

Abstract
Merosin deficient Congenital Muscular Dystrophy (MDC1A), or LAMA2-related muscular dystrophy (LAMA2-RD), is a recessive disorder resulting from mutations in the LAMA2 gene, encoding for the alpha-2 chain of laminin-211. The disease is predominantly characterized by progressive muscular dystrophy affecting patient motor function and reducing life expectancy. However, LAMA2-RD also comprises a developmentally-associated dysmyelinating neuropathy that contributes to the disease progression, in addition to brain abnormalities; the latter often underappreciated. In this brief review, we present data supporting the impact of peripheral neuropathy in the LAMA2-RD phenotype, including both mouse models and human studies. We discuss the molecular mechanisms underlying nerve abnormalities and involved in the laminin-211 pathway, which affects axon sorting, ensheathing and myelination. We conclude with some final considerations of consequences on nerve regeneration and potential therapeutic strategies.
Keywords: LAMA2 gene, neuropathy, human, mousemodel, laminin
Introduction
LAMA2 encodes the α2 chains of the laminin-211 (also known as merosin), a major component of the basal lamina of Schwann cells and skeletal muscles (Ehrig et al., 1990). Indeed, loss of function mutations of the LAMA2 gene in humans, and the Lama2 gene in rodents, results in muscular dystrophy, dysmyelinating neuropathy, and brain abnormalities. This results in Merosin-deficient Congenital Muscular Dystrophy (MDC1A, OMIM #607855) also known as LAMA2-related muscular dystrophy (LAMA2-RD).
LAMA2 Neuropathy in Human Being
Shortly after the identification of the LAMA2 gene in 1995 (Helbling-Leclerc et al., 1995), abnormalities in the nerve conduction studies of children affected by LAMA2-RD were reported (Shorer et al., 1995). Over two decades later, the clinical significance and pathophysiology of such alterations are yet to be clarified.
The first neurophysiological studies conducted on genetically confirmed patients outlined the high prevalence of mild-to-moderate motor demyelinating neuropathy: deep peroneal nerve motor conduction velocity ranged from 27 to 42 m/s in patients older than 2 years (normal values >42 m/s; 43–57 m/s; Shorer et al., 1995). Although initial reports suggested that compound motor action potential (CMAP) amplitudes and sensory fibers were both preserved in LAMA2-RD, subsequent studies disproved these findings.
Neurophysiological evidence of demyelinating sensorimotor neuropathy can be present as early as 1–6 months of age. With growth, conduction velocities may progressively decrease (Mercuri et al., 1996; Quijano-Roy et al., 2004) along with a reduction of CMAP amplitudes, consistent with a combined axonal and demyelinating polyneuropathy (Brett et al., 1998; Fujii et al., 2011; Verma et al., 2018). Conversely, conduction blocks have not been reported in other studies (Di Muzio et al., 2003; Quijano-Roy et al., 2004; Verma et al., 2018). Although the presence of residual merosin in muscle usually correlates with a milder clinical phenotype and lesser muscle involvement, there is no proven relation with peripheral nerve damage. This may be due to either discordant expression of the laminin-α2 chain in the basement membrane surrounding myofibers and Schwann cells, or the role of compensatory tissue-specific laminin isoforms (see paragraphs below on animal models; Vainzof et al., 1995; Mora et al., 1996; Prelle et al., 1997; Di Muzio et al., 2003).
Muscle and skin biopsies of patients affected by LAMA2-RD display absence of laminin α2 in intramuscular motor nerves (a finding not observed in patients with secondary merosin deficiency), and in skin neural structures, respectively (Tomé et al., 1994; Hayashi et al., 1995; Osari et al., 1996; Marbini et al., 1997; Sewry et al., 1997; Chan et al., 2014).
Morphological data from sensory sural nerve biopsies have been scarcely described in the literature. The few studies available show a reduced number of fibers, especially those of larger caliber (>6–7 μm), and variable myelin diameter. In particular, both focally thickened myelin (tomacula like), and thinner and uncompacted myelin have been reported; the former predominantly in small fibers and possibly at paranodes. Associated findings were shorter internodes and wider nodes of Ranvier (>5 μm), suggesting a disorder in myelinogenesis that resembles murine models (Shorer et al., 1995; Mercuri et al., 1996; Deodatoa et al., 2002; Di Muzio et al., 2003; Quijano-Roy et al., 2004; North et al., 2014). Whilst demyelination and onion bulbs were not observed in sural nerve biopsies, post-mortem pathology of the cauda equina showed clear evidence of ongoing segmental demyelination and remyelination in one case (Hissong et al., 2016). Moreover, one case report described a marked reduction in the number of myelinated axons together with naked axons and increased collagen deposition on electron microscopy (Brett et al., 1998).
It is still not clear to which extent peripheral neuropathy contributes to muscle weakness in patients affected by LAMA2-RD. Absent deep tendon reflexes, distal muscle atrophy and weakness, neurophysiology, and neuropathology studies are consistent with a predominantly dysmyelinating sensory-motor polyneuropathy with some axonal involvement (Mora et al., 1996; Deodatoa et al., 2002; Di Muzio et al., 2003; Verma et al., 2018). However, studies are reporting the preservation of CMAP amplitudes and the absence of neurogenic changes on electromyography (EMG), suggesting that axonal degeneration may be negligible in some patients (Quijano-Roy et al., 2004). It is of course possible that neurogenic defects are somehow masked in these patients by the predominant muscular dystrophy phenotype, or that nerve involvement is prevalent in some mutation types.
Neurophysiology revealed reduced sensory action potentials (SAP) in a few cases (Di Muzio et al., 2003; Quijano-Roy et al., 2004). However, tactile sensation, proprioception, and vibration are usually normal (Chan et al., 2014) or mildly altered (Mora et al., 1996) at clinical assessment.
Overall, the main neuropathophysiological feature of LAMA2-RD seems to be an abnormal maturation of myelin sheets accompanied by segmental demyelination. In murine models, the secondary axonal loss has been extensively described; yet, this is less commonly reported in humans (Brett et al., 1998). Neuropathy could be still a potentially detrimental contributor to disease burden, particularly in patients with partial deficiency where the overall clinical picture is not overshadowed by severe muscle involvement. Finally, although we lack consistent data suggesting progressive axonal loss and clinically significant neuropathy in patients with LAMA2-RD, future therapies might reveal new phenotypes.
LAMA2 Neuropathy in Mouse Models
The prototype of LAMA2-RD was first reported in mice of the Bar Harbor 129 Re strain in 1955 (dystrophic mice Lama2dy/dy); this was however limited to the recessive inherited muscular dystrophy phenotype (Michelson et al., 1955). In the 1970s, nerve conduction studies showed functional abnormalities of the peripheral nerve (Papapetropoulos and Bradley, 1972; Bradley, 2008), which was confirmed by subsequent morphological studies. In 1971, Harris observed a reduced number of intramuscular myelinated fibers in Lama2dy/dy mice (Harris and Wilson, 1971). This data was confirmed and quantified in the tibialis nerve in 1972 (Harris et al., 1972), and further extended to other nerves and roots by Bradley and Jenkison (1973). Similar findings were described a year later in Lama2dy2J/dy2J (Biscoe et al., 1974). The genetic characterization of dystrophic mice was achieved two decades after this, when mutations of the Laminin chain α2 gene (Lama2) were reported (Xu et al., 1994; Sunada et al., 1995). Recently, mouse engineering by homologous recombination generated further mutants that have almost complete (Lama2dyW/dyW; Kuang et al., 1998) or a complete lack (Lama2dy3K/dy3K; Miyagoe et al., 1997) of laminin-211 expression. Finally, a further mutant was generated by ENU-induced point mutation C79R, called Lama2dynfm417/dynfm417 (Patton et al., 2008). This mouse mutant shows typical Lama2 muscle and nerve pathology in the presence of normal expression (but not function) of laminin-211. This is likely due to defective high-level organization (3D interactions between different laminin-211 heterotrimers; see Yurchenco and McKee, 2019) and missing interaction with other specific molecular partners.
