

Abstract
BACKGROUND
Glucose-6-phosphate dehydrogenase (G6PD) deficiency decreases the ability of red blood cells (RBCs) to withstand oxidative stress. Refrigerated storage of RBCs induces oxidative stress. We hypothesized that G6PD-deficient donor RBCs would have inferior storage quality for transfusion as compared with G6PD-normal RBCs.
METHODS
Male volunteers were screened for G6PD deficiency; 27 control and 10 G6PD-deficient volunteers each donated 1 RBC unit. After 42 days of refrigerated storage, autologous 51-chromium 24-hour posttransfusion RBC recovery (PTR) studies were performed. Metabolomics analyses of these RBC units were also performed.
RESULTS
The mean 24-hour PTR for G6PD-deficient subjects was 78.5% ± 8.4% (mean ± SD), which was significantly lower than that for G6PD-normal RBCs (85.3% ± 3.2%; P = 0.0009). None of the G6PD-normal volunteers (0/27) and 3 G6PD-deficient volunteers (3/10) had PTR results below 75%, a key FDA acceptability criterion for stored donor RBCs. As expected, fresh G6PD-deficient RBCs demonstrated defects in the oxidative phase of the pentose phosphate pathway. During refrigerated storage, G6PD-deficient RBCs demonstrated increased glycolysis, impaired glutathione homeostasis, and increased purine oxidation, as compared with G6PD-normal RBCs. In addition, there were significant correlations between PTR and specific metabolites in these pathways.
CONCLUSION
Based on current FDA criteria, RBCs from G6PD-deficient donors would not meet the requirements for storage quality. Metabolomics assessment identified markers of PTR and G6PD deficiency (e.g., pyruvate/lactate ratios), along with potential compensatory pathways that could be leveraged to ameliorate the metabolic needs of G6PD-deficient RBCs.
TRIAL REGISTRATION
ClinicalTrials.gov NCT04081272.
FUNDING
The Harold Amos Medical Faculty Development Program, Robert Wood Johnson Foundation grant 71590, the National Blood Foundation, NIH grant UL1 TR000040, the Webb-Waring Early Career Award 2017 by the Boettcher Foundation, and National Heart, Lung, and Blood Institute grants R01HL14644 and R01HL148151.
Keywords: Hematology
Keywords: Monogenic diseases

Introduction
Red blood cell (RBC) transfusion is one of the most common therapeutic interventions for hospitalized patients (1). In particular, chronic transfusions are essential for patients with sickle cell disease and thalassemia, due to intrinsic RBC defects and/or ineffective erythropoiesis in these patients. Accelerated clearance of transfused RBCs contributes to some adverse effects associated with chronic RBC transfusion therapy, such as iron overload, alloimmunization, and, possibly, increased susceptibility to infection (2–6). As such, multiple efforts are directed toward providing optimal RBC products. The Food and Drug Administration (FDA) sets acceptability criteria for RBC units at the end of their maximum allowable storage period (e.g., 42 days), which are based primarily on a 24-hour posttransfusion recovery (PTR) of at least 75% on average (i.e., 75% of the transfused RBCs should still be circulating 24 hours later) and an in vitro hemolysis rate of less than 1%. Furthermore, the one-sided, lower limit of the 95% confidence interval for the proportion of successful PTRs (defined as PTR ≥75%) must be at least 70% (i.e., no more than 2 failed PTRs of <75% among a cohort of 20 healthy volunteer blood donors). Interestingly, PTRs vary substantially among blood donors (7) and these differences are specific and repeatable for each donor, suggesting that some donors are good storers and some are poor storers (8). In vitro analyses of stored RBCs identified interdonor metabolic heterogeneity, which can affect the metabolic age of a stored RBC unit at least as much as the chronological age of the unit (5, 9). In addition, because RBC storage quality is heritable (10–12), genetic factors may underlie at least some of this variation.
Glucose-6-phosphate dehydrogenase (G6PD) deficiency, an X-linked disorder, is the most common human enzymopathy, affecting approximately 400 million people worldwide (13). In addition, G6PD is the rate-limiting enzyme of the pentose phosphate pathway (PPP), which generates reduced nicotinamide adenine dinucleotide phosphate (NADPH), a cofactor that drives multiple antioxidant pathways in RBCs (14). Indeed, NADPH is critical for recycling oxidized glutathione (i.e., GSSG) to its reduced form (i.e., GSH) by glutathione reductase. In addition, it fuels multiple NADPH-dependent antioxidant enzymes (e.g., glutathione peroxidase, catalase, peroxiredoxins, glutaredoxins, the thioredoxin reductase system, biliverdin reductase B, and the ascorbate-tocopherol axis) (15). Because G6PD-deficient RBCs have a diminished capacity for producing NADPH (16), they are susceptible to damage and destruction by increased oxidative stress, such as that caused by medications, infection, or diet (17).
Oxidative stress biomarkers increase during refrigerated storage (18, 19), suggesting that storage itself may cause oxidative stress. In addition, when RBCs are stored under hypoxic conditions (20) or in the presence of the antioxidant ascorbic acid (21), which decrease oxidative stress, PTR markedly improves in humans and mice, respectively. Clearly, RBCs did not evolve to cope with the oxidant stress arising during refrigerated storage. However, RBCs have developed mechanisms to cope with oxidant stress during aging in vivo, and some of these are activated during routine blood bank storage. For example, studies with stable isotope-labeled tracers suggest that storage-induced oxidation of Cys152 of the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH) causes a shift in glucose metabolism toward the oxidative phase of the PPP; this phenomenon is mitigated or aggravated by hypoxic or hyperoxic storage, respectively (22). Because G6PD is the key enzyme in the oxidative phase of the PPP, G6PD-deficiency limits NADPH production in RBCs, thereby decreasing their ability to regenerate GSH and prevent the accumulation of peroxidation/inflammatory products (23). Indeed, glutathione homeostasis and antioxidant defenses are altered in units obtained from G6PD-deficient donors (23, 24). However, these studies were only performed with donors carrying the Class II Mediterranean G6PD variant (<10% residual activity) and not with donors affected by the milder, but more common, Class III A- variant (10%–60% residual activity); the latter affects approximately 10% of the African-American donor population (25). Moreover, no study to date has evaluated the correlation of metabolomics changes in stored human G6PD-deficient RBCs to the gold-standard PTR outcome.
Although American blood donors are not routinely screened for G6PD deficiency, some blood collection agencies will exclude a prospective donor who reports a history of being G6PD-deficient. In a study from 1970, the PTR of stored G6PD-deficient RBCs was decreased in comparison with RBCs from G6PD-normal donors (26). However, given the advent of modern additive solutions, which enhance RBC storage duration, and the introduction of leukoreduction, which mitigates the RBC storage lesion (27), it is not known whether G6PD deficiency remains a source of variation in donor RBC storage quality. Because G6PD-deficient RBCs are part of the routine inventory of transfusion services (at a prevalence of ~0.3% ), and because chronically transfused patients with sickle cell disease require antigen-matched RBCs that frequently come from donors of African descent who have a high frequency of G6PD deficiency, the latter are more likely to receive G6PD-deficient RBC units (i.e., up to 12.3% of units transfused) (25). Therefore, transfusions of G6PD-deficient RBCs into these patients may have reduced therapeutic efficacy (28). Indeed, several case reports document adverse clinical outcomes following transfusion of G6PD-deficient RBCs (29–31), raising the question of whether donors should be screened for G6PD deficiency (32). The current study examined the storage quality of RBCs from G6PD-deficient and G6PD-normal donors, using 24-hour PTR as the primary outcome measure. Furthermore, correlations between metabolomics findings, G6PD activity, and PTR are provided.
Results
Study participant demographics and recruitment.
A total of 145 male volunteers were screened for G6PD deficiency. To enrich for subjects with a greater probability of being G6PD-deficient, subjects of African, Asian, Hispanic, Mediterranean, and Middle Eastern descent, based upon maternal origin, were specifically recruited. Of the screened volunteers, 42 (29%) were of African descent, 32 (22.1%) were of Asian descent, 30 (20.7%) were Hispanic, 26 (17.9%) were of Mediterranean descent, 7 (4.8%) were of Middle Eastern descent, and 8 (5.5%) were White. Fifteen volunteers (10%) were G6PD deficient and 130 (90%) were G6PD normal. Of the G6PD-deficient volunteers, 10 were of African descent, 3 were of Mediterranean descent, and 2 were Hispanic. Ten G6PD-deficient (deficient) and 30 G6PD-normal (control) volunteers agreed to participate in the study and donated one unit of leukocyte-reduced RBCs stored in additive solution 3 (AS-3) (Figure 1). Two volunteers from the control group then withdrew from the study and 1 volunteer from the control group was excluded due to positive viral serologic testing following blood donation. Therefore, 27 control and 10 G6PD-deficient subjects completed the entire study; the demographics of these subjects are shown in Table 1.
Figure 1. CONSORT diagram.

Number of subjects who were screened or excluded, dropped out, or completed the study.
Table 1. Demographic characteristics of subjects completing the study.
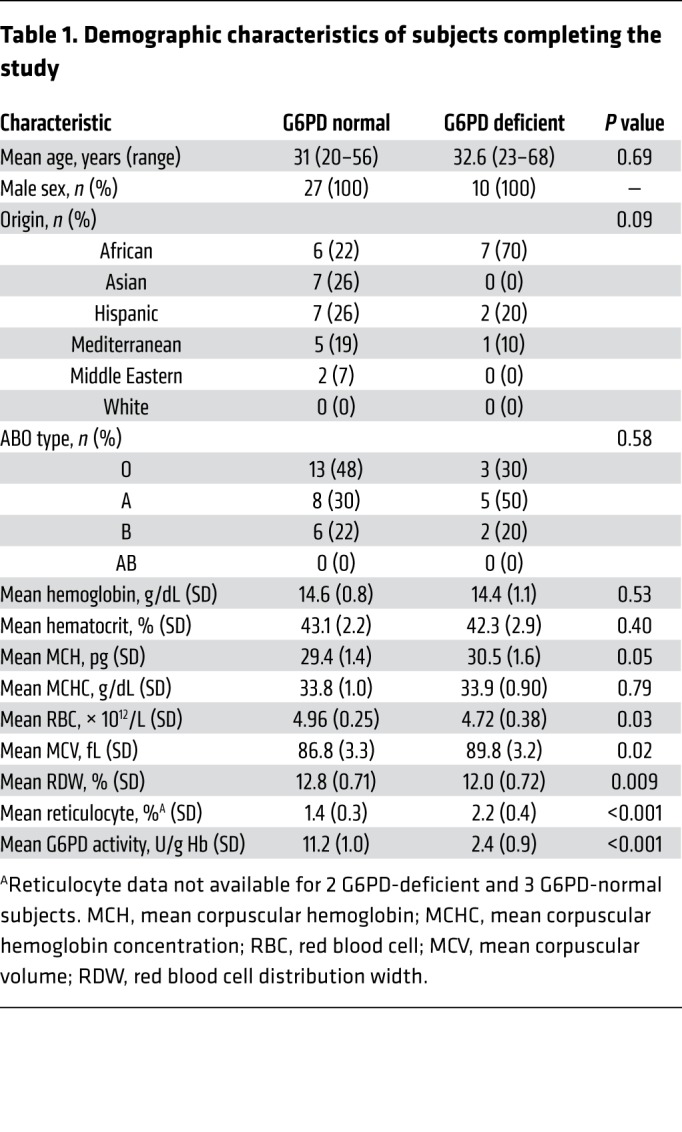
G6PD enzyme activity levels for participating subjects are provided in Supplemental Figure 1 (supplemental material available online with this article; https://doi.org/10.1172/JCI133530DS1). G6PD activity of at least 9.9 U/g Hb was considered normal based on the referral laboratory reference range. One control subject had a G6PD activity 12% less than the reference range (8.7 U/g Hb), but G6PD exon sequencing did not identify any mutations. Although the presence of a rare splicing, promoter, or untranslated region mutation was not ruled out for this particular volunteer (33), he was considered to be in the control, G6PD-normal group due to the proximity of his G6PD enzymatic activity to the lower limit of the reference range. Furthermore, a sensitivity analysis categorizing this subject as G6PD-deficient did not change the primary outcome (data not shown). Overall, 9 G6PD-deficient subjects were identified with the A- variant (Class III) and 1 with the Mediterranean variant (Class II). There were no significant differences in the mean baseline hemoglobin of control (14.6 ± 0.8 g/dL) and deficient (14.4 ± 1.1 g/dL) (P = 0.53) subjects, mean baseline hematocrit of control (43.1% ± 2.2%) and deficient (42.3% ± 2.9%) (P = 0.40) subjects, mean baseline mean corpuscular hemoglobin (MCH) of control (29.4 ± 1.4 pg) and deficient (30.5 ± 1.6 pg) (P = 0.05) subjects, and mean baseline mean corpuscular hemoglobin concentration (MCHC) of control (33.8 ± 1.0 g/dL) and deficient (33.9 ± 0.9 g/dL) (P = 0.79) subjects. In contrast, the mean RBC count was significantly lower in deficient (4.72 ± 0.38 × 1012/L) than normal (4.96 ± 0.25 × 1012/L) (P = 0.03) subjects. The mean reticulocyte count of deficient subjects was significantly higher (2.2% ± 0.8%) than control subjects (1.4% ± 0.3%) (P < 0.001). However, it was still below the upper limit of normal for our laboratory’s reference range (2.26%). In addition, the mean corpuscular volume (MCV) was significantly higher in deficient (89.8 ± 3.2 fL) than normal (86.8 ± 3.3 fL) (P = 0.02) subjects and the red blood cell distribution width (RDW) was significantly lower in deficient (12.0% ± 0.72%) compared with normal (12.8% ± 0.71%) (P = 0.009) subjects. As expected, the mean G6PD activity of deficient subjects (2.4 ± 0.9 U/g Hb) was significantly lower than for controls (11.2 ± 1.0 U/g Hb) (P < 0.001; Table 1).
G6PD deficiency decreases PTR.
The 24-hour PTR for the 27 G6PD-normal subjects was 85.3% ± 3.2% (mean ± SD), as compared with 78.5% ± 8.4% for the 10 G6PD-deficient subjects (P = 0.0009; Figure 2A). Although none of the RBC units in the control group (0/27) had a 24-hour PTR less than 75%, the key FDA criterion for stored RBC quality, 3 G6PD-deficient RBC units (3/10) had PTRs below 75% (63.7%, 68.0%, and 74.2%). Thus, based on this number of failures (3/10), RBC units collected from G6PD-deficient subjects, as a group, do not meet current FDA criteria for acceptability (7). The PTRs stratified among subjects of African descent (86.6% ± 2.0% and 78.5% ± 8.8% for G6PD-normal and G6PD-deficient, respectively; P < 0.05; Figure 2B) and non–African descent (85.0% ± 3.4% and 78.4% ± 9.3% for G6PD-normal and G6PD-deficient, respectively; P < 0.05; Figure 2C) were similar. Finally, although there was an association between G6PD enzyme activity and PTR across all subjects (R2 = 0.27, P = 0.001; Figure 2D), there was no significant association when adjusted for G6PD-status (i.e., variation in G6PD activity does not predict PTR among G6PD-deficient or G6PD-normal subjects when examined separately). Of note, the single subject with the Mediterranean variant and G6PD activity of 0.3 U/g Hb had a relatively normal PTR (81.4%) despite having the lowest G6PD enzyme activity in the study population.
Figure 2. G6PD deficiency reduces 24-hour PTR.

(A) The 51-chromium PTRs for G6PD-normal (n = 27) and G6PD-deficient (n = 10) subjects are shown. The dotted gray line denotes the FDA criterion for acceptability (at outdate, on average >75% of transfused RBCs should still be circulating for 24 hours). (B) The 51-chromium PTRs for G6PD-normal (n = 6) and G6PD-deficient (n = 7) subjects of African origin and (C) non–African origin are shown (n = 21 and n = 3 for G6PD-normal and G6PD-deficient, respectively). Mean and SD are shown. Statistical significance calculated using an unpaired t test. *P < 0.05, ***P < 0.001. (D) Correlation between G6PD enzyme activity and PTR is shown. Results of linear regression for all subjects (N = 37) is shown with the 95% CI in dashed gray lines (R2 = 0.27; P = 0.001). Within-group results for G6PD-deficient (red) and G6PD-normal (blue) volunteers are depicted along with the results of the corresponding linear regression (red and blue lines, respectively), which are not statistically significant by Pearson correlation. The unfilled red circle represents the single subject with the Mediterranean variant.
G6PD deficiency decreases methemoglobin, but does not affect in vitro storage hemolysis.
Both control and G6PD-deficient RBCs had in vitro storage hemolysis rates of less than 1% throughout storage (0.29% ± 0.18% and 0.24% ± 0.07% at 6 weeks, respectively; adjusted P = 0.79 for difference; Figure 3A), thereby meeting another important FDA criterion of RBC storage quality. However, as compared with G6PD-normal units, a trend toward decreasing methemoglobin was observed over time in G6PD-deficient stored units (P = 0.086); these differences were statistically significant after 3 and 6 weeks of storage (Figure 3B). Although there was also an increasing trend in lactate, and a decreasing trend in pH, glucose, and sodium during storage, there were no differences between G6PD-deficient and control units (Figure 3, C–F).
Figure 3. Standard in vitro measures of RBC units throughout storage.

(A) Spontaneous hemolysis, (B) methemoglobin, (C) lactate, (D) pH, (E) glucose, and (F) sodium were measured in aliquots obtained from each unit at the designated storage time. G6PD-normal (blue; n = 27) and G6PD-deficient (red; n = 10) are represented. Mean and SD are shown. Statistical significance was calculated using 2-way repeated-measures ANOVA with Sidak’s multiple comparisons test; *P < 0.05. Thick dotted red line denotes the FDA criterion for the allowable spontaneous hemolysis rate during storage.
G6PD-deficiency increases glycolysis.
At baseline (i.e., before refrigerated storage), metabolomics analyses of RBCs from G6PD-deficient volunteers show evidence of increased glycolysis as compared with G6PD-normal RBCs (Figure 4). Glycolytic intermediates (Figure 4A), including bisphosphoglycerate (BPG), 2/3-phosphoglycerate (2/3PG), phosphoenolpyruvate (PEP), and lactate, were significantly increased in G6PD-deficient RBCs (all P < 0.01; Figure 4, B–D and F). Consistent with these increases in glycolytic intermediates, G6PD-deficient RBCs also had significantly increased ATP levels (P < 0.01; Figure 4G) as compared with controls. Pyruvate levels did not differ significantly between G6PD-deficient and G6PD-normal RBCs (Figure 4E).
Figure 4. Differences in glycolysis between G6PD-deficient and G6PD-normal RBCs at baseline.

(A) Schematic of the glycolytic pathway. The levels of glycolytic intermediates were measured in RBCs collected directly from volunteers and measured before storage. (B) bisphosphoglycerate, (C) 2/3 phosphoglycerate, (D) phosphoenolpyruvate, (E) pyruvate, (F) lactate, and (G) ATP in G6PD-normal (blue; n = 27) and G6PD-deficient (red; n = 10) RBCs are shown. Median with interquartile range represented. Unfilled red circles represent the single subject with the Mediterranean variant. Statistical significance was assessed by the Mann Whitney U test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
G6PD-deficient individuals cannot upregulate the oxidative branch of the PPP.
RBCs from G6PD-deficient individuals demonstrate a defective oxidative branch of the PPP (data are reported in tabulated form in Supplemental Tables 1 and 2, and in vectorial form in Supplemental Figures 2–4). These findings were confirmed by tracing experiments with [1,2,3-13C3]-glucose in the presence or absence of methylene blue, a PPP-stimulating agent (Figure 5A). Thus, by tracing heavy carbon moieties into glycolytic and PPP intermediates and by-products (including isotopologues M+2 and M+3 of lactate), we confirmed that fresh G6PD-normal RBCs, but not fresh G6PD-deficient RBCs, could activate the oxidative branch of the PPP (Figure 5). Specifically, methylene blue treatment did not change [13C3]-glucose phosphate levels in fresh or stored G6PD-deficient and G6PD-normal RBCs, suggesting there was no difference in flux through the glycolytic pathway (Figure 5B). Nonetheless, methylene blue treatment increased [13C2]-lactate levels and the ratio of [13C2]-lactate/[13C3]-glucose phosphate levels only in fresh G6PD-normal RBCs, suggesting increased flux through the PPP at baseline, but only in G6PD-normal subjects (Figure 5, C and D; P < 0.05). Stored G6PD-deficient and G6PD-normal RBCs were unable to activate the oxidative phase of the PPP (Figure 5C). Finally, in G6PD-deficient RBCs at baseline, pentose phosphates were derived more from glycolytic intermediates (e.g., increased [13C3]-pentose phosphate; P < 0.0001), than from the oxidative phase of the PPP (Figure 5, E and F). Therefore, before refrigerated storage, G6PD-deficient RBCs are characterized by increased glycolysis and decreased ability to activate the oxidative phase of the PPP, the latter of which is important for producing NADPH.
Figure 5. Defective activation of the PPP in response to methylene blue stimulation in G6PD-deficient RBCs.

(A) Fresh (unstored) or 6-week-stored RBC lysates were incubated with [1,2,3-13C3]-glucose and then treated with methylene blue to stimulate the oxidative phase of the PPP. The presence of 3-carbon moieties was measured and isotopologues compared. 13C2 3-carbon species indicate metabolism by the PPP and 13C3 species indicate metabolism by glycolysis. (B) Glycolytic pathway activity, as measured by [13C3]-glucose phosphate levels, (C) PPP pathway activity, as measured by [13C2]-lactate levels, (D) the ratio of [13C2]-lactate levels/[13C3]-glucose phosphate levels indicating the ratio of PPP/glycolysis, and (E) [13C3]-pentose phosphate demonstrating the pentose phosphate derived from glycolysis pathway activity in fresh and stored (as labeled) G6PD-normal (blue; n = 27) and G6PD-deficient (red; n = 10) RBCs, with either vehicle (circles) or methylene blue treatment (squares), were all determined. Medians with interquartile ranges are represented. Unfilled symbols represent the single subject with the Mediterranean variant. Statistical significance calculated using 2-way ANOVA with Sidak’s multiple comparisons test; *P < 0.05, ****P < 0.0001. (F) Schematic demonstrating that pentose phosphates are derived more from the contributions of glycolytic intermediates, rather than the oxidative phase of the PPP, in G6PD-deficient RBCs.
Metabolomics during storage.
Principal component analysis (PCA) of metabolite differences in the glycolytic and PPP pathways (Figure 6A) comparing G6PD-normal and G6PD-deficient RBCs during 6 weeks of refrigerated storage revealed changes that evolve related to storage time and G6PD status (Figure 6B). There were no significant differences in glucose levels (Figure 6C) and fructose 1,6-bisphosphate levels (Figure 6F) between G6PD-deficient and G6PD-normal RBCs. However, the levels of glucose 6-phosphate (G6P) and 6-phosphogluconolactone (6-PGL) exhibited a significant interaction with G6PD status over time (Figure 6, D and E), with median levels crossing each other at 2 to 4 weeks of storage. Similar to baseline, G6PD-deficient RBCs had increased glycolysis during storage, as compared with G6PD-normal RBCs, with significantly higher 2/3 PG and pyruvate levels throughout (Figure 6, I and J). The latter are products of the 2 ATP-producing reactions during glycolysis (Figure 6A), which are important for maintaining RBC ATP levels.
Figure 6. G6PD-deficient RBCs are characterized by altered glycolysis and PPP activity during storage.

(A) Schematic of glycolysis with branching to the PPP. Metabolite levels were measured in RBCs during weeks 1 to 6 of refrigerated storage. (B) PCA of the metabolites throughout storage. (C) Glucose, (D) glucose-6-phosphate, (E) 6-phosphogluconolactone, (F) fructose 1,6 bisphosphate, (G) glyceraldehyde-3-phosphate, (H) bisphosphoglycerate, (I) 2/3-phosphoglycerate, (J) pyruvate, (K) lactate, (L) pyruvate/lactate ratio, (M) ATP. Medians with interquartile ranges are shown. G6PD-normal (blue; n = 27) and G6PD-deficient (red; n = 10) are represented. The dotted red line with the unfilled circle represents the single subject with the Mediterranean variant. Statistical significance was calculated using 2-way repeated-measures ANOVA with Sidak’s multiple comparisons test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Significance for the effect of storage time and the interaction between G6PD status and time are as labeled, or not significant if not shown. Significant time points are shown and significant differences between G6PD-deficient and G6PD-normal subjects are denoted with a bracket to the right of the curves. When the interaction term was significant, the significance of the main effects was assessed in MetaboAnalyst.
In contrast to results at baseline, at which point RBCs have not yet been exposed to the oxidant stress of storage, there are only minor differences between G6PD-deficient and G6PD-normal RBCs in lactate (Figure 6K) and ATP (Figure 6M) throughout storage. Nonetheless, similar to pyruvate, the pyruvate/lactate ratio (Figure 6L) is significantly higher in G6PD-deficient RBCs throughout storage. Of note, the reaction catalyzed by lactate dehydrogenase (LDH) converts pyruvate to lactate and consumes NADH in the process. However, higher oxidant stress in G6PD-deficient RBCs promotes the activation of NADH-dependent cytochrome-b5 reductase, which would compete with LDH for free NADH. Our results suggest that G6PD-deficient RBCs limit the LDH-catalyzed reaction during storage to favor the activity of cytochrome-b5 reductase, consistent with our previous observation that this enzyme is quantitatively more abundant in RBCs from G6PD-deficient subjects in comparison to controls (23). Thus, G6PD-deficient RBCs have increased glycolysis as compared with G6PD-normal RBCs during storage, but they are also characterized by significantly higher (P < 0.0001) pyruvate/lactate ratios.
Differences in NADPH- and NADH-dependent reactions.
Consistent with the expected defect from G6PD deficiency, G6PD-deficient RBCs demonstrated a decrease in NADPH-dependent reactions and an increase in reactions that can produce NADPH (Figure 7). For example, reduction of GSSG to GSH is an NADPH-dependent reaction and, consistent with this, G6PD-deficient RBCs had significantly lower levels of GSH throughout storage (Figure 7A). Similarly, reducing biliverdin to bilirubin consumes NADPH, and G6PD-deficient RBCs had significantly increased biliverdin throughout storage (Figure 7B), suggesting decreased activity in this pathway. Converting glucose to hexose sugar alcohols (e.g., sorbitol) is also an NADPH-dependent process that may be downregulated in G6PD-deficient RBCs, as evidenced by significantly decreased hexose sugar alcohols throughout storage (Figure 7C). Furthermore, dihydroxynonene (DHN), which is produced from 4-hydroxynonenal (HNE) by an NADPH-consuming reaction, is significantly decreased in G6PD-deficient, as compared with G6PD-normal, RBCs during storage (Figure 7D).
Figure 7. NADPH-dependent/generating metabolic pathways affected by G6PD deficiency in stored RBCs.

Metabolite levels were measured in RBCs during weeks 1 to 6 of refrigerated storage. Schematic of the NADPH-dependent/generating pathway shown with graphs of relevant metabolites throughout storage to the right in each figure panel. (A) Reduction of oxidized glutathione (GSSG) to reduced glutathione (GSH) and glutathionylation of cysteine residues (S-glutathionyl-Cys), (B) conversion of biliverdin to bilirubin, (C) conversion of glucose to hexose sugar alcohol, (D) conversion of 4-hydroxynonenal (HNE) to dihydroxynonene (DHN), and (E) conversion of malate to pyruvate. Medians with interquartile ranges are shown. G6PD-normal (blue; n = 27) and G6PD-deficient (red; n = 10) are represented. The dotted red lines with the unfilled circles represent the single subject with the Mediterranean variant. Statistical significance was calculated using 2-way repeated-measures ANOVA with Sidak’s multiple comparisons test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Significance for the effect of storage time and the interaction between G6PD status and time are as labeled, or not significant if not shown. Significant time points are shown and significant differences between G6PD-deficient and G6PD-normal subjects are denoted with a bracket to the right of the curves. When the interaction term was significant, the significance of the main effects was assessed in MetaboAnalyst.
Tricarboxylic acid (TCA) cycle enzymes are found in RBCs, along with their corresponding substrates and products (34, 35). As examples, converting isocitrate to α-ketoglutarate produces NADH, whereas converting malate to pyruvate via malic enzyme, which is present and active in RBCs (36), produces NADPH. Consistent with the hypothesis that G6PD-deficient RBCs conserve NADH and increase NADPH production through non-G6PD catalyzed reactions, they have significantly higher levels of α-ketoglutarate (Figure 7E) and pyruvate (Figure 6J) throughout storage, as compared with G6PD-normal RBCs. Taken together, although steady-state measurements do not allow us to conclude directionality of the aforementioned pathways (i.e., we cannot conclude whether we are observing a decrease in the activation of the NADH/NADPH-dependent reaction or an increase in the NADH/NADPH-generating reaction), these results suggest that G6PD-deficient RBCs are characterized by alterations of several NADH- and NADPH-dependent pathways.
Metabolites in the nonoxidative phase of the PPP.
The nonoxidative phase of the PPP is connected to the oxidative phase, through 6-phosphogluconate (6PG) dehydrogenase, which produces ribulose 5-phosphate (37). Glycolysis also contributes to the nonoxidative phase of the PPP through reversible interactions involving glyceraldehyde 3-phosphate (G3P) and fructose 6-phosphate (F6P) via the activity of transketolase and transaldolase (Figure 8A). At baseline, G6PD-deficient RBCs demonstrate increased nonoxidative PPP activity, as compared with G6PD-normal RBCs, evidenced by significantly increased concentrations of sedoheptulose phosphate (SP) (P = 0.005) and erythrose 4-phosphate (E4P) (P = 0.004) (Figure 8, C and D). Furthermore, during storage there is a trend toward higher ribose phosphate (RP) and SP in G6PD-deficient RBCs (Figure 8, B and C). Therefore, despite having a compromised oxidative phase of the PPP, G6PD-deficient RBCs maintain high levels of nonoxidative phase intermediates as compared with G6PD-normal RBCs. Data from the tracing experiments (Figure 5, E and F) suggest that glycolysis provides much of the nonoxidative phase intermediates in G6PD-deficient RBCs. These results may also be consistent with inefficient nucleotide salvage, adversely affecting ATP levels, which decline to favor ATP breakdown into AMP and its oxidation to IMP and hypoxanthine.
Figure 8. The nonoxidative phase of the PPP is maintained in G6PD-deficient RBCs.

(A) Schematic of the nonoxidative phase of the PPP and its connection with glycolysis in gray. Metabolite levels were measured in RBCs before storage (fresh) and during weeks 1 to 6 of refrigerated storage. The metabolites (B) ribose phosphate, (C) sedoheptulose phosphate, and (D) erythrose 4-phosphate are shown. Medians with interquartile ranges are shown. G6PD-normal (blue; n = 27) and G6PD-deficient (red; n = 10) are represented. The dotted red lines with unfilled red circles represent the single subject with the Mediterranean variant. Statistical significance was calculated by Mann Whitney U test (fresh) or by 2-way repeated-measures ANOVA with Sidak’s multiple comparisons test (storage); *P < 0.05; **P < 0.01, ****P < 0.0001. Significance for the effect of time and the interaction between G6PD status and time are as labeled, or not significant if not shown. Significant differences between G6PD-deficient and G6PD-normal subjects are denoted with a bracket to the right of the curves.
Pathways and metabolites correlating with RBC G6PD activity and PTR.
Metabolic markers predictive of PTR, particularly in the context of G6PD deficiency, have not yet been reported in the literature. To this end, we correlated our PTR data to discovery-mode metabolomics analyses of RBCs from G6PD-deficient and G6PD-normal subjects. Thus, PTR measurements were correlated to metabolomics data from the total study population, G6PD deficient subjects, or G6PD-normal subjects at baseline (Figure 9, A–C, respectively) or at the end of storage (week 6; Figure 9, D–F). Correlation analyses are reported in the form of circos plots, in which each metabolite or other variable (e.g., PTR or G6PD activity) is a node. Nodes are connected with an edge if the module of their linear correlation (Spearman) is above 0.4. Highlighted metabolites represent the most significant variables that correlate with either G6PD activity or PTR in the total population of subjects at baseline (Figure 9A) or at week 6 (Figure 9D), only in the G6PD-deficient subjects at baseline (Figure 9B) or at week 6 (Figure 9E), or in the G6PD-normal subjects at baseline (Figure 9C) or at week 6 (Figure 9F). These results highlight a significant linkage between G6PD activity and PTR only when the total subject population is analyzed in a combined fashion. Top correlates to PTR and G6PD activity are identified, including metabolic intermediates of the PPP (e.g., ribose phosphate, erythrose phosphate), oxidation reactions and nucleotide salvage (e.g., hypoxanthine, IMP), transamination (α-ketoglutarate, pyridoxal), lipid remodeling (several acyl-carnitines and octenoyl-carnitine), tryptophan metabolites (formyl-kynurenine), and conjugated bile acids (glyco- and taurocholate). Overall, metabolites involved in lipid remodeling, energy and redox metabolism, and nucleotide salvage had the best correlation with PTR (Figure 9, G–L). For example, octenoyl-carnitine (Figure 9G), involved in lipid remodeling, ribose phosphate (Figure 9H), in the nonoxidative phase of the PPP, and epiandrosterone, a steroid hormone, at baseline had some of the best correlations with PTR. In addition, α-ketoglutarate (Figure 9J), produced by an NADH-generating reaction, hypoxanthine (Figure 9K), involved in purine salvage reactions, and octenoyl-carnitine (Figure 9L) measured after 6 weeks of storage had some of the strongest correlations with PTR.
Figure 9. Metabolic alterations at baseline and end of storage in G6PD-deficient RBCs correlate with G6PD activity and PTR.

Circos plots of metabolic correlates to PTR and G6PD activity in the total population of subjects at baseline (A) or after 6 weeks of storage (D), in only G6PD-deficient subjects at baseline (B) or week 6 (E), or only G6PD-normal subjects at baseline (C) or week 6 (F). Metabolites are represented as nodes and connected by an edge if their linear correlation (Spearman) is higher than 0.4. Highlighted nodes represent the most significant correlates (Spearman, FDR adjusted P values < 0.05) with PTR (black spheres) or G6PD activity (blue spheres). The correlation of baseline levels of octenoyl-carnitine (G), ribose phosphate (H), and epiandrosterone (I); and week 6 levels of α-ketoglutarate (J), hypoxanthine (K), and octenoyl-carnitine (L) in the total subject population, or limited to only G6PD-deficient or G6PD-normal subjects, respectively. *P < 0.05, **P < 0.01. G6PD-normal (blue; n = 27) and G6PD-deficient (red; n = 10) subjects are represented. The unfilled red circle represents the single subject with the Mediterranean variant.
Discussion
Our results demonstrate that, after 40 to 42 days of storage, leukoreduced, G6PD-deficient RBCs have a decreased 24-hour PTR as compared with G6PD-normal RBCs. These results with G6PD-normal subjects are similar to other published 24-hour PTR studies using AS-3 preserved units after 6 weeks of storage (38). In contrast, if analyzed separately, RBC units from G6PD-deficient donors would not pass the current FDA criteria for clinical transfusion acceptability (7). Chronically transfused patients may be adversely affected by nonoptimal RBC transfusions, and patients with sickle cell disease, in particular, are exposed to a significant number of transfusions from G6PD-deficient donors (i.e., up to 12%) (25). Thus, the current results raise concerns regarding potential harm to patients transfused with G6PD-deficient RBCs.
Previous studies have demonstrated significantly increased reticulocytes and MCV in G6PD-deficient individuals with the A- and Mediterranean variants (39–42). The results of studies comparing hemoglobin and hematocrit in deficient and normal subjects have varied, with studies specifically of blood donors showing no difference in hemoglobin and hematocrit between deficient and normal subjects (23, 39), and others demonstrating lower hemoglobin and/or hematocrit in deficient subjects (41, 42). In addition, decreased RBC count in G6PD-deficient subjects has been observed in studies with blood donors (39), as well as other healthy individuals (41, 42). The findings of increased reticulocyte count and decreased RBC count in G6PD-deficient subjects in the current study, in the setting of no differences in hemoglobin and hematocrit, suggest the presence of a subclinical, compensated hemolysis in deficient subjects at baseline.
The only previous study examining 24-hour PTR in G6PD-deficient and G6PD-normal RBCs also noted significant differences (26). However, these nonleukoreduced RBC units were stored in either acid citrate dextrose–adenine (ACD-A) or citrate phosphate dextrose adenine 1 (CPDA-1) solutions and, under both conditions, G6PD-deficient RBCs exhibited lower 24-hour PTRs (67.2% vs 74.4% in ACD-A and 58.1% vs 72% in CPDA-1). At that time, the criteria for the suitability of stored RBCs included an average 24-hour PTR greater than 70%. Similar to our results, a greater proportion of the G6PD-deficient RBC units failed this criterion. In addition, they found no correlation between G6PD enzyme activity and PTR within a given G6PD-status group (i.e., separately assessing the correlation between PTR and enzyme activity for G6PD-normal RBCs and G6PD-deficient RBCs; Figure 2D and ref. 26). We also observed significant variability in the PTR among subjects with G6PD-deficiency (SD = 8.4%), suggesting that there are other factors (e.g., genetic or environmental) that affect storage quality, independent of G6PD activity. Notably, the storage solution affected the quality of the G6PD-deficient, but not G6PD-normal, RBCs. Thus, although PTR for G6PD-deficient RBCs stored in CPDA-1 (58.1%) was lower than in ACD-A (67.2%), there was no significant difference in PTR for G6PD-normal RBCs stored in either preservative (26). Because the type of storage solution affects RBC metabolism (9), these results suggest that G6PD-deficient RBCs may vary in quality based on storage solution composition.
We found no significant differences in storage hemolysis in vitro (Figure 3A), another important FDA criterion for assessing RBC quality, between G6PD-deficient and G6PD-normal RBCs. In addition, prior studies comparing G6PD status in various additive solutions, for example ACD-A, CPDA-1 (26), and SAGM (23), or following irradiation (43), did not reveal differences in storage hemolysis in vitro. Although the determinants of in vitro hemolysis during refrigerated storage are not well understood, factors such as sex, age, ethnicity (44), and other unknown heritable traits (12) may be important.
G6PD-deficient RBCs maintained lower methemoglobin levels during storage (Figure 3B). Methemoglobin, which contains oxidized iron (Fe3+), releases oxygen poorly to tissues. Thus, reduction of methemoglobin is important for maintaining optimal RBC function; to this end, cytochrome-b5 reductase uses NADH for this purpose. Stored G6PD-deficient RBCs have substantially increased cytochrome-b5 reductase levels as compared with G6PD-normal RBCs (23). During glycolysis, NADH is produced by GAPDH when converting G3P to BPG. Our data suggest that G6PD-deficient RBCs maximize NADH production, as evidenced by significant decreases in glyceraldehyde 3-phosphate and significant increases in BPG, the substrate and product of GAPDH, respectively (Figure 6, G and H, and Figure 4B). G6PD-deficient RBCs also appear to preserve NADH by limiting the conversion of pyruvate to lactate, an NADH-consuming reaction catalyzed by LDH, as evidenced by the significantly increased pyruvate/lactate ratios throughout storage (Figure 6, J–L). Thus, G6PD-deficient RBCs demonstrate alterations in glycolysis that enable them to maintain NADH levels throughout storage. Although the reduced methemoglobin levels observed in refrigerator-stored G6PD-deficient RBCs may be advantageous upon transfusion from an oxygen delivery perspective, it is likely that the increased clearance observed with G6PD-deficient RBCs overshadows this relatively small difference in methemoglobin levels.
In the current and prior studies (23, 45), G6PD-deficient RBCs, whether fresh or stored, demonstrated increased glycolysis. Since G6P is at the bifurcation point of the PPP and glycolysis, more G6P should be available for glycolysis in G6PD-deficient RBCs due to their defect in the oxidative branch of the PPP. Important products of glycolysis include ATP and NADH. Therefore, increased glycolysis is expected to allow G6PD-deficient RBCs to maintain ATP levels. Consistent with this, fresh G6PD-deficient RBCs had significantly increased ATP as compared with G6PD-normal RBCs (Figure 4G). Furthermore, G6PD-deficient RBCs maintained ATP at levels similar to G6PD-normal RBCs during storage (Figure 6M).
The PPP contains oxidative and nonoxidative branches. The oxidative branch results in oxidation, catalyzed by G6PD, of G6P to 6-PGL, followed by decarboxylation, catalyzed by 6PG dehydrogenase, to produce ribulose-5-phosphate. Tracing experiments with [1,2,3-13C3]-glucose, in the presence or absence of the PPP-stimulating agent methylene blue, demonstrate that fresh G6PD-normal, but not G6PD-deficient, RBCs activate the oxidative phase of the PPP (Figure 5). However, following 6 weeks of storage, both G6PD-deficient and G6PD-normal RBCs were unable to activate the oxidative branch of the PPP following methylene blue treatment, suggesting that this pathway ultimately degrades during storage, even in G6PD-normal donors, as has been previously observed (46).
G6PD deficiency decreases the ability of RBCs to produce NADPH (16). As a possible compensatory mechanism for maintaining NADPH, G6PD-deficient RBCs increased activity in alternative reactions and pathways that generate NADPH. Thus, because of the expected defect in NADPH production by the oxidative branch of the PPP due to G6PD deficiency (Figure 5), G6PD-deficient RBCs have storage-induced decreases in GSH, hexose sugar alcohol, and DHN, which are produced by NADPH-consuming reactions (Figure 7). G6PD-deficient RBCs had significantly lower levels of GSH throughout storage, likely due to a combination of decreased GSSG reduction to GSH (a NADPH-dependent reaction), export of GSSG from RBCs (47), and increased protein glutathionylation in response to oxidative stress. In addition, biliverdin was significantly increased in G6PD-deficient RBCs during storage, consistent with decreased NADPH-driven conversion to bilirubin. G6PD-deficient RBCs also had significant increases in TCA cycle intermediates, such as succinate, fumarate, and malate (Figure 7). The conversion of malate to pyruvate is an NADPH-producing reaction, raising the possibility that the increasing pyruvate levels during storage in G6PD-deficient RBCs (Figure 6) are not only due to conversion from PEP during glycolysis, but may also be a by-product of NADPH formation from malate. Therefore, G6PD-deficient RBCs may compensate for their decreased ability to produce NADPH through the PPP by increasing activity in alternative pathways that are independent of G6PD.
The strongest correlations to PTR were metabolites related to pathways that (a) produce NADH (e.g., α-ketoglutarate), (b) salvage nucleotides (e.g., hypoxanthine and IMP), (c) are intermediates of the PPP (e.g., erythrose phosphate and ribose phosphate), (d) are involved in lipid remodeling (e.g., octenoyl-carnitine), (e) that maintain glutathione homeostasis (e.g., glutathione disulfide and S-glutathionylcysteine), and (f) are a steroid hormone (e.g., epiandrosterone; Figure 9). Consistent with seminal studies on RBC metabolism and PTR, maintenance of energy metabolism and, in particular, ATP levels were positively correlated with PTR (46). Our results further confirm and expand on the negative correlation of ATP breakdown and oxidant stress–induced deamination (i.e., hypoxanthine and IMP) to PTR (48, 49). Consistently, the levels of reduced GSH, a critically important antioxidant, positively correlated with PTR, in keeping with classic studies (46). Decreases in S-glutathionylcysteine and GSH during storage are more marked in G6PD-deficient RBCs, consistent with likely increases in cysteine glutathionylation as a compensatory protection against oxidative stress in G6PD-deficient subjects (50). Finally, the correlation of epiandrosterone, a steroid hormone shown to affect glucose metabolism and PPP activity in vitro (51), and PTR from samples at baseline is interesting given the potential link between sex hormones and RBC storage quality (44, 52, 53).
The current study has some limitations. For example, the primary outcome is the 24-hour PTR, as opposed to other markers of RBC destruction, such as indirect bilirubin or iron parameters, which may correlate better with hemolysis in vivo (54). This primary outcome was chosen a priori because of the precautionary principle; that is, the entire unit was not transfused in this study because we wished to prevent G6PD-deficient donors from experiencing severe hemolysis in the event that storage of their RBCs led to severe damage. In addition, although PTR was quantified in this study, RBC life span was not evaluated beyond the 24-hour time point. Nonetheless, prior studies showed no differences in the posttransfusion life span of G6PD-deficient and G6PD-normal RBCs (26). Also, only the extreme of storage was examined (i.e., 6 weeks); thus, future studies could examine effects at shorter storage intervals. Furthermore, in a study using autologous recipients, one could not determine whether protective compensatory mechanisms in the plasma of G6PD-deficient subjects could compensate for their poor redox status, as previously reported (55), or whether the difference in PTR is due to an intrinsic defect of G6PD-deficient RBCs as opposed to extrinsic effects of G6PD-deficiency on RBC clearance. It is also possible that plasma factors in G6PD-normal recipients could improve the survival of G6PD-deficient RBCs (55). However, this effect would need to be studied in the context of allogeneic transfusions, which were not the focus of this autologous PTR study in which the primary outcome measure was the FDA gold-standard test. Also, the chromium labeling efficiency and elution rate may be different in G6PD-deficient, as compared with G6PD-normal, RBCs. However, the chromium labeling efficiency in damaged RBCs is minimally affected (56), and the variability seen in chromium elution rates among patients with hematologic diseases are not sufficient to explain the overall differences in PTR observed in G6PD-deficient donors (57). In addition, although there was a statistically significant decrease in the PTR for G6PD-deficient RBCs, this study was performed in healthy volunteers. Thus, it remains unknown if this PTR difference would be greater in transfused patients, such as those with sickle cell disease, who experience increased oxidative stress at baseline (58). For example, chronically transfused pediatric patients with sickle cell disease have poorer suppression of reticulocytosis and higher hemoglobin S levels between transfusion episodes when receiving G6PD-deficient donor RBCs (28). It is also possible that results would vary if donor RBCs were stored in different additive solutions. Thus, all RBC units in this study were stored in AS-3. However, prior studies found a substantial difference in PTR when G6PD-deficient RBCs were stored in ACD-A (67.2%) or CPDA (58.1%) (26). Finally, some of our correlative results are limited by an appreciation that the lowest PTRs were observed with G6PD-deficient RBCs. As such, metabolites correlating with G6PD activity per se (or not) also significantly correlated with PTR. Although further studies in larger cohorts are necessary to validate the present findings, there is growing interest in the potential effects of RBC donor genetics on transfusion outcomes (59).
Experts in the field of transfusion medicine are focusing significant effort on determining the genetic factors underlying the observed donor variability in RBC storage (60–62). The current study provides unequivocal evidence that one such genetic factor, G6PD deficiency, significantly (P = 0.0009) decreases 24-hour PTR after 6 weeks of storage. In addition, RBC units from donors with G6PD deficiency, if studied as a group, would not meet the current FDA criterion for transfusion quality. The clinical impact of this finding for all transfused patients, and for chronically transfused patients with sickle cell disease, in particular (25), should be further investigated. In addition, understanding the metabolic mechanism(s) by which G6PD deficiency impairs storage quality will yield valuable information for not only improving the storage of G6PD-deficient RBCs, but also for improving RBC transfusion efficacy overall.
Methods
Participant recruitment.
Male volunteers were recruited using flyers, person-to-person communication, and email, between November 2012 and August 2017. Screening was limited to males because G6PD deficiency is X-linked; thus, heterozygous females are not expected to have the same severity of enzyme deficiency as hemizygous males, and homozygous females are rare in the population (63). Furthermore, advertisements focused on ethnicities known to have a higher prevalence of G6PD deficiency, which included African, Mediterranean, Middle Eastern, Hispanic/Latino, and Asian origin. Inclusion criteria were male sexual phenotype, weight greater than 110 pounds, and hemoglobin greater than 11.5 g/dL. Participants were also screened for microcytosis (Sysmex XN9000, Sysmex) and for hemoglobin variants (Bio-Rad Variant II; Bio-Rad). G6PD activity was measured by a Clinical Laboratory Improvement Amendments–certified (CLIA-certified) reference laboratory (Associated Regional and University Pathologists Laboratories). All 13 exons of the G6PD gene of G6PD-deficient volunteers were analyzed by next-generation sequencing in the CLIA-certified Personalized Genomics Laboratory at Columbia University Irving Medical Center, New York, New York, to identify the specific G6PD variant present (64).
Blood donation and storage.
Following screening and confirming G6PD activity, 10 G6PD-deficient and 30 G6PD-normal males donated 1 unit of whole blood at the New York Blood Center (New York, New York), each of which was processed into packed RBCs, leukoreduced, and stored in AS-3. RBC units were refrigerator stored for up to 42 days under standard blood banking conditions. Using sterile docking procedures, 25-mL aliquots were removed during each week of storage (i.e., weeks 1, 2, 3, 4, 5, 6) for in vitro analyses. Each 25-mL aliquot was divided as follows: 3 mL for blood gas analysis; 7 mL distributed into 250-μL aliquots and frozen in liquid nitrogen; and 15 mL centrifuged at 3900g for 7 minutes at 4°C. The supernatant of the latter was transferred to 1.5-mL microcentrifuge tubes and the remaining RBCs were centrifuged again at 3900g for 7 minutes at 4°C. Supernatant was again transferred to 1.5-mL microcentrifuge tubes and all supernatants were centrifuged at 12,600g for 6 minutes. All separated RBCs and supernatants were separately distributed into 1.5-mL microcentrifuge tubes in 250-μL aliquots and frozen in liquid nitrogen. All frozen samples were stored at –80°C until metabolomics analysis. At week 5, an additional sample was submitted for aerobic and anaerobic blood culture to confirm the absence of bacterial contamination during storage.
Conventional measurements in vitro.
At each week of storage, pH, methemoglobin, sodium, glucose, and lactate were measured using a clinically validated ABL 800 Flex Blood Gas Analyzer (Radiometer). Hemoglobin and hematocrit were measured on a clinically validated Sysmex XN9000. Supernatant hemoglobin was measured weekly, as follows: supernatant was obtained by centrifuging 500 μL of RBC concentrate at 2000g for 10 minutes and hemoglobin quantified using the Drabkin method; the hemolysis rate was then calculated as previously described (65).
Metabolomics.
At study completion, samples were shipped on dry ice to the University of Colorado Denver–Anschutz Medical Campus, where they were either untreated or incubated with 100 μM methylene blue for 1 hour at 37°C in the presence of 5 mM [1,2,3-13C3]-glucose. Samples were then extracted with a mixture of ice-cold methanol, acetonitrile, and water (5:3:2 vol/vol/vol, respectively) at a 1:10 ratio for RBCs (15 μL sample with 135 μL extraction solvent) or a 1:25 ratio for RBC supernatant. Extractions were performed by vigorous vortexing for 30 minutes at 4°C; insoluble materials were then pelleted by centrifugation for 10 minutes at 4°C; centrifugation speeds were 18,000g for RBC extractions and 12,000g for RBC supernatant extractions. Clarified extraction supernatants were analyzed by ultra-high-pressure liquid chromatography coupled online to high-resolution mass spectrometry (UHPLC-MS) using a Thermo Vanquish UHPLC coupled to a Thermo Q Exactive MS. Samples were randomized and run in positive and negative ion modes (separate runs). Injection volumes were 10 μL for RBC samples and 20 μL for RBC supernatant samples. UHPLC phases were water (A) and acetonitrile (B) supplemented with 0.1% formic acid for positive mode runs and 1 mM ammonium acetate for negative mode runs. Metabolites were separated on a Kinetex C18 column (2.1 × 150 mm, 1.7 μm; Phenomenex), equipped with a guard column, using a 5-minute gradient under the following conditions: flow rate 0.45 mL/min; column temperature 45°C; sample compartment temperature 7°C; solvent gradient: 0–0.5 minutes 5% B, 0.5–1.1 minutes 5%–95% B, 1.1–2.75 minutes hold at 95% B, 2.75–3 minutes 95%–5% B, 3–5 minutes hold at 5% B (66). The mass spectrometer was operated in full MS mode at a resolution of 70,000, scan range 65–900 m/z, maximum injection time 200 ms, microscans 2, automatic gain control (AGC) 3 × 106 ions, electrospray source voltage 4.0 kV, capillary temperature 320°C, and sheath gas 45, auxiliary gas 15, and sweep gas 0 (all nitrogen). Raw data files were converted to mzXML format using MassMatrix (Case Western Reserve University) and analyzed using Maven (Princeton University). Hierarchical clustering analysis (HCA) and partial least squares-discriminant analysis (PLS-DA) were performed with the MetaboAnalyst 4.0 and GENE E (Princeton) freeware. Instrument stability and quality control were assessed using replicate injections of a technical mixture every 15 runs, as previously described (67, 68).
Tracing experiments with [1,2,3-13C3]-glucose.
Tracing experiments with [1,2,3-13C3]-glucose were performed as previously described (22), with fresh RBCs collected directly from donors (fresh) or RBCs stored for 6 weeks (stored).
24-hour PTR studies.
After 40 to 42 days of storage (week 6), 24-hour PTR studies were performed as previously described (69). Briefly, 25 mL RBCs were labeled with 20 μCi sodium chromate (Mallinckrodt Pharmaceuticals), followed by infusion of the labeled RBCs over 1 minute. Blood samples were collected before infusion and at 5, 7.5, 10, 12.5, 15, 30, 60, and 120 minutes after infusion; a final sample was obtained at 24 hours after infusion. Radioactivity and hemoglobin were measured in each sample on a Gamma Counter (Packard Cobra II Auto-Gamma, Perkin Elmer) and a Sysmex XN9000 analyzer, respectively. The 24-hour PTR was then calculated using all timed blood draws for estimating the 51-chromium per gram hemoglobin circulating at time zero (70).
Statistics.
The primary outcome difference in 24-hour PTR between G6PD-deficient and G6PD-normal subjects was calculated using an independent 2-sided Student’s t test. Historical studies suggested that the difference in RBC recovery would be relatively large (72.2% ± 9.18% and 58.1% ± 9.81% in G6PD-normal and G6PD-deficient subjects, respectively) (26). However, we expected a smaller difference and assumed a 6% SD for our primary outcome measure based on studies using modern storage solutions (7). Thus, to provide sufficient power to explore whether specific in vitro storage metabolomics parameters could predict PTR in vivo, we recruited 10 G6PD-deficient subjects and 30 G6PD-normal controls. Using an independent Student’s t test, this provided greater than 78% power to detect a 6% absolute difference in PTR. Secondary analyses of differences in metabolite levels over time were performed using 2-way repeated-measures ANOVA and Sidak’s multiple comparisons test (or mixed-effects model if missing values) using Prism version 8 (GraphPad Software, Inc.), or MetaboAnalyst version 4.0. Significance of main effects was determined using 2-factor time-series analysis in MetaboAnalyst with a false discovery rate (FDR) of 0.10. Subsequently, Prism was used to graph the significant findings and final P values determined using 2-way repeated-measures ANOVA. In addition, metabolic correlations (Spearman) were calculated and plotted as circos plots through MetaboScape version 4.0 (Bruker) for the network building software Cytoscape version 3.7 (National Resource for Network Biology). In this analysis, metabolites are plotted as nodes and connected with an edge if the module of their correlation (Spearman) is higher than 0.4. Highlighted nodes represent top ranking metabolites by the module of their linear Spearman’s correlations (filtered by FDR < 0.10) for each analysis (total sample, G6PD-deficient or G6PD-normal groups, either at baseline or after 6 weeks of storage).
Study approval.
The study was conducted according to the Declaration of Helsinki and in accordance with good clinical practice guidelines. The Columbia University (protocol AAAJ6862) and New York Blood Center (protocol 401165) IRBs approved this study. All research participants were provided with written, informed consent before study participation.
Author contributions
ROF, EAH, and SLS designed the clinical trial. ROF led the clinical trial. FR, FLC, and TT assisted with data collection. RY, EC, AS, MS, and CD assisted with the PTR studies. EAH, AD, JCZ, and ROF analyzed the clinical data. DK, YG, and BHS assisted with blood donation procedures. AE and JS assisted with blood banking processes. SG, JAR, TN, and AD performed metabolomics analyses. ROF and AD drafted the first version of the manuscript. All authors edited and approved the final manuscript.
Supplementary Material
Acknowledgments
This work was supported by the Harold Amos Medical Faculty Development Program, Robert Wood Johnson Foundation grant 71590, the National Blood Foundation, National Heart, Lung, and Blood Institute grant R01HL148151, and the National Center for Advancing Translational Sciences, NIH, through grant UL1 TR000040. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. AD was supported by funds from the Webb-Waring Early Career Award 2017 by the Boettcher Foundation, the National Heart, Lung, and Blood Institute grants R01HL14644 and R01HL148151, and the National Institute of General Medical Sciences grant RM1GM131968. The authors thank the inpatient nurses and staff of the Irving Center for Clinical and Translational Research for their outstanding patient care and support along with the dedicated blood bank staff at the Columbia University Irving Medical Center-New York Presbyterian Hospital. We also thank Connie Zheng (University of Colorado) for technical assistance with the metabolomics analysis. Finally, we thank the human volunteers who participated in the study.
Version 1. 01/21/2020
In-Press Preview
Version 2. 03/23/2020
Electronic publication
Version 3. 05/01/2020
Print issue publication
Footnotes
Conflict of interest: AD and TN are founders of Omix Technologies, Inc. and Altis Biosciences, LLC. SLS is a consultant for Tioma, Inc. AD and SLS are members of the Scientific Advisory Board of Hemanext, Inc.
Role of funding source: The sponsor was not involved in the study design, collection, analysis, and interpretation of data, writing of the manuscript, or decision to submit the manuscript for publication.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(5):2270–2285.https://doi.org/10.1172/JCI133530.
Contributor Information
Richard O. Francis, Email: rof3@cumc.columbia.edu.
Angelo D’Alessandro, Email: ANGELO.DALESSANDRO@UCDENVER.EDU.
Andrew Eisenberger, Email: abe6@cumc.columbia.edu.
Mark Soffing, Email: ms4138@cumc.columbia.edu.
Randy Yeh, Email: yehr@mskcc.org.
Esther Coronel, Email: esther.coronel@gmail.com.
Arif Sheikh, Email: Arif.Sheikh@mountsinai.org.
Francesca Rapido, Email: francesca.rapido@gmail.com.
Francesca La Carpia, Email: fl2404@cumc.columbia.edu.
Julie A. Reisz, Email: JULIE.HAINES@CUANSCHUTZ.EDU.
Sarah Gehrke, Email: SARAH.GEHRKE@CUANSCHUTZ.EDU.
Travis Nemkov, Email: TRAVIS.NