

Abstract
BACKGROUND
Mirabegron is a β3-adrenergic receptor (β3-AR) agonist approved only for the treatment of overactive bladder. Encouraging preclinical results suggest that β3-AR agonists could also improve obesity-related metabolic disease by increasing brown adipose tissue (BAT) thermogenesis, white adipose tissue (WAT) lipolysis, and insulin sensitivity.
METHODS
We treated 14 healthy women of diverse ethnicities (27.5 ± 1.1 years of age, BMI of 25.4 ± 1.2 kg/m2) with 100 mg mirabegron (Myrbetriq extended-release tablet, Astellas Pharma) for 4 weeks in an open-label study. The primary endpoint was the change in BAT metabolic activity as measured by [18F]-2-fluoro-d-2-deoxy-d-glucose (18F-FDG) PET/CT. Secondary endpoints included resting energy expenditure (REE), plasma metabolites, and glucose and insulin metabolism as assessed by a frequently sampled intravenous glucose tolerance test.
RESULTS
Chronic mirabegron therapy increased BAT metabolic activity. Whole-body REE was higher, without changes in body weight or composition. Additionally, there were elevations in plasma levels of the beneficial lipoprotein biomarkers HDL and ApoA1, as well as total bile acids. Adiponectin, a WAT-derived hormone that has antidiabetic and antiinflammatory capabilities, increased with acute treatment and was 35% higher upon completion of the study. Finally, an intravenous glucose tolerance test revealed higher insulin sensitivity, glucose effectiveness, and insulin secretion.
CONCLUSION
These findings indicate that human BAT metabolic activity can be increased after chronic pharmacological stimulation with mirabegron and support the investigation of β3-AR agonists as a treatment for metabolic disease.
TRIAL REGISTRATION
Clinicaltrials.gov NCT03049462.
FUNDING
This work was supported by grants from the Intramural Research Program of the NIDDK, NIH (DK075112, DK075116, DK071013, and DK071014).
Keywords: Endocrinology, Metabolism
Keywords: Adipose tissue, Cholesterol, Glucose metabolism

Introduction
The rising rates of obesity are a global health epidemic. In the US alone, by 2030 it is predicted that 86% of adults will be overweight or obese (1). Comorbidities include impaired glucose tolerance, dyslipidemia, hypertension, and a proinflammatory state (2). Addressing these problems with behavioral interventions alone has been largely unsuccessful, highlighting the need in many patients for adjunct therapy to maintain long-term improvements in obesity-related metabolic disease (3). One complementary approach has been pharmacological treatment to increase fatty acid and glucose oxidation. For several decades, an attractive target has been the β3-adrenergic receptor (β3-AR), whose activation in rodents leads to increased energy expenditure and improved glucose tolerance (4). The mechanism has been thought to include a combination of increased brown adipose tissue (BAT) thermogenesis and white adipose tissue (WAT) capacity for glucose uptake (4, 5).
Translation of the β3-AR rodent studies to humans has not been straightforward. Species differences in drug selectivity, oral bioavailability, and gene expression were limiting factors for achieving weight loss (6, 7). Nevertheless, early-phase clinical trials showed improved glucose tolerance (8) and increased fatty acid oxidation (9). Using the β3-AR agonist mirabegron (Myrbetriq, extended-release tablet, Astellas Pharma), which is approved for the treatment of overactive bladder, we recently showed that a single 200-mg dose stimulated BAT glucose uptake and WAT lipolysis and altered bile acid metabolism in healthy, lean men (7). To our knowledge, neither the acute nor the chronic effects of mirabegron on adult women have been examined, so the current study addressed the hypothesis that 4 weeks of treatment with mirabegron could increase BAT metabolic activity.
Results
Fourteen healthy young women (27.5 ± 1.1 years of age) of mixed ethnicity (5 Blacks, 8 Whites, 1 American Indian/Alaska Native; 1 Hispanic, 13 non-Hispanics) initiated the study (Figure 1A and Table 1). Each subject was treated for 4 weeks with a daily oral dose of 100 mg mirabegron (Figure 1B). This dosage is higher than the maximum approved dosage (50 mg daily) and was chosen because it was more likely to stimulate BAT (7). By day 14 of treatment, we presumed that the subjects’ β3-ARs were being stimulated continuously, as trough plasma concentrations of mirabegron were nearly at the Ki of 55 nM (10): 40.3 ± 5.0 and 45.2 ± 4.5 nM on days 14 and 28, respectively (Figure 2 and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI131126DS1). Upon completion of the trial (day 28), there were no changes in weight, fat mass, fat-free mass, or reported food intake (Table 1).
Figure 1. Flow diagram and study design.

(A) Flow diagram describing the numbers and disposition of the study subjects. (B) On day 0 and day 27, the subjects underwent metabolic testing consisting of a FSIGT test, a liver scan, an exercise tolerance test (VO2max), and DXA. The subjects then entered the metabolic chamber at 1800 hours and remained overnight and through 1400 hours on day 1 and day 28, after which they were transported to the PET/CT suite. Blood was drawn to measure metabolites before treatment on day 1 and day 28 at 0800 hours and then just prior to 18F-FDG administration in the chamber at 1300 hours. Mirabegron was administered at 0900 hours. The black bars above the diagrams refer to the 30-minute still periods during which the REE was measured. The black arrows indicate the time points at which blood was drawn for pharmacokinetic measurements.
Table 1. Anthropometric values at study initiation and completion.
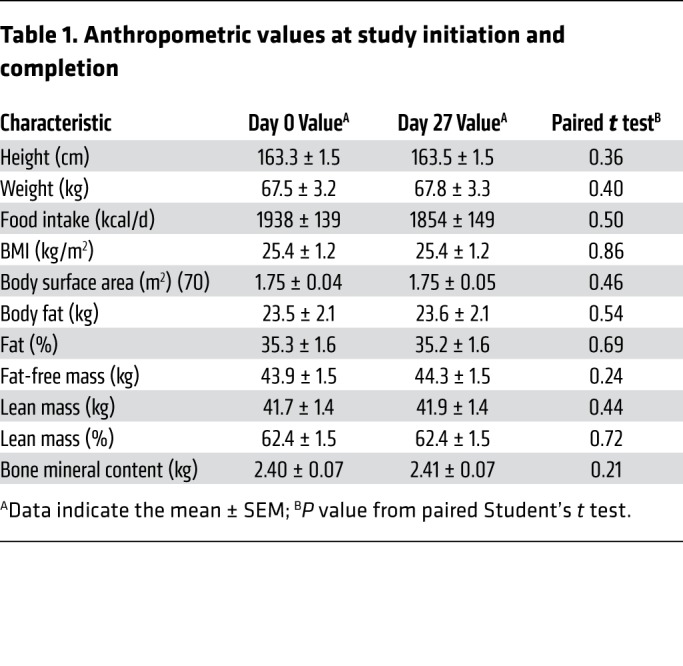
Figure 2. Mirabegron pharmacokinetics.

Plasma concentration of mirabegron during day 1 (black circles, dashed black line), on interim visit day 14 (blue triangle), and during day 28 (green squares, green line). The Ki of mirabegron is 55 nM. Values represent the mean ± SEM. n = 12. * P < 0.05, ** P < 0.01, and *** P < 0.001, by paired Student’s t test.
The prespecified primary endpoint, the subjects’ detectable BAT metabolic activity as measured via [18F]-2-fluoro-d-2-deoxy-d-glucose (18F-FDG) PET/CT, significantly increased (Figure 3, A and B, and Supplemental Figure 1), with a median of 195 to 473 mL•g/mL (P = 0.039). We observed similar proportional increases in BAT volume (Figure 3C), with a median of 72 to 149 mL (P = 0.036), and maximum metabolic activity (Figure 3D), with a median of 10 to 29 g/mL (P = 0.017). The extent of changes in BAT activity and volume were not the same across the group. The women who had less BAT on day 1 had larger increases than did those who started with more (R2 = 0.65 and 0.71, respectively, for activity and volume, both P < 0.001) (Supplemental Figure 2, A and B). These patterns suggest that chronic mirabegron treatment is particularly effective at increasing BAT activity in subjects who had little BAT before treatment, but there may also be an upper threshold in its efficacy. Of note, although daily outdoor temperatures have been found to affect the activity of BAT in response to cold stimulation (11), we found no relationship between the change in log10 BAT activity and the change in mean outdoor temperature, using linear regression analysis (R2 = 0.11, P = 0.35).
Figure 3. Effects of chronic mirabegron treatment on BAT, skeletal muscle, and scWAT.

(A) PET images on day 1 and day 28 for 2 representative subjects; magenta arrowheads point to the supraclavicular BAT depot with low (top) and high (bottom) initial BAT 18F-FDG uptake after an acute dose of mirabegron. Detectable BAT (B) metabolic activity and (C) volume in subjects on day 1 (black circles) and day 28 (black squares). Both y axes are shown using a log10 scale. P values correspond to the paired Student’s t test on the log10-transformed data, which was the prespecified analysis. SUVmax in (D) BAT, (E) erector spinae skeletal muscle, and (F) dorsolumbar scWAT from PET/CT scans of subjects performed on day 1 and day 28. Individual volume was measured on day 1 (black circles) and day 28 (black squares); red bars represent group medians for BAT and means for skeletal muscle and WAT. P values were determined using a paired Student’s t test. n = 14.
The PET/CT imaging also allowed us to measure metabolic activities of other tissues that can contribute to thermogenesis. In contrast to BAT, 18F-FDG uptake in erector spinae skeletal muscle was unchanged (–0.01 ± 0.05 g/mL, P = 0.77) and was lower in the dorsal-lumbar depot of subcutaneous WAT (scWAT) (–0.15 ± 0.04 g/mL, P = 0.006) (Figure 3, E and F). The reason for the assessment of scWAT glucose uptake in particular was to determine whether there had been a detectable increase in thermogenic adipocytes in this very large depot. Since we did not detect an increase in muscle or scWAT activity, it suggests that the major targets of chronic exposure to a β3-AR are the adipose depots previously identified (12) (Supplemental Figure 3).
Human thermogenic adipocytes can originate from 2 distinct lineages: constitutive “brown” adipocytes in the cervical and supraclavicular regions and recruitable “beige/brite” adipocytes in the supraclavicular and abdominal depots as well as other, smaller sites (13–18). Without biopsies, we were unable to make a direct distinction between these 2 cell types or determine whether the increased metabolic activity was due to hypertrophy or hyperplasia. Given the wide distribution of activation, it is likely that both brown and beige/brite adipocytes contributed to the higher metabolic activity (19). The increase was not uniformly distributed, with the mediastinal, paraspinal, and abdominal depots showing the largest gains (Supplemental Figure 4) (7).
To evaluate how mirabegron affected whole-body metabolism, we used a repeated-measures ANOVA to determine the effects of the day of study, the time, and their interaction on resting energy expenditure (REE) (Figure 4A and Supplemental Table 2). We observed a significant effect of both time of day (P < 0.001) and the interaction between the day of study and the time of day (P = 0.001): the initial dose of mirabegron on day 1 increased the REE by 10.7% (+6.4 ± 1.2 kcal/h, P < 0.001), yet the day-28 dose of mirabegron did not further increase the REE above the day-28 pre-dose baseline (0.8% = 0.5 ± 1.2 kcal/h, P = 0.70). However, the baseline REE (at 0800 hours) on day 28 was 5.8% higher than the baseline REE prior to drug exposure on day 1 (+82 kcal/d, P = 0.01).
Figure 4. Effects of chronic mirabegron treatment on REE and RQ.

Changes in (A) REE and (B) RQ in response to 100 mg oral mirabegron on day 1 (circles) and day 28 (squares) as measured in a metabolic chamber during 20-minute still periods at 0800 hours (white circles and squares) and 1300 hours (black circles and squares). Red bars represent group means. A repeated-measures ANOVA was used to determine the effects of the day of study, the time, and their interaction. n = 14.
The respiratory quotient (RQ) reflects the substrate source. The acute dose of mirabegron on both day 1 and day 28 lowered the RQ (–0.069 ± 0.007 and –0.051 ± 0.007, respectively; both P < 0.001) (Figure 4B and Supplemental Table 3), indicating a net increase in fat oxidation. In contrast to the results for REE, the baseline RQ on day 28 was not different from the RQ on day 1 (P = 0.15), and the P value for the interaction between the day of study and the time of day was higher (P = 0.06). These changes in REE and RQ were likely due to both prolonged fasting and mirabegron itself, but the lack of a placebo group precluded us from determining the contributions from each. We also measured energy expenditure and the RQ for the entire chamber stay, from evening until the next afternoon, and found no difference in either parameter: 74.4 ± 8.4 kcal/h to 75.0 ± 8.0 kcal/h (P = 0.57) and 0.850 ± 0.023 to 0.854 ± 0.025 (P = 0.51), respectively. There were also no changes in total sleep duration or sleep efficiency: 391.8 ± 76.6 minutes to 354.5 ± 65.3 minutes (P = 0.11), and 89.8% ± 8.0% to 91.5% ± 4.6% (P = 0.27), respectively. In addition, the average core temperature was 0.11°C ± 0.02°C higher on day 28 (Supplemental Figure 5).
A concern related to chronic treatment with adrenergic agonists is pathologic overstimulation of the cardiovascular system. Here, the resting heart rate (HR), systolic blood pressure (SBP), and rate-pressure product (RPP), a correlate of myocardial oxygen consumption (20), all increased acutely on day 1 in response to mirabegron, and baseline values were higher on day 28 (Supplemental Table 4). Since, to our knowledge, there is no published evidence yet to support direct binding of mirabegron to either the β1-AR or the β2-AR, these findings lend further support to the notion of a mechanism whereby mirabegron is taken up by sympathetic nerve terminals and then causes the release of norepinephrine to bind to cardiac β1-ARs (21). Such cardiovascular stimulation is similar to what we previously reported at a dose of 200 mg mirabegron in men (7, 22) and demonstrates why dosages higher than 50 mg are not used clinically for overactive bladder. We noted a diurnal variation in the change in HR such that mirabegron increased the HR more overnight than when the subjects were awake and moving: +6.2 ± 1.7 bpm between 1900 and 2200 hours versus +8.4 ± 1.5 bpm between 0 and 0300 hours (P < 0.001). None of the changes in HR or RPP correlated with the changes in REE (both P > 0.05, not shown). According to self-administered questionnaires, 2 subjects reported occasionally experiencing some of the anticipated mild treatment-emergent adverse effects (23): palpitations, headaches, bowel habit changes, and tachycardia during exercise; none of these effects were higher than grade 1. The dose of mirabegron on day 28 did not increase these cardiovascular parameters any further. The changes were not long-lasting: 2 weeks after stopping treatment, plasma mirabegron levels were 5.4 ± 0.1 nM, and HRs returned to the values seen at screening (Supplemental Figure 6). Mirabegron treatment had no effect on exercise tolerance, with no change in the maximal oxygen uptake (VO2max), maximum HR, or maximum wattage (Supplemental Figure 7, A–C). Chronic mirabegron treatment also did not lead to changes in measures of liver stiffness, liver steatosis, or gallbladder volume (Supplemental Figure 7, D–F).
The effects of acute and chronic mirabegron treatment on plasma metabolites and hormones could be separated into 3 distinct patterns (Supplemental Tables 5–8 and Supplemental Tables 11–13). Most metabolites demonstrated (a) no effect or changes resulting from fasting, independent of mirabegron. However, 1 subset showed (b) acute increases in nonesterified fatty acids (NEFAs) and β-hydroxybutyrate on day 28 that were blunted compared with changes on day 1. Plasma level increases of these metabolites were likely due to β3-AR stimulation of WAT. It remains to be determined whether chronic mirabegron treatment leads to reduced responsiveness of WAT or if there is increased consumption of the metabolites. The final pattern (c) of metabolite changes in response to chronic mirabegron treatment has the greatest potential to improve metabolic health. We observed increased fasting levels of the following: HDL (+8%), apolipoprotein A1 (ApoA1) (+12%), ApoE (+7%), total bile acids (+49%), total glucose-dependent insulinotropic polypeptide (GIP) (+31%), and adiponectin (+35%). In addition, we observed a reduction in the ApoB100/ApoA1 ratio, a biomarker of cardiovascular risk (24). We found higher levels of both total chenodeoxycholic acid (CDCA) (1003 ± 254 to 1641 ± 359 nM, P = 0.02) and the unconjugated form (212 ± 64 to 542 ± 159 nM, P = 0.02), the latter of which may be able to further boost BAT thermogenesis (25).
Without altering body weight, the initial clinical trials using non–FDA-approved β3-AR agonists provided some evidence for improved glucose tolerance (8) and insulin sensitivity (9). In addition, rodent studies showed that β3-AR–mediated activation of WAT can increase pancreatic β cell insulin secretion (26–28). To assess both glucose metabolism and β cell function in our study using mirabegron, we performed frequently sampled intravenous glucose tolerance (FSIGT) tests (Figure 5, A and B) and quantified metabolic parameters using Bergman’s minimal model (Supplemental Table 9) (29). Modeling showed that the parameters associated with insulin sensitivity, insulin-independent glucose metabolism, and insulin secretion all increased substantially (Figure 5, C–F). Whole-body insulin sensitivity (SI), reflecting insulin’s action in both stimulating glucose uptake and suppressing endogenous glucose production, increased by 36% (P = 0.026). Although SI includes the actions of insulin in both stimulating glucose uptake and suppressing endogenous glucose production, the effects on tissue glucose uptake are dominant (30). Glucose effectiveness (SG), the ability of glucose to mediate its own net disappearance, independent of the elevated insulin response, increased by 34% (P = 0.002).
Figure 5. FSIGT outcomes.

Plasma (A) glucose and (B) insulin responses during a FSIGT test on day 0 (circles, black line) and day 27 (squares, green line). Error bars indicate the SEM. The inset in A shows the levels from 0 to 10 minutes after injection of glucose. Individual changes in (C) SG, (D) whole-body SI, (E) AIRG, and (F) the DI. Red bars represent group means. Comparisons for each time point in A and B (* P < 0.05) and between day 0 (white circles) and day 27 (white squares) in C–F were made using a paired Student’s t test. n = 12.
Regarding β cell function, the acute insulin response to glucose (AIRG) increased by 37% (P = 0.039). The disposition index (DI), an overall measure of the ability of β cells to secrete insulin when normalized to the degree of insulin resistance, increased by 82% (P = 0.005). Despite the increases in both SI and AIRG, we found no relationship between the changes in these 2 measures for any of the subjects (P = 0.39). The change in homeostatic model assessment of insulin resistance (HOMA-IR), a measure of insulin resistance (31), was not significant after chronic mirabegron treatment (0.09 ± 0.81, P = 0.72).
Discussion
The labyrinthine paths toward developing pharmacological activators of β3-ARs and BAT thermogenesis to treat human metabolic disease have much in common. Both have been bolstered by decades of highly encouraging preclinical studies (4, 5, 8, 9), yet, to date, neither has achieved its goal. A major step forward for the β3-AR was the recent approval of mirabegron, the first highly selective agonist to treat overactive bladder (23). A single dose of mirabegron stimulates BAT glucose uptake (22), a marker of thermogenesis, along with increases in WAT lipolysis, REE, and changes in plasma bile acids (7). In this study, we report the physiological effects of chronic daily oral treatment with mirabegron. The numerous potentially beneficial metabolic responses now require determination of which responses are directly attributable to mirabegron and which are downstream of the initial effects.
Chronic mirabegron treatment increased BAT metabolic activity, the primary endpoint, and this approach now joins chronic cold exposure as another way to augment BAT thermogenesis (32–34). Two distinct facets of our findings relate to where and in whom BAT increased. The preferential amplification of the perirenal depots is particularly noteworthy, given the recent identification there of unilocular, dormant brown adipocytes (35). These cells express the β3-AR at much higher levels than are seen in scWAT and respond to treatment with adrenergic agonists by increasing their thermogenic capacity. In addition, these dormant cells have less sympathetic innervation, which could make them comparatively more responsive to pharmacological activation through the blood than through cold exposure, which relies on sympathetic neurons. Additional studies are needed to validate this model and determine whether it applies to the mediastinal and paraspinal depots as well.
The favorable changes in multiple lipoproteins and bile acids may also be connected to BAT activation. A potential mechanism comes from a series of recent studies in mice showing that when BAT is stimulated chronically, it consumes triglyceride-rich (TG-rich) lipoproteins and subsequently generates HDL cholesterol. HDL is then taken up and converted in the liver to bile acids, particularly via the alternative pathway that preferentially synthesizes CDCA (36–39). The plasma bile acids then bind to farnesoid X receptor (FXR) and G protein–coupled bile acid receptor 1 (TGR5) receptors and exert a combination of metabolically beneficial effects through the liver, intestine, microbiome, and mononuclear phagocytic system, including the production of incretins such as GIP (40, 41). Future studies are required to corroborate the mechanisms by which the observed changes in lipoproteins, adipokines, and bile acids are achieved and to conclusively demonstrate whether these effects represent new, physiologically relevant roles for BAT in human metabolism.
Besides effects on BAT, HDL cholesterol, and bile acids, mirabegron also produced substantial improvements in glucose and insulin metabolism. In context, the increases we observed in SG, SI, AIRG, and the DI, between 30% and 90%, were generally in the range of interventions associated with improvements in insulin sensitivity or pancreatic β cell function: an acute dose of intravenous GLP-1 (SI +4%–29%, AIRG +48%–66%) (42, 43); chronic metformin treatment in women with polycystic ovary syndrome (SG +30%, SI +2.5%, AIRG +43%, DI +72%) (44); 9–15 months after bariatric surgery (SI +116%, AIRG –56%) (45); and 6 weeks of mild exercise training in healthy adults (SG +28%, SI +32%) (46). The improvements in glucose metabolism with mirabegron treatment were not associated with changes in either fasting glucose or insulin. Similarly, intravenous GLP-1 (42, 43) and exercise (46) did not raise or lower either fasting glucose or insulin levels, whereas metformin lowered fasting glucose levels (44). Therefore, preliminary evidence suggests that the effects of mirabegron on glucose metabolism, HDL cholesterol, and bile acids resemble those achieved through mild exercise (47, 48).