All of these Lama2 mutants are characterized by progressive muscle wasting and consequent motor impairment, ranging from a less severe phenotype and almost normal lifespan of Lama2dy2J/dy2J (and likely Lama2dynfm417/dynfm417) to the most severe form Lama2dy3K/dy3K, which die within 3–4 weeks after birth. Peripheral nerves display typical morphological features that are considered prototypic abnormalities of Lama2 neuropathy. The morphological hallmark is the presence of defective axonal sorting during nerve development resulting in bundles of “naked” unsorted axons (Figures 1, 2 and Supplementary Table S1). This finding is spanning from spinal roots (more consistently) to peripheral nerves, including cranial nerves (Biscoe et al., 1974). These bundles contain several axons of mixed caliber, as well as those larger than 1 μm that should be sorted out and myelinated. Axons are tightly packed and often completely unsheathed or only partially surrounded by Schwann cell protrusions (Figure 3). Axonal sorting is a process that in rodents is completed within a few weeks, starting a couple of weeks before birth and ending around postnatal day 10 (P10). A similar process likely occurs in all vertebrates. In this process, immature Schwann cells surrounding bundles of mixed caliber axons start a physiological process of axon “docking” and “locking” to sort them out of the bundle, as well described and illustrated by Henry Webster in the 1970s and recently reviewed (Feltri et al., 2016). Only axons with a diameter larger than 1 μm are sorted, although the molecular mechanism that sustains their selection (docking) and engagement (locking) is still mostly unclear. In parallel, immature Schwann cells proliferate to match the axon number and to organize a continuous basal lamina around them (Webster et al., 1973; Jessen and Mirsky, 2005). In Lama2 models, many axon bundles are devoid of Schwann cell processes, although in other Schwann cell processes are visible between axons (Stirling, 1975; Yu et al., 2001; Yang et al., 2005). This also reflects the mouse age as they tend to reduce in size and number with age (at least in long-living Lama2dy2J/dy2J.mice), suggesting that radial sorting may last longer in mutant mice (Yang et al., 2005 and S. C. Previtali personal observation).
Figure 1.
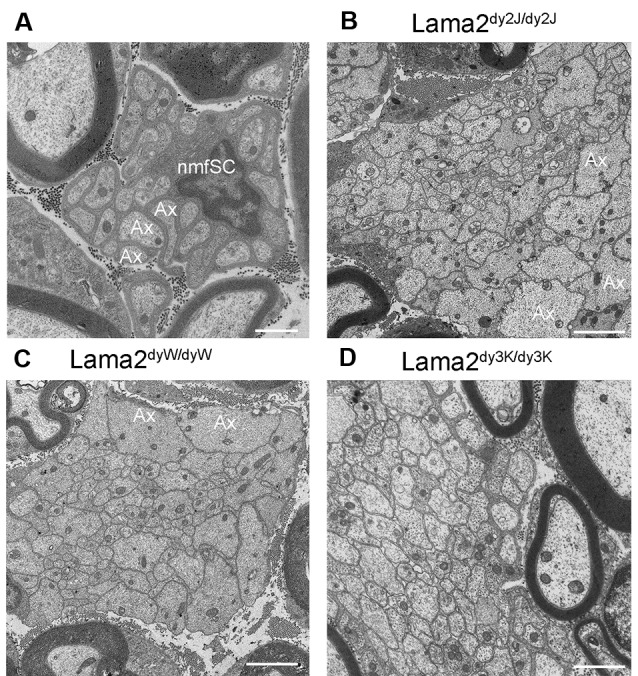
Bundle of unsorted axons in Lama2 mutants. (A) Normal Remak fiber, formed by a non-myelin forming Schwann cell (nmfSC) with unmyelinated axons (Ax); axons are smaller than 1 μm and are well separated and ensheathed by a single non-myelin forming Schwann cell. (B–D) Bundle of unsorted, non-ensheathed, and tightly packed axons of different caliber (some are larger than 1 μm, as Ax in figure B and C), are similarly present in Lama2dy2J/dy2J (B), Lama2dyW/dyW (C) and Lama2dy3K/dy3K (D) mice. Bar = 1 μm in (A), 2 μm in (B,C), 1 μm in (D).
Figure 2.

Schematic representation of unsorted axons in Lama2 nerves. Each axon along its length can be myelinated at one internode and belong to a bundle of unsorted axons in the subsequent one. The electron microscope (E.M.) constitutes a hypothetical cross-section of the nerve fibers in the scheme; unsorted axons are pseudo-colored in yellow, myelinated fibers in blue.
Figure 3.
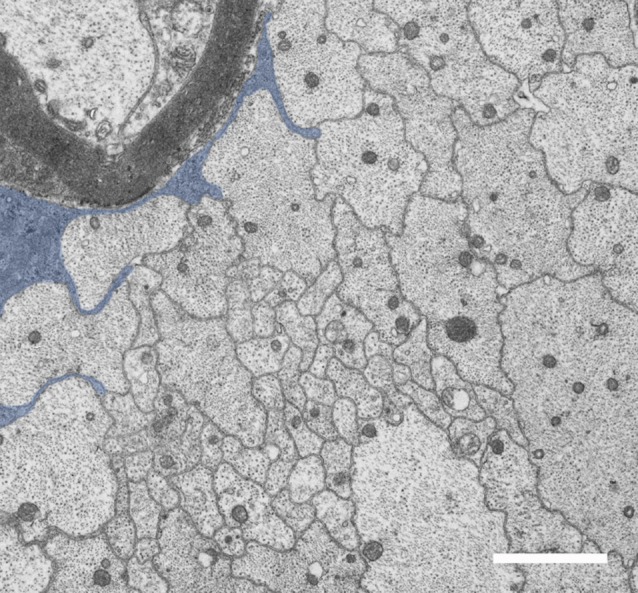
Lama2 Schwann cells fail to sort axons. Unsorted axons are surrounded by immature Schwann cells that fail to sort them. In this E.M. photograph from Lama2dy2J/dy2J sciatic nerve, the axon protrusions of one immature Schwann cell are pseudo-colored in blue. Bar = 1 μm.
Other nerve abnormalities include a reduced number of myelinated fibers as already reported in the original description of Lama2dy/dy mice, including ventral and dorsal roots, tibial and sciatic nerves (Harris et al., 1972; Bradley and Jenkison, 1973; Salafsky and Stirling, 1973). Axon diameters are significantly reduced in Lama2dy2J/dy2J (Gawlik et al., 2006) along with a reduced number of myelinated fibers in Lama2dy3K/dy3K mice (Yu et al., 2001). Occasional degenerating axons are described in Lama2dy/dy mice (Bradley and Jenkison, 1973).
Myelin thickness is reported normal or reduced in many myelinated fibers of Lama2dy/dy and Lama2dy2J/dy2J mice (Gawlik et al., 2006), in which few fibers (usually of small diameter) may have thicker myelin sheath (Ghidinelli et al., 2017). In Lama2dy3K/dy3K nerves, fibers are significantly thinner (Yu et al., 2001).
Nodes of Ranvier are reported wider in dystrophic Lama2dy/dy and Lama2dy2J/dy2J (Madrid et al., 1975; Jaros and Bradley, 1979; Occhi et al., 2005), whereas they are narrowed in Lama2dy3K/dy3K (Yu et al., 2001; Gawlik et al., 2006). Internodes are diffusely shorter in almost all the Lama2 subtypes (Court et al., 2009 and E. Porrello and S.C. Previtali, unpublished results).
Finally, sensory nerves are sometimes reported as morphologically less affected than motor nerves (Jaros and Jenkison, 1983), although our experience revealed similar findings in motor and sensory roots and nerves (S.C. Previtali, unpublished results).
With regards to other laminin isoforms described in peripheral nerves of Lama2 mice, the α1 chain (generating laminin-111) is not expressed in wild type nerves, whereas it is reported to be upregulated in sciatic nerves of Lama2dy2J/dy2J mice (Previtali et al., 2003b), but absent in Lama2dy3K/dy3K (Gawlik et al., 2006). The α4 chains, generating the main laminin isoform in embryonic nerve development laminin-411, is reported to be upregulated in the nerves of Lama2dy/dy, Lama2dy2J/dy2J and Lama2dy3K/dy3K mice (Patton et al., 1997; Ringelmann et al., 1999; Yu et al., 2001; Gawlik et al., 2006; Domi et al., 2015). The α5 chain (generating laminin-511) is also modestly upregulated in nerves of Lama2dy/dy, Lama2dy2J/dy2J and Lama2dy3K/dy3K mice (Patton et al., 1997; Ringelmann et al., 1999; Gawlik et al., 2006; Domi et al., 2015). Moreover, β1 and γ1 chains, necessary to generate all the laminin isoforms, are normally expressed in all the Lama2 mutants (Patton et al., 1997; Gawlik et al., 2006).
Accordingly, minor defects in radial sorting are described when Laminin-411 is deleted (Yang et al., 2005), whereas a severe defect is observed when both α2 and α4 chains are ablated (Yang et al., 2005). Similarly, targeted deletion of γ1 chain, impeding the formation of all the above-mentioned laminin isoforms (laminin-111, -211, -411 and -511) results in the most severe and widespread neuropathy characterized by complete lack of myelination and sorting defect (Chen and Strickland, 2003).
Molecular Mechanism of LAMA2 Neuropathy
Several laminin-211 receptors have been described so far in peripheral nerves, which activate downstream signaling pathways necessary for proper nerve development, function, and maintenance. Among them, the expression of integrins α6β1, α7β1, α6β4 and dystroglycan has been reported as timely and spatially regulated (Previtali et al., 2003b; Berti et al., 2006), and their effective function confirmed or denied by specifically targeted gene disruption. The G-protein-coupled receptor 126 (GPR126) has been more recently included in this list (Petersen et al., 2015).
Expression studies (Previtali et al., 2003b) showed that β1 integrins are the first ones expressed in nerve development, since early embryonic stages, likely associated with the α6 subunits. Dystroglycan appears later on, around the time of birth. Finally, α7β1 and α6β4 appear in post-natal development. Accordingly, the deletion of the integrin β1 chain resulted in severe neuropathy with axonal sorting defects and dysmyelination (Feltri et al., 2002), thus confirming the role of β1 integrins in this pathway and the pathogenesis of the neuropathy. The Schwann cell deletion of dystroglycan, instead, only mildly impacted nerve development (Saito et al., 2003; Berti et al., 2011), whereas the deletion of both β1 integrins and dystroglycan almost completely impaired axonal sorting and myelination (Berti et al., 2011). Not surprisingly, loss of β4 integrin did not affect nerve development, while it may confer stability to mature myelin in peripheral nerves (Nodari et al., 2008).
More intriguingly, neither loss of α6 nor α7 integrin chains in Schwann cells affected nerve development and function (Previtali et al., 2003a; Pellegatta et al., 2013). While the loss of α7 could be compensated by the presence of other (redundant) β1 integrins (likely α6β1) in post-natal development, it was unexpected that deletion of α6, expressed since early embryonic nerve development, did not affect nerve formation. It was shown that α7 is upregulated, thereby compensating for the absence of α6 during nerve development and that both α6 and α7 integrins should be deleted to impair the ability of Schwann cells to spread and bind laminin-211 or -411 (Pellegatta et al., 2013). However, double α6/α7 integrin mutants showed only a mild phenotype suggesting that other Schwann cell-β1 integrins might also contribute to radial sorting during peripheral nerve development (Pellegatta et al., 2013). Integrin β1 can couple with other α chains described in the peripheral nerve, such as α1, α2, α3, α4, α5, α8, α9, α10 and αv (Lefcort et al., 1992; Milner et al., 1997; Stewart et al., 1997; Previtali et al., 2003b). These β1-integrins bind other extracellular matrix components (i.e., collagen, fibronectin, vitronectin, tenascin, and partially laminins) and may participate in radial sorting of axons or partially compensate for the loss of α6 and α7 in mutant mice. Accordingly, loss of collagen XV aggravates radial sorting defects in laminin-411 null mice (Rasi et al., 2010).
Downstream signaling pathways originating from integrins and/or dystroglycan laminin-211 receptors have been widely, although not exhaustively, investigated. They involve molecules mainly regulating cytoskeleton rearrangement for proper cell polarization and formation of Schwann cell protrusions necessary for axon recognition, sorting, and ensheathment. Among others, they include integrin-linked kinase (ILK), focal adhesion kinase (FAK), Rho (Rac1, Cdc42) and Ras (RalA/B) -GTPases, Profilin, Merlin/neurofibromin (Nf2) and neuronal Wiskott-Aldrich syndrome protein (N-WASP; Benninger et al., 2007; Nodari et al., 2007; Pereira et al., 2009; Jin et al., 2011; Novak et al., 2011; Guo et al., 2013; Grove and Brophy, 2014; Montani et al., 2014; Ommer et al., 2019). Other laminin-211 downstream molecules are instead involved in regulating cell cycle and survival for proper matching of axon-Schwann cell units, such as Cdc42, FAK, Wingless-Integrated (Wnt)/α-catenin, and Jab1 (Benninger et al., 2007; Grove et al., 2007; Grigoryan et al., 2013; Porrello et al., 2014). Most of these molecules and associated pathways have been previously reviewed (Monk et al., 2015; Feltri et al., 2016).
More recently, a strict interaction between laminin-211 and neuregulin 1 type III (NRG1 III), the main signal for peripheral nerve myelination (Nave and Salzer, 2006), has been discovered (Ghidinelli et al., 2017). During nerve development, laminin-211 limits NRG1 III function through the inhibition of protein kinase A (PKA). Loss of laminin-211 would result in overactivation of the NRG1 III pathway resulting in defective radial sorting, inappropriate/premature myelination causing polyaxonal myelination or thicker myelin sheath (Ghidinelli et al., 2017). This would explain the occurrence of hypermyelinated small-caliber fibers in LAMA2 patients (Shorer et al., 1995; Di Muzio et al., 2003) and mice (Ghidinelli et al., 2017). Whether this effect is mediated by different laminin-211 receptors, and possibly α6α4, remains elusive (Heller et al., 2014).
Finally, studies in zebrafish and mouse mutants showed that GPR126 is required in Schwann cells for myelin expression (Monk et al., 2009) and radial sorting of axons (Monk et al., 2011; Mogha et al., 2013). GPR126 acts as a collagen IV and laminin-211 receptor (Paavola et al., 2014; Petersen et al., 2015), whose interaction promotes receptor cleavage into N-terminal fragment (NTF) and seven-transmembrane containing C-terminal fragment (CTF; Langenhan et al., 2013). The NTF fragment is necessary to guide radial sorting of axons and is generated by the interaction of GPR126 with a sort of (more) “immature” laminin-211 (i.e., low polymerization state), thus keeping the GPR126 receptor “inactive” for myelination (Petersen et al., 2015). Laminin-211 maturation (i.e., polymerization and interaction with other ECM components) switches to GPR126 in “active” state and through CTF can promote cAMP elevation, PKA activation, and thus myelination (Petersen et al., 2015).
Regeneration in LAMA2 Neuropathies
Intact Schwann cell basal lamina and correct formation of regenerating tracks of transdifferentiated Schwann cells (known as Bungner bands) is a prerequisite to preserve Schwann cell-axon interaction in successful nerve regeneration after damage (Jessen and Mirsky, 2019). Thus, matrix components of the basal lamina, such as laminin-211, would constitute key elements for nerve regeneration. Accordingly, the expression of laminin-211 and -411 (either as protein or mRNA) are upregulated after nerve damage (Wallquist et al., 2002). Moreover, laminin-211 is well known to promote neurite growth and nerve regeneration (Anton et al., 1994), even as a substrate of artificial nerve graft (Seo et al., 2013).
It is therefore not surprising that Lama2dy/dy Schwann cells provide a poor environment for neurite growth in vitro (Uziyel et al., 2000). Accordingly, nerve or spinal root damage in Lama2dy/dy mice resulted in defective axon regeneration and remyelination (Bray et al., 1983; Uziyel et al., 2000). Defective reinnervation was also observed in Lama2dy2J/dy2J mice (Parry and Melenchuk, 1981; S.C. Previtali and E. Porrello, unpublished results), and have been described in mice with conditional inactivation of the laminin γ1 chain, disrupting both laminin-211 and -411 (Chen and Strickland, 2003). Finally, the deletion of the laminin α4 chain did not affect nerve regeneration, suggesting that only laminin-211 (not -411) is necessary for the nerve to regenerate (Wallquist et al., 2005).
In conclusion, although there is no direct evidence in human LAMA2-RD patients, data from animal models suggest that defective nerve regeneration may contribute to the progression of LAMA2 neuropathy.
Laminin α2 Chain and Neuromuscular Junctions
At the basal lamina of neuromuscular junctions (NMJs), the laminin α2 chain assembles in trimers with β2 and γ1 forming laminin-221 (Sanes et al., 1990; Patton et al., 1997). Therein, other laminin isoforms are also present, including laminin-421 and -521 (Patton et al., 1997). These three laminin isoforms are essential in establishing and maintaining the structure of NMJs and the alignment of the presynaptic zone (Rogers and Nishimune, 2017). Thus, potentially, loss of laminin chain α2 might affect NMJ formation and function, contributing to the motor phenotype of LAMA2/Lama2.
There were no reports in the literature of NMJ abnormalities in LAMA2 patients, neither in terms of symptoms nor as neurophysiological findings typical of the myasthenic syndrome. Single fiber EMG was reported in one case and described as unremarkable (Chan et al., 2014). Lama2 mice have been investigated at NMJs. Lama2 mutants (at least Lama2dy/dy and Lama2dy2J/dy2J) develop smaller post-synaptic junctional folds, partial axon detachment and minor Schwann cell infiltration of the synaptic cleft (Gilbert et al., 1973; Banker et al., 1979; Law et al., 1983; Desaki et al., 1995). However, they normally assemble the presynaptic active zone and properly appose to the acetylcholine receptors (Gilbert et al., 1973). More severe effects on NMJs at pre and post-synaptic zone are instead a consequence of the deletion of laminin chain α4 and/or α5, and particularly in mice devoid of β2 chain (reviewed in Rogers and Nishimune, 2017).
Laminin chain α2 has been also described in the assembly and clustering of acetylcholine receptors, through the interaction with agrin, perlecan, and MuSK (Smirnov et al., 2005). However, acetylcholine receptors seem to be preserved in Lama2dy/dy and Lama2dy2J/dy2J mice (Banker et al., 1979) and are most likely regulated by laminin α4 and α5 chains (Nishimune et al., 2008).
Loss of laminin α2 in Lama2dy/dy does not affect α4 and α5 expression at the NMJ (Ringelmann et al., 1999). α4 and α5 was instead upregulated in Lama2dy3K/dy3K mice, which otherwise showed normal expression of other NMJ components such as neuronal cell adhesion molecule and utrophin (Miyagoe et al., 1997). Finally, NMJs of Lama2dyNmf417/dyNmf417 mice showed normal expression of laminin chain α2 (as well as other components) suggesting normal assembly and possibly function (Patton et al., 2008).
All these data suggest that loss of laminin-221 at the NMJ is mostly compensated by laminin-421 and -521, although it cannot be excluded that minor abnormalities described in Lama2 NMJs might contribute to the motor phenotype and axonal neuropathy in these mutants.
Potential Treatments for LAMA2/LAMA2 Neuropathy
Lack of the α2 chains of the laminin-211 in peripheral nerves is responsible for peripheral neuropathy in LAMA2 disorder. The obvious mechanism to repair this genetic defect would involve gene replacing and/or gene editing, still not feasible therapeutically so far. Gene replacement is mainly limited by the size of the LAMA2 gene (around 9 Kb), too large to be inserted in useful viral vectors. Gene editing, instead, has been successfully used to repair Lama2 mutations with the contemporary rescue of the peripheral neuropathy (Kemaladewi et al., 2017). Here, the main limitation is related to the off-target effects of the technique (Tsai et al., 2015).
Other strategies have been used to counteract or prevent the neuropathy. One major finding was the observation that the expression of α1 chain in peripheral nerves improved the neuropathy in Lama2dy2J/dy2J mice, including axonal sorting and myelination (Gawlik et al., 2006). This was recently confirmed by employing the CRISPR/Cas9 technology targeting the Lama1 gene promoter delivered by adeno-associated virus (AAV9; Kemaladewi et al., 2019). Laminin-111 protein was upregulated in muscle and nerves of Lama2dy2J/dy2J mice, and specifically, in peripheral nerve, it rescued myelination and nerve conduction velocities (Kemaladewi et al., 2019).
A further strategy for treating LAMA2/Lama2 disorder is the use of linker proteins mini-agrin and αLNNd. The first one was able to reconnect orphan laminin-211 receptors to the other laminin isoforms expressed in muscle and nerves and the second one to allow laminin polymerization (Yurchenco et al., 2018). These proteins are sufficiently small to be packed into AAV vectors. Accordingly, mini-agrin delivery with AAV9 was able to reach the peripheral nerve promoting the amelioration of axonal sorting and myelination in Lama2dyW/dyW treated mice (Qiao et al., 2018). Cell delivery of mini-agrin by mesoangioblasts showed instead efficacy in skeletal muscle but not in peripheral nerves, as these cells could not enter the endoneurium and were stopped in the perineurium of treated mice (Domi et al., 2015). Finally, αLNNd was proven to be effective in promoting myelination in the presence of non-polymerizing laminin isoforms (McKee et al., 2012).
Apoptosis was shown to play a role in the pathology of Lama2 mice (Girgenrath et al., 2004; Dominov et al., 2005), while doxycycline, as well as other tetracycline derivatives, had been reported to inhibit apoptosis in mammalian cells (Davies et al., 2005). Thus, doxycycline was investigated in Lama2dyW/dyW mutants where it showed amelioration of muscle and nerve pathology (Girgenrath et al., 2009; Homma et al., 2011). Although the doxycycline mechanism of action in nerves of Lama2 mutants remains vague, it might be linked to reduced cell death of immature Schwann cells and amelioration of Schwann cell differentiation (Homma et al., 2011). Moreover, is possible that doxycycline acts through different mechanisms in different tissues.
Glatiramer acetate (GA), an agent for immune modulation, has been shown to significantly improve mobility and muscle strength in the Lama2dy2J/dy2J mice (Dadush et al., 2010). Nerve conduction velocities were also reported significantly increased in these treated mice, suggesting a valuable effect of the drug on Lama2 neuropathy (Rabie et al., 2019).
Author Contributions
SP and AZ wrote the manuscript and prepared figures.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to Angelo Quattrini, Giorgia Dina, Paola Podini and Emanuela Porrello (IRCCS Ospedale San Raffaele) for E.M. images; to Markus Ruegg and Judith Reinhard (Biozentrum, University of Basel, Switzerland) for Lama2dyW/dyW nerve samples; and to Abdel-Mannan Omar for critical reading.
Footnotes
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2020.00060/full#supplementary-material.
References
- Anton E. S., Sandrock A. W., Jr., Matthew W. D. (1994). Merosin promotes neurite growth and schwann cell migration in vitro and nerve regeneration in vivo: evidence using an antibody to merosin, ARM-1. Dev. Biol. 164, 133–146. 10.1006/dbio.1994.1186 [DOI] [PubMed] [Google Scholar]
- Banker B. Q., Hirst N. S., Chester C. S., Fok R. Y. (1979). Histometric and electron cytochemical study of muscle in the dystrophic mouse. Ann. N Y Acad. Sci. 317, 115–131. 10.1111/j.1749-6632.1979.tb37331.x [DOI] [PubMed] [Google Scholar]
- Benninger Y., Thurnherr T., Pereira J. A., Krause S., Wu X., Chrostek-Grashoff A., et al. (2007). Essential and distinct roles for cdc42 and rac1 in the regulation of Schwann cell biology during peripheral nervous system development. J. Cell Biol. 177, 1051–1061. 10.1083/jcb.200610108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti C., Bartesaghi L., Ghidinelli M., Zambroni D., Figlia G., Chen Z. L., et al. (2011). Non-redundant function of dystroglycan and β 1 integrins in radial sorting of axons. Development 138, 4025–4037. 10.1242/dev.065490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti C., Nodari A., Wrabetz L., Feltri M. L. (2006). Role of integrins in peripheral nerves and hereditary neuropathies. Neuromolecular Med. 8, 191–204. 10.1385/nmm:8:1-2:191 [DOI] [PubMed] [Google Scholar]
- Biscoe T. J., Caddy K. W. T., Pallot D. J., Pehrson U. M. M., Stirling C. A. (1974). The neurological lesion in the dystrophic nouse. Brain Res. 76, 534–536. 10.1016/0006-8993(74)90830-0 [DOI] [PubMed] [Google Scholar]
- Bradley W. G. (2008). Nerve, muscle, and muscular dystrophy. Dev. Med. Child Neurol. 13, 528–531. 10.1111/j.1469-8749.1971.tb03062.x [DOI] [PubMed] [Google Scholar]
- Bradley W. G., Jenkison M. (1973). Abnormalities of peripheral nerves in murine muscular dystrophy. J. Neurol. Sci. 18, 227–247. 10.1016/0022-510x(73)90009-9 [DOI] [PubMed] [Google Scholar]
- Bray G. M., David S., Carlstedt T., Aguayo A. J. (1983). Effects of crush injury on the abnormalities in the spinal roots and peripheral nerves of dystrophic mice. Muscle Nerve 6, 497–503. 10.1002/mus.880060705 [DOI] [PubMed] [Google Scholar]
- Brett F. M., Costigan D., Farrell M. A., Heaphy P., Thornton J., King M. D. (1998). Merosin-deficient congenital muscular dystrophy and cortical dysplasia. Eur. J. Paediatr. Neurol. 2, 77–82. 10.1016/s1090-3798(98)80045-7 [DOI] [PubMed] [Google Scholar]
- Chan S. H. S., Foley A. R., Phadke R., Mathew A. A., Pitt M., Sewry C., et al. (2014). Limb girdle muscular dystrophy due to LAMA2 mutations: diagnostic difficulties due to associated peripheral neuropathy. Neuromuscul. Disord. 24, 677–683. 10.1016/j.nmd.2014.05.008 [DOI] [PubMed] [Google Scholar]
- Chen Z. L., Strickland S. (2003). Laminin γ1 is critical for Schwann cell differentiation, axon myelination, and regeneration in the peripheral nerve. J. Cell Biol. 163, 889–899. 10.1083/jcb.200307068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court F. A., Hewitt J. E., Davies K., Patton B. L., Uncini A., Wrabetz L., et al. (2009). A laminin-2, dystroglycan, utrophin axis is required for compartmentalization and elongation of myelin segments. J. Neurosci. 29, 3908–3919. 10.1523/JNEUROSCI.5672-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadush O., Aga-Mizrachi S., Ettinger K., Tabakman R., Elbaz M., Fellig Y., et al. (2010). Improved muscle strength and mobility in the dy2J/dy2J mouse with merosin deficient congenital muscular dystrophy treated with Glatiramer acetate. Neuromuscul. Disord. 20, 267–272. 10.1016/j.nmd.2010.02.002 [DOI] [PubMed] [Google Scholar]
- Davies J. E., Wang L., Garcia-Oroz L., Cook L. J., Vacher C., O’Donovan D. G., et al. (2005). Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat. Med. 11, 672–677. 10.1038/nm1242 [DOI] [PubMed] [Google Scholar]
- Deodatoa F., Sabatellib M., Ricci E., Mercurid E., Muntonid F., Sewryd C., et al. (2002). Hypermyelinating neuropathy, mental retardation and epilepsy in a case of merosin deficiency. Neuromuscul. Disord. 12, 392–398. 10.1016/s0960-8966(01)00312-1 [DOI] [PubMed] [Google Scholar]
- Desaki J., Matsuda S., Sakanaka M. (1995). Morphological changes of neuromuscular junctions in the dystrophic (dy) mouse: a scanning and transmission electron microscopic study. J. Electron Microsc. Tokyo. 44, 59–65. [PubMed] [Google Scholar]
- Di Muzio A., De Angelis M. V., Di Fulvio P., Ratti A., Pizzuti A., Stuppia L., et al. (2003). Dysmyelinating sensory-motor neuropathy in merosin-deficient congenital muscular dystrophy. Muscle Nerve 27, 500–506. 10.1002/mus.10326 [DOI] [PubMed] [Google Scholar]
- Domi T., Porrello E., Velardo D., Capotondo A., Biffi A., Tonlorenzi R., et al. (2015). Mesoangioblast delivery of miniagrin ameliorates murine model of merosin-deficient congenital muscular dystrophy type 1A. Skelet. Muscle 5:30. 10.1186/s13395-015-0055-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominov J. A., Kravetz A. J., Ardelt M., Kostek C. A., Beermann M. L., Miller J. B. (2005). Muscle-specific BCL2 expression ameliorates muscle disease in laminin α2-deficient, but not in dystrophin-deficient, mice. Hum. Mol. Genet. 14, 1029–1040. 10.1093/hmg/ddi095 [DOI] [PubMed] [Google Scholar]
- Ehrig K., Leivo I., Argraves W. S., Ruoslahti E., Engvall E. (1990). Merosin, a tissue-specific basement membrane protein, is a laminin-like protein. Proc. Natl. Acad. Sci. U S A 87, 3264–3268. 10.1073/pnas.87.9.3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri M. L., Poitelon Y., Previtali S. C. (2016). How schwann cells sort axons: new concepts. Neuroscientist 22, 252–265. 10.1177/1073858415572361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri M. L., Porta D. G., Previtali S. C., Nodari A., Migliavacca B., Cassetti A., et al. (2002). Conditional disruption of β1 integrin in Schwann cells impedes interactions with axons. J. Cell Biol. 156, 199–209. 10.1083/jcb.200109021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii Y., Sugiura C., Fukuda C., Maegaki Y., Ohno K. (2011). Sequential neuroradiological and neurophysiological studies in a Japanese girl with merosin-deficient congenital muscular dystrophy. Brain Dev. 33, 140–144. 10.1016/j.braindev.2010.02.003 [DOI] [PubMed] [Google Scholar]
- Gawlik K. I., Li J. Y., Petersén Å., Durbeej M. (2006). Laminin α1 chain improves laminin α2 chain deficient peripheral neuropathy. Hum. Mol. Genet. 15, 2690–2700. 10.1093/hmg/ddl201 [DOI] [PubMed] [Google Scholar]
- Ghidinelli M., Poitelon Y., Shin Y. K., Ameroso D., Williamson C., Ferri C., et al. (2017). Laminin 211 inhibits protein kinase A in Schwann cells to modulate neuregulin 1 type III-driven myelination. PLoS Biol. 15:e2001408. 10.1371/journal.pbio.2001408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J. J., Steinberg M. C., Banker B. Q. (1973). Ultrastructural alterations of the motor end plate in myotonic dystrophy of the mouse (dy2J/dy2J). J. Neuropathol. Exp. Neurol. 32, 345–364. 10.1097/00005072-197307000-00001 [DOI] [PubMed] [Google Scholar]
- Girgenrath M., Beermann M. L., Vishnudas V. K., Homma S., Miller J. B. (2009). Pathology is alleviated by doxycycline in a laminin-α2-null model of congenital muscular dystrophy. Ann. Neurol. 65, 47–56. 10.1002/ana.21523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girgenrath M., Dominov J. A., Kostek C. A., Miller J. B. (2004). Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J. Clin. Invest. 114, 1635–1639. 10.1172/jci22928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan T., Stein S., Qi J., Wende H., Garratt A. N., Nave K. A., et al. (2013). Wnt/Rspondin/β-catenin signals control axonal sorting and lineage progression in Schwann cell development. Proc. Natl. Acad. Sci. U S A 110, 18174–18179. 10.1073/pnas.1310490110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove M., Brophy P. J. (2014). FAK is required for schwann cell spreading on immature basal lamina to coordinate the radial sorting of peripheral axons with myelination. J. Neurosci. 34, 13422–13434. 10.1523/JNEUROSCI.1764-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove M., Komiyama N. H., Nave K. A., Grant S. G., Sherman D. L., Brophy P. J. (2007). FAK is required for axonal sorting by Schwann cells. J. Cell Biol. 176, 277–282. 10.1083/jcb.200609021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L., Moon C., Zheng Y., Ratner N. (2013). Cdc42 regulates schwann cell radial sorting and myelin sheath folding through NF2/merlin-dependent and independent signaling. Glia 61, 1906–1921. 10.1002/glia.22567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. B., Wilson P. (1971). Mechanical properties of dystrophic mouse muscle. J. Neurol. Neurosurg. Psychiatry 34, 512–520. 10.1136/jnnp.34.5.512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. B., Wallace C., Wing J. (1972). Myelinated nerve fibre counts in the nerves of normal and dystrophic mouse muscle. J. Neurol. Sci. 15, 245–249. 10.1016/0022-510x(72)90067-6 [DOI] [PubMed] [Google Scholar]
- Hayashi Y. K., Koga R., Tsukahara T., Ishii H., Matsuishi T., Yamashita Y., et al. (1995). Deficiency of laminin α2-Chain mRNA in muscle in a patient with merosin-negative congenital muscular dystrophy. Muscle Nerve 18, 1027–1030. 10.1002/mus.880180918 [DOI] [PubMed] [Google Scholar]
- Helbling-Leclerc A., Zhang X., Topaloglu H., Cruaud C., Tesson F., Weissenbach J., et al. (1995). Mutations in the laminin α2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet. 11, 216–218. 10.1038/ng1095-216 [DOI] [PubMed] [Google Scholar]
- Heller B. A., Ghidinelli M., Voelkl J., Einheber S., Smith R., Grund E., et al. (2014). Functionally distinct PI 3-kinase pathways regulate myelination in the peripheral nervous system. J. Cell Biol. 204, 1219–1236. 10.1083/jcb.201307057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hissong E., Salvatore S., Tanji K., Lavi E. (2016). Congenital muscular dystrophy, cardiomyopathy and peripheral neuropathy due to merosin deficiency: peripheral nerve histology of cauda equina. Hum. Pathol. Case Rep. 4, 4–8. 10.1016/j.ehpc.2015.06.003 [DOI] [Google Scholar]
- Homma S., Beermann M. L., Miller J. B. (2011). Peripheral nerve pathology, including aberrant Schwann cell differentiation, is ameliorated by doxycycline in a laminin-α2-deficient mouse model of congenital muscular dystrophy. Hum. Mol. Genet. 20, 2662–2672. 10.1093/hmg/ddr168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaros E., Bradley W. G. (1979). Atypical axon-schwann cell relationships in the common peroneal nerve of the dystrophic mouse: an ultrastructural study. Neuropathol. Appl. Neurobiol. 5, 133–147. 10.1111/j.1365-2990.1979.tb00666.x [DOI] [PubMed] [Google Scholar]
- Jaros E., Jenkison M. (1983). Quantitative studies of the abnormal axon-Schwann cell relationship in the peripheral motor and sensory nerves of the dystrophic mouse. Brain Res. 258, 181–196. 10.1016/0006-8993(83)91141-1 [DOI] [PubMed] [Google Scholar]
- Jessen K. R., Mirsky R. (2005). The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 6, 671–682. 10.1038/nrn1746 [DOI] [PubMed] [Google Scholar]
- Jessen K. R., Mirsky R. (2019). The success and failure of the schwann cell response to nerve injury. Front. Cell. Neurosci. 13:33. 10.3389/fncel.2019.00033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F., Dong B., Georgiou J., Jiang Q., Zhang J., Bharioke A., et al. (2011). N-WASp is required for Schwann cell cytoskeletal dynamics, normal myelin gene expression and peripheral nerve myelination. Development 138, 1329–1337. 10.1242/dev.058677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemaladewi D. U., Bassi P. S., Erwood S., Al-Basha D., Gawlik K. I., Lindsay K., et al. (2019). A mutation-independent approach for muscular dystrophy via upregulation of a modifier gene. Nature 572, 125–130. 10.1038/s41586-019-1430-x [DOI] [PubMed] [Google Scholar]
- Kemaladewi D. U., Maino E., Hyatt E., Hou H., Ding M., Place K. M., et al. (2017). Correction of a splicing defect in a mouse model of congenital muscular dystrophy type 1A using a homology-directed-repair-independent mechanism. Nat. Med. 23, 984–989. 10.1038/nm.4367 [DOI] [PubMed] [Google Scholar]
- Kuang W., Xu H., Vachon P. H., Engvall E. (1998). Disruption of the lama2 gene in embryonic stem cells: laminin α2 is necessary for sustenance of mature muscle cells. Exp. Cell Res. 241, 117–125. 10.1006/excr.1998.4025 [DOI] [PubMed] [Google Scholar]
- Langenhan T., Aust G., Hamann J. (2013). Sticky signalingxxxxxxadhesion class g protein-coupled receptors take the stage. Sci. Signal. 6:re3. 10.1126/scisignal.2003825 [DOI] [PubMed] [Google Scholar]
- Law P. K., Saito A., Fleischer S. (1983). Ultrastructural changes in muscle and motor end-plate of the dystrophic mouse. Exp. Neurol. 80, 361–382. 10.1016/0014-4886(83)90289-3 [DOI] [PubMed] [Google Scholar]
- Lefcort F., Venstrom K., McDonald J. A., Reichardt L. F. (1992). Regulation of expression of fibronectin and its receptor, α5β1, during development and regeneration of peripheral nerve. Development 116, 767–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid R. E., Jaros E., Cullen M. J., Bradley W. G. (1975). Genetically determined defect of Schwann cell basement membrane in dystrophic mouse. Nature 257, 319–321. 10.1038/257319a0 [DOI] [PubMed] [Google Scholar]
- Marbini A., Bellanova M. F., Ferrari A., Lodesani M., Gemignani F. (1997). Immunohistochemical study of merosin-negative congenital muscular dystrophy: laminin α2 deficiency in skin biopsy. Acta Neuropathol. 94, 103–108. 10.1007/s004010050680 [DOI] [PubMed] [Google Scholar]
- McKee K. K., Yang D. H., Patel R., Chen Z. L., Strickland S., Takagi J., et al. (2012). Schwann cell myelination requires integration of laminin activities. J. Cell Sci. 125, 4609–4619. 10.1242/jcs.107995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri E., Pennock J., Goodwin F., Sewry C., Cowan F., Dubowitz L., et al. (1996). Sequential study of central and peripheral nervous system involvement in an infant with merosin-deficient congenital muscular dystrophy. Neuromuscul. Disord. 6, 425–429. 10.1016/s0960-8966(96)00383-5 [DOI] [PubMed] [Google Scholar]
- Michelson A. N., Russell E. S., Pinckney J. (1955). Dystrophia muscularis: a hereditary primary myopathy in the house mouse. Proc. Natl. Acad. Sci. U S A 41, 1079–1084. 10.1073/pnas.41.12.1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner R., Wilby M., Nishimura S., Boylen K., Edwards G., Fawcett J., et al. (1997). Division of labor of Schwann cell integrins during migration on peripheral nerve extracellular matrix ligands. Dev. Biol. 185, 215–228. 10.1006/dbio.1997.8547 [DOI] [PubMed] [Google Scholar]
- Miyagoe Y., Hanaoka K., Nonaka I., Hayasaka M., Nabeshima Y., Arahata K., et al. (1997). Laminin α2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 415, 33–39. 10.1016/s0014-5793(97)01007-7 [DOI] [PubMed] [Google Scholar]
- Mogha A., Benesh A. E., Patra C., Engel F. B., Schöneberg T., Liebscher I., et al. (2013). Gpr126 functions in schwann cells to control differentiation and myelination via G-protein activation. J. Neurosci. 33, 17976–17985. 10.1523/JNEUROSCI.1809-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk K. R., Feltri M. L., Taveggia C. (2015). New insights on schwann cell development. Glia 63, 1376–1393. 10.1002/glia.22852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk K. R., Naylor S. G., Glenn T. D., Mercurio S., Perlin J. R., Dominguez C., et al. (2009). A G protein-coupled receptor is essential for schwann cells to initiate myelination. Science 325, 1402–1405. 10.1126/science.1173474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk K. R., Oshima K., Jörs S., Heller S., Talbot W. S. (2011). Gpr126 is essential for peripheral nerve development and myelination in mammals. Development 138, 2673–2680. 10.1242/dev.062224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montani L., Buerki-Thurnherr T., de Faria J. P., Pereira J. A., Dias N. G., Fernandes R., et al. (2014). Profilin 1 is required for peripheral nervous system myelination. Development 141, 1553–1561. 10.1242/dev.101840 [DOI] [PubMed] [Google Scholar]
- Mora M., Moroni I., Uziel G., di Blasi C., Barresi R., Farina L., et al. (1996). Mild clinical phenotype in a 12-year-old boy with partial merosin deficiency and central and peripheral nervous system abnormalities. Neuromuscul. Disord. 6, 377–381. 10.1016/0960-8966(96)00359-8 [DOI] [PubMed] [Google Scholar]
- Nave K.-A., Salzer J. L. (2006). Axonal regulation of myelination by neuregulin 1. Curr. Opin. Neurobiol. 16, 492–500. 10.1016/j.conb.2006.08.008 [DOI] [PubMed] [Google Scholar]
- Nishimune H., Valdez G., Jarad G., Moulson C. L., Müller U., Miner J. H., et al. (2008). Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J. Cell Biol. 182, 1201–1215. 10.1083/jcb.200805095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodari A., Previtali S. C., Dati G., Occhi S., Court F. A., Colombelli C., et al. (2008). xxx6xxx4 integrin and dystroglycan cooperate to stabilize the myelin sheath. J. Neurosci. 28, 6714–6719. 10.1523/JNEUROSCI.0326-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodari A., Zambroni D., Quattrini A., Court F. A., D’Urso A., Recchia A., et al. (2007). β1 integrin activates Rac1 in Schwann cells to generate radial lamellae during axonal sorting and myelination. J. Cell Biol. 177, 1063–1075. 10.1083/jcb.200610014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- North K. N., Wang C. H., Clarke N., Jungbluth H., Vainzof M., Dowling J. J., et al. (2014). Approach to the diagnosis of congenital myopathies. Neuromuscul. Disord. 24, 97–116. 10.1016/j.nmd.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak N., Bar V., Sabanay H., Frechter S., Jaegle M., Snapper S. B., et al. (2011). N-WASP is required for membrane wrapping and myelination by Schwann cells. J. Cell Biol. 192, 243–250. 10.1083/jcb.201010013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Occhi S., Zambroni D., Del Carro U., Amadio S., Sirkowski E. E., Scherer S. S., et al. (2005). Both laminin and Schwann cell dystroglycan are necessary for proper clustering of sodium channels at nodes of Ranvier. J. Neurosci. 25, 9418–9427. 10.1523/JNEUROSCI.2068-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ommer A., Figlia G., Pereira J. A., Datwyler A. L., Gerber J., DeGeer J., et al. (2019). Ral GTPases in schwann cells promote radial axonal sorting in the peripheral nervous system. J. Cell Biol. 218, 2350–2369. 10.1083/jcb.201811150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osari S. I., Kobayashi O., Yamashita Y., Matsuishi T., Goto M., Tanabe Y., et al. (1996). Basement membrane abnormality in merosin-negative congenital muscular dystrophy. Acta Neuropathol. 91, 332–336. 10.1007/s004010050433 [DOI] [PubMed] [Google Scholar]
- Paavola K. J., Sidik H., Zuchero J. B., Eckart M., Talbot W. S. (2014). Type IV collagen is an activating ligand for the adhesion G protein-coupled receptor GPR126. Sci. Signal. 7:ra76. 10.1126/scisignal.2005347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos T. A., Bradley W. G. (1972). Spinal motor neurones in murine muscular dystrophy and spinal muscular atrophy. A quantitative histological study. J. Neurol. Neurosurg. Psychiatry 35, 60–65. 10.1136/jnnp.35.1.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry D. J., Melenchuk S. (1981). Rate and extent of functional reinnervation in fast-twitch and slow-twitch muscles of the dystrophic mouse (C57Bl 6J dy2j dy2j). Exp. Neurol. 72, 446–461. 10.1016/0014-4886(81)90235-1 [DOI] [PubMed] [Google Scholar]
- Patton B. L., Miner J. H., Chiu A. Y., Sanes J. R. (1997). Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J. Cell Biol. 139, 1507–1521. 10.1083/jcb.139.6.1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton B. L., Wang B., Tarumi Y. S., Seburn K. L., Burgess R. W. (2008). A single point mutation in the LN domain of LAMA2 causes muscular dystrophy and peripheral amyelination. J. Cell Sci. 121, 1593–1604. 10.1242/jcs.015354 [DOI] [PubMed] [Google Scholar]
- Pellegatta M., De Arcangelis A., D’Urso A., Nodari A., Zambroni D., Ghidinelli M., et al. (2013). α6β1 and α7β1 integrins are required in Schwann cells to sort axons. J. Neurosci. 33, 17995–18007. 10.1523/JNEUROSCI.3179-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira J. A., Benninger Y., Baumann R., Gonçalves A. F., Özçelik M., Thurnherr T., et al. (2009). Integrin-linked kinase is required for radial sorting of axons and Schwann cell remyelination in the peripheral nervous system. J. Cell Biol. 185, 147–161. 10.1083/jcb.200809008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen S. C., Luo R., Liebscher I., Giera S., Jeong S. J., Mogha A., et al. (2015). The adhesion GPCR GPR126 Has distinct, domain-dependent functions in schwann cell development mediated by interaction with laminin-211. Neuron 85, 755–769. 10.1016/j.neuron.2014.12.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello E., Rivellini C., Dina G., Triolo D., Del Carro U., Ungaro D., et al. (2014). Jab1 regulates Schwann cell proliferation and axonal sorting through p27. J. Exp. Med. 211, 29–43. 10.1084/jem.20130720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prelle A., Comi G. P., Rigoletto C., Turconi A., Felisari G., Ciscato P., et al. (1997). An atypical case of partial merosin deficiency congenital muscular dystrophy. J. Neurol. 244, 391–395. 10.1007/s004150050110 [DOI] [PubMed] [Google Scholar]
- Previtali S. C., Dina G., Nodari A., Fasolini M., Wrabetz L., Mayer U., et al. (2003a). Schwann cells synthesize α7β1 integrin which is dispensable for peripheral nerve development and myelination. Mol. Cell. Neurosci. 23, 210–218. 10.1016/s1044-7431(03)00014-9 [DOI] [PubMed] [Google Scholar]
