

Abstract
We previously established that global deletion of the enhancer of trithorax and polycomb (ETP) gene, Asxl2, prevents weight gain. Because proinflammatory macrophages recruited to adipose tissue are central to the metabolic complications of obesity, we explored the role of ASXL2 in myeloid lineage cells. Unexpectedly, mice without Asxl2 only in myeloid cells (Asxl2ΔLysM) were completely resistant to diet-induced weight gain and metabolically normal despite increased food intake, comparable activity, and equivalent fecal fat. Asxl2ΔLysM mice resisted HFD-induced adipose tissue macrophage infiltration and inflammatory cytokine gene expression. Energy expenditure and brown adipose tissue metabolism in Asxl2ΔLysM mice were protected from the suppressive effects of HFD, a phenomenon associated with relatively increased catecholamines likely due to their suppressed degradation by macrophages. White adipose tissue of HFD-fed Asxl2ΔLysM mice also exhibited none of the pathological remodeling extant in their control counterparts. Suppression of macrophage Asxl2 expression, via nanoparticle-based siRNA delivery, prevented HFD-induced obesity. Thus, ASXL2 controlled the response of macrophages to dietary factors to regulate metabolic homeostasis, suggesting modulation of the cells’ inflammatory phenotype may impact obesity and its complications.
Keywords: Metabolism
Keywords: Adipose tissue, Macrophages, Obesity

Introduction
Obesity is characterized by chronic low-grade inflammation and macrophage accumulation in white adipose tissue (WAT). These cells regulate metabolism by producing proinflammatory cytokines, such as TNF-α, that compromise glucose homeostasis (1). The concept of macrophage-mediated inflammation impairing adipose tissue health is fortified by the observation that macrophages in WAT of obese patients generally polarize to the M1 phenotype, while lean fat contains an abundance of M2 cells (2). This concept raises the possibility that promoting a predominance of hypoinflammatory macrophages in patients may reduce pathological remodeling of WAT and as such, diminish the complications of obesity. In fact, ablation of CD11c-expressing cells normalizes insulin sensitivity in obese mice (3). Absence of proinflammatory macrophages, however, does not influence body weight, underscoring the uncertainty of the cell’s precise effect on obesity.
We previously reported that global deletion of the enhancer of trithorax and polycomb (ETP) gene, Asxl2, completely protects mice from HFD-induced weight gain while inducing lipodystrophy and hepatic steatosis (4). Reasoning that this phenotype is likely mediated by altered function of metabolic tissues, specifically hepatocytes and adipocytes, we conditionally deleted the gene in liver or fat. Unlike their globally deleted counterparts, however, these mice exhibited no liver or adipose tissue abnormalities and gained weight on HFD equal to that of controls.
Many studies have established that macrophages are central to adipose tissue homeostasis and their phenotype is distinct in obese versus lean fat. With this in mind, we deleted Asxl2 exclusively in myeloid lineage cells (Asxl2ΔLysM). Whereas HFD-fed control mice increased body mass by 50% in 8 weeks, surprisingly those without Asxl2, targeted only in myeloid cells, were completely resistant to weight gain despite increased food intake and similar activity and fecal fat. Moreover, while HFD-fed control mice were insulin resistant, those with myeloid-depleted Asxl2 were metabolically normal. Importantly, myeloid-specific Asxl2 deletion markedly diminished HFD-stimulated macrophage abundance in fat, and its associated inflammatory cytokine expression. Likely due to preservation of catecholamines, HFD Asxl2ΔLysM BAT was protected from lipid-mediated UCP1 suppression typically attending obesity. Thus, resting energy expenditure was 45% greater in HFD-fed Asxl2ΔLysM mice than control mice. WAT of HFD-fed Asxl2ΔLysM mice also exhibited none of the pathological remodeling extant in their control counterparts. Finally, siRNA-mediated Asxl2 suppression in vivo, using nanoparticles (NPs) specifically targeting inflammatory macrophages, also prevented HFD-induced obesity. These experiments establish that targeting a single myeloid lineage gene can eliminate diet-induced obesity and its complications.
Results
HFD-fed Asxl2ΔLysM mice are resistant to metabolic challenge.
We previously reported that global deletion of Asxl2 effects glucose and lipid metabolism (4). Unlike their WT counterparts, Asxl2–/– mice fed a HFD fail to gain weight, indicating a possible role for ASXL2 in metabolically active tissue. Given these observations and the fact that ASXL2 controls PPARγ-dependent gene expression (4), which is required for adipogenesis, we hypothesized that global Asxl2 deficiency prevents adipogenesis by disrupting PPARγ in fat. To test this hypothesis, we mated Asxl2fl/fl mice with those expressing adiponectin-Cre to selectively delete Asxl2 in adipocytes (Asxl2ΔAdipoq) (5). Contrary to our hypothesis, Asxl2 deficiency in adipose tissue did not affect body weight at steady state nor in response to HFD (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI128687DS1). The same holds regarding liver-selective Asxl2 deletion using albumin-Cre (Asxl2ΔAlb) (Supplemental Figure 1B). Additionally, glucose tolerance of HFD-fed Asxl2ΔAdipoq and Asxl2ΔAlb mice mirrored their Cre-negative counterparts (Supplemental Figure 1, C and D)
Given that adipocyte and hepatic expression of ASXL2 does not regulate its metabolic properties we turned to myeloid cells, particularly macrophages, because of their established role in the biology of obesity. Thus, we deleted Asxl2 in the myeloid lineage using LysM-Cre (Asxl2ΔLysM) and characterized the transcriptomic profiles of bone marrow–derived macrophages (BMMs) by performing unbiased RNA sequencing (RNA-seq). Principal component analysis of all expressed genes (median counts per million > 1.0) in Asxl2ΔLysM compared with Asxl2fl/fl macrophages revealed 766 significantly downregulated and 950 significantly upregulated genes (|fold change| > 1.3, FDR P value < 0.001) (Figure 1A). Of these, the 15 most upregulated and downregulated genes are detailed in a heatmap (Figure 1B). Enrichr analysis of differentially expressed genes (6, 7) revealed substantial downregulation of those associated with extracellular matrix (ECM) organization, collagen formation, cytokine-cytokine interaction, and inflammatory response (Figure 1C), including genes encoding Tnfa, Mmps, and Cxcls, in Asxl2ΔLysM BMMs. In contrast, cell cycle– and mitosis-related genes were upregulated. The fact that most of the dominant downregulated genes are also implicated in modulating adipose tissue inflammation and fibrosis supports the concept that genetic deletion of Asxl2 in myeloid cells may modify obesity. Therefore, we fed normal chow or HFD to Asxl2fl/fl and Asxl2ΔLysM mice. As expected, body weights of both control and Asxl2ΔLysM mice were similar on chow diet, while those of HFD-fed control mice (Asxl2fl/fl) increased approximately 50% after 8 weeks (Figure 1D). Surprisingly, those lacking Asxl2 exclusively in myeloid lineage cells were completely impervious to diet-induced weight gain, mirroring their chow-fed counterparts. Consistent with these observations, inguinal WAT (iWAT) and gonadal WAT (gWAT) weights, as well as adipocyte size, were substantially less in Asxl2ΔLysM mice compared with Asxl2fl/fl controls on an HFD (Figure 1, E and F). Dual-energy x-ray absorptiometry (DXA) scans showed markedly increased visceral and subcutaneous adiposity in HFD-fed Asxl2fl/fl controls but only a modest gain in analogous Asxl2ΔLysM mice (Figure 1, G and H). Asxl2ΔLysM mice were also protected from HFD-induced glucose and insulin intolerance (Figure 1, I and J) and Asxl2ΔLysM mice, but not Asxl2fl/fl mice, did not develop diet-induced hepatic steatosis (Figure 1K). Thus, diet-induced metabolic syndrome is prevented by ASXL2 inactivation in myeloid lineage cells.
Figure 1. ASXL2 regulates weight gain and metabolic homeostasis.

(A–C) RNA-seq analysis of bone marrow macrophages derived from Asxl2fl/fl and Asxl2ΔLysM mice. (A) Principal component analysis of all differentially expressed genes. Shaded ellipses are 95% confidence intervals for each group. (B) Heatmap of the top 30 most differentially expressed genes in macrophages based on log(fold change) > 1.3 with adjusted P < 0.001. (C) Gene Ontology (GO) term analysis of all genes significantly downregulated (negative enrichment, blue bar) or upregulated (positive enrichment, red bar) in Asxl2ΔLysM macrophages. (D–K) Two-month-old control and Asxl2ΔLysM mice were fed chow diet or HFD for 8 weeks. (D) Body weight with time. (E) Weight of gonadal WAT (gWAT) and inguinal WAT (iWAT) depots at sacrifice. (F) Size of WAT adipocytes at sacrifice. (G) DXA scans at time of sacrifice. (H) DXA-determined percentage body fat at sacrifice. (I) Glucose tolerance test performed before sacrifice. (J) Insulin tolerance test performed before sacrifice. (K) Hematoxylin and eosin–stained liver of control and Asxl2ΔLysM mice after 8 weeks on HFD. Scale bar: 400 μm. Data are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; as determined by 2-way ANOVA with Holm-Sidak post hoc analysis for multiple comparisons (D–F and H–J).
