of Oth-NpuDnaBN/Oth-NpuDnaBC at the canonical split site and tested trans-splicing activity (Fig. 5C). Unlike the covalently connected cis-splicing Oth-NpuDnaB intein, trans-splicing between Oth-NpuDnaBN/Oth-NpuDnaBC derived from Oth-NpuDnaB intein was drastically impaired (Fig. 5C, 5% yield). Whereas the combination of CI-NpuDnaBN/Oth-NpuDnaBC did not yield any trans-spliced product (0% yield), the pair of CI-NpuDnaBN/NpuDnaBC could produce the ligated product (Fig. 5D, 100% yield (reference sample)). Oth-NpuDnaBC could still react further with the wild-type NpuDnaBN fragment (23% yield), confirming that it is functional (Fig. 5D). These observations confirm that NpuDnaBC and Oth-NpuDnaBC fragments have become orthogonal with CI-NpuDnaBN. Engineering of the charge network in the region corresponding to β3 and β6 in the gp41–1 intein was indeed sufficient to create orthogonal split inteins from a cis-splicing intein, at least with the test case of the NpuDnaB mini-intein.
Fig. 5.

Engineering of the cis-splicing NpuDnaB mini-intein toward an orthogonal split intein pair. (A) Schematic comparison between the original and engineered NpuDnaB mini-inteins in the regions corresponding to β3 and β6 in the gp41–1 intein structure. Solid lines indicate possible favored charge interactions. Dotted red lines indicate possible unfavored charge interactions. Pairs of the split inteins tested in panels B and C are indicated by arrows and lines, where broken lines indicate poor or no trans-splicing. (B) Cis-splicing of the charge-introduced CI-NpuDnaB and the designed orthogonal Oth-NpuDnaB mini-inteins by SDS-PAGE analysis. Cis-spliced products and excised inteins are indicated by red and black arrows, respectively. M, 0h, 3h, and E above the lanes indicate molecular markers, before induction, 3 hours after induction, and elution fraction from Ni-NTA columns. The gel of a single experiment is shown. (C) Trans-splicing of split inteins derived from the NpuDnaB intein (left), Charge-Introduced (CI)-NpuDnaB intein (middle), and the designed orthogonal (Oth)-NpuDnaB intein (right) by splitting at the canonical split site. The results of one to three individual experiments for each combination are shown. (D) Test for orthogonality between split inteins derived from the NpuDnaB intein. SDS-PAGE analysis of trans-splicing for the pairs of CI-NpuDnaBN/NpuDnaBC (left), CI-NpuDnaBN/Oth-NpuDnaBC (middle), and NpuDnaBN/Oth-NpuDnaBC (right) indicated by arrows with solid and broken lines in panel A. The results of one or two individual experiments per combination are shown. For panels C and D, N and C with arrows indicate the bands for the N- and C-terminal precursors, respectively. L indicates the ligated product by trans-splicing. M, 0h, I, A, I+A, and E stand for molecular markers, before induction, IPTG induction, arabinose induction, both IPTG and arabinose induction, and elution fraction from Ni-NTA spin columns, respectively.