- Previtali S. C., Nodari A., Taveggia C., Pardini C., Dina G., Villa A., et al. (2003b). Expression of laminin receptors in Schwann cell differentiation: evidence for distinct roles. J. Neurosci. 23, 5520–5530. 10.1523/jneurosci.23-13-05520.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao C., Dai Y., Nikolova V. D., Jin Q., Li J., Xiao B., et al. (2018). Amelioration of muscle and nerve pathologyin LAMA2 muscular dystrophy by AAV9-Mini-Agrin. Mol. Ther. Methods Clin. Dev. 9, 47–56. 10.1016/j.omtm.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quijano-Roy S., Renault F., Romero N., Guicheney P., Fardeau M., Estournet B. (2004). EMG and nerve conduction studies in children with congenital muscular dystrophy. Muscle Nerve 29, 292–299. 10.1002/mus.10544 [DOI] [PubMed] [Google Scholar]
- Rabie M., Yanay N., Fellig Y., Konikov-Rozenman J., Nevo Y. (2019). Improvement of motor conduction velocity in hereditary neuropathy of LAMA2-CMD dy2J/dy2J mouse model by glatiramer acetate. Clin. Neurophysiol. 130, 1988–1994. 10.1016/j.clinph.2019.07.029 [DOI] [PubMed] [Google Scholar]
- Rasi K., Hurskainen M., Kallio M., Staven S., Sormunen R., Heape A. M., et al. (2010). Lack of collagen XV impairs peripheral nerve maturation and, when combined with laminin-411 deficiency, leads to basement membrane abnormalities and sensorimotor dysfunction. J. Neurosci. 30, 14490–14501. 10.1523/JNEUROSCI.2644-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringelmann B., Röder C., Hallmann R., Maley M., Davies M., Grounds M., et al. (1999). Expression of laminin α1, α2, α4 and α5 chains, fibronectin, and tenascin-C in skeletal muscle of dystrophic 129ReJ dy/dy mice. Exp. Cell Res. 246, 165–182. 10.1006/excr.1998.4244 [DOI] [PubMed] [Google Scholar]
- Rogers R. S., Nishimune H. (2017). The role of laminins in the organization and function of neuromuscular junctions. Matrix Biol. 57–58, 86–105. 10.1016/j.matbio.2016.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito F., Moore S. A., Barresi R., Henry M. D., Messing A., Ross-Barta S. E., et al. (2003). Unique role of dystroglycan in peripheral nerve myelination, nodal structure, and sodium channel stabilization. Neuron 38, 747–758. 10.1016/s0896-6273(03)00301-5 [DOI] [PubMed] [Google Scholar]
- Salafsky B., Stirling C. A. (1973). Altered neural protein in murine muscular dystrophy. Nat. New Biol. 246, 126–128. 10.1038/newbio246126a0 [DOI] [PubMed] [Google Scholar]
- Sanes J. R., Engvall E., Butkowski R., Hunter D. D. (1990). Molecular heterogeneity of basal laminae: isoforms of laminin and collagen IV at the neuromuscular junction and elsewhere. J. Cell Biol. 111, 1685–1699. 10.1083/jcb.111.4.1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S. Y., Min S. K., Bae H. K., Roh D., Kang H. K., Roh S., et al. (2013). A laminin-2-derived peptide promotes early-stage peripheral nerve regeneration in a dual-component artificial nerve graft. J. Tissue Eng. Regen. Med. 7, 788–800. 10.1002/term.1468 [DOI] [PubMed] [Google Scholar]
- Sewry C. A., D’Alessandro M., Wilson L. A., Sorokin L. M., Naom I., Bruno S., et al. (1997). Expression of laminin chains in skin in merosin-deficient congenital muscular dystrophy. Neuropediatrics 28, 217–222. 10.1055/s-2007-973703 [DOI] [PubMed] [Google Scholar]
- Shorer Z., Philpot J., Muntoni F., Sewry C., Dubowitz V. (1995). Demyelinating peripheral neuropathy in merosin-deficient congenital muscular dystrophy. J. Child Neurol. 10, 472–475. 10.1177/088307389501000610 [DOI] [PubMed] [Google Scholar]
- Smirnov S. P., Barzaghi P., McKee K. K., Ruegg M. A., Yurchenco P. D. (2005). Conjugation of LG domains of agrins and perlecan to polymerizing laminin-2 promotes acetylcholine receptor clustering. J. Biol. Chem. 280, 41449–41457. 10.1074/jbc.m508939200 [DOI] [PubMed] [Google Scholar]
- Stewart H. J., Turner D., Jessen K. R., Mirsky R. (1997). Expression and regulation of α1α1 integrin in Schwann cells. J. Neurobiol. 33, 914–928. 10.1002/(sici)1097-4695(199712)33:7<914::aid-neu4>3.0.co;2-b [DOI] [PubMed] [Google Scholar]
- Stirling C. A. (1975). Abnormalities in Schwann cell sheaths in spinal nerve roots of dystrophic mice. J. Anat. 119, 169–180. [PMC free article] [PubMed] [Google Scholar]
- Sunada Y., Bernier S. M., Utani A., Yamada Y., Campbell K. P. (1995). Identification of a novel mutant transcript of laminin α2 chain gene responsible for muscular dystrophy and dysmyelination in dy2J mice. Hum. Mol. Genet. 4, 1055–1061. 10.1093/hmg/4.6.1055 [DOI] [PubMed] [Google Scholar]
- Tomé F. M. S., Evangelista T., Leclerc A., Sunada Y., Manole E., Estournet B., et al. (1994). Congenital muscular dystrophy with merosin deficiency. C. R. Acad. Sci. III 317, 351–357. [PubMed] [Google Scholar]
- Tsai S. Q., Zheng Z., Nguyen N. T., Liebers M., Topkar V. V., Thapar V., et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197. 10.1038/nbt.3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziyel Y., Hall S., Cohen J. (2000). Influence of laminin-2 on Schwann cell-axon interactions. Glia 32, 109–121. 10.1002/1098-1136(200011)32:2<109::aid-glia10>3.0.co;2-1 [DOI] [PubMed] [Google Scholar]
- Vainzof M., Marie S. K. N., Reed U. C., Schwartzmann J. S., Pavanello R. C. M., Passos-Bueno M. R., et al. (1995). Deficiency of merosin (laminin M or α2) in congenital muscular dystrophy associated with cerebral white matter alterations. Neuropediatrics 26, 293–297. 10.1055/s-2007-979777 [DOI] [PubMed] [Google Scholar]
- Verma S., Goyal P., Guglani L., Peinhardt C., Pelzek D., Barkhaus P. E. (2018). COL6A and LAMA2 mutation congenital muscular dystrophy: a clinical and electrophysiological study. J. Clin. Neuromuscul. Dis. 19, 108–116. 10.1097/cnd.0000000000000198 [DOI] [PubMed] [Google Scholar]
- Wallquist W., Patarroyo M., Thams S., Carlstedt T., Stark B., Cullheim S., et al. (2002). Laminin chains in rat and human peripheral nerve: distribution and regulation during development and after axonal injury. J. Comp. Neurol. 454, 284–293. 10.1002/cne.10434 [DOI] [PubMed] [Google Scholar]
- Wallquist W., Plantman S., Thams S., Thyboll J., Kortesmaa J., Lännergren J., et al. (2005). Impeded interaction between Schwann cells and axons in the absence of laminin α4. J. Neurosci. 25, 3692–3700. 10.1523/jneurosci.5225-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster H. D. F., Martin J. R., O’Connell M. F. (1973). The relationships between interphase Schwann cells and axons before myelination: a quantitative electron microscopic study. Dev. Biol. 32, 401–416. 10.1016/0012-1606(73)90250-9 [DOI] [PubMed] [Google Scholar]
- Xu H., Wu X. R., Wewer U. M., Engvall E. (1994). Murine muscular dystrophy caused by a mutation in the laminin α2 (Lama2) gene. Nat. Genet. 8, 297–302. 10.1038/ng1194-297 [DOI] [PubMed] [Google Scholar]
- Yang D., Bierman J., Tarumi Y. S., Zhong Y. P., Rangwala R., Proctor T. M., et al. (2005). Coordinate control of axon defasciculation and myelination by laminin-2 and -8. J. Cell Biol. 168, 655–666. 10.1083/jcb.200411158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A. C. H., Wong H. K., Yung H. W., Lau L. T. (2001). Schwann cell myelination occurred without basal lamina formation in laminin α2 chain-null mutant (dy3K/dy3K) mice. Glia 35, 101–110. 10.1002/glia.1075 [DOI] [PubMed] [Google Scholar]
- Yurchenco P. D., McKee K. K. (2019). Linker protein repair of lama2 dystrophic neuromuscular basement membranes. Front. Mol. Neurosci. 12:305. 10.3389/fnmol.2019.00305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenco P. D., McKee K. K., Reinhard J. R., Rüegg M. A. (2018). Laminin-deficient muscular dystrophy: molecular pathogenesis and structural repair strategies. Matrix Biol. 71–72, 174–187. 10.1016/j.matbio.2017.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.