Obesity resistance of myeloid-specific Asxl2 deletion reflects relatively increased energy expenditure.
Despite failure to gain weight, food intake of HFD-fed Asxl2ΔLysM mice, normalized to body weight, surpassed that of their obese control counterparts, while fecal fat content was the same (Figure 2, A and B). These observations raised the prospect that resistance to diet-induced weight gain of Asxl2ΔLysM mice reflects relatively enhanced energy utilization. To address this possibility, control and Asxl2ΔLysM mice were placed in metabolic cages for 48 hours, 8 weeks after initiation of HFD. Despite equivalent physical activity (Figure 2C), energy expenditure, normalized to body weight, was approximately 45% greater, through light and dark cycles, in Asxl2ΔLysM mice than similarly fed controls (Figure 2D). The respiratory exchange ratio (RER) was also greater in HFD Asxl2ΔLysM mice (Figure 2E). Thus, the obesity resistance of Asxl2ΔLysM mice reflects a relative increase in energy utilization.
Figure 2. ASXL2 regulates energy expenditure.

(A) Food intake normalized to body weight of HFD-fed Asxl2fl/fl and Asxl2ΔLysM mice. (B) Fecal fat of HFD-fed Asxl2fl/fl and Asxl2ΔLysM mice. HFD-fed Asxl2fl/fl and Asxl2ΔLysM mice were placed in metabolic cages for 48 hours with 12-hour light/dark cycles. (C) Activity, (D) energy expenditure, and (E) respiratory exchange rate (RER) at 48 hours. Data are presented as mean ± SD. *P < 0.05, as determined by unpaired t test (A and E).
BAT is protected in Asxl2ΔLysMmice. The energy expenditure of HFD-fed Asxl2ΔLysM mice raised the possibility that it reflects uncoupling of ATP and respiration, as induced by UCP1. Thus, we explored beiging of iWAT. Like their control counterparts, chow-fed Asxl2ΔLysM mice contained numerous foci of characteristic, UCP1-expressing beige fat foci that were absent in HFD-fed control animals (Supplemental Figure 2A). Despite their normal metabolic phenotype, iWAT of HFD-fed Asxl2LysM mice surprisingly mirrored that of their control counterparts, as it was also devoid of UCP1-producing adipocytes. Thus, the robust energy expenditure induced by myeloid-specific Asxl2 deletion likely does not represent iWAT beiging.
Similarly to WAT, the appearance of brown adipose tissue (BAT) of chow-fed Asxl2ΔLysM mice was indistinguishable from their control counterparts (Figure 3A). As previously noted, HFD induces whitening of BAT in Asxl2fl/fl mice in that adipocytes contain large lipid droplets reminiscent of those in WAT (8, 9). Although BAT of HFD-fed Asxl2ΔLysM mice exhibited some evidence of whitening compared with that of chow-fed animals, it was minimal relative to that of HFD-fed Asxl2fl/fl mice. Consequent to avoidance of diet-induced lipid accumulation, the weight of HFD Asxl2ΔLysM BAT was lower than that of control (Figure 3B). In keeping with evidence that prevention of HFD-induced whitening preserves BAT activity (10), UCP1 expression determined by immunostaining, quantitative real-time PCR (qPCR), and immunoblot, was relatively enhanced in HFD Asxl2ΔLysM mice (Figure 3, C–E, and Supplemental Figure 2B). Furthermore, PET imaging documented greater palmitate metabolism by HFD Asxl2ΔLysM than Asxl2fl/fl BAT, in vivo (Figure 3F). Although fludeoxyglucose metabolism by HFD Asxl2ΔLysM BAT trended higher relative to that of Asxl2fl/fl counterparts, the difference was not significant (Figure 3G). Circulating free fatty acids (FFAs) were increased in HFD control mice but not those with myeloid Asxl2 deletion, likely reflecting maintenance of their robust metabolism in BAT (Figure 3H and ref. 11). In keeping with previous data, serum triglycerides were unaltered in each cohort of mice regardless of diet (Figure 3I).
Figure 3. BAT is protected in Asxl2ΔLysM mice.

(A) Hematoxylin and eosin–stained histological sections of BAT of Asxl2fl/fl and Asxl2ΔLysM mice after 8 weeks on chow diet or HFD. Scale bar: 300 μm. (B) BAT weight of HFD-fed Asxl2fl/fl and Asxl2ΔLysM mice. (C) UCP1-immunostained histological sections of BAT of Asxl2fl/fl and Asxl2ΔLysM mice after 8 weeks on chow diet or HFD. Scale bar: 200 μm. (D) qPCR analysis of Ucp1 mRNA abundance in Asxl2fl/fl and Asxl2ΔLysM mice after 8 weeks on HFD. (E) Immunoblot of UCP1 abundance in Asxl2fl/fl and Asxl2ΔLysM mice after 8 weeks on HFD. (F and G) PET scan determination of (F) palmitate and (G) fludeoxyglucose metabolism in BAT of Asxl2fl/fl and Asxl2ΔLysM mice after 8 weeks on HFD. (F) Serum free fatty acids (H) and triglycerides (I) of Asxl2fl/fl and Asxl2ΔLysM mice after 8 weeks on chow diet or HFD. (J) Core body temperature of Asxl2fl/fl and Asxl2ΔLysM mice maintained at room temperature (RT, 23°C) or 4° for 30 minutes, after 8 weeks on chow diet or HFD. (K) Two-month-old Ucp1–/–, Asxl2fl/fl, Asxl2ΔLysM, and Asxl2ΔLysM Ucp1–/– mice were fed HFD for 8 weeks at neutral thermal temperature (30°C); body weight change was measured with time. *P < 0.05 for comparisons between Ucp1–/– and Asxl2ΔLysM Ucp1–/–; #P < 0.05 for comparisons between Asxl2ΔLysM and Asxl2ΔLysM Ucp1–/– at 8 weeks of HFD.Data are presented as mean ± SD. *P < 0.05; **P < 0.01; as determined by unpaired t test (B and D) or 2-way ANOVA with Holm-Sidak post hoc analysis for multiple comparisons (F, H, J, and K).
As in mice, human fat mass and BAT activity are negatively correlated, whereas BAT activity and energy expenditure are positively related (12). Hence, the obesity resistance of Asxl2ΔLysM mice may represent their preserved UCP1-mediated energy expenditure. In keeping with the hypothesis that their failure to gain weight represents preservation of UCP1-mediated ATP uncoupled respiration, core body temperature of HFD Asxl2ΔLysM mice, maintained at 4°C, was significantly higher than that of control (Figure 3J). Asxl2ΔLysM mice on an HFD, housed at 30°C, also had lower body weight than Asxl2fl/fl controls (Figure 3K). Indicating that UCP1 contributes to obesity resistance of the myeloid-mutant mice at thermoneutrality, the percentage increase in body weight of HFD-fed Asxl2ΔLysM mice, globally lacking Ucp1 (Asxl2ΔLysM Ucp1–/–), mirrored that of Asxl2fl/fl and was significantly greater than Asxl2ΔLysM (Figure 3K). Thus, UCP1 contributes to the obesity resistance of Asxl2ΔLysM mice at room temperature and thermoneutrality (13).
Catecholamines are increased in ASXL2ΔLysM BAT.
Macrophages are proposed to influence weight gain by altering uncoupled respiration. A common hypothesis holds that alternatively activated myeloid cells express catecholamines that promote UCP1 expression by beige adipocytes, thereby enhancing energy expenditure (14). Recent data, however, challenge this postulate and it has been concluded that alternatively activated macrophages do not express catecholamines (15). Consistent with this conclusion, naive and palmitate- or IL-4–treated Asxl2fl/fl and Asxl2ΔLysM BMMs expressed little tyrosine hydroxylase mRNA (Th), whose product catalyzes a rate-limiting step in catecholamine synthesis (Figure 4A). It is likely, therefore, that macrophages do not produce catecholamines that contribute to the obesity resistance of Asxl2ΔLysM mice. Despite a paucity of tyrosine hydroxylase expression in macrophages, norepinephrine was unexpectedly approximately twice as abundant in HFD-fed Asxl2ΔLys BAT as control (Figure 4B). Because processing of triglycerides to FFAs and glycerol is regulated by norepinephrine, we measured lipolysis in BAT explants derived from HFD-fed control and Asxl2ΔLysM mice. Corroborating palmitate uptake and confirming the functional significance of catecholamine abundance, lipolysis in Asxl2ΔLysM BAT explants was increased, both in the basal state and when stimulated with isoproterenol (Figure 4C). Thus, despite their likely failure to produce norepinephrine, Asxl2-deficient macrophages may regulate BAT metabolism by affecting catecholamine signaling.
Figure 4. Catecholamines are relatively increased in Asxl2ΔLysM BAT.

(A) Tyrosine hydroxylase mRNA expression by naive and IL-4– or palmitate-treated Asxl2fl/fl and Asxl2ΔLysM BMMs. Adrenal tissue served as positive control. (B) Norepinephrine (NE) content of BAT of HFD-fed control and Asxl2ΔLysM mice. (C) Glycerol released from BAT explants derived from HFD-fed control and Asxl2ΔLysM mice. (D) Maoa mRNA abundance in BAT stromal vascular fraction of chow- or HFD-fed control and Asxl2ΔLysM mice. Data are presented as mean ± SD. *P < 0.05, as determined by unpaired t test (B) or 2-way ANOVA with Holm-Sidak’s post hoc analysis for multiple comparisons (C and D).
Although a subset of macrophages associated with sympathetic neurons do not express catecholamines, they incorporate and degrade norepinephrine, thus preventing its accumulation in BAT (16, 17). These sympathetic neuron–associated macrophages reduce catecholamines in BAT via expression of solute carrier member 2 (Slc6a2) norepinephrine transporter and/or monoamine oxidase A (Maoa), an amine-degrading mitochondrial enzyme. Indicating that impaired catecholamine degradation likely contributes to the preservation of BAT in HFD-fed Asxl2ΔLysM mice, Maoa in the stromal vascular fraction (SVF) of BAT was diminished relative to their Asxl2fl/fl counterparts and mirrored that of chow-fed mice (Figure 4D).
Absence of Asxl2 in myeloid lineage cells prevents accumulation of macrophages in WAT and BAT in response to HFD.
Adipose tissues contain diverse myeloid cell types, including abundant eosinophils and macrophages. In the lean state, eosinophils produce IL-4 to sustain an antiinflammatory program in WAT macrophages (18). In obesity, however, eosinophils in WAT are reduced and macrophages upregulate expression of proinflammatory cytokines such as TNF-α that impair glucose homeostasis (19, 20). We therefore asked whether ASXL2 expression in myeloid lineage cells determines eosinophil or macrophage responses to HFD in WAT and BAT.
We identified CD45+SiglecF+SSChi eosinophils and CD45+F4/80+CD64+ macrophages in WAT and compared their abundance in Asxl2fl/fl and Asxl2ΔLysM mice (Supplemental Figure 3). Eosinophils were decreased in WAT of HFD-fed mice irrespective of genotype, suggesting that ASXL2 does not determine eosinophil responses to diet-induced obesity (Supplemental Figure 4). In contrast, WAT macrophages were increased in frequency and number following an HFD in Asxl2fl/fl mice, but not in Asxl2ΔLysM mice (Figure 5, A and B). A similar pattern occurred regarding macrophage frequencies and numbers in BAT (Figure 5, C and D). Consistent with this observation and confirming previous RNA-seq data, HFD induced expression of inflammatory cytokine mRNAs, including Il1b and Tnfa, in both BAT (Figure 5E) and WAT (Supplemental Figure 5) SVF of HFD-fed Asxl2fl/fl but not Asxl2ΔLysM mice. Additionally, macrophages expressing Adgre1 (F4/80), Cd68, and Fcgr1a (CD64) were decreased in Asxl2ΔLysM BAT SVF (Figure 5F). To determine if the decreased macrophage infiltration in BAT is a consequence of myeloid deletion of Asxl2 or of obesity resistance, we fed mice an HFD for 4 weeks, at which time body weight of WT mice had not yet increased (Figure 5G). Confirming that the relative paucity of macrophages in HFD-fed Asxl2ΔLysM mice is not due to their failure to gain weight, expression of the macrophage markers Adgre1 (F4/80), Tnfa, and Il1b was significantly lower in BAT SVF of 4-week HFD-fed Asxl2ΔLysM mice than similarly fed, nonobese Asxl2fl/fl controls (Figure 5H).
Figure 5. ASXL2 expression in myeloid cells is required for macrophage accumulation in WAT and BAT in obesity.

Asxl2fl/fl and Asxl2ΔLysM mice were fed either a chow diet or HFD. (A) Frequencies and (B) numbers of F4/80+CD64+ macrophages in gonadal WAT: pregated on singlet, live, CD45+ cells. (C) Frequencies and (D) numbers of F4/80+CD64+ macrophages in BAT: pregated on singlet, live, CD45+ cells. (E) Inflammatory cytokine and chemokine mRNA expression in stromal vascular fraction of BAT of Asxl2fl/fl or Asxl2ΔLysM mice after 8 weeks fed with chow diet or HFD. (F) Macrophage marker mRNA expression in stromal vascular fraction of BAT of Asxl2fl/fl or ASXL2ΔLysM mice after 8 weeks fed with chow diet or HFD. (G) Body weight and brown or gonadal fat pad weight of Asxl2fl/fl and Asxl2ΔLysM mice after 4 weeks of HFD. (H) Adgre1 (F4/80) and inflammatory cytokine mRNA expression in stromal vascular fraction of BAT of Asxl2fl/fl or Asxl2ΔLysM mice after 4 weeks on HFD. (I) IL-1β and TNF-α secretion by bone marrow–derived macrophages of Asxl2fl/fl or Asxl2ΔLysM mice stimulated with 100 ng/mL LPS for 3 hours, followed by 15 μM nigericin for 1 hour. (J–L) Histological sections of WAT of HFD-fed control or Asxl2ΔLysM mice stained to identify (J) fibrosis (hematoxylin and eosin), (K) hypoxic adipocytes, and (L) crown-like structures (F4/80). Scale bars: 1 mm (J), 400 μm (K), and 200 μm (L). (M) ECM gene mRNA expression in gonadal WAT stromal vascular fraction of chow- or HFD-fed Asxl2fl/fl or Asxl2ΔLysM mice. Data are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; as determined by unpaired t test (H and I) or 2-way ANOVA with Holm-Sidak post hoc analysis for multiple comparisons (B, D–F, and M).
Induction of the NLRP3 inflammasome enhances MAOA abundance (16). The paucity of IL-1β mRNA, expressed by Asxl2ΔLysM relative to Asxl2fl/fl macrophages, therefore raised the possibility that preservation of BAT catecholamines reflects suppressed inflammasome activity. Expression of Nlrp3 by Asxl2ΔLysM and Asxl2fl/fl BMMs, in response to LPS or TNF-α was, however, similar, suggesting that NLRP3 is not regulated by ASXL2 (Supplemental Figure 6A). Additionally, LPS- and nigericin-induced inflammasome activation, reflected by cellular foci upon binding of the FLICA probe to active caspase-1 (21), yielded no difference between Asxl2ΔLysM and Asxl2fl/fl macrophages (Supplemental Figure 6B). Interestingly, LPS-stimulated secretion of IL-1β by Asxl2ΔLysM BMMs was decreased, suggesting that ASXL2 inactivation dampens pathways involved in the secretion, but not maturation, of IL-1β (Figure 5I). Likewise, TNF-α expression by mutant cells was decreased (Figure 5I). It is therefore likely that absence of ASXL2 in macrophages preserves catecholamines by their limited infiltration into BAT and diminished expression and secretion of proinflammatory cytokines. Supporting this conclusion, no differences in Maoa expression were observed in Asxl2ΔLysM and control BMMs (Supplemental Figure 6C).
Obesity is characterized by destructive changes in WAT, including hypovascularity-induced hypoxia leading to adipocyte death (19, 22) and eventuating in peri-adipocyte fibrosis that compromises lipid uptake and, therefore, metabolic function. The fibrosis of obese WAT enhances the inflammatory state of pre-adipocytes and macrophages, thus invoking a cycle of ECM production, inflammation, and pathological adipose tissue remodeling (23). Like their gross phenotype, the microscopic appearance of WAT of chow-fed Asxl2ΔLysM mice was indistinguishable from their control counterparts (not shown). As expected, WAT of Asxl2fl/fl mice, maintained on HFD, contained abundant fibrosis, a plethora of hypoxic cells, and numerous crown-like structures in which macrophages phagocytose lipid droplets (Figure 5, J–L). In contrast, adipocytes of HFD-fed Asxl2ΔLysM mice exhibited no evidence of fibrosis, crown-like structures, or hypoxic cells. In keeping with these morphological observations, HFD promoted expression of selected collagen synthesis genes in WAT SVF in Asxl2fl/fl mice but not Asxl2ΔLysM counterparts (Figure 5M). These observations indicate that ASXL2 is required for macrophage accumulation and inflammation in WAT and BAT in the context of diet-induced obesity and suggest that ASXL2 expression in macrophages may contribute to pathological remodeling of fat.