Discussion
Intein-based technologies gave rise to a broad range of widely applied methods in both biotechnology and basic research. Many of these methods utilize naturally occurring split inteins and their homologs because artificially split inteins derived from cis-splicing inteins are usually less efficient and/or require denaturing/refolding due to poor solubility [15,26,28,44,45]. The poor solubility of precursor fragments particularly limits their application in vitro. Previous attempts to derive artificially split inteins from several cis-splicing inteins have not been hugely successful, resulting in less productive trans-splicing [15,28]. The poor solubility of artificially split inteins has recently been alleviated in part by using a salt-inducible split intein. The highly soluble split intein was derived from inteins from extreme halophilic archaea, but requires salt-addition to induce the protein splicing reaction [46]. Having two or more robust and orthogonal split inteins could widen the applications of PTS, for example, in multiple-fragment ligation for segmental isotopic labeling, bioorthogonal protein conjugations by semi-synthesis (e.g. antibody-drug conjugate), and protein-based logic gates [21,23,24,25,26]. Naturally split inteins, such as the cyanobacterial DnaE inteins, are not only rare but also often cross-active to each other; however, they exhibit a robust splicing activity and high tolerance of variations at the splice junctions [32,35,47]. The gp41–1 intein is another fragmented split intein exhibiting robust splicing activity [20] and could be used as an orthogonal intein with other split DnaE inteins. However, the splicing activity of the gp41–1 intein turned out to be rather sensitive to variations at the splice junctions (Fig. 2). We determined the crystal structure of the cis-splicing gp41–1 intein at 1.0 Å, which is hitherto the highest resolution available for intein structures. The structure shed light on the features common between the two naturally occurring split inteins, providing the structural basis to guide the engineering of natural-like split inteins from prevalent cis-splicing inteins. The three-dimensional structures of both the gp41–1 and NpuDnaE inteins highlighted an extended charge network on the strands corresponding to β3 and β6 in the gp41–1 intein. This network is absent in most cis-splicing inteins and could play an essential role in efficient trans-splicing. Charge swapping between the corresponding β3 and β6 regions in the NpuDnaE intein did not affect cis- nor trans-splicing, suggesting that the charge network in β3 and β6 regions alone cannot sufficiently account for orthogonality within the naturally split NpuDnaE intein fragments.
In contrast, we successfully demonstrated an orthogonal design by charge engineering of the split NpuDnaB mini-intein, derived from a cis-splicing intein, in the same β3 and β6 regions. Trans-splicing of the NpuDnaB mini-intein in vivo can be as efficient as the NpuDnaE intein and has a high tolerance of variations at the splicing junction [15,42,43]. Split inteins engineered from NpuDnaB mini-intein are new additions to the protein engineering toolbox using protein trans-splicing and contribute to overcoming the junction sequence and extein dependencies that currently complicate PTS applications. More than 1500 inteins or intein-like domains have been identified from the sequence databases [48]. Not all cis-splicing inteins can be converted into active mini-inteins by deleting the inserted homing endonuclease regions due to a mutualism developed between HINT and homing endonuclease domains [31,49]. However, a few hundred mini-inteins in the intein database that carry various junction sequences remain experimentally untested and unexplored. The common structural features found among naturally split inteins could be exploited to convert many other naturally occurring cis-splicing inteins into natural-like split inteins with robust trans-splicing activity. This process would yield new orthogonal split inteins with desired features such as optimal junction sequences and a high tolerance of foreign extein sequences. The resulting engineered inteins expand the applicability of protein trans-splicing in protein engineering, chemical biology, and synthetic biology, in particular when applications require scar-less protein ligation.
Methods
Plasmid constructions
All plasmids used and designed in this study are listed and summarized in Supplementary Table S1, including the oligonucleotide sequences used. The gp41–1 intein variant for crystallization was cloned in plasmid pBHRSF38 as a SUMO fusion protein with an inactivating C1A substitution and a stop codon after the last residue of Asn125 for purification [50]. Cis-splicing gp41–1 intein variants with loop or junction variations are encoded in plasmids pADHDuet21, pBHDuet37, pBHDuet321, pHBDuet021, pHBDuet087, pHBDuet088, and pHBDuet182. pHBDuet093 is a cis-splicing vector with the charge-swapped NpuDnaE intein. Cis-splicing vectors containing the charge-introduced and orthogonal NpuDnaB mini-intein variants are pHBDuet139 and pHBDuet140, respectively. A dual vector system using the pair of pHBDuet095 and pHBBAD106, derived from pHBDuet093, was used for testing trans-splicing of the CS-NpuDnaE intein, in which the N- and C-terminal fragments can be induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) and arabinose, respectively [51]. Plasmids pSKDuet01 (Addgene #12172) and pSKBAD2 (Addgene #15335) encoding the natural NpuDnaEN and NpuDnaEC intein fragment were used to test orthogonality of CS-NpuDnaEC and CS-NpuDnaEN, respectively [35]. Two previously described plasmids pSADuet259 (Addgene #121910) and pSABAD250 (Addgene #45612) encoding NpuDnaBΔ283ΔC39 and NpuDnaBC39, respectively, were used as a reference for trans-splicing of the NpuDnaB mini-intein [15]. A pair of two precursor fragments with CI-NpuDnaBΔ290ΔC39 (pHBDuet148, Addgene #121911) and CI-NpuDnaBC39 (pHBBAD113, Addgene #121912) were derived from plasmid pHBDuet139 (Addgene #121913). Split intein fragments derived from Oth-NpuDnaB, i.e., Oth-NpuDnaBΔ290ΔC39 and Oth-NpuDnaBC39, were encoded in pHBDuet116 (Addgene #121915) and pHBBAD168 (Addgene #121916), respectively, which were derived from pHBDuet140 (Addgene #121914). The plasmids with Addgene numbers are available from www.addgene.org (Addgene, Watertown, MA, USA).
Protein production and purification
All recombinant proteins were produced in E. coli T7 Express (New England Biolabs, Ipswich, MA, USA). For small-scale expression and purification of amounts sufficient to analyze protein splicing in cis and trans, 5 mL LB medium cultures supplemented with 25 μg mL−1 kanamycin, 100 μg mL−1 ampicillin, or both were grown at 37°C until an OD600 of 0.6 was reached. Cultures to express a precursor protein containing a cis-splicing intein were then induced with a final concentration of 1 mM IPTG for 3 hours. For co-expression of N- and C-terminal precursors for trans-splicing, 0.04%-arabinose induction was followed by IPTG addition with a delay of 30 min. The co-expression lasted for a total time of 4 hours at 30°C. The cell cultures were harvested by centrifugation at 4700 xg for 10 min, 4°C and lysed using 400 μL B-PER bacterial protein extraction reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the instructions of the manufacturer. Elution fractions from IMAC purification using Ni2+-NTA spin columns (Qiagen, Hilden, NRW, Germany) were analyzed by 16.5% polyacrylamide SDS-PAGE gels stained with Coomassie Blue. Cis-splicing efficiency in percent was determined by gel band intensity quantification of the IMAC elution fractions from the depicted gels using Image J 2.0.0 [52] and estimated from 100 × [Spliced product]/ ([Precursor] + [C-cleavage products]). Trans-splicing was evaluated from the gel pictures by determination of the relative IMAC-purification yield from each combination of split inteins, that is, the band intensity for the eluted trans-spliced product normalized to the highest yielding combination of split pairs (reference sample), as previously described [15]. In order to compare trans-splicing results among different gels, the intensities were normalized to the sum of three marker bands (14.4 kDa, 18.4 kDa, and 25 kDa). Inactive gp41–1 intein with C1A mutation utilized in structural studies was produced in 2 L LB medium supplemented with 25 μg mL−1 kanamycin by induction with a final concentration of 1 mM IPTG at an OD600 of 0.6 for 3 hours. The cells were harvested by centrifugation and lyzed in Buffer A (50 mM sodium phosphate pH 8.0, 300 mM NaCl) by continuous passaging through an EmulsiFlex-C3 homogenizer (Avestin, Ottawa, ON, Canada) at 15000 psi for 10 min, 4°C. The cell lysate was cleared by centrifugation at 38000 xg for 60 min, 4°C, and loaded on a HisTrap column (GE Healthcare, Chicago, IL, USA) for purification. The protein was purified by following the previously described two-step protocol, including the removal of the hexahistidine tag and SUMO fusion domain [50]. The purified protein contained an additional sequence “SGG” as the N-terminal extein sequence of the gp41–1 intein. For crystallization, the protein was dialyzed against deionized water and concentrated to a final concentration of 45 mg/mL using an ultrafiltration device.
Crystallization, data collection, and structure solution
Diffracting crystals of the fusion protein comprising the N- and C-terminal gp41–1 intein fragments were obtained at room temperature by mixing 100 nL concentrated protein with 100 nL mother liquor (100 mM citric acid, pH 3.5, 100 mM magnesium sulfate, and 30% w/v polyethylene glycol (PEG) 3350). 30% PEG 3350 was sufficient as a cryoprotectant. Data were collected at beamline i02 at Diamond Light Source (Didcot, UK) equipped with a Pilatus 6MF detector. Data were processed using HKL3000 [53] at the nominal resolution of 1.02 Å (Table 1). The structure was solved by molecular replacement with Phaser [54] using the NpuDnaE intein (PDB ID: 4kl5) [55] as the starting model. The structure was rebuilt with Coot [56] and refined with REFMAC5 [57]. Although data completeness in the outermost shell (1.04 – 1.02 Å) was only 37%, the <I/σI> ratio was quite significant at 2.8. Since the completeness in the 1.08–1.06 Å shell was 74% and <I/σI> 3.6, we could safely claim the effective resolution of at least 1.06 Å. However, all data were used in refinement, with almost 2800 reflections present beyond this effective resolution limit.
The protein chain could be traced in the electron density without breaks for all 128 residues (125 intein residues and three residues of the amino acid sequence “SGG” preceding the first intein residue). Alternate conformations were modeled for 22 protein residues, extending to the main chain for 18 of them. A non-canonical cis peptide bond was modeled between Lys87 and Glu88 based on unambiguous electron density for this part of the main chain (the electron density is also unambiguous for the side chain of Lys87, whereas the side chain of Glu88 appears to be partially disordered). Final validation was performed with MolProbity [58], showing an acceptable quality of the model (score 1.8, 35th percentile). The coordinates and structure factors were deposited in the Protein Data Bank with the accession code 6qaz.
Structures were drawn using PyMOL (The PyMOL Molecular Graphics System, Version 2.2.0, Schrödinger, LLC.). Sequence alignments were generated using Clustal Omega 1.2.4 (Conway Institute, University College Dublin, Ireland).
Supplementary Material
Plasmids and oligonucleotides used in this study.
Acknowledgments
We thank B. Haas, S. Jääskeläinen, AD. Hietikko for their technical help in the preparation of proteins and plasmids. We thank Dr. K. Kogan for his assistance at the crystallization facility. This work was supported in part by the Academy of Finland (137995, 277335), Novo Nordisk Foundation (NNF17OC0025402 to HMB., NNF17OC0027550 to HI), Sigrid Jusélius Foundation and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, as well as with Federal funds from the National Cancer Institute, NIH, under Contract No. HHSN261200800001E (to ML). The crystallization and NMR facilities have been supported by Biocenter Finland and HiLIFE-INFRA.
The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views or policies of the U. S. Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U. S. Government.
Abbreviations
- CI
charge-introduced
- CS
charge-swapped
- DnaB
bacterial helicase
- DnaE
catalytic α subunit of DNA polymerase III
- GB1
B1 domain of the Streptococcus sp. IgG binding protein G
- HINT
Hedgehog/INTein
- IntC
C-terminal intein split fragment
- IntN
N-terminal intein split fragment
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- Npu
Nostoc punctiforme
- NTA
nitrilotriacetic acid
- Oth
orthogonal
- PDB
protein data bank
- PEG
polyethylene glycol
- PTS
protein trans-splicing
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SUMO
yeast small ubiquitin-like modifier domain
Footnotes
Declaration of Interests
The authors declare no competing interests.
Database: Structural data are available in the RCSB Protein Data Bank under the accession number 6qaz
References
- 1.Hirata R, Ohsumk Y, Nakano A, Kawasaki H, Suzuki K & Anraku Y (1990) Molecular structure of a gene, VMA1, encoding the catalytic subunit of H(+)-translocating adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae. J. Biol. Chem. 265, 6726–33. [PubMed] [Google Scholar]
- 2.Kane PM, Yamashiro CT, Wolczyk DF, Neff N, Goebl M & Stevens TH (1990) Protein splicing converts the yeast TFP1 gene product to the 69-kD subunit of the vacuolar H(+)-adenosine triphosphatase. Science 250, 651–7. [DOI] [PubMed] [Google Scholar]
- 3.Paulus H (2000) Protein splicing and related forms of protein autoprocessing. Annu. Rev. Biochem. 69, 447–96. [DOI] [PubMed] [Google Scholar]
- 4.Perler FB, Davis EO, Dean GE, Gimble FS, Jack WE, Neff N, Noren CJ, Thorner J & Belfort M (1994) Protein splicing elements: inteins and exteins--a definition of terms and recommended nomenclature. Nucleic Acids Res. 22, 1125–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belfort M (2017) Mobile self-splicing introns and inteins as environmental sensors. Curr. Opin. Microbiol. 38, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper AA, Chen YJ, Lindorfer MA & Stevens TH (1993) Protein splicing of the yeast TFP1 intervening protein sequence: a model for self-excision. EMBO J. 12, 2575–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Topilina NI & Mills K V (2014) Recent advances in in vivo applications of intein-mediated protein splicing. Mob. DNA 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarmiento C & Camarero JA (2019) Biotechnological Applications of Protein Splicing. Curr. Protein Pept. Sci. 20, 408–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Volkmann G & Iwaï H (2010) Protein trans-splicing and its use in structural biology: opportunities and limitations. Mol. Biosyst. 6, 2110. [DOI] [PubMed] [Google Scholar]
- 10.Mills K V, Johnson MA & Perler FB (2014) Protein splicing: how inteins escape from precursor proteins. J. Biol. Chem. 289, 14498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buskirk AR, Ong Y-C, Gartner ZJ & Liu DR (2004) Directed evolution of ligand dependence: small-molecule-activated protein splicing. Proc. Natl. Acad. Sci. U. S. A. 101, 10505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peck SH, Chen I & Liu DR (2011) Directed evolution of a small-molecule-triggered intein with improved splicing properties in mammalian cells. Chem. Biol. 18, 619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thiel I V, Volkmann G, Pietrokovski S & Mootz HD (2014) An atypical naturally split intein engineered for highly efficient protein labeling. Angew. Chem. Int. Ed. Engl. 53, 1306–10. [DOI] [PubMed] [Google Scholar]
- 14.Stevens AJ, Sekar G, Shah NH, Mostafavi AZ, Cowburn D & Muir TW (2017) A promiscuous split intein with expanded protein engineering applications. Proc. Natl. Acad. Sci. U. S. A. 114, 8538–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aranko AS, Oeemig JS, Zhou D, Kajander T, Wlodawer A & Iwaï H (2014) Structure-based engineering and comparison of novel split inteins for protein ligation. Mol. BioSyst. 10, 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oeemig JS, Zhou D, Kajander T, Wlodawer A & Iwaï H (2012) NMR and crystal structures of the Pyrococcus horikoshii RadA intein guide a strategy for engineering a highly efficient and promiscuous intein. J. Mol. Biol. 421, 85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizutani R et al. (2002) Protein-splicing Reaction via a Thiazolidine Intermediate: Crystal Structure of the VMA1-derived Endonuclease Bearing the N and C-terminal Propeptides. J Mol Biol 316, 919–929 [DOI] [PubMed] [Google Scholar]
- 18.Wu H, Hu Z & Liu XQ (1998) Protein trans-splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. U. S. A. 95, 9226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dassa B, London N, Stoddard BL, Schueler-Furman O & Pietrokovski S (2009) Fractured genes: a novel genomic arrangement involving new split inteins and a new homing endonuclease family. Nucleic Acids Res. 37, 2560–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carvajal-Vallejos P, Pallissé R, Mootz HD & Schmidt SR (2012) Unprecedented rates and efficiencies revealed for new natural split inteins from metagenomic sources. J. Biol. Chem. 287, 28686–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otomo T, Ito N, Kyogoku Y & Yamazaki T (1999) NMR observation of selected segments in a larger protein: central-segment isotope labeling through intein-mediated ligation. Biochemistry 38, 16040–4. [DOI] [PubMed] [Google Scholar]
- 22.Shi J & Muir TW (2005) Development of a tandem protein trans-splicing system based on native and engineered split inteins. J. Am. Chem. Soc. 127, 6198–206. [DOI] [PubMed] [Google Scholar]
- 23.Busche AEL, Aranko AS, Talebzadeh-Farooji M, Bernhard F, Dötsch V & Iwaï H (2009) Segmental isotopic labeling of a central domain in a multidomain protein by protein trans-splicing using only one robust DnaE intein. Angew. Chem. Int. Ed. Engl. 48, 6128–31. [DOI] [PubMed] [Google Scholar]
- 24.Shah NH, Vila-Perelló M & Muir TW (2011) Kinetic control of one-pot trans-splicing reactions by using a wild-type and designed split intein. Angew. Chem. Int. Ed. Engl. 50, 6511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Möhlmann S; et al. (2011) Site-specific modification of ED-B-targeting antibody using intein-fusion technology. BMC Biotechnology, 11: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lohmueller JJ, Armel TZ, Silver PA. (2012) A tunable zinc finger-based framework for Boolean logic computation in mammalian cells. Nucleic Acids Res. 40, 5180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun W, Yang J & Liu X-Q (2004) Synthetic two-piece and three-piece split inteins for protein trans-splicing. J. Biol. Chem. 279, 35281–6. [DOI] [PubMed] [Google Scholar]
- 28.Aranko AS, Wlodawer A & Iwaï H (2014) Nature’s recipe for splitting inteins. Protein Eng. Des. Sel. 27, 263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holm L & Laakso LM (2016) Dali server update. Nucleic Acids Res. 44, W351–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall TM, Porter JA, Young KE, Koonin E V, Beachy PA & Leahy DJ (1997) Crystal structure of a Hedgehog autoprocessing domain: homology between Hedgehog and self-splicing proteins. Cell 91, 85–97. [DOI] [PubMed] [Google Scholar]
- 31.Iwaï H, Mikula KM, Oeemig JS, Zhou D, Li M & Wlodawer A (2017) Structural Basis for the Persistence of Homing Endonucleases in Transcription Factor IIB Inteins. J. Mol. Biol. 429, 3942–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dassa B, Amitai G, Caspi J, Schueler-Furman O & Pietrokovski S (2007) Trans protein splicing of cyanobacterial split inteins in endogenous and exogenous combinations. Biochemistry 46, 322–30. [DOI] [PubMed] [Google Scholar]
- 33.Shah NH, Eryilmaz E, Cowburn D & Muir TW (2013) Naturally split inteins assemble through a “capture and collapse” mechanism. J. Am. Chem. Soc. 135, 18673–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walther TH, Gottselig C, Grage SL, Wolf M, Vargiu A V, Klein MJ, Vollmer S, Prock S, Hartmann M, Afonin S, Stockwald E, Heinzmann H, Nolandt O V, Wenzel W, Ruggerone P & Ulrich AS (2013) Folding and self-assembly of the TatA translocation pore based on a charge zipper mechanism. Cell 152, 316–26. [DOI] [PubMed] [Google Scholar]
- 35.Iwai H, Züger S, Jin J & Tam P-H (2006) Highly efficient protein trans-splicing by a naturally split DnaE intein from Nostoc punctiforme. FEBS Lett. 580, 1853–8. [DOI] [PubMed] [Google Scholar]
- 36.Aranko AS, Züger S, Buchinger E & Iwaï H (2009) In vivo and in vitro protein ligation by naturally occurring and engineered split DnaE inteins. PLoS One 4, e5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Aboye T, Breindel L, Shekhtman A & Camarero JA (2016) Efficient recombinant expression of SFTI-1 in bacterial cells using intein-mediated protein trans-splicing. Biopolymers 106, 818–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jagadish K, Borra R, Lacey V, Majumder S, Shekhtman A, Wang L & Camarero JA (2013) Expression of fluorescent cyclotides using protein trans-splicing for easy monitoring of cyclotide-protein interactions. Angew. Chem. Int. Ed. Engl. 52, 3126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bi T, Li Y, Shekhtman A & Camarero JA (2018) In-cell production of a genetically-encoded library based on the θ-defensin RTD-1 using a bacterial expression system. Bioorg. Med. Chem. 26, 1212–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muona M, Aranko AS & Iwai H (2008) Segmental isotopic labelling of a multidomain protein by protein ligation by protein trans-splicing. Chembiochem 9, 2958–61. [DOI] [PubMed] [Google Scholar]
- 41.Oeemig JS, Aranko AS, Djupsjöbacka J, Heinämäki K & Iwaï H (2009) Solution structure of DnaE intein from Nostoc punctiforme: structural basis for the design of a new split intein suitable for site-specific chemical modification. FEBS Lett. 583, 1451–6. [DOI] [PubMed] [Google Scholar]
- 42.Iwaï H & Aranko AS (2017) Protein Ligation by HINT Domains In Chemical Ligation pp. 421–445. John Wiley & Sons, Inc., Hoboken, NJ, USA [Google Scholar]
- 43.Ellilä S, Jurvansuu JM & Iwaï H (2011) Evaluation and comparison of protein splicing by exogenous inteins with foreign exteins in Escherichia coli. FEBS Lett. 585, 3471–7. [DOI] [PubMed] [Google Scholar]
- 44.Southworth MW, Adam E, Panne D, Byer R, Kautz R & Perler FB (1998) Control of protein splicing by intein fragment reassembly. EMBO J. 17, 918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otomo T, Teruya K, Uegaki K, Yamazaki T & Kyogoku Y (1999) Improved segmental isotope labeling of proteins and application to a larger protein. J. Biomol. NMR 14, 105–14. [DOI] [PubMed] [Google Scholar]
- 46.Ciragan A, Aranko AS, Tascon I & Iwaï H (2016) Salt-inducible Protein Splicing in cis and trans by Inteins from Extremely Halophilic Archaea as a Novel Protein-Engineering Tool. J. Mol. Biol. 428, 4573–4588. [DOI] [PubMed] [Google Scholar]
- 47.Cheriyan M, Pedamallu CS, Tori K & Perler F (2013) Faster protein splicing with the Nostoc punctiforme DnaE intein using non-native extein residues. J. Biol. Chem. 288, 6202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Novikova O, Jayachandran P, Kelley DS, Morton Z, Merwin S, Topilina NI & Belfort M (2016) Intein Clustering Suggests Functional Importance in Different Domains of Life. Mol. Biol. Evol. 33, 783–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hiraga K, Derbyshire V, Dansereau JT, Van Roey P & Belfort M (2005) Minimization and stabilization of the Mycobacterium tuberculosis recA intein. J. Mol. Biol. 354, 916–26. [DOI] [PubMed] [Google Scholar]
- 50.Guerrero F, Ciragan A & Iwaï H (2015) Tandem SUMO fusion vectors for improving soluble protein expression and purification. Protein Expr. Purif. 116, 42–9. [DOI] [PubMed] [Google Scholar]
- 51.Züger S & Iwai H (2005) Intein-based biosynthetic incorporation of unlabeled protein tags into isotopically labeled proteins for NMR studies. Nat. Biotechnol. 23, 736–40. [DOI] [PubMed] [Google Scholar]
- 52.Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET & Eliceiri KW (2017) ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minor W, Cymborowski M, Otwinowski Z & Chruszcz M (2006) HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr. D. Biol. Crystallogr. 62, 859–66. [DOI] [PubMed] [Google Scholar]
- 54.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC & Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aranko AS, Oeemig JS, Kajander T & Iwaï H (2013) Intermolecular domain swapping induces intein-mediated protein alternative splicing. Nat. Chem. Biol. 9, 616–22. [DOI] [PubMed] [Google Scholar]
- 56.Emsley P, Lohkamp B, Scott WG & Cowtan K (2010) Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murshudov GN, Vagin AA & Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. Biol. Crystallogr. 53, 240–55. [DOI] [PubMed] [Google Scholar]
- 58.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS & Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weiss MS (2001) Global indicators of X-ray data quality. J. Appl. Crystallogr. 34, 130–135. [Google Scholar]
- 60.Brünger AT (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Plasmids and oligonucleotides used in this study.