

Abstract
Motivation
Single-cell RNA-sequencing (scRNA-seq) has enabled studies of tissue composition at unprecedented resolution. However, the application of scRNA-seq to clinical cancer samples has been limited, partly due to a lack of scRNA-seq algorithms that integrate genomic mutation data.
Results
To address this, we present
CONICS
COpy-Number analysis In single-Cell RNA-Sequencing. CONICS is a software tool for mapping gene expression from scRNA-seq to tumor clones and phylogenies, with routines enabling: the quantitation of copy-number alterations in scRNA-seq, robust separation of neoplastic cells from tumor-infiltrating stroma, inter-clone differential-expression analysis and intra-clone co-expression analysis.
Availability and implementation
CONICS is written in Python and R, and is available from https://github.com/diazlab/CONICS.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Single-cell RNA-sequencing (scRNA-seq) is being rapidly adopted to model expression kinetics during dynamic biological processes. However, there are unaddressed challenges to applying scRNA-seq to clinical cancer samples. Firstly, distinguishing neoplastic cells (daughter cells of the tumor-initiating cell) from tumor-infiltrating stromal and immune cells is an open problem. Secondly, while sub-populations of cells from non-malignant tissue are typically defined by their ontogeny, differentiation status, or unique expression profile, the sub-populations of interest in the context of cancer are sub-clones defined by DNA mutations.
The inability to separate neoplastic cells from stroma is a significant barrier to the use of scRNA-seq on clinical samples. In non-malignant tissue, transcriptomic clustering and dimensionality-reduction techniques are often employed to identify sub-populations. However, separating cells by gene expression alone is not satisfactory in tumor samples, since neoplastic cells often express gene programs that are similar to infiltrating stroma.
Several studies have used expressed point mutations to stratify neoplastic cells (e.g. Kim et al., 2015). However, point mutations can be challenging to quantify in individual cells, due to variability in coverage (Tirosh et al., 2016) and emerging evidence suggests that large-scale copy-number variants (CNVs) are robustly detectible in scRNA-seq (Müller et al., 2017; Venteicher et al., 2017). COpy-Number analysis In single-Cell RNA-Sequencing (CONICS) implements algorithms to identify large-scale CNVs in scRNA-seq. This provides a rigorous way to separate neoplastic cells for downstream analysis.
Sequencing only the 3’ ends of genes is often used as a cost-saving measure, to increase the throughput of cells interrogated (e.g. mRNA capture-bead protocols). Expressed point mutations in the 5’ ends of genes may not be covered by 3’ sequencing. However, large-scale CNVs can be identified without full-transcript coverage.
CONICS includes algorithms to triage cells from a scRNA-seq assay, based on the presence of CNVs detected in an orthogonal DNA sequencing experiment. CONICS integrates tumor-normal fold-changes with the minor-allele frequencies of point mutations, to estimate false-discovery rates (FDRs) in CNV classification. Additionally, CONICS includes routines to perform downstream phylogeny assessment and gene co-expression analysis.
2 Results
2.1 Quantification of copy-number alterations in scRNA-seq
To illustrate the use of CONICS, we performed scRNA-seq and exome sequencing (exome-seq) on a glioblastoma biopsy (SF10281), and a patient-matched blood control (Supplementary Material). This produced 96 novel scRNA-seq libraries, and exome-wide DNA sequencing data (EGAD00001003114). The expression of an individual gene may not correlate with its copy-number status, but we and others have shown that CNV status and average gene-expression levels do strongly correlate for megabase-sized alterations, in single cells (Hou et al., 2016; Müller et al., 2016; Venteicher et al., 2017).
CONICS exploits this result to triage single cells, based on CNV calls from an orthogonal platform, such as exome-seq. The inputs for CONICS are a scRNA-seq dataset to be tested for CNVs, a scRNA-seq dataset to use as a control, as well as annotations of CNV regions and point mutations to be quantified in single cells.
CONICS includes routines for estimating the global correlations between CNV status and gene expression in single cells (Fig. 1A, top-left). Moreover, CONICS estimates the CNV status of a given test cell, at a given significance threshold, via comparison to the control scRNA-seq dataset. In our example, non-malignant adult-human brain scRNA-seq (Darmanis et al., 2015) was used as a control (Fig. 1A, top-right).
Fig. 1.