Myeloid-specific BAP1 deletion arrests HFD-induced weight gain.
BAP1 is a tumor suppressor and deubiquitinase that complexes with and stabilizes ASXL2 (24). This relationship raised the possibility that, like ASXL2, absence of BAP1 in myeloid lineage cells impacts HFD-induced weight gain. Thus, mirroring our approach to ASXL2, we mated Bap1fl/fl and LysM-Cre mice. Similarly to Asxl2ΔLysM mice, those lacking Bap1 exclusively in myeloid lineage cells failed to gain weight on an HFD and maintain glucose and insulin sensitivity (Figure 6, A–C). Like Asxl2ΔLysM mice, HFD-fed Bap1ΔLysM mice had diminished Maoa and arrested lipolysis in BAT (Supplemental Figure 7). Given its essential role in stabilizing ASXL2, the obesity-preventing properties of myeloid-specific deletion of Bap1 is in keeping with the similar phenotype of Asxl2ΔLysM mice.
Figure 6. siRNA-mediated Asxl2 suppression in macrophages prevents diet-induced obesity.

(A) Body weight, (B) glucose tolerance test, and (C) insulin tolerance test of Bap1fl/fl and Bap1ΔLysM mice fed HFD for 6 weeks. (D) WT BMMs were incubated with GFP-siRNA– or Asxl2-siRNA–associated nanoparticles. Asxl2 mRNA was measured by qPCR and compared to that of Asxl2ΔLysM BMMs. Colocalization of macrophages (F4/80) and Asxl2-siRNA–associated nanoparticles in (E) spleen and (F) gonadal WAT of HFD-fed WT mice. Scale bars: 100 μm (E) and 30 μm (F). (G) Body weight of WT HFD-fed mice administered GFP-siRNA– or Asxl2-siRNA–associated nanoparticles. Data are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; as determined by 1-way ANOVA with Holm-Sidak post hoc analysis for multiple comparisons (A–C and G).
siRNA-mediated ASXL2 suppression in macrophages prevents diet-induced obesity.
Our data raise the possibility that therapeutic suppression of ASXL2 expression in macrophages may obviate diet-induced obesity. To explore this prospect, we turned to Asxl2 siRNA formulated with a biocompatible cationic amphipathic peptide (p5RHH) to create 55-nm NPs that are phagocytosed by macrophages, followed by endosomolysis and siRNA release to engage the RISC complex (25–27). The clinical potential of this strategy is underscored by the fact that NP-mediated ablation of specific subsets of myeloid lineage cells positively affects conditions such as stroke and sepsis (28) and the same macrophage-targeting NPs, used here, combined with NF-κB p65 siRNA, prevent inflammatory and osteoarthritis (29, 30). We therefore asked if a similar strategy to suppress macrophage ASXL2 expression would arrest diet-induced obesity. To this end, we used a Cy5.5-labeled p5RHH-Asxl2 siRNA, designed and synthesized by Sigma-Aldrich, multiplexed in unitary peptidic NPs, which suppressed Asxl2 mRNA expression in isolated BMMs by approximately 50% (Figure 6D). We administered 0.5 mg/kg to mice, twice per week while maintaining them on an HFD for 8 weeks.
Distribution of the p5RHH-siRNA complex in mice with collagen-induced arthritis appears limited to inflamed joints and kidneys. In contrast to the relatively restricted localization in inflammatory arthritis and perhaps reflecting the systemic inflammation accompanying obesity (22), Asxl2-siRNA–associated NPs were distributed in kidneys, spleen, bone, muscle, lung, and subcutaneous and visceral fat depots (Supplemental Figure 8). Confirming that they target macrophages, Asxl2-siRNA–associated NPs localized exclusively in cells expressing F4/80 in spleen and targeted the same cells in gWAT of HFD-fed WT mice (Figure 6, E and F). Administration of this NP complex to HFD-fed WT mice completely prevented weight gain relative to those receiving GFP-siRNA–associated NPs (Figure 6G). Thus, pharmacological arrest of Asxl2 expression in macrophages has antiobesity properties. Furthermore, prevention of diet-induced weight gain by macrophage-specific siRNA-associated NPs fortifies the myeloid specificity of LysM-mediated Asxl2 ablation.
Discussion
Resistance of Asxl2ΔLysM mice to diet-induced weight gain suggests obesity can be positively affected by manipulation of myeloid lineage cells. Furthermore, evidence that abundant fat intake may prolong longevity in humans and mice buttresses the potential importance of possibly limiting obesity in the face of HFD (31, 32).
This study was initiated with the expectation that absence of the gene in classical metabolism-regulating tissues, namely liver or fat, mediates the obesity resistance of global Asxl2 deletion. Although that proved not to be the case, mice bearing myeloid-specific deletion of the ETP gene mirrored their globally deficient counterparts in that they failed to gain HFD-induced weight. Importantly, unlike those with global deletion, insulin and glucose homeostases of Asxl2ΔLysM mice on HFD remain preserved. Fortifying the concept that myeloid-expressed ASXL2 regulates diet-induced obesity, similar conditional deletion of Bap1, which stabilizes the ETP protein and enables its epigenetic properties, also prevents HFD-induced weight gain and its metabolic consequences (24, 33).
In the face of enhanced food consumption, unaltered activity, and normal intestinal absorption of HFD-fed Asxl2ΔLysM mice, it is likely their obesity resistance reflects enhanced basal energy expenditure relative to similarly fed Asxl2fl/fl controls. Furthermore, the relative increase in BAT metabolic activity, as determined by fatty acid incorporation and UCP1 abundance, suggests that protection of BAT from the effects of obesity by myeloid-specific deletion of Asxl2 is substantially responsible for increased energy expenditure of the mutant mice. On the other hand, Asxl2ΔLysM macrophage–mediated prevention of steatosis, which reduces hepatocyte β-oxidation, may also contribute to the relatively robust energy expenditure of the conditionally deleted mice (34).
Morphologically, the most impressive difference between HFD control and Asxl2ΔLysM BAT is accumulation of large lipid droplets in the former. This whitening diminishes BAT efficacy. Given their reduced basal energy expenditure, it is possible that lipid droplet accumulation in BAT may contribute to the predisposition of previously obese individuals to regain weight. Although prevention of whitening of Asxl2ΔLysM BAT is likely central to maintaining energy expenditure, the means by which lipid accumulation in this tissue is prevented in our mutant is enigmatic.
In normal HFD obesity, hypoxia-induced necrotic adipocytes recruit and activate macrophages which, in turn, induce a proinflammatory phenotype in WAT (19). This induction of inflammation is the product of secreted cytokines such as TNF-α, which promote ECM expression by adipocytes, further enhancing their inflammatory state and systemic insulin resistance. Surviving adipocytes, while increased in size, are mechanically restricted, thus compromising their capacity to incorporate fatty acids in the form of triglycerides. This process likely prompts spillover of fatty acids from inflamed, dysfunctional WAT adipocytes. In consequence, large lipid droplets accumulate in other tissues such as liver, muscle, and BAT, which participate in triglyceride clearance, thus compromising insulin sensitivity and decreasing energy expenditure due to reduced UCP1 (9, 35). The contention that myeloid-specific Asxl2 deletion protects BAT by preventing HFD-induced lipid spillover is supported by the simultaneous prevention of hepatic steatosis. In the absence of ASXL2, macrophages are less prone to inflammatory activation and ECM expression and as such, pathological WAT remodeling is limited which may prevent lipid spillover and restrict whitening of BAT.
On the other hand, BAT effectively incorporates triglycerides and metabolizes fatty acids, a process mediated by thermogenic activation involving UCP1 induction (9). Incorporation of triglycerides by BAT is enhanced in circumstances of increased dietary fat and restricted hepatic uptake, as occurs in steatosis. Thus, it is possible that prevention of HFD-induced BAT whitening by Asxl2ΔLysM macrophages reflects arrest of dietary hyperlipidemia, thus limiting fatty acid incorporation.
Ultimately, however, catecholamines are central to maintenance of BAT morphology and function, raising the possibility that Asxl2 deletion prompts their production by myeloid lineage cells. Although norepinephrine abundance is approximately twice that of control in Asxl2ΔLysM BAT, tyrosine hydroxylase expression by WT BMMs and those lacking ASXL2 is undetectable, consistent with reports challenging the concept that alternatively activated macrophages produce catecholamines. The abundance of Maoa in the SVF of Asxl2ΔLysM BAT indicates that accumulation of norepinephrine reflects its retarded degradation, which prevents whitening (17). Asxl2 deletion in myeloid cells reduces stimulated secretion of TNF-α, which promotes Maoa expression (36), suggesting that Asxl2ΔLysM macrophages may preserve catecholamines by the cell’s limited infiltration into BAT, regardless of weight gain and their diminished expression of proinflammatory cytokines. RNA-seq analysis revealed no difference in catecholamine-regulating protein expression by Asxl2ΔLysM and Asxl2fl/fl BMMs, supporting the concept that restricted macrophage infiltration, rather than diminished MAOA synthesis, is the dominant cause of BAT preservation. Confirmation of this conclusion, however, requires direct measurement of MAOA and SLC6A2 in BAT-residing macrophages, particularly those that are associated with sympathetic neurons, which are difficult to obtain in purity (17). Additionally, future studies will determine if prevention of macrophage accumulation in Asxl2ΔLysM BAT reflects modified proliferation of resident macrophages or dampened recruitment of circulating monocytes.
Because alternatively activated macrophages produce collagens and other matrix components, their ECM proteins may hypothetically contribute to the pathological remodeling of obese fat (37, 38). This being the case, macrophages that prevent obesity may express less ECM protein than control. In fact, the RNA-seq profile of Asxl2ΔLysM BMMs reveals a unique phenotype relative to classical alternatively activated cells in which collagen production is increased to enable tissue healing (39–41). Although crown-like structures typically contain M1-polarized macrophages, those associated with adipocyte fibrosis are predominantly M2 cells (42). The role that macrophage-secreted ECM proteins play in fibrotic remodeling of obesity-associated WAT is unknown but the paucity of these molecules, expressed by Asxl2ΔLysM macrophages, may contribute to their protection against diet-induced weight gain and insulin resistance.
Although UCP1 activity in WT BAT is robust at room temperature, mice lacking the uncoupling enzyme, in this circumstance, have normal body mass, indicating cold-associated compensatory mechanisms (i.e., non-UCP1) preventing weight gain (43, 44). At thermoneutrality, however, chow-fed Ucp1–/– mice develop obesity (45) and in WT mice, HFD partially activates the enzyme (45, 46). Additionally, human BAT incorporates more glucose and releases more lactate than WAT in warm conditions (47). These observations indicate that HFD can stimulate BAT metabolism even at thermoneutrality and raise the possibility that preservation of BAT may diminish diet-induced weight gain in the absence of thermal stress. In fact, such appears to be the case regarding Asxl2ΔLysM mice.
Collectively, these data highlight the role of ASXL2 in altering the gene expression profile of macrophages. Further, deletion of Asxl2 may protect adipose tissue homeostasis by locally influencing macrophage accumulation and function. Thus, the capacity of myeloid deletion of ASXL2 to prevent inflammatory remodeling of fat may represent a partnership of decreased recruitment of macrophages and suppression of their proinflammatory and ECM pathway genes.
To our knowledge, these findings are the first to identify ASXL2 as a master regulator of macrophage function to control systemic metabolic homeostasis. In addition, targeting ASXL2 in macrophages may represent a novel therapeutic strategy against obesity. Nevertheless, caution is warranted, as Asxl2 mutations are associated with macrocephaly and dysmorphic features (48) as well as hematopoietic malignancies. At least in mice, however, development of these leukemic disorders appears to require inactivation of the gene in hematopoietic stem cells that are not targeted by LysM-Cre (49). Thus, in contrast to mice lacking germline Asxl2, those in which the gene is absent only in myeloid lineage cells exhibit no splenomegaly or other evidence of leukemia. Furthermore, the fact that both LysM-Cre–mediated deletion and NP-associated Asxl2 siRNA prevent obesity adds further support for potential clinical relevance and indicates that postprandial arrest of the ETP protein is effective. Therefore, ASXL2 may have evolved to serve as a metabolic switch that determines macrophage responses to environmental stimuli such as dietary lipids.
Methods
Animals.
LysM-Cre, Adipoq-Cre, Albumin-Cre (Alb-Cre), and Ucp1–/– mice were obtained from The Jackson Laboratory. Bap1fl/fl mice were generously provided by Genentech Inc. (50). All animals were housed in the animal care unit of Washington University School of Medicine, where they were maintained according to guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care.
Generation of Asxl2-deficient mice.
Asxl2tm1a(EUCOMM)Wtsi (clone ID: HEPD0751_1_D05) embryonic stem cells were purchased from the European Conditional Mouse Consortium (EUCOMM) and were microinjected into B6(Cg)-Tyrc-2J/J blastocysts to generate chimeric mice at the Washington University Pathology/Immunology Micro-Injection Core. Highly chimeric mice were bred with B6(Cg)-Tyrc-2J/J females to select for germline transmission. To remove the LacZ-neomycin–resistance cassette, the mice were first crossed with transgenic mice expressing Flp recombinase under the control of the actin promoter (The Jackson Laboratory). The Flp transgene was removed by crossing the mice with WT C57BL6/J mice. Asxl2fl/+ mice were intercrossed to generate Asxl2fl/fl mice. Asxl2fl/fl, Asxl2fl/WT, and Asxl2WT littermate mice were genotyped by PCR with primers ASXL2-5′-arm (5′-CCCACACGCTCAGTCTCTACTCGC-3′), ASXL2-3′-arm (5′-TCTCCCTTCTTTGTTGCTGCCACC-3′), and LAR3 (5′-CAACGGGTTCTTCTGTTAGTCC-3′) using the following parameters: 95°C for 3 minutes, followed by 35 cycles of 95°C for 30 seconds, 62°C for 30 seconds, and 72°C for 30 seconds, and then 72°C for 5 minutes. The WT allele was detected as a band at 828 bp, whereas the floxed allele was detected as a band at 642 bp. Asxl2fl/fl mice generated were subsequently crossed with LysM-Cre mice (The Jackson Laboratory), Adipoq-Cre, and Alb-Cre. After Cre recombination, the floxed and WT alleles were confirmed by PCR with primers (ASXL2-F, 5′-ACTTTCCTCAGACCATCAGCTTCC-3′; ASXL2-R, 5′-CCCCACGCCCCTCTCTCA-3′; and ASXL2-loxP, 5′-TGAACTGATGGCGAGCTCAGACC-3′) using the same parameters as above. The WT allele was detected as a band at 320 bp, whereas the floxed allele was detected as a band at 378 bp.
RNA extraction and qPCR.
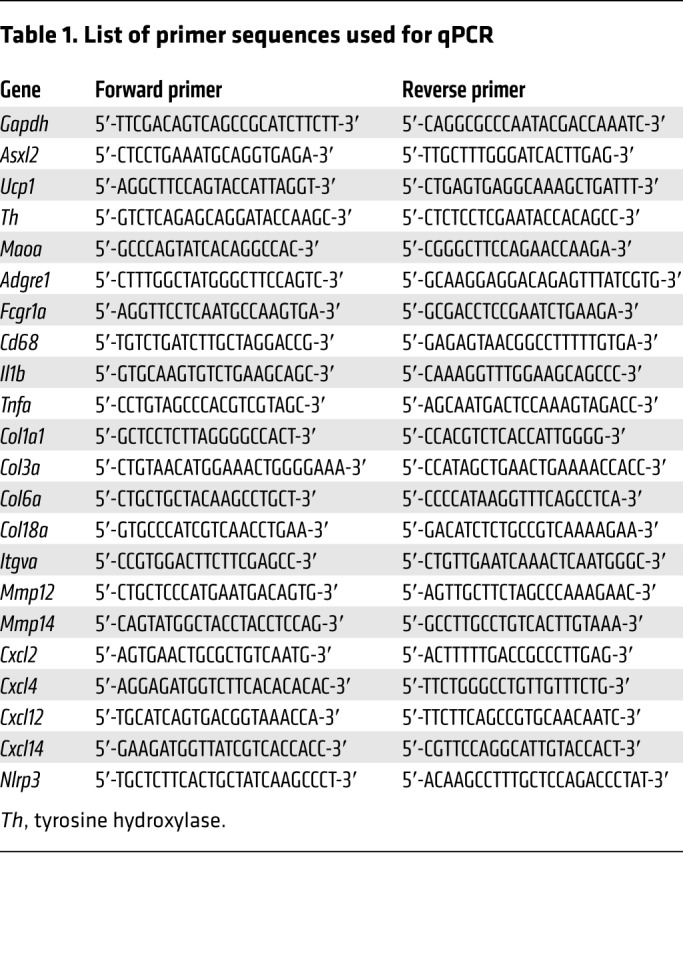
RNA from cultured cells was isolated and purified using the RNeasy RNA purification kit (Qiagen); RLT lysis buffer was supplemented with β-mercaptoethanol (1%). RNA from tissues was isolated using the TRIzol method. Purified RNA was treated with DNase I (Invitrogen) before reverse transcription. cDNA was synthesized from RNA (1 μg) using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). Real-time PCR was performed using the SYBR Green Master Mix kit and gene-specific primers. The qPCR reaction was performed on an ABI PRISM 7500 sequence detection system (Applied Biosystems). All reactions were performed in triplicate and relative mRNA levels were calculated by the comparative threshold cycle method using GAPDH as an internal control. Detailed sequences of the oligonucleotides used in qPCR analyses are listed in Table 1.
Table 1. List of primer sequences used for qPCR.
HFD feeding studies.
Mice were housed in a pathogen-free barrier facility with unrestricted access to water and standard mouse chow containing 6% fat, which is designated as standard chow (chow) herein. For dietary intervention studies, mice were fed chow until age 8 weeks or 4 weeks and thereafter were randomized into groups that were fed either chow or an HFD (Research Diets, catalog D12492). Food intake and weight were monitored weekly. At the end of the study, serum was collected and tissues were snap-frozen in liquid nitrogen and stored at –80°C.
Body composition analysis.
Body composition was measured using DXA with a Faxitron UltraFocus according to the manufacturer’s instructions. Body fat mass and lean mass were measured between 2 pm and 3 pm.
Indirect calorimetry.
Indirect calorimetry was performed as described previously (51). O2 consumption (VO2) and CO2 production (VCO2) were measured for 48 hours to determine energy expenditure. Mean relative VO2 (expressed in mL/kg/min) and respiratory quotient (RQ; calculated as VCO2/VO2) were determined.
GTT and ITT.
Glucose tolerance tests (GTTs) were performed on 16- to 20-week-old male mice by placing them in clean cages and starving 6 hours with free access to water. Mice were weighed and a small amount of blood was obtained from the lateral saphenous vein for baseline (time 0) glucose measurements. Mice were then injected intraperitoneally with 50% sterile dextrose (1 mg/g body weight). Tail blood glucose was determined at 15, 30, 60, and 120 minutes after challenge using a Bayer Contour glucometer. For insulin tolerance tests (ITTs), 16- to 20-week-old male mice were placed in clean cages without food but free access to water. Following a 6-hour fast, the mice were weighed and baseline glucose readings were taken using a Bayer Contour glucometer. Mice were injected intraperitoneally with human insulin (Humulin, Eli Lilly) at a dose of 0.5 U/kg body weight and blood glucose measured at 15, 30, 45, and 60 minutes afterward.
Thermoneutrality studies.
Ucp1–/– mice were bred with Asxl2-Cre mice to generate Asxl2ΔLysM on a Ucp1–/– background. These mice, along with the controls (Asxl2fl/fl, Ucp1–/–, and Asxl2ΔLysM) were housed at thermoneutrality. Mice were fed chow until age 8 weeks and thereafter were randomized into groups that were fed an HFD. Body weights were monitored weekly.
In vivo metabolic imaging.
Mice were secured in a custom-designed acrylic restraining device and placed inside the field of view (FOV) of cross-calibrated Siemens Inveon PET/CT or Focus F220 scanners. A 30-minute dynamic positron emission tomography (PET) image acquisition was initiated immediately preceding a bolus injection of [18F]fludeoxyglucose (0.2–0.5 mCi) or [11C]palmitate via the tail vein. Dynamic images were reconstructed using the ordered subsets expectation maximization (OSEM) algorithm and 40 frames per imaging session. Images were analyzed by drawing regions of interest (ROIs) on BAT to ascertain in vivo measures of glucose (via fludeoxyglucose) and fatty acid (via [11C]-palmitate) metabolism time course. Data derived from ROIs were normalized to standardized uptake values (SUV = activity × [weight of mouse]/[injected dose]) to account for differences in injected dose and weight of mice.
p5RHH-siRNA NP preparation.
p5RHH peptide (provided by Genscript) was dissolved at 10 mM in DNase-, RNase-, and protease-free sterile purified water (Cellgro) and stored in 10-μL aliquots at −80°C before use. The Cy5.5-labeled or nonlabeled siRNAs, scrambled siRNA (target sequence, GACGUAAACGGCCACAAGCC), and Asxl2 siRNA (target sequence, CCUAGGAGUUGCAGACUUG) were procured from Sigma-Aldrich, dissolved at 100 μM in 1× siRNA buffer (Thermo Fisher Scientific), and stored in 10-μL aliquots at −80°C before use. The p5RHH-siRNA NPs were prepared by mixing equal volumes of the aforementioned p5RHH peptide and siRNA at a peptide/siRNA ratio of 100:1 in 200 μL HBSS with Ca2+ and Mg2+ (Gibco and Life Technologies) and incubated at 10 minutes on ice before i.v. injection (26). For NP localization experiments, mice were sacrificed 24 hours after injection and tissues of interest were harvested and stored at –80°C. For body weight experiments, mice were i.v. injected with NPs twice per week after 3 weeks of HFD.
For in vitro experiments, the p5RHH-siRNA NPs were prepared by mixing equal volumes of the aforementioned p5RHH peptide and siRNA at a peptide/siRNA ratio of 100:1 in HBSS with Ca2+ and Mg2+ and incubated at 37°C for 40 minutes and then added to the macrophage culture.
Morphometric analysis of WAT.
Epididymal WAT was fixed in 10% formalin for 2 days and transferred to 70% ethanol. Sections were stained with hematoxylin and eosin and images acquired using ImageJ (NIH).
Fecal fat determination.
Fecal fat content was determined gravimetrically as described previously (52). Briefly, 0.2 g of dried feces was solubilized in 1.6 mL water and homogenized using 1-mm glass beads. Feces were extracted in 6 mL chloroform/methanol (2:1) and the organic phase was transferred to a pre-weighed vial, dried under nitrogen, and reweighed to determine lipid mass.
Isolation of immune cells and flow cytometry.
Murine epididymal WAT, iWAT, and BAT were harvested and digested with 0.1% collagenase type II (Sigma-Aldrich) at 37°C with shaking at 200 rpm for 60 minutes. Digested tissues were filtered through a 100-μm nylon mesh and centrifuged at 500 g for 5 minutes. Floating adipocytes were removed, and the SVF pellet was resuspended in red blood cell lysis buffer (ACK RBC Lysis Buffer). Recovered cells were washed and stained with Zombie UV (1:600; BioLegend) according to the manufacturer’s protocol, followed by surface staining for flow cytometric analysis with fluorochrome-conjugated antibodies. The following antibodies were used: rat anti–mouse CD45–BUV395 (BD Horizon, clone 30-F11; 1:200), rat anti–mouse CD304–BV421 (BioLegend, clone 3E12; 1:300), rat anti–mouse CD86–BV605 (BioLegend, clone GL-1; 1:300), rat anti–mouse/human CD11b–BV650 (BioLegend, clone M1/70; 1:400), hamster anti–mouse CD11c–BV711 (BioLegend, clone N418; 1:300), rat anti–mouse Ly6G–BV785 (BioLegend, clone 1A8; 1:300), rat anti–mouse F4/80–APC (BioLegend, clone BM8; 1:300), rat anti–mouse MHC-II–Alexa Fluor 700 (BioLegend, clone M5/114.15.2; 1:300), rat anti–mouse CD14-APC/Cy7 (BioLegend, clone Sa14-2; 1:200), rat anti–mouse SiglecF–PE (BD Pharmingen, clone E50-2440; 1:400), rat anti–mouse CD206–PE/Cy7 (BioLegend, clone C068C2; 1:200), mouse anti–mouse CD64–PE-Dazzle594 (BioLegend, clone X54-5/7.1; 1:300), and rat anti–mouse Ly6C–FITC (BioLegend, clone HK1.4; 1:300) in Brilliant Stain Buffer (BD Biosciences) containing 10 μg/mL Fc Block (purified unconjugated rat anti–mouse CD16/32; clone 2.4G2, BD Pharmingen). Cells were incubated in the staining cocktail on ice for 30 minutes. Stained cells were washed 3 times in 200 μL FACS buffer (PBS containing 2.5% heat-inactivated FBS and 2.5 mM EDTA) before data acquisition on a BD X20 flow cytometer. Cells were counted using CountBright Absolute Counting Beads (Invitrogen) for flow cytometry according to the manufacturer’s instructions. Cell frequencies were defined as percentage of the parent gate or as percentage of live cells, as indicated.
Lipolysis assay.
For ex vivo lipolysis assay, 20 mg of BAT was collected and cultured in 200 μL of low-glucose DMEM supplemented with 2% fatty acid–free BSA with or without 10 μM isoproterenol in a 96-well plate for 60 minutes at 37°C. Media were collected and glycerol release assays (MilliporeSigma) were performed per the manufacturer’s protocol. Tissue samples were harvested for protein quantification using Bradford reagent.
Immunohistochemical staining.
Paraffin sections (5 μm) were rehydrated and treated with 0.3% hydrogen peroxide in methanol for 15 minutes to suppress the endogenous peroxidase activity. Antigen retrieval was achieved by microwaving the sections in 10 mM citrate buffer for 10 minutes followed by gradual cooling to room temperature. Sections were incubated overnight at 4°C. Immunostaining was detected using the Histostain-SP Broad Spectrum (DAB) kit (Thermo Fisher Scientific, 95-9643).
Hypoxyprobe labeling.
HFD-fed control or Asxl2ΔLysM mice were injected intraperitoneally with 60 mg/kg hypoxyprobe-1 (pimonidazole HCl, Hypoxyprobe, Inc.) in PBS. Gonadal WAT was isolated 75 minutes later and fixed overnight at 4°C. Mice that did not receive hypoxyprobe were analyzed in parallel to serve as a negative control for hypoxyprobe antibody specificity. Detection of hypoxyprobe binding was performed using the Hypoxyprobe-1 Plus Kit (Hypoxyprobe, Inc.).
Macrophage isolation.
Primary BMMs were prepared as described previously (53) with slight modification. Marrow was extracted from femora and tibiae of 6- to 8-week-old mice with α-MEM and cultured in α-MEM containing 10% inactivated FBS, 100 IU/mL penicillin, and 100 μg/mL streptomycin (α-10 medium) with conditioned media (1:10) from CMG12-14 cells (murine M-CSF–producing cell line; ref. 54) on plastic petri dishes. Cells were incubated at 37°C in 6% CO2 for 3 days and then washed with PBS and lifted with 1× trypsin/EDTA in PBS.
NLRP3 inflammasome activation.
Cultured BMMs were primed for 4 hours with LPS (100 ng/mL; Sigma-Aldrich) or TNF-α (10 ng/mL) alone, and then exposed to nigericin (15 μM) for an additional hour to activate the NLRP3 inflammasome. Media were collected for IL-1β ELISA and cells harvested for either Western blot or gene expression analysis. To study NLRP3 inflammasome activation based on caspase-1 activity, cells were cultured on coverslips and primed with LPS for 4 hours. Cells were stimulated with nigericin for 30 minutes followed by incubation with FLICA FAM-YVAD-FMK probe (ImmunoChemistry Technology LLC) for an additional 30 minutes and analyzed by fluorescence microscopy.
Western blot analysis and immunoprecipitation.
Cultured cells were washed twice with ice-cold PBS and lysed in radioimmunoprecipitation assay (RIPA) buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM NaF, and 1× protease inhibitor mixture (Roche Applied Science). Total cell lysates were prepared by homogenizing the tissue in TNET lysis buffer consisting of 1% Triton X-100, 150 mM NaCl, 50 mM Tris pH 7.4, and 2 mM EDTA supplemented with protease and phosphatase inhibitors. After incubation on ice for 10 minutes, cell lysates were clarified by centrifugation at 21,000 g for 10 minutes. Total lysates (40 μg protein) were resolved by 8%–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto PVDF membranes. Filters were blocked in 0.1% casein in PBS for 1 hour and incubated with primary antibodies at 4°C overnight followed by probing with fluorescently labeled secondary antibodies (Jackson ImmunoResearch Laboratories). Proteins were detected with the Odyssey Infrared Imaging System (LI-COR Biosciences).
RNA-seq library preparation and sequencing.
Total RNA integrity was determined using an Agilent Bioanalyzer or 4200 Tapestation. Library preparation was performed with 10 μg of total RNA with a Bioanalyzer RIN score greater than 8.0. Ribosomal RNA was removed by poly-A selection using oligo-dT beads (mRNA Direct kit, Life Technologies). mRNA was then fragmented in buffer containing 40 mM Tris acetate pH 8.2, 100 mM potassium acetate, and 30 mM magnesium acetate and heated to 94°C for 150 seconds. mRNA was reverse transcribed to yield cDNA using SuperScript III RT enzyme (Life Technologies, per the manufacturer’s instructions) and random hexamers. A second-strand reaction was performed to yield double-stranded cDNA. cDNA was blunt ended, had an A base added to the 3′ ends, and then had Illumina sequencing adapters ligated to the ends. Ligated fragments were then amplified for 12 cycles using primers incorporating unique index tags. Fragments were sequenced on an Illumina HiSeq 2500 using single reads extending 50 bases.
RNA-seq data acquisition, quality control, and processing.
RNA-seq reads were aligned to the GRCm38.76 assembly from Ensembl with STAR version 2.0.4b (55). Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 1.4.5. Transcript counts were produced by Sailfish version 0.6.3. Sequencing performance was assessed for total number of aligned reads, total number of uniquely aligned reads, genes and transcripts detected, ribosomal fraction known junction saturation, and read distribution over known gene models with RSeQC version 2.3. All gene-level and transcript counts were then imported into the R/Bioconductor package EdgeR and TMM normalized to adjust for differences in library size. Genes or transcripts not expressed in any sample were excluded from further analysis. Performance of the samples was assessed with a Spearman correlation matrix and multidimensional scaling plots. Generalized linear models with robust dispersion estimates were created to test for gene/transcript level differential expression. The fit of the trended and tagwise dispersion estimates were then plotted to confirm proper fit of the observed mean-to-variance relationship where the tagwise dispersions are equivalent to the biological coefficients of variation of each gene. Differentially expressed genes and transcripts were then filtered for FDR-adjusted P values less than or equal to 0.05.
To enhance the biological interpretation of the large set of transcripts, grouping of genes/transcripts based on functional similarity was achieved using the R/Bioconductor packages GAGE and Pathview. GAGE and Pathview were also used to perform pathway maps on known signaling and metabolism pathways curated by KEGG. The raw data have been deposited to the NCBI’s Gene Expression Omnibus (GEO) repository (GSE144180).
Statistics.
Statistical significance was determined using unpaired, 2-tailed Student’s t test, or 1-way or 2-way ANOVA with the Holm-Sidak multiple-comparisons test. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001 indicate the level of significance in all experiments.
Study approval.
All animal experimentation was approved by the Animal Studies Committee of Washington University School of Medicine.
Author contributions
WZ, NR, JRB, KIS, GM, SAW, NAA, and SLT were responsible for conceptualization. WZ, NR, JRB, JRM, JWW, KIS, SAW, NAA, and SLT developed methodology. SLT validated the data. RDH performed formal analysis. WZ, NR, JRB, YL, JWW, YA, HP, TAP, EPN, and KIS performed investigations. AD, SAW, and NAA acquired resources. RDH, SAW, NAA, and SLT curated data. WZ, NR, and SLT wrote the original draft of the manuscript, which GM, KIS, SAW, and NAA reviewed and edited. NOD, GJR, NAA, and SLT supervised the study. SLT administered the project and acquired funding. WZ and NR are sequentially listed as first authors. Our decision in this regard was based on the following: WZ developed the hypothesis that myeloid deletion of ASXL2 would prevent diet-induced weight gain. He also made the initial discovery that his hypothesis was correct and that the mechanism involved preservation of brown adipose tissue. NR discovered the role of altered energy expenditure and differences in processing catecholamines and suggested and supervised RNA-seq analysis of BMMs. She also brought the BAP1-based experiments to the study. Given that WZ’s contribution was initiating the project and establishing key observations, he has been listed first.
Supplementary Material
Acknowledgments
This study was supported by the following grants from the NIH: K99 HL1388163 (to JWW), AR064755 (to GM), AR068972 (to GM), AR070975 (to GM), HL38180 (to NOD), DK56260 (to NOD), P30 DK52574 (to NOD), P41 EB025815 (to KIS), HL073646 (to SAW), DK102691 (to SAW), AI019653 (to GJR), DK109668 (to GJR), DK056341 (to NAA), AR046523 (to SLT), DK111389 (to SLT), and P30 AR074992 (to SLT and GM). JRB is supported by the Physician-Scientist Training Program at Washington University School of Medicine and the Children’s Discovery Institute. We thank the Cyclotron and the Preclinical PET/CT Imaging facilities at Mallinckrodt Institute of Radiology (MIR) for production of radiopharmaceuticals and imaging studies.
Version 1. 04/20/2020
Electronic publication
Version 2. 05/01/2020
Print issue publication
Footnotes
JWW’s present address is: Department of Integrative Biology and Physiology, University of Minnesota Medical School, Minneapolis, Minnesota, USA.
Conflict of interest: SAW is equity holder in Trasir Therapeutics, Inc. GM is a consultant for Aclaris Therapeutics, Inc.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(5):2644–2656.https://doi.org/10.1172/JCI128687.
Contributor Information
Wei Zou, Email: weizou@wustl.edu.
Nidhi Rohatgi, Email: nidhirohatgi@wustl.edu.
Jonathan R. Brestoff, Email: brestoff@wustl.edu.
John R. Moley, Email: johnmoley@wustl.edu.
Yongjia Li, Email: liyongjia@wustl.edu.
Jesse W. Williams, Email: jww@umn.edu.
Yael Alippe, Email: yaelalippe@wustl.edu.
Hua Pan, Email: huapan@health.usf.edu.
Terri A. Pietka, Email: tpietka@dom.wustl.edu.
Gabriel Mbalaviele, Email: gmbalaviele@wustl.edu.
Elizabeth P. Newberry, Email: epnewberry@wustl.edu.
Nicholas O. Davidson, Email: nod@wustl.edu.
Anwesha Dey, Email: dey.anwesha@gene.com.
Kooresh I. Shoghi, Email: shoghik@mir.wustl.edu.
Richard D. Head, Email: rhead@path.wustl.edu.
Samuel A. Wickline, Email: wickline@usf.edu.
Gwendalyn J. Randolph, Email: gjrandolph@wustl.edu.
Nada A. Abumrad, Email: nabumrad@dom.wustl.edu.
Steven L. Teitelbaum, Email: teitelbs@wustl.edu.
References
- 1.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8(4):301–309. doi: 10.1016/j.cmet.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Izawa T, et al. ASXL2 regulates glucose, lipid, and skeletal homeostasis. Cell Rep. 2015;11(10):1625–1637. doi: 10.1016/j.celrep.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee KY, et al. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes. 2013;62(3):864–874. doi: 10.2337/db12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen EY, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, et al. Exosomal microRNA miR-92a concentration in serum reflects human brown fat activity. Nat Commun. 2016;7:11420. doi: 10.1038/ncomms11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartelt A, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17(2):200–205. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- 10.Shimizu I, et al. Vascular rarefaction mediates whitening of brown fat in obesity. J Clin Invest. 2014;124(5):2099–2112. doi: 10.1172/JCI71643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blondin DP, et al. Dietary fatty acid metabolism of brown adipose tissue in cold-acclimated men. Nat Commun. 2017;8:14146. doi: 10.1038/ncomms14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoneshiro T, et al. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest. 2013;123(8):3404–3408. doi: 10.1172/JCI67803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eisinger K, Liebisch G, Schmitz G, Aslanidis C, Krautbauer S, Buechler C. Lipidomic analysis of serum from high fat diet induced obese mice. Int J Mol Sci. 2014;15(2):2991–3002. doi: 10.3390/ijms15022991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu Y, et al. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell. 2014;157(6):1292–1308. doi: 10.1016/j.cell.2014.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer K, et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat Med. 2017;23(5):623–630. doi: 10.1038/nm.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Camell CD, et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature. 2017;550(7674):119–123. doi: 10.1038/nature24022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pirzgalska RM, et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med. 2017;23(11):1309–1318. doi: 10.1038/nm.4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu D, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332(6026):243–247. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121(6):2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. 2017;127(1):1–4. doi: 10.1172/JCI92035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alippe Y, et al. Bone matrix components activate the NLRP3 inflammasome and promote osteoclast differentiation. Sci Rep. 2017;7(1):6630. doi: 10.1038/s41598-017-07014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strissel KJ, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56(12):2910–2918. doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- 23.Divoux A, et al. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes. 2010;59(11):2817–2825. doi: 10.2337/db10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daou S, et al. The BAP1/ASXL2 histone H2A deubiquitinase complex regulates cell proliferation and is disrupted in cancer. J Biol Chem. 2015;290(48):28643–28663. doi: 10.1074/jbc.M115.661553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou KK, Pan H, Lanza GM, Wickline SA. Melittin derived peptides for nanoparticle based siRNA transfection. Biomaterials. 2013;34(12):3110–3119. doi: 10.1016/j.biomaterials.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hou KK, Pan H, Ratner L, Schlesinger PH, Wickline SA. Mechanisms of nanoparticle-mediated siRNA transfection by melittin-derived peptides. ACS Nano. 2013;7(10):8605–8615. doi: 10.1021/nn403311c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou KK, Pan H, Schlesinger PH, Wickline SA. A role for peptides in overcoming endosomal entrapment in siRNA delivery - A focus on melittin. Biotechnol Adv. 2015;33(6 pt 1):931–940. doi: 10.1016/j.biotechadv.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang CY, Dong X, Gao J, Lin W, Liu Z, Wang Z. Nanoparticle-induced neutrophil apoptosis increases survival in sepsis and alleviates neurological damage in stroke. Sci Adv. 2019;5(11):eaax7964. doi: 10.1126/sciadv.aax7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou HF, et al. Peptide-siRNA nanocomplexes targeting NF-κB subunit p65 suppress nascent experimental arthritis. J Clin Invest. 2014;124(10):4363–4374. doi: 10.1172/JCI75673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan H, et al. Suppression of NF-κB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Proc Natl Acad Sci U S A. 2016;113(41):E6199–E6208. doi: 10.1073/pnas.1608245113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dehghan M, et al. Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): a prospective cohort study. Lancet. 2017;390(10107):2050–2062. doi: 10.1016/S0140-6736(17)32252-3. [DOI] [PubMed] [Google Scholar]
- 32.Roberts MN, et al. A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab. 2017;26(3):539–546.e5. doi: 10.1016/j.cmet.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baughman JM, et al. NeuCode proteomics reveals Bap1 regulation of metabolism. Cell Rep. 2016;16(2):583–595. doi: 10.1016/j.celrep.2016.05.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hatori M, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012;15(6):848–860. doi: 10.1016/j.cmet.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao X, Salomon C, Freeman DJ. Extracellular vesicles from adipose tissue-a potential role in obesity and type 2 diabetes? Front Endocrinol (Lausanne) 2017;8:202. doi: 10.3389/fendo.2017.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta V, Khan AA, Sasi BK, Mahapatra NR. Molecular mechanism of monoamine oxidase A gene regulation under inflammation and ischemia-like conditions: key roles of the transcription factors GATA2, Sp1 and TBP. J Neurochem. 2015;134(1):21–38. doi: 10.1111/jnc.13099. [DOI] [PubMed] [Google Scholar]
- 37.Cai J, Li B, Liu K, Li G, Lu F. Macrophage infiltration regulates the adipose ECM reconstruction and the fibrosis process after fat grafting. Biochem Biophys Res Commun. 2017;490(2):560–566. doi: 10.1016/j.bbrc.2017.06.078. [DOI] [PubMed] [Google Scholar]
- 38.Schnoor M, et al. Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity. J Immunol. 2008;180(8):5707–5719. doi: 10.4049/jimmunol.180.8.5707. [DOI] [PubMed] [Google Scholar]
- 39.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 40.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4(8):583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lech M, Anders HJ. Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta. 2013;1832(7):989–997. doi: 10.1016/j.bbadis.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 42.Spencer M, et al. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am J Physiol Endocrinol Metab. 2010;299(6):E1016–E1027. doi: 10.1152/ajpendo.00329.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowell BB, Spiegelman BM. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000;404(6778):652–660. doi: 10.1038/35007527. [DOI] [PubMed] [Google Scholar]
- 44.Golozoubova V, Gullberg H, Matthias A, Cannon B, Vennström B, Nedergaard J. Depressed thermogenesis but competent brown adipose tissue recruitment in mice devoid of all hormone-binding thyroid hormone receptors. Mol Endocrinol. 2004;18(2):384–401. doi: 10.1210/me.2003-0267. [DOI] [PubMed] [Google Scholar]
- 45.Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009;9(2):203–209. doi: 10.1016/j.cmet.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 46.Kazak L, et al. UCP1 deficiency causes brown fat respiratory chain depletion and sensitizes mitochondria to calcium overload-induced dysfunction. Proc Natl Acad Sci U S A. 2017;114(30):7981–7986. doi: 10.1073/pnas.1705406114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weir G, et al. Substantial metabolic activity of human brown adipose tissue during warm conditions and cold-induced lipolysis of local triglycerides. Cell Metab. 2018;27(6):1348–1355.e4. doi: 10.1016/j.cmet.2018.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shashi V, et al. De novo truncating variants in ASXL2 are associated with a unique and recognizable clinical phenotype. Am J Hum Genet. 2016;99(4):991–999. doi: 10.1016/j.ajhg.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J, et al. Loss of Asxl2 leads to myeloid malignancies in mice. Nat Commun. 2017;8:15456. doi: 10.1038/ncomms15456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dey A, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012;337(6101):1541–1546. doi: 10.1126/science.1221711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Funai K, et al. Muscle lipogenesis balances insulin sensitivity and strength through calcium signaling. J Clin Invest. 2013;123(3):1229–1240. doi: 10.1172/JCI65726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Newberry EP, Xie Y, Kennedy SM, Luo J, Davidson NO. Protection against Western diet-induced obesity and hepatic steatosis in liver fatty acid-binding protein knockout mice. Hepatology. 2006;44(5):1191–1205. doi: 10.1002/hep.21369. [DOI] [PubMed] [Google Scholar]
- 53.Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106(12):1481–1488. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;(8):1477–1488. doi: 10.1359/jbmr.2000.15.8.1477. [DOI] [PubMed] [Google Scholar]
- 55.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.