As mentioned above, the changes in SI and AIRG were not correlated. This discordance may have been because mirabegron probably affects insulin sensitivity and β cell function via different mechanisms that are not directly connected. One reason for significant improvements in SI and SG, but not the HOMA-IR, may have been that the subjects here had near-normal HOMA-IR at study initiation (1.75 ± 0.69), and HOMA-IR does not provide a precise estimate of peripheral insulin action in this population (49). Any significant effects of mirabegron on glucose metabolism would therefore likely be detected only when glucose homeostasis was strongly perturbed, such as in the setting of the FSIGT test (50, 51).
How mirabegron improves glucose metabolism is not known. Contributions could have been from the higher plasma adiponectin (52), a WAT-derived adipokine that is associated with higher insulin sensitivity in skeletal muscle (53) and liver (54). The elevated levels of the incretin GIP can also improve glucose-stimulated insulin secretion (55). A third, parallel mechanism may involve the β cells themselves. Although we did not detect β3-AR mRNA in human islets (data not shown), there is evidence that WAT lipolysis (26, 27, 56), followed by activation of the β cell fatty acid receptor GPR40, can stimulate insulin release (57). In summary, beneficial metabolic changes caused by chronic mirabegron treatment may come from stimulation of the β3-AR on human BAT and WAT.
These findings must be considered in the context of the principal limitations of this study: it involved a small group of young, healthy women of diverse ethnicities with a narrow BMI range, and each was treated with active drug and served as her own control. Future studies using placebo controls are needed to better determine whether the changes in BAT and other physiological markers are dependent on mirabegron and how mirabegron contributed to the changes seen versus time and day. In addition, studies are needed in other populations, such as the elderly and patients with metabolic disease. Also, although the FSIGT test is a useful tool for initial inquiries into insulin sensitivity and β cell function, further studies are required to identify tissue sites of improved metabolism. Finally, the beneficial effects of mirabegron were seen at a dosage higher than that approved by the FDA, so the outcomes here cannot yet be applied to patients. In particular, the elevations in HR, SBP, and myocardial oxygen consumption with this dosage of mirabegron may confer too great a cardiovascular risk for clinical treatment of metabolic dysfunction in obese patients. However, there are other β3-AR agonists in late-stage clinical trials that may have greater β3/β1-AR selectivity (58, 59). Such drugs may improve metabolism with an acceptable side-effect profile.
In conclusion, we demonstrated that chronic treatment for 4 weeks with the β3-AR agonist mirabegron in healthy, young women with a range of BMIs led to increases in BAT metabolic activity, REE, plasma HDL and associated lipoproteins, insulin sensitivity, and pancreatic β cell insulin secretion. These metabolic benefits occurred without changes in the subjects’ weight, fat mass, or lean body mass. Therefore, chronic activation of the β3-AR may be an effective way to treat metabolic disease.
Methods
Sample size calculation.
Our previous studies showed that BAT activity is not normally distributed; rather, it is right skewed (7, 22). Thus, our sample-size determination was based on log10BAT activity. Since there were no published studies assessing the effects of mirabegron on log10BAT activity, we studied a similar cohort exposed to chronic cold exposure (32) and found changes in log10 BAT volume of +0.21 ± 0.25 mL. Since BAT volume correlates with BAT activity (12), we then determined that a sample size of 14 women was necessary to detect whether a change in BAT metabolic activity was different from a null hypothesis of 0.00 with 80% power using a paired Student’s t test with a significance level of 0.05 and a correlation (R) of 0.50. Treatment was nonblinded for both the staff and study participants.
Subject information and protocol design.
The inclusion criteria included generally healthy women between the age 18 and 40 years. Exclusion included were as described (https://clinicaltrials.gov/ct2/show/NCT03049462). Only women were recruited, because they were not included in our previous studies, which used a 200-mg mirabegron dose that can cause QT prolongation above 450 ms in women but not men (60). The protocol included a screening visit at which we conducted a medical history and physical examination, measured metabolites and hormones, and assessed the heart rhythm via ECG; 2 study visits (days 0–1 and days 27–28); and a follow-up safety visit to reassess HR and rhythm, inquire about any adverse effects, and measure plasma mirabegron, as described below and in Figure 1A. In practice, day 28 was 28 ± 0.4 days after day 0, and the follow-up safety visit was 16 ± 0.7 days after day 28. Mean daily temperatures in Bethesda, Maryland, for the 20 months of the study were obtained from the National Oceanic and Atmospheric Administration (https://www.ncdc.noaa.gov/cdo-web/search) and are displayed in Supplemental Figure 8 in relation to the subjects’ individual study days. When a specific day’s temperature was not available, we used the day that was closest in time to the actual study day.
Twenty-two subjects were enrolled in the study, but 1 was ineligible to participate, and 7 either withdrew or were lost to follow-up prior to initiation. All 14 subjects who initiated the study completed it. Four of the 14 subjects reported travel outside of the Bethesda, Maryland, area during the study period and could not be included in subsequent analyses related to outdoor temperature. Subjects were asked to keep consistent weight maintenance diets and exercise habits over the course of the study. All 14 subjects who initiated the study completed it. There was high daily adherence to medication administration, which was monitored using Nomi SMRxT Real-time medication adherence technology and also assessed by measuring trough concentrations of mirabegron 2 weeks and 4 weeks after study day 1.
Metabolic testing on day 0 and day 27.
Healthy volunteers who passed screening were admitted to the Metabolic Clinical Research Unit (MCRU) at the NIH Hatfield Clinical Research Center the evening before testing at initiation (day 0) and completion (day 27) of the study. The goal was to study all subjects on days 0–1 and days 27–28, on the same day of their menstrual cycles. In practice, the difference in days of the cycle between days 0 and 1 and days 27 and 28 was –0.6 ± 2.3 days. Of the 14 subjects, 12 were studied within the first 3 weeks of their menstrual cycles. During the evenings prior to the FSIGT test and the BAT imaging, the subjects ate a weight-maintenance, caffeine-free dinner of 716 ± 111 kcal with a macronutrient distribution of 55% carbohydrate, 15% protein, and 30% fat (61, 62). Prior to 0000 hours, the subjects were provided a high-carbohydrate, high-protein snack that was 251 ± 49 kcal. Inpatient rooms were kept at 24°C, and the subjects were asked to go to sleep at the same time each night. Clothing was standardized during each metabolic chamber stay as described previously (12).
The subjects fasted from 0000 hours until completion of testing later that day (day 0 and day 27). The subjects first underwent a FSIGT test from 0800–1100 hours. At time 0, the participants received an intravenous bolus of glucose (0.3 g/kg body weight). At 20 minutes, a bolus of insulin (0.03 U/kg body weight) was administered. Blood samples (~2 mL) for glucose and insulin were taken at –10, –1, 0, 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 14, 16, 20, 22, 23, 24, 25, 27, 30, 40, 50, 60, 70, 80, 90, 120, 150, and 180 minutes. Afterwards, the subjects met with a dietician to review food intake records and collect anthropometric measurements (neck, waist, hip, arm, and leg circumference and length) in triplicate. Liver stiffness and steatosis were assessed using vibration-controlled transient elastography (VCTE) and continuous attenuation parameter (CAP) measurements, respectively, with the FibroScan device (Echosens) and the M probe. Ten valid measurements were obtained, and the median result was used. Afterwards, the participants ate an isocaloric lunch and underwent ECG testing. At 1500 hours, the subjects performed a VO2max test to measure volitional fatigue on a cycle ergometer under a 15- to 20-W/min continuous ramp protocol. At 1700 hours, a dual-energy x-ray absorptiometry (DXA) scan was performed (iDXA, GE Healthcare). At 1800 hours, the subjects entered the metabolic chamber.
Quantification of metabolic activity, physiological, and clinical measurements.
The subjects stayed in the metabolic chamber for 20 hours (1800–1400 hours), during which energy expenditure and respiratory quotient were measured via indirect calorimetry from oxygen consumption and carbon dioxide production, and the ECG was recorded by a Holter monitor (Del Mar-Reynolds). Energy expenditure and RQ were calculated for the entire 20-hour chamber stay, and the first and last 30 minutes were excluded in determining the overall energy expenditure and RQ. At 1800 hours, the participants were provided an isocaloric dinner and snack; they fasted from 0 hours until exiting the chamber the following day (day 1 and day 28). Thirteen of the 14 enrolled subjects completed the full overnight stay in the metabolic chamber. One subject entered the metabolic chamber at 0800 hours on day 1 and day 28 instead of 1800 hours the prior night because of scheduling constraints. HR and blood pressure were determined as described previously(7, 63). Core temperature was measured by the participant using a handheld infrared tympanic thermometer (PRO4000, Braun).
The relative humidity of each chamber was maintained between 30% and 50%. The chamber’s temperature was set to 26°C to ensure that BAT was not activated by cold exposure. While in the chamber, the subjects were asked to minimize their physical activity and stay awake other than during their nightly sleep period. Sleep actigraphy was measured using triaxial accelerometers (Actigraph GT3X+, Actigraph) worn on the wrist. The Cole-Kripke algorithm implemented in ActiLife software (version 6.12.0) was used to detect time in bed, sleep onset, and awakenings and recorded total minutes in bed, total sleep time, and sleep efficiency, which is the ratio of the 2 (64).
Between 0825 and 0855 hours and 1310 and 1340 hours, the participants sat upright and still, without any physical activity. These inactive periods provided motion-free data to calculate REE and resting HR. The study subjects were administered 100 mg mirabegron at 0900 hours (+0 minutes). Blood samples used to measure blood metabolites were obtained 60 minutes prior to mirabegron dosing (0800 hours) and then 60 minutes prior to exiting the metabolic chamber (1300 hours). Blood samples used to test for plasma mirabegron concentrations were obtained at +0, +30, +60, +120, +180, +210, +240, +270, +300, and +360 minutes relative to the time of drug administration (0900 hours). Four hours after mirabegron administration (1300 hours), the subjects were injected with a 185-MBq (5-mCi) bolus of 18F-FDG for PET/CT scanning, after which they spent an additional 60 minutes inside the chamber with limited physical activity to allow for uptake of the radioactive tracer.
PET/CT images were acquired and analyzed as described previously (12, 65). In brief, PET/CT images were reconstructed into image voxels of 1.45 × 1.45 × 1.5 mm for PET and 0.98 × 0.98 × 1.5 mm for CT and uploaded into ImageJ (NIH) for image processing (66). The PET/CT Viewer plug-in with features customized for BAT quantification was used in each of the subsequent analyses. Specific CT density ranges were used to identify fat (–300 to –10 HU) from air and other tissues. 18F-FDG uptake (g/mL) in each PET image voxel was quantified as a standardized uptake value (SUV) initially normalized to the individual’s lean body mass. Both PET SUV and CT HU criteria were met to identify metabolically active adipose tissue.
BAT metabolic activity, defined as BAT volume multiplied by the SUV mean, where the SUV mean is the average radioactivity concentration in BAT divided by the injected 18F-FDG dose per body weight. BAT metabolic activity along with BAT volume; BAT, skeletal muscle, and scWAT SUVmax; and gallbladder volume were quantified as previously described (7, 12, 65). One region of interest (ROI) was created on each axial slice, avoiding regions that were not metabolically active fat in order to minimize false-positive detection. ROI selection began at the slice corresponding to vertebra C3 and continued inferiorly until the umbilicus. All axial ROIs were summed to calculate total body BAT volume and activity, and SUVs were averaged to determine the mean (SUVmean). The study parameters are also summarized in Supplemental Tables 10–13 according to the Brown Adipose Reporting Criteria in Imaging STudies (BARCIST 1.0) criteria (67).
Measurement of plasma mirabegron and individual bile acid concentrations via UPLC-MS/MS.
Detection and quantification of mirabegron were achieved by ultraperformance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS) utilizing a Thermo Scientific Vanquish UPLC with a Thermo Scientific ALTIS triple quadrupole mass spectrometer as described previously (7). Individual bile acid detection and quantification were achieved using a Thermo Scientific Vanquish UPLC with a Thermo Scientific ALTIS triple quadrupole mass spectrometer and heated electrospray ionization (HESI-II) in negative ion mode (2500 V). Bile acid quantitation and the internal standard were based on the retention time and m/z.
Calibration stock solutions and internal standard 2H4-CDCA (10 ng/nL) were prepared in MeOH and stored at 4°C. Calibration stock standards (50 μL) were mixed with 50 μL H2O and 150 μL internal standard for LC-MS analysis, and plasma samples were prepared by protein precipitation with 50 μL plasma mixed with 50 μL MeOH and 150 μL internal standard, vortexed, and then centrifuged at 4°C, 14,000 rpm for 15 minutes. The supernatant was transferred into a LC-MS vial. The injection volume was 2 μL.
HPLC-grade solvents and LC-MS modifiers were purchased from Sigma-Aldrich. A Waters Acquity UPLC BEH C18, 2.1 × 100 mm, 1.7-μm column was maintained at 40°C. Solvent A contained H2O, 5 mM NH4OAc with 0.012% FA, and solvent B contained MeOH, 5 mM NH4OAc with 0.012% FA. The flow rate was 400 μL/min, the gradient was started at 30% B from 0 minutes to 0.5 minutes, increased to 95% B at 12 minutes, maintained at 95% B to 14 minutes, and then returned to 30% B at 15 minutes to 18 minutes. Bile acid standards were analyzed, and the calibration curve had a minimum R2 of 0.99 or greater with 1/× weighting.
Measurement of metabolites, hormones, and adipokines.
Glucose, insulin, NEFAs, glucagon, growth hormone, total T3, free T4, thyroid-stimulating hormone (TSH), adrenocorticotropic hormone (ACTH), parathyroid hormone (PTH), total protein, creatine kinase, pyruvate, lactate, cortisol, norepinephrine, epinephrine, and dopamine were measured at the NIH’s Department of Laboratory Medicine. For epinephrine and dopamine concentrations that were below the assay detection limit, the half-minimum value was imputed. Lipid profile (total cholesterol, HDL cholesterol [HDL-C], LDL-C, and TGs), were measured using the Roche Cobas 6000 analyzer (Roche Diagnostics).
Metabolic proteins and hormones were measured at the NIDDK Clinical Core Lab. A colorimetric assay kit was used to measure β-hydroxybutyrate (Cayman Chemical). Total bile acids were measured in plasma using the NBT kit from Diazyme. FGF-19, leptin, and adiponectin were measured using Quantikine ELISA kits (R&D Systems). Active glucagon-like peptide 1 (GLP-1), active GIP, active ghrelin, total peptide YY (PYY), total GIP, and FGF21 were measured in plasma containing dipeptidyl peptidase IV (DPP-IV) and protease inhibitors from Sigma-Aldrich using immunoassay kits from Meso Scale Discovery. ApoA1, ApoE, ApoB100, and ApoC3 were measured in plasma samples using the MILLIPLEX Human Apolipoprotein Magnetic Bead Panel (MilliporeSigma) based on Luminex xMAP technology.
Human islet studies.
Islets from previously nondiabetic or type 2 diabetic human cadaver donor pancreases were obtained via the NIDDK-sponsored Integrated Islet Distribution Program as described before (68). mRNA was extracted and quantified via quantitative PCR as described previously (7).
Statistics.
After completing the study of 14 subjects with detectable BAT, data were analyzed with JMP 13.0.0 software (SAS Institute) and GraphPad Prism 7.0 (GraphPad Software). To evaluate the primary endpoint, a paired Student’s t test was applied to log-transformed data, which was the prespecified analysis. All other paired comparisons were performed using either a paired Student’s t test or a repeated-measures ANOVA, with the assumption that the underlying distributions were normal. Simple linear regression was used to determine how the difference in mean outdoor temperature between day 1 and day 28 affected log10 BAT activity. All P values were 2 tailed, with statistical significance set at a P value of 0.05 or less for comparisons associated with the anthropometric, pharmacokinetic, bioenergetic, and glucometabolic measures. For the behaviors of the 38 metabolites and hormones in Supplemental Table 5, the critical P value was determined using a Benjamini-Hochberg FDR of Q = 0.25 (69).
Study approval.
This clinical trial was registered with Clinicaltrials.gov (NCT03049462) and has the FDA Investigational New Drug registration number 116246. This study was approved by the Human Studies Institutional Review Board of the NIDDK and the NIH Radiation Safety Committee. Healthy volunteers were recruited by word of mouth or through the Patient Recruitment and Public Liaison Office of the Clinical Center and provided written informed consent according to Declaration of Helsinki principles.
Author contributions
AEOM, JWJ, JDL, RJB, ASB, KYC, and AMC were responsible for the experimental design. AEOM, JWJ, HC, PJW, and AMC wrote the manuscript. AEOM, JWJ, LAF, YSF, NK, ASB, BPL, CMM, WD, and PH quantified tissue metabolic activity. AEOM, JWJ, JDL, RJB, SM, LAF, TMC, KYC, and AMC performed the physiological measurements. DK and YR assessed liver fat and stiffness. HC, NBJ, and PJW measured plasma mirabegron and bile acid levels. MW developed and interpreted the assays of plasma metabolites. JWJ, FP, MA, RNB, and AMC performed and analyzed the FSIGT tests. CC and ZAS quantified human pancreatic β cell mRNA expression. JWJ and AMC performed the biostatistics. All authors contributed to editing of the manuscript. AEOM and JWJ contributed equally to this work; AEOM was primarily responsible for organizing and interpreting the clinical data and is therefore listed first.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the NIDDK, NIH (DK075112, DK075116, DK071013, and DK071014). We are grateful for the excellent support provided by the NIH Metabolic Clinical Research Unit nursing team; the NIH Clinical Center Nutrition Department; the NIDDK Clinical Laboratory Core Lab; the NIH Department of Laboratory Medicine; the research pharmacy; and the PET technologists. We thank Douglas Rosing for acting as the independent internal data safety monitor. We also wish to thank Wouter van Marken Lichtenbelt for sharing his team’s data so that we could perform our sample-size calculations; Sushil Rane and his laboratory for the gift of human islets; Paul Wakim and Sungyoung Auh for their biostatistical advice; Marc Reitman, Jack Yanovski, and Clifton Bogardus for their discussions about the data and their implications; Anne Sumner, Sushil Rane, and Gordon Weir for their guidance in interpreting the FSIGT data; and David Sacks and Jeff Basilio for their advice on measuring tissue glycemia. We are especially grateful to our volunteers for their commitment to this study.
Version 1. 01/21/2020
In-Press Preview
Version 2. 03/23/2020
Electronic publication
Version 3. 05/01/2020
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(5):2209–2219.https://doi.org/10.1172/JCI131126.
See the related Commentary at Might β3-adrenergic receptor agonists be useful in disorders of glucose homeostasis?.
Contributor Information
James W. Johnson, Email: jameswheelerjohnson@gmail.com.
Joyce D. Linderman, Email: lindermanj@niddk.nih.gov.
Robert J. Brychta, Email: brychtar@niddk.nih.gov.
Suzanne McGehee, Email: mcgeheems@niddk.nih.gov.
Laura A. Fletcher, Email: laura.a.fletcher@gmail.com.
Yael A. Fink, Email: yaelifink99@gmail.com.
Devika Kapuria, Email: devika.kapuria@gmail.com.
Thomas M. Cassimatis, Email: thomas.cassimatis@nih.gov.
Nathan Kelsey, Email: nk36363@gmail.com.
Cheryl Cero, Email: cheryl.cero@nih.gov.
Francesca Piccinini, Email: Francesca.Piccinini@cshs.org.
Alison S. Baskin, Email: alisonbaskin1@gmail.com.
Brooks P. Leitner, Email: brooksleitner@gmail.com.
Hongyi Cai, Email: hongyi.cai@nih.gov.
Corina M. Millo, Email: millocm@cc.nih.gov.
William Dieckmann, Email: WDieckmann@cc.nih.gov.
Mary Walter, Email: waltermf@niddk.nih.gov.
Norman B. Javitt, Email: norman.javitt@med.nyu.edu.
Yaron Rotman, Email: rotmany@niddk.nih.gov.
Peter J. Walter, Email: walterpj@niddk.nih.gov.
Marilyn Ader, Email: Marilyn.Ader@cshs.org.
Richard N. Bergman, Email: Richard.Bergman@cshs.org.
Peter Herscovitch, Email: PHerscovitch@cc.nih.gov.
Kong Y. Chen, Email: chenkong@niddk.nih.gov.
Aaron M. Cypess, Email: aaron.cypess@nih.gov.