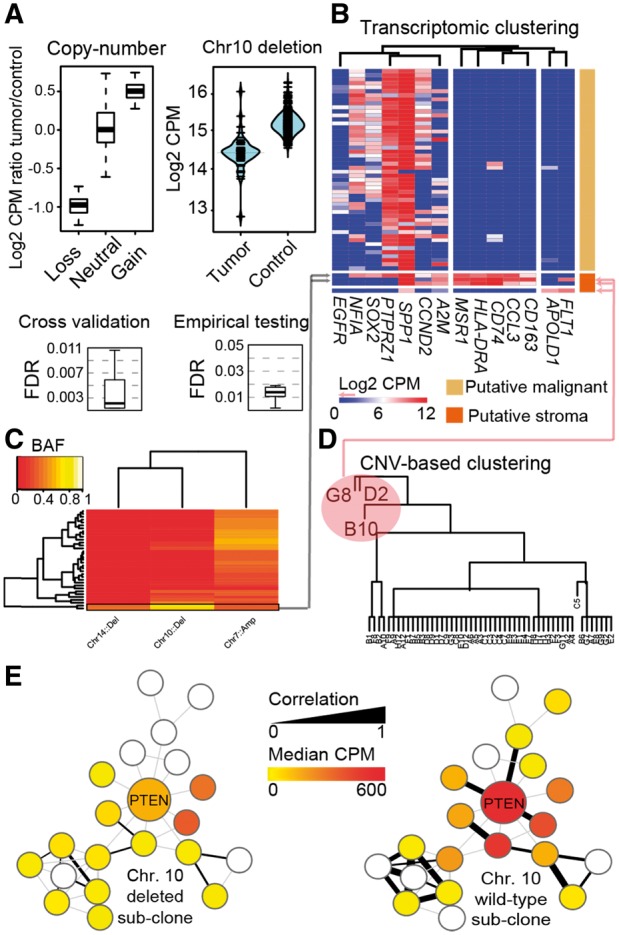
An example of CONICS analysis on scRNA-seq and exome-seq of a glioblstoma biopsy. (A) CONICS quantifies CNVs in single cells with a controlled error rate: ScRNA-seq read-count correlations with CNV status (top left); scRNAseq read-count distributions for an example CNV (top right); FDR estimates in assigning CNV status to individual cells, computed via cross validation (bottom left) and via comparison to a control dataset (bottom right). (B–D) CONICS estimates CNV allele frequency, which, when compared to the expression of canonical markers and clustering of CNV status, enables the rigorous separation of stromal /immune cells from neoplastic cells. (E) Co-expression network of PTEN, produced by CONICS, compared between cells with a chr. 10 loss and wild-type
For users who do not have DNA sequencing data and/or may not have a control scRNA-seq dataset, we also provide CONICSmat (Supplementary Material). CONICSmat is a separate R package that provides some of the functionality of CONICS. However, CONICSmat requires fewer inputs and software dependencies.
2.2 FDR estimation and validation
To estimate the FDR of CNV assignments, CONICS provides a routine to perform 10-fold cross-validation of CNV classification. CONICS also provides a routine to estimate FDR via an empirical test, using a gold-standard scRNA-seq experiment, if available. For example, we used non-malignant fetal-human brain scRNA-seq (Diaz et al., 2016) to estimate FDRs of CNV calls in our glioblastoma scRNA-seq (Fig. 1A, bottom).
Additionally, CONICS compares average allele frequencies of point mutations on CNV regions. Taken together with a clustering based on gene expression, these metrics enable the robust separation of neoplastic cells from tumor-infiltrating stromal and immune cells (Fig. 1B).
2.3 Mapping gene expression to sub-clones and phylogenies
CONICS contains routines to facilitate phylogeny and co-expression network analysis, based on clones inferred from CNV calls. In particular, CONICS implements the Fitch–Margoliash method to build phylogenies from inferred CNV calls. Other phylogenic techniques can alternatively be employed, using the CNV and point-mutation incidence matrices produced by CONICS as a starting point. CONICS also provides code to estimate co-expression networks within a given clone. SCDE (Kharchenko et al., 2014) is used to adjust correlation coefficients for cell-dropout rates. From this, CONICS produces local co-expression networks which can then be compared between inferred clones (Fig. 1C).
Supplementary Material
Funding
This work was supported by a Cancer League Research Grant, a NCI Helen Diller Family Comprehensive Cancer Center support grant (P30-CA82103-18) and a UCSF Brain Tumor SPORE Career Development Award (P50-CA097257-13: 7017) and a gift from the Dabbiere Family to A.D.
Conflict of Interest: none declared.
References
- Darmanis S. et al. (2015) A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci., 112, 7285–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz A. et al. (2016) SCell: integrated analysis of single-cell RNA-seq data. Bioinformatics, 32, 2219–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y. et al. (2016) Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res., 26, 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko P.V. et al. (2014) Bayesian approach to single-cell differential expression analysis. Nat. Methods, 11, 740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.-T. et al. (2015) Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol., 16, 127.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S. et al. (2016) Single‐cell sequencing maps gene expression to mutational phylogenies in PDGF‐ and EGF‐driven gliomas. Mol. Syst. Biol., 12, 889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S. et al. (2017) Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol., 18, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I. et al. (2016) Large-scale single-cell RNA-seq reveals a developmental hierarchy in human oligodendroglioma. Nat. Publ. Gr., 539, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher A.S. et al. (2017) Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science (80-.), 355, eaai8478.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.