

Abstract
Pancreatic ductal adenocarcinoma is a particularly difficult cancer to treat due to a lack of effective screening or treatment. Pancreatic cancer cells exhibit high proliferating cell nuclear antigen (PCNA) expression, which is associated with poor prognosis. PCNA, an important nuclear DNA replication and repair protein, regulates a myriad of proteins via the interdomain connector loop. Within this region, amino acids 126–133 are critical for PCNA interactions in cancer cells. Here, we investigate the ability of a decoy cell-penetrating peptide, R9-caPeptide, that mimics the interdomain connector loop region of PCNA to disrupt PCNA-protein interactions in pancreatic cancer cells. Our data suggest that R9-caPeptide causes dose-dependent toxicity in a panel of pancreatic cancer cell lines by inhibiting DNA replication fork progression and PCNA-regulated DNA repair, ultimately causing lethal DNA damage. Overall, these studies lay the foundation for novel therapeutic strategies that target PCNA in pancreatic cancer.
Keywords: Pancreatic Cancer, PCNA, DNA repair, Cell-penetrating peptide, DNA damage, Replication Stress, Proliferating cell nuclear antigen, Cell death, DNA damage response, Peptide therapeutics
Graphical Abstract
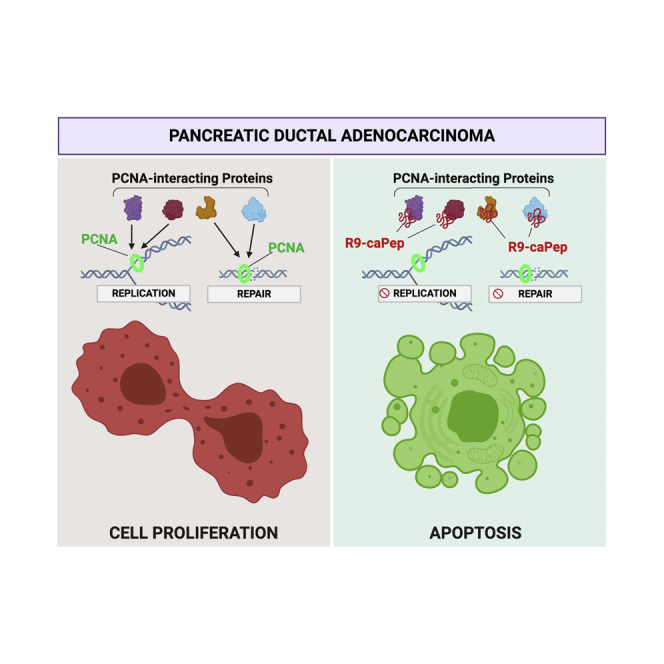
Proliferating cell nuclear antigen (PCNA), an important DNA replication and repair protein, is associated with poor prognosis in pancreatic cancer. Here, the authors demonstrate the ability of a decoy cell-penetrating peptide in targeting PCNA by inhibiting DNA replication fork progression and PCNA-regulated DNA repair, ultimately causing lethal DNA damage.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the third leading cause of cancer-related deaths in the United States, with approximately 54,000 newly diagnosed cases in this calendar year alone and almost 44,000 resulting deaths.1 Due to a lack of early detection and effective therapies, pancreatic cancer is projected to become the second leading cause of cancer-related deaths within the next 20 years.2 The poor outlook on PDAC is largely due to the advanced disease state at diagnosis.3
A typical pancreatic tumor is molecularly heterogeneous, harboring approximately 63 genetic alterations.4 Over one-third of all cases carry deleterious somatic mutations to the DNA damage response genes.5 About 50% of all PDAC tumors show KRAS activating mutations, as well as loss-of-function mutations of TP53, SMAD4, and CDKN2A.2 Additionally, a high frequency of genetic alterations in DNA maintenance genes, including ATM, FANCM, XRCC4, and XRCC6, have been identified.2 PDAC is known to have a mutator phenotype because of an accumulation of mutations resulting from the dysregulation of DNA damage repair.6 The extensive damage tolerance of these tumors appears to be a protective factor, because most patients do not respond to traditional chemotherapies and those that do often develop resistance rapidly.7,8
Proliferating cell nuclear antigen (PCNA), often referred to as the “ring-master of the genome,”9 is a ring-shaped clamp that encircles the DNA strand acting as a replication scaffold to regulate proteins during replication and repair processes. The main interaction site of PCNA with other molecules is the interdomain connector loop (IDCL), spanning amino acids (aa) M121 to Y133.10 The IDCL is recognized by several key “docking” proteins, including p21 (CDKN1A),11 DNA polymerase δ (Pol δ),12 flap endonuclease 1 (FEN1),13 DNA methyltransferase 1 (DNMT1),14 and DNA ligase 1 (LIGI).15 PCNA is known to be indispensable to pancreatic cancer growth and survival, because the cells demonstrate a higher dependence on PCNA-regulated DNA repair in comparison with normal cells.16, 17, 18
We have previously shown that cancer cells replicate in a significantly more error-prone manner compared with normal cells, partially because of the differential association of DNA repair proteins with PCNA.19 We showed that these differential interactions were due to previously unidentified post-translational modifications to PCNA in tumor cells, which cause PCNA to be more acidic, as opposed to the basic isoform prevalent in normal cells.20 After further characterization, we identified a region within IDCL that is critical for this differential interaction and developed a peptide to mimic this region of specificity (aa 126–133), which we named caPeptide. We showed that caPeptide inhibited in vitro DNA replication due to its ability to disrupt specific PCNA interactions with various DNA replication components, including Pol δ and FEN1.21,22 Furthermore, when attached to an R9 peptide (nine arginines) for cellular delivery, the R9-caPeptide exhibited cytotoxicity in breast and neuroblastoma cancer cell lines. It was also shown that R9-caPeptide is non-toxic to non-malignant cell lines, such as human peripheral blood mononuclear cells, human neural crest stem cells, and human mammary epithelial cells.21,23
Because DNA damage repair pathways are activated in pancreatic cancer and based on previous data demonstrating the ability of R9-caPeptide to effectively disrupt PCNA-protein interactions in cancer cells, we hypothesized that this unique targeting ability of R9-caPeptide can also be exploited in pancreatic cancer. In the current study, we investigated the role of R9-caPeptide in a panel of five pancreatic cancer cell lines and found that this peptide targets PCNA-interacting proteins, leading to DNA damage and ultimately apoptotic cell death.
Results
Enhanced PCNA Expression Is Associated with Poor Survival in PDAC
To determine relative PCNA expression in PDAC and normal pancreas, we extracted RNA sequencing (RNA-seq) data from TCGA (The Cancer Genome Atlas: https://www.cancer.gov/tcga) and G-TEx (Genotype-Tissue Expression: https://gtexportal.org/home/) databases. We observed that PCNA is significantly overexpressed in pancreatic cancers compared with normal pancreas as shown in Figure 1A (p < 0.0001). Furthermore, survival analysis using the Kaplan-Meier Plotter showed that enhanced PCNA expression was associated with worse survival in TCGA cohort (Figure 1B). The median overall survival for the PCNA-low cohort was 35.3 months, whereas the survival in the PCNA-high cohort was 17.3 months. These analyses demonstrate that there is enhanced PCNA expression in PDAC compared with normal pancreas, and that high expression of PCNA is associated with poor survival.
Figure 1.

Enhanced PCNA Expression Is Associated with Poor Survival in PDAC
(A) mRNA expression levels from normal pancreas (G-Tex dataset) and pancreatic ductal adenocarcinoma (PDAC; TCGA dataset) are compared. p value is calculated using the Mann-Whitney test. (B) PDAC patients from TCGA cohort (N = 177) were divided into two groups based on PCNA mRNA expression. The cutpoint was determined using the Kaplan-Meier Plotter (KM-Plotter). Overall survival is plotted using the Kaplan-Meier method. The p value is calculated using the log rank test.
R9-caPeptide Is Cytotoxic to Pancreatic Cancer Cell Lines
Because we showed that R9-caPeptide had a cytotoxic effect in breast and neuroblastoma cancer cell lines, we wanted to investigate whether this effect could be extended to pancreatic cancer cells. To test this, we treated a panel of five pancreatic cancer cell lines (MIA PaCa-2, PANC-1, UPN3, Capan-1, and BxPC-3) with increasing concentrations of R9-caPeptide, similar to conditions described previously.21,23 As illustrated in Figure 2A, the pancreatic cancer cell lines display a range of sensitivity to R9-caPeptide treatment, with BxPC-3 exhibiting the highest sensitivity (∼10 μM) and PANC-1 exhibiting the highest resistance (∼40 μM). These values are consistent with those previously observed in other cancer types.21,23
Figure 2.

R9-caPeptide Is Cytotoxic to Pancreatic Cancer Cell Lines
(A) Pancreatic cancer cell lines were treated with increasing concentrations (0–200 μM) of R9-caPeptide (R9-caPep) for 48 h, then assayed for cell death using the CellTiter Glo assay. (B) Pancreatic cancer cell lines were treated with 50 μM R9-caPep or scrambled peptide (R9-scrPep) for 72 h, then assayed for cell death using the CellTiter Glo assay. Data are represented as mean ± SD.
To confirm that the observed cytotoxicity is dependent on the R9-caPeptide sequence, we generated a scrambled R9 peptide (R9-scrPeptide) containing the same amino acids as R9-caPeptide.21,23 As shown in Figure 2B, for the three pancreatic cancer cell lines tested, R9-caPeptide was significantly more toxic compared with R9-scrPeptide. These data suggest that R9-caPeptide is capable of inducing cytotoxicity in pancreatic cancer cell lines consistent with its effects observed previously in other cancers.21,23
R9-caPeptide Inhibits Replication Fork Progression
To determine the mechanism by which R9-caPeptide induces cytotoxicity in pancreatic cancer cell lines, we investigated the effect of R9-caPeptide on replication fork progression. The MIA PaCa-2 cell line was exposed to increasing concentrations of R9-caPeptide, and resulting DNA fiber lengths were measured using DNA fiber analysis. To establish baseline replication fork progression, we labeled DNA (with chlorodeoxyuridine [CldU]) for 15 min prior to the introduction of R9-caPep or DMSO (control). CldU was washed, and a second label (iododeoxyuridine [IdU]) was added to track replication fork progression in the presence of varying concentrations of R9-caPeptide (Figure 3A). As shown in Figure 3B, it was observed that R9-caPeptide resulted in a dose-dependent decrease in replication fork progression. These data suggest that R9-caPeptide inhibits replication fork progression in the MIA PaCa-2 pancreatic cancer cell line.
Figure 3.

R9-caPep Inhibits Replication Fork Progression
(A) Schematic showing the experimental design for the DNA fiber analysis. Replicating DNA was labeled with CldU for 15 min, followed by IdU for 30 min. R9-caPep was added with IdU labeling. (B) Analysis: CldU and IdU tract lengths were measured (in μm) at each concentration of R9-caPep and were normalized to label exposure time to calculate replication speed (1 μm = 2.59 kb). One-way ANOVA demonstrated a significant decrease in replication speed during R9-caPep exposure. ∗∗∗p < 0.0001 for Dunnett’s multiple comparison test (comparison was performed with CldU violin plot at 0 μM).
R9-caPeptide Induces DNA Damage in Pancreatic Cancer Cell Lines
Given the role of PCNA not only as a replication scaffold but also in repairing DNA damage, we investigated the effect of R9-caPeptide on DNA damage in pancreatic cancer. The panel of pancreatic cancer cell lines was treated with 50 μM R9-caPeptide for up to 48 h. Cells were harvested at 0, 4, 24, and 48 h for western blot analysis. DNA damage was assessed by probing for Ser139 phosphorylation of H2AX (γ-H2AX), a commonly used biomarker for DNA damage associated with double-strand breaks. As shown in Figure 4, γ-H2AX was upregulated in a time-dependent manner in three of the four cell lines tested: UPN3, PANC-1, and MIA PaCa-2. H2AX levels remained constant at all time points, indicating an increase in H2AX phosphorylation was not due to an increase in total protein content. These data suggest that R9-caPeptide induces DNA damage in pancreatic cancer cell lines.
Figure 4.

R9-caPep Induces DNA Damage in Pancreatic Cancer Cell Lines
Pancreatic cancer cells were treated with 50 μM R9-caPep for up to 48 h. Representative western blot analysis was performed to analyze both phosphorylated γ-H2AX and unphosphorylated H2AX. Actin was used as the loading control.
R9-caPeptide Induces G1/G0 Cell-Cycle Arrest with Significant Cell Death in Pancreatic Cancer
We further characterized the observed cytotoxicity of R9-caPeptide. MIA PaCa-2 cells were treated with increasing concentrations of R9-caPeptide (0–100 μM) for 24 h and processed for the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay that fluorescently labels the 3′ end of DNA double-strand breaks in apoptotic cells. We observed an increase in the labeling intensity at high concentrations of R9-caPeptide, indicative of higher apoptotic cell death (Figure 5A). Flow cytometry analysis with a similar treatment scheme indicated that R9-caPeptide stalls cells in the G1/G0 phase of the cell cycle, with a significant increase in sub-G0 phase representative of apoptotic cells (Figure 5B). Overall, these data suggest that R9-caPeptide interferes with the cell cycle of pancreatic cancer cells, causing significant cell death.
Figure 5.

Cell Death Due to R9-caPep Is Largely Apoptotic in Pancreatic Cancer
MIA PaCa-2 cells were treated with increasing concentrations (0–100 μM) of R9-caPep for 24 h. (A) Treated MIA PaCa-2 cells were assayed for double-strand DNA breaks using the TUNEL assay. (B) Treated MIA PaCa-2 cells were labeled with BrdU for cell-cycle analysis. Data are represented as mean ± SD.
Discussion
The PCNA protein has a homo-trimeric configuration, allowing it to act as a replication scaffold for a myriad of proteins, and plays a central role in DNA replication and repair processes. The majority of PCNA-interacting proteins dock at its IDCL region; however, mechanisms of these interactions remain poorly understood. In efforts to study the differential interactions of PCNA in normal and cancer cells, we previously identified a region within the IDCL of PCNA (aa 126–133) that is of particular importance to protein-protein interactions in cancer cells, more so than in normal cells.23 We previously generated a peptide that mimics this region of PCNA to target these critical protein interactions, effectively killing various cancer cell types.21,23 In the current study, we extend these observations and characterize the cytotoxicity of R9-caPeptide in pancreatic cancer cells. The acute relevance of this study is supported by recent observations that dysregulated DNA repair is a key targetable vulnerability in pancreatic cancer, with multiple early-phase ongoing clinical trials that aim to target dysregulated DNA repair pathways in pancreatic cancer (ClinicalTrials.org: NCT03601923, NCT03553004, NCT03404960, NCT02677038, NCT04005690, NCT02632448, and NCT03682289).
In this study, we show that R9-caPeptide inhibits replication fork progression, causes DNA damage, and ultimately leads to apoptosis of pancreatic cancer cells.21, 22, 23 Importantly, R9-caPeptide is minimally toxic to normal or non-cancerous immortalized cells as reported in our previous work.19,21 The observed inhibition of replication progression is likely mediated through disruption of PCNA-FEN1 and PCNA-LIG1 interactions that are important in the maturation of Okazaki fragments, compromising replication fork progression, as shown previously.21 Stalled replication forks are restarted by using recombination machinery, enabling repair or bypass of the blocking lesion in a non-mutagenic manner.24 PCNA regulates recombination-dependent repair of stalled replication forks, and failure to resolve such stalled forks leads to lethal DNA damage.21 Consistent with these observations, we found that R9-caPeptide caused significant DNA damage due to an increase in double-strand breaks leading to apoptosis in three out of four pancreatic cancer cell lines tested.
We have previously demonstrated the in vivo efficacy of R9-caPeptide following intra-tumoral injection.21 However, direct intratumoral injection is likely not a clinically feasible approach for pancreatic cancer that presents with metastases. Even patients with clinically localized pancreatic cancer are thought to harbor clinically occult micro-metastases. Our lab is actively exploring strategies for systemic delivery of R9-caPeptide. One limitation in the systemic delivery of peptides for cancer therapeutics is their rapid clearance by plasma and tissue peptidases. Peptide modification by applying medicinal chemistry approaches can overcome this problem, as has been demonstrated for numerous peptide therapeutics used in the clinic today.25 Another challenge is the ability to target intracellular or nuclear proteins using peptide therapeutics. In this study, we demonstrated that R9 modification was sufficient to target PCNA interactions and result in cell death.
By demonstrating that PCNA interactions that are of particular importance in cancer cells can be targeted using a decoy peptide that mimics a region of IDCL, our study lays the foundation for future work that aims to target IDCL in pancreatic cancer. Further studies are required to explore the targeting of PCNA-regulated replication and repair in combination with currently used therapies (Figure 6). Ongoing studies are exploring optimization of R9-caPeptide, as well as the development of small-molecule inhibitors for translation to clinical trials in pancreatic cancer.
Figure 6.

Conceptual Model of PCNA Targeting in Pancreatic Cancer
A schematic model of various mechanisms that lead to DNA damage and possible interventions with PCNA-targeted therapy that could be applied in pancreatic cancer.
Materials and Methods
Peptide Production
R9-caPeptide was synthesized and isolated to >95% purity by AnaSpec (San Jose, CA, USA) and provided in powder form. Peptides were dissolved in phosphate-buffered saline (PBS; Corning, Corning, NY, USA), aliquoted, lyophilized, and stored at −20°C. Prior to use, R9-caPeptide aliquots were reconstituted to a concentration of 10 mM.
Cell Culture
All cancer cell lines were cultured according to procedures established by the American Type Culture Collection (ATCC). MIA PaCa-2, PANC-1, and UPN cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Corning, Corning, NY, USA), supplemented with 10% fetal bovine serum (FBS; Omega Scientific, Tarzana, CA, USA) and 1% penicillin/streptomycin (P/S; Corning, Corning, NY, USA). Capan-1 cell lines were cultured in Iscove’s modified Dulbecco’s medium (IMDM; Corning, Corning, NY, USA), supplemented with 20% FBS and 1% P/S. BxPC-3 cell lines were cultured in RPMI-1640 (Corning, Corning, CA, USA), modified to contain 10 mM (Corning, Corning, CA, USA), 1 mM sodium pyruvate (Corning, Corning, CA, USA), and 4,500 mg/L glucose (Corning, Corning, CA, USA), and supplemented with 10% FBS and 1% P/S. All cell cultures were maintained at 37°C in 5% CO2.
In Vitro Cytotoxicity of R9-caPeptide
Exponentially growing (1 × 103 to 5 × 103, depending on cell doubling time) pancreatic cancer cells described above were seeded in 96-well plates. Increasing concentrations of R9-caPeptide (0–200 μM) were added to each well in quadruplicate and incubated for 48 h at 37°C in 5% CO2. Cell viability was determined using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA), according to the manufacturer’s instructions. Activity was calculated as the % of cells alive/concentration versus control cells with no R9-caPeptide treatment, where 100% indicates no cell death (high ATP levels) and 0% indicates complete cell death (low or no ATP levels). Data were analyzed, and IC50 values were determined following the guidelines described in Sebaugh26 and using the sigmoidal dose-response equation in GraphPad Prism 5 software (La Jolla, CA, USA).
Cell Proliferation Assay Using Flow Cytometry
MIA PaCa-2 cells were treated with increasing concentrations of R9-caPeptide (0–100 μM) for 24 h, then pulse-labeled with 10 μM bromodeoxyuridine (BrdU) in DMEM supplemented with 10% FBS and 1% P/S for 1 h at 37°C. Untreated MIA PaCa-2 cells were used as the control. Pulse-labeled cells were then recovered, washed, and processed for BrdU staining using the protocol specified in the BD Pharmingen BrdU Flow Kit (BD Life Sciences, San Jose, CA, USA). In brief, the cells were fixed, permeabilized, stained with anti-BrdU, and counter-stained with fluorescein isothiocyanate-conjugated (FITC) goat anti-mouse IgG1. Following 7-aminoactinomycin D (7-AAD) staining, cellular data were acquired using the BD FACSDiva software with the BD LSRFORTESSA (San Jose, CA, USA). Further analysis of the data was subsequently performed using FlowJo v.10 software (Ashland, OR, USA).
Cytofluorometric Analysis of Nuclear Apoptosis by TUNEL Assay
MIA PaCa-2 cells were treated with increasing concentrations of R9-caPeptide (0–100 μM, 0 as control) for 24 h, and nuclear apoptosis was assessed by TUNEL. This assay was performed using the In Situ Cell Death Detection Kit, TMR red kit (Roche, Basel, Switzerland) according to the manufacturer’s protocol. In brief, PBS-washed MIA PaCa-2 cells (2 × 107 cells/mL) were fixed with 2% paraformaldehyde (Millipore, Sigma, St. Louis, MO, USA) for 60 min at 15°C–25°C. Cells were washed twice in PBS followed by permeabilization with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice. After washing with PBS, cells were incubated with 50 μL TUNEL reaction mixture for 1 h at 37°C in a dark, humidified atmosphere. Terminal deoxynucleotidyl transferase (TdT) enzyme was not added to the negative control. All samples were washed twice in PBS prior to analysis. Cellular data were acquired using the BD FACSDiva software with the BD LSRFORTESSA (San Jose, CA, USA).
Western Blot Analysis
Pancreatic cancer cells were seeded in 100-mm tissue culture-treated culture dishes and treated with 50 μM R9-caPeptide. At the specified treatment time points (4, 24, and 48 h), cells were washed three times in ice-cold TBS (20 mM Tris [pH 7.6] and 137 mM NaCl) and scraped into TBS with 1× Thermo Scientific Halt phosphatase inhibitor cocktail, 1× Halt protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, USA), and 5 mM EDTA. Cells were pelleted in a swinging bucket centrifuge at 1,500 rpm for 5 min, the supernatant was removed, and the pellets were stored at −80°C prior to processing. Pellets were then sonicated in SDS buffer (50 mM Tris [pH 6.8], 2% SDS, 10% glycerol, 1× Thermo Scientific Halt phosphatase inhibitor cocktail, 1× Halt protease inhibitor cocktail, and 5 mM EDTA) at 30% amplitude in 5-s intervals until no longer viscous with the tubes in ice water. After heating at 95°C for 5 min and then cooling to room temperature, the total protein concentration was determined by DC Protein Assay (Bio-Rad, Hercules, CA, USA) with BSA as a standard. DTT and bromophenol blue were added to a final concentration of 90 mM DTT and ∼0.08% bromophenol blue, respectively. After resolving 0.025 mg protein extract for each lane by SDS-PAGE, it was transferred to a nitrocellulose membrane using the Pierce G2 Fast Blotter (Thermo Fisher Scientific, Waltham, MA, USA) for 5 min. The blots were blocked in 5% nonfat dried milk, TBS, and 0.05% Tween 20 for 1 h. Antibodies recognizing H2AX (Cell Signaling Technology, Beverly, MA, USA), phospho-histone H2AX Ser139 (Millipore, Burlington, MA, USA), and actin (Abcam, Cambridge, UK) were detected by ECL prime or AzureSpectra 800 secondary antibodies and imaged on the Azure c600 (Azure Biosystems, Dublin, CA, USA).
DNA Fiber Assay
DNA fiber assays were performed using a modified version of the technique described before.27 In brief, actively dividing MIA PaCa-2 cells (150,000 cells/well of a six-well dish) were pulse labeled with 100 μM CldU (Sigma-Aldrich, St. Louis, MO, USA) for 15 min at 37°C and 5% CO2 in complete media. The CldU was subsequently removed from the cells by washing three times with 1× PBS (Corning, Tewksbury, MA, USA). The cells were then pulse labeled with 200 μM IdU (Sigma-Aldrich, St. Louis, MO, USA) for 30 min in the presence of 0, 25, 50, or 100 μM R9-caPeptide at 37°C, 5% CO2. After labeling, the cells were washed three times with PBS and collected by trypsinization and centrifugation at 500 × g for 5 min. The pelleted cells were resuspended in PBS and counted with a Beckman Coulter Z2 particle counter (Brea, CA, USA). Two thousand cells were spread on a microscope slide (Leica, Wetzlar, Germany) and lysed by layering lysis buffer (0.5% SDS, 200 mM Tris-HCl [pH 7.4], 400 mM NaCl, 0.2% Nonidet P-40 [NP-40]) on the cells. Slides were made in quadruplicate for each experimental condition. After a 10-min lysis period, the slides were placed at an angle to allow the DNA fibers to spread down the slide. After 2 h the slides were fixed with 3:1 methanol/acetic acid and the DNA denatured with 2.5M HCl. Following a blocking step, the DNA fibers were hybridized with an antibody specific to CldU (Abcam, Cambridge, UK) (derived from rat) and an antibody specific to IdU (BD Biosciences, San Jose, CA, USA) (derived from mouse). The primary antibodies were then detected using secondary antibodies conjugated to a fluorophore: goat anti-rat IgG conjugated to Alexa Fluor 488 (Thermo Fisher Scientific, Waltham, MA, USA) and rabbit anti-mouse IgG conjugated to Alexa Fluor 594 (Thermo Fisher Scientific, Waltham, MA, USA). After washing, coverslips were mounted on the slides, and DNA fibers were imaged by fluorescent microscopy (Zeiss Observer II widefield light microscope; Carl Zeiss, Oberkochen, DE, USA). The ImageJ program (NIH, Bethesda, MD, USA) was used to measure green and red fluorescing lengths of DNA fibers. The fibers chosen for measurement were those with a clearly defined section of a green fluorescence followed immediately by a section of red fluorescence, and only fibers with similar lengths of green fluorescence (as judged typical for the experiment) were scored.
Statistical Analysis
For continuous endpoints, a two-sample t test was used. When comparing multiple groups, we used one-way ANOVA with Dunnett’s multiple comparison test. For survival data, the Kaplan-Meier method and the log-rank test were applied. Analyses were conducted using GraphPad Prism software (Version 8).
Author Contributions
Study Concept and Design: S.J.S., L.M., and M.R. Data Collection, Analysis, and Interpretation: all authors. Manuscript Preparation and Critical Revision: all authors. Final Approval of Manuscript: all authors.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors thank Supriya Deshpande, PhD, for assistance with manuscript editing. This work was supported in part by the American College of Surgeons—Research Fellowship, American Cancer Society Research Grant Pilot Project, and The City of Hope Young Innovator Award (to M.R.). In addition, this work was supported by research awards to L.H.M. from the Department of Defense, United States (W81XWH-11-1-0786); National Institutes of Health, United States, National Cancer Institute (R01 CA121289); St. Baldrick's Foundation, United States (https://www.stbaldricks.org/); the Alex Lemonade Stand Foundations (Rich Award), United States; and the ANNA Fund, United States (http://annafund.com/).
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Waddell N., Pajic M., Patch A.M., Chang D.K., Kassahn K.S., Bailey P., Johns A.L., Miller D., Nones K., Quek K., Australian Pancreatic Cancer Genome Initiative Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ying H., Dey P., Yao W., Kimmelman A.C., Draetta G.F., Maitra A., DePinho R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016;30:355–385. doi: 10.1101/gad.275776.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbas S. Molecular biology of adenocarcinoma of the pancreatic duct, current state and future therapeutic avenues. Surg. Oncol. 2013;22:69–76. doi: 10.1016/j.suronc.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Jones S., Zhang X., Parsons D.W., Lin J.C., Leary R.J., Angenendt P., Mankoo P., Carter H., Kamiyama H., Jimeno A. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loeb L.A. Human Cancers Express a Mutator Phenotype: Hypothesis, Origin, and Consequences. Cancer Res. 2016;76:2057–2059. doi: 10.1158/0008-5472.CAN-16-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conroy T., Hammel P., Hebbar M., Ben Abdelghani M., Wei A.C., Raoul J.L., Choné L., Francois E., Artru P., Biagi J.J., Canadian Cancer Trials Group and the Unicancer-GI–PRODIGE Group FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018;379:2395–2406. doi: 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 8.Von Hoff D.D., Ervin T., Arena F.P., Chiorean E.G., Infante J., Moore M., Seay T., Tjulandin S.A., Ma W.W., Saleh M.N. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paunesku T., Mittal S., Protić M., Oryhon J., Korolev S.V., Joachimiak A., Woloschak G.E. Proliferating cell nuclear antigen (PCNA): ringmaster of the genome. Int. J. Radiat. Biol. 2001;77:1007–1021. doi: 10.1080/09553000110069335. [DOI] [PubMed] [Google Scholar]
- 10.Krishna T.S., Kong X.P., Gary S., Burgers P.M., Kuriyan J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell. 1994;79:1233–1243. doi: 10.1016/0092-8674(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 11.Waga S., Hannon G.J., Beach D., Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 12.Ducoux M., Urbach S., Baldacci G., Hübscher U., Koundrioukoff S., Christensen J., Hughes P. Mediation of proliferating cell nuclear antigen (PCNA)-dependent DNA replication through a conserved p21(Cip1)-like PCNA-binding motif present in the third subunit of human DNA polymerase delta. J. Biol. Chem. 2001;276:49258–49266. doi: 10.1074/jbc.M106990200. [DOI] [PubMed] [Google Scholar]
- 13.Warbrick E., Lane D.P., Glover D.M., Cox L.S. Homologous regions of Fen1 and p21Cip1 compete for binding to the same site on PCNA: a potential mechanism to co-ordinate DNA replication and repair. Oncogene. 1997;14:2313–2321. doi: 10.1038/sj.onc.1201072. [DOI] [PubMed] [Google Scholar]
- 14.Chuang L.S., Ian H.I., Koh T.W., Ng H.H., Xu G., Li B.F. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997;277:1996–2000. doi: 10.1126/science.277.5334.1996. [DOI] [PubMed] [Google Scholar]
- 15.Levin D.S., McKenna A.E., Motycka T.A., Matsumoto Y., Tomkinson A.E. Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Curr. Biol. 2000;10:919–922. doi: 10.1016/s0960-9822(00)00619-9. [DOI] [PubMed] [Google Scholar]
- 16.Aaltomaa S., Lipponen P., Syrjänen K. Proliferating cell nuclear antigen (PCNA) immunolabeling as a prognostic factor in axillary lymph node negative breast cancer. Anticancer Res. 1993;13:533–538. [PubMed] [Google Scholar]
- 17.Chu J.S., Huang C.S., Chang K.J. Proliferating cell nuclear antigen (PCNA) immunolabeling as a prognostic factor in invasive ductal carcinoma of the breast in Taiwan. Cancer Lett. 1998;131:145–152. doi: 10.1016/s0304-3835(98)00118-9. [DOI] [PubMed] [Google Scholar]
- 18.Tahan S.R., Neuberg D.S., Dieffenbach A., Yacoub L. Prediction of early relapse and shortened survival in patients with breast cancer by proliferating cell nuclear antigen score. Cancer. 1993;71:3552–3559. doi: 10.1002/1097-0142(19930601)71:11<3552::aid-cncr2820711115>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 19.Sekowski J.W., Malkas L.H., Schnaper L., Bechtel P.E., Long B.J., Hickey R.J. Human breast cancer cells contain an error-prone DNA replication apparatus. Cancer Res. 1998;58:3259–3263. [PubMed] [Google Scholar]
- 20.Malkas L.H., Herbert B.S., Abdel-Aziz W., Dobrolecki L.E., Liu Y., Agarwal B., Hoelz D., Badve S., Schnaper L., Arnold R.J. A cancer-associated PCNA expressed in breast cancer has implications as a potential biomarker. Proc. Natl. Acad. Sci. USA. 2006;103:19472–19477. doi: 10.1073/pnas.0604614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu L., Smith S., Li C., Hickey R.J., Stark J.M., Fields G.B., Lang W.H., Sandoval J.A., Malkas L.H. A PCNA-derived cell permeable peptide selectively inhibits neuroblastoma cell growth. PLoS ONE. 2014;9:e94773. doi: 10.1371/journal.pone.0094773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith S.J., Hickey R.J., Malkas L.H. Validating the disruption of proliferating cell nuclear antigen interactions in the development of targeted cancer therapeutics. Cancer Biol. Ther. 2016;17:310–319. doi: 10.1080/15384047.2016.1139247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith S.J., Gu L., Phipps E.A., Dobrolecki L.E., Mabrey K.S., Gulley P., Dillehay K.L., Dong Z., Fields G.B., Chen Y.R. A Peptide mimicking a region in proliferating cell nuclear antigen specific to key protein interactions is cytotoxic to breast cancer. Mol. Pharmacol. 2015;87:263–276. doi: 10.1124/mol.114.093211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heller R.C., Marians K.J. Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell Biol. 2006;7:932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 25.Lau J.L., Dunn M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018;26:2700–2707. doi: 10.1016/j.bmc.2017.06.052. [DOI] [PubMed] [Google Scholar]
- 26.Sebaugh J.L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011;10:128–134. doi: 10.1002/pst.426. [DOI] [PubMed] [Google Scholar]
- 27.Frum R.A., Deb S., Deb S.P. Use of the DNA fiber spreading technique to detect the effects of mutant p53 on DNA replication. Methods Mol. Biol. 2013;962:147–155. doi: 10.1007/978-1-62703-236-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]