References
- 1.Wang Y, Beydoun MA, Liang L, Caballero B, Kumanyika SK. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity (Silver Spring) 2008;16(10):2323–2330. doi: 10.1038/oby.2008.351. [DOI] [PubMed] [Google Scholar]
- 2.Heymsfield SB, Wadden TA. Mechanisms, pathophysiology, and management of obesity. N Engl J Med. 2017;376(15):1492. doi: 10.1056/NEJMc1701944. [DOI] [PubMed] [Google Scholar]
- 3.American College of Cardiology/American Heart Association Task Force on Practice Guidelines, Obesity Expert Panel, 2013 Expert Panel Report: Guidelines (2013) for the management of overweight and obesity in adults. Obesity (Silver Spring) 2014;22 Suppl 2:S41–410. doi: 10.1002/oby.20821. [DOI] [PubMed] [Google Scholar]
- 4.Liu X, Pérusse F, Bukowiecki LJ. Mechanisms of the antidiabetic effects of the beta 3-adrenergic agonist CL-316243 in obese Zucker-ZDF rats. Am J Physiol. 1998;274(5):R1212–R1219. doi: 10.1152/ajpregu.1998.274.5.R1212. [DOI] [PubMed] [Google Scholar]
- 5.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84(1):277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 6.Arch JR. Challenges in β(3)-adrenoceptor agonist drug development. Ther Adv Endocrinol Metab. 2011;2(2):59–64. doi: 10.1177/2042018811398517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baskin AS, et al. Regulation of human adipose tissue activation, gallbladder size, and bile acid metabolism by a β3-adrenergic receptor agonist. Diabetes. 2018;67(10):2113–2125. doi: 10.2337/db18-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cawthorne MA, Sennitt MV, Arch JR, Smith SA. BRL 35135, a potent and selective atypical beta-adrenoceptor agonist. Am J Clin Nutr. 1992;55(1 Suppl):252S–257S. doi: 10.1093/ajcn/55.1.252s. [DOI] [PubMed] [Google Scholar]
- 9.Weyer C, Tataranni PA, Snitker S, Danforth E, Ravussin E. Increase in insulin action and fat oxidation after treatment with CL 316,243, a highly selective beta3-adrenoceptor agonist in humans. Diabetes. 1998;47(10):1555–1561. doi: 10.2337/diabetes.47.10.1555. [DOI] [PubMed] [Google Scholar]
- 10.Vrydag W, Alewijnse AE, Michel MC. Do gene polymorphisms alone or in combination affect the function of human beta3-adrenoceptors? Br J Pharmacol. 2009;156(1):127–134. doi: 10.1111/j.1476-5381.2008.00014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senn JR, et al. Outdoor temperature influences cold induced thermogenesis in humans. Front Physiol. 2018;9:1184. doi: 10.3389/fphys.2018.01184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leitner BP, et al. Mapping of human brown adipose tissue in lean and obese young men. Proc Natl Acad Sci USA. 2017;114(32):8649–8654. doi: 10.1073/pnas.1705287114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150(2):366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharp LZ, et al. Human BAT possesses molecular signatures that resemble beige/brite cells. PLoS ONE. 2012;7(11):e49452. doi: 10.1371/journal.pone.0049452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lidell ME, et al. Evidence for two types of brown adipose tissue in humans. Nat Med. 2013;19(5):631–634. doi: 10.1038/nm.3017. [DOI] [PubMed] [Google Scholar]
- 16.Cypess AM, et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med. 2013;19(5):635–639. doi: 10.1038/nm.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jespersen NZ, et al. A classical brown adipose tissue mRNA signature partly overlaps with brite in the supraclavicular region of adult humans. Cell Metab. 2013;17(5):798–805. doi: 10.1016/j.cmet.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 18.Sacks HS, et al. Adult epicardial fat exhibits beige features. J Clin Endocrinol Metab. 2013;98(9):E1448–E1455. doi: 10.1210/jc.2013-1265. [DOI] [PubMed] [Google Scholar]
- 19.Finlin BS, et al. Human adipose beiging in response to cold and mirabegron. JCI Insight. 2018;3(15):121510. doi: 10.1172/jci.insight.121510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gobel FL, Norstrom LA, Nelson RR, Jorgensen CR, Wang Y. The rate-pressure product as an index of myocardial oxygen consumption during exercise in patients with angina pectoris. Circulation. 1978;57(3):549–556. doi: 10.1161/01.CIR.57.3.549. [DOI] [PubMed] [Google Scholar]
- 21.Mo W, Michel MC, Lee XW, Kaumann AJ, Molenaar P. The β3 -adrenoceptor agonist mirabegron increases human atrial force through β1-adrenoceptors: an indirect mechanism? Br J Pharmacol. 2017;174(16):2706–2715. doi: 10.1111/bph.13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cypess AM, et al. Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell Metab. 2015;21(1):33–38. doi: 10.1016/j.cmet.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sacco E, Bientinesi R. Mirabegron: a review of recent data and its prospects in the management of overactive bladder. Ther Adv Urol. 2012;4(6):315–324. doi: 10.1177/1756287212457114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandhu PK, et al. Lipoprotein biomarkers and risk of cardiovascular disease: a laboratory medicine best practices (LMBP) systematic review. J Appl Lab Med. 2016;1(2):214–229. doi: 10.1373/jalm.2016.021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broeders EP, et al. The bile acid chenodeoxycholic acid increases human brown adipose tissue activity. Cell Metab. 2015;22(3):418–426. doi: 10.1016/j.cmet.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Grujic D, et al. Beta3-adrenergic receptors on white and brown adipocytes mediate beta3-selective agonist-induced effects on energy expenditure, insulin secretion, and food intake. A study using transgenic and gene knockout mice. J Biol Chem. 1997;272(28):17686–17693. doi: 10.1074/jbc.272.28.17686. [DOI] [PubMed] [Google Scholar]
- 27.Gavrilova O, Marcus-Samuels B, Reitman ML. Lack of responses to a beta3-adrenergic agonist in lipoatrophic A-ZIP/F-1 mice. Diabetes. 2000;49(11):1910–1916. doi: 10.2337/diabetes.49.11.1910. [DOI] [PubMed] [Google Scholar]
- 28.Heine M, et al. Lipolysis triggers a systemic insulin response essential for efficient energy replenishment of activated brown adipose tissue in mice. Cell Metab. 2018;28(4):644–655.e4. doi: 10.1016/j.cmet.2018.06.020. [DOI] [PubMed] [Google Scholar]
- 29.Boston RC, Stefanovski D, Moate PJ, Sumner AE, Watanabe RM, Bergman RN. MINMOD Millennium: a computer program to calculate glucose effectiveness and insulin sensitivity from the frequently sampled intravenous glucose tolerance test. Diabetes Technol Ther. 2003;5(6):1003–1015. doi: 10.1089/152091503322641060. [DOI] [PubMed] [Google Scholar]
- 30.Bergman RN, Prager R, Volund A, Olefsky JM. Equivalence of the insulin sensitivity index in man derived by the minimal model method and the euglycemic glucose clamp. J Clin Invest. 1987;79(3):790–800. doi: 10.1172/JCI112886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muniyappa R, Lee S, Chen H, Quon MJ. Current approaches for assessing insulin sensitivity and resistance in vivo: advantages, limitations, and appropriate usage. Am J Physiol Endocrinol Metab. 2008;294(1):E15–E26. doi: 10.1152/ajpendo.00645.2007. [DOI] [PubMed] [Google Scholar]
- 32.van der Lans AA, et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest. 2013;123(8):3395–3403. doi: 10.1172/JCI68993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoneshiro T, et al. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest. 2013;123(8):3404–3408. doi: 10.1172/JCI67803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanssen MJ, et al. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat Med. 2015;21(8):863–865. doi: 10.1038/nm.3891. [DOI] [PubMed] [Google Scholar]
- 35.Jespersen NZ, et al. Heterogeneity in the perirenal region of humans suggests presence of dormant brown adipose tissue that contains brown fat precursor cells. Mol Metab. 2019;24:30–43. doi: 10.1016/j.molmet.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartelt A, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17(2):200–205. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- 37.Berbée JF, et al. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nat Commun. 2015;6:6356. doi: 10.1038/ncomms7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dijk W, et al. ANGPTL4 mediates shuttling of lipid fuel to brown adipose tissue during sustained cold exposure. Elife. 2015;4:e08428. doi: 10.7554/eLife.08428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Worthmann A, et al. Cold-induced conversion of cholesterol to bile acids in mice shapes the gut microbiome and promotes adaptive thermogenesis. Nat Med. 2017;23(7):839–849. doi: 10.1038/nm.4357. [DOI] [PubMed] [Google Scholar]
- 40.Chávez-Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology. 2017;152(7):1679–1694.e3. doi: 10.1053/j.gastro.2017.01.055. [DOI] [PubMed] [Google Scholar]
- 41.Kuhre RE, et al. Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol Metab. 2018;11:84–95. doi: 10.1016/j.molmet.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan TM, et al. Combination of peptide YY3-36 with GLP-1(7-36) amide causes an increase in first-phase insulin secretion after IV glucose. J Clin Endocrinol Metab. 2014;99(11):E2317–E2324. doi: 10.1210/jc.2014-2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D’Alessio DA, Kahn SE, Leusner CR, Ensinck JW. Glucagon-like peptide 1 enhances glucose tolerance both by stimulation of insulin release and by increasing insulin-independent glucose disposal. J Clin Invest. 1994;93(5):2263–2266. doi: 10.1172/JCI117225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pau CT, Keefe C, Duran J, Welt CK. Metformin improves glucose effectiveness, not insulin sensitivity: predicting treatment response in women with polycystic ovary syndrome in an open-label, interventional study. J Clin Endocrinol Metab. 2014;99(5):1870–1878. doi: 10.1210/jc.2013-4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vidal J, et al. Long-term effects of roux-en-Y gastric bypass surgery on plasma glucagon-like peptide-1 and islet function in morbidly obese subjects. J Clin Endocrinol Metab. 2009;94(3):884–891. doi: 10.1210/jc.2008-1620. [DOI] [PubMed] [Google Scholar]
- 46.Nishida Y, et al. Effect of moderate exercise training on peripheral glucose effectiveness, insulin sensitivity, and endogenous glucose production in healthy humans estimated by a two-compartment-labeled minimal model. Diabetes. 2004;53(2):315–320. doi: 10.2337/diabetes.53.2.315. [DOI] [PubMed] [Google Scholar]
- 47.Kodama S, et al. Effect of aerobic exercise training on serum levels of high-density lipoprotein cholesterol: a meta-analysis. Arch Intern Med. 2007;167(10):999–1008. doi: 10.1001/archinte.167.10.999. [DOI] [PubMed] [Google Scholar]
- 48.Morville T, et al. Divergent effects of resistance and endurance exercise on plasma bile acids, FGF19, and FGF21 in humans. JCI Insight. 2018;3(15):122737. doi: 10.1172/jci.insight.122737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reaven GM. What do we learn from measurements of HOMA-IR? Diabetologia. 2013;56(8):1867–1868. doi: 10.1007/s00125-013-2948-3. [DOI] [PubMed] [Google Scholar]
- 50.Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27(6):1487–1495. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- 51.Ader M, et al. Failure of homeostatic model assessment of insulin resistance to detect marked diet-induced insulin resistance in dogs. Diabetes. 2014;63(6):1914–1919. doi: 10.2337/db13-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116(7):1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stefan N, et al. Plasma adiponectin concentration is associated with skeletal muscle insulin receptor tyrosine phosphorylation, and low plasma concentration precedes a decrease in whole-body insulin sensitivity in humans. Diabetes. 2002;51(6):1884–1888. doi: 10.2337/diabetes.51.6.1884. [DOI] [PubMed] [Google Scholar]
- 54.Stefan N, et al. Plasma adiponectin and endogenous glucose production in humans. Diabetes Care. 2003;26(12):3315–3319. doi: 10.2337/diacare.26.12.3315. [DOI] [PubMed] [Google Scholar]
- 55.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132(6):2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 56.MacPherson RE, Castellani L, Beaudoin MS, Wright DC. Evidence for fatty acids mediating CL 316,243-induced reductions in blood glucose in mice. Am J Physiol Endocrinol Metab. 2014;307(7):E563–E570. doi: 10.1152/ajpendo.00287.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pang Z, et al. GPR40 is partially required for insulin secretion following activation of beta3-adrenergic receptors. Mol Cell Endocrinol. 2010;325(1-2):18–25. doi: 10.1016/j.mce.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 58.Michel MC. How β3 -adrenoceptor-selective is mirabegron? Br J Pharmacol. 2016;173(3):429–430. doi: 10.1111/bph.13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edmondson SD, et al. Discovery of vibegron: a potent and selective β3 adrenergic receptor agonist for the treatment of overactive bladder. J Med Chem. 2016;59(2):609–623. doi: 10.1021/acs.jmedchem.5b01372. [DOI] [PubMed] [Google Scholar]
- 60.Malik M, et al. Proarrhythmic safety of repeat doses of mirabegron in healthy subjects: a randomized, double-blind, placebo-, and active-controlled thorough QT study. Clin Pharmacol Ther. 2012;92(6):696–706. doi: 10.1038/clpt.2012.181. [DOI] [PubMed] [Google Scholar]
- 61.Mifflin MD, St Jeor ST, Hill LA, Scott BJ, Daugherty SA, Koh YO. A new predictive equation for resting energy expenditure in healthy individuals. Am J Clin Nutr. 1990;51(2):241–247. doi: 10.1093/ajcn/51.2.241. [DOI] [PubMed] [Google Scholar]
- 62.Malandrino N, et al. Visceral fat does not contribute to metabolic disease in lipodystrophy. Obes Sci Pract. 2019;5(1):75–82. doi: 10.1002/osp4.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen KY, et al. Brown fat activation mediates cold-induced thermogenesis in adult humans in response to a mild decrease in ambient temperature. J Clin Endocrinol Metab. 2013;98(7):E1218–E1223. doi: 10.1210/jc.2012-4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cole RJ, Kripke DF, Gruen W, Mullaney DJ, Gillin JC. Automatic sleep/wake identification from wrist activity. Sleep. 1992;15(5):461–469. doi: 10.1093/sleep/15.5.461. [DOI] [PubMed] [Google Scholar]
- 65.Kim K, et al. Whole body and regional quantification of active human brown adipose tissue using 18F-FDG PET/CT. J Vis Exp. 2019;(146):e58469. doi: 10.3791/58469. [DOI] [PubMed] [Google Scholar]
- 66.Barbaras L, Tal I, Palmer MR, Parker JA, Kolodny GM. Shareware program for nuclear medicine and PET/CT PACS display and processing. AJR Am J Roentgenol. 2007;188(6):W565–W568. doi: 10.2214/AJR.06.1058. [DOI] [PubMed] [Google Scholar]
- 67.Chen KY, et al. Brown adipose reporting criteria in imaging studies (BARCIST 1.0): recommendations for standardized FDG-PET/CT experiments in humans. Cell Metab. 2016;24(2):210–222. doi: 10.1016/j.cmet.2016.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim SY, et al. Loss of cyclin-dependent kinase 2 in the pancreas links primary β-cell dysfunction to progressive depletion of β-cell mass and diabetes. J Biol Chem. 2017;292(9):3841–3853. doi: 10.1074/jbc.M116.754077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19(3):368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- 70.Du Bois D, Du Bois EF. A formula to estimate the approximate surface area if height and weight be known. 1916. Nutrition. 1989;5(5):303–311; discussion 312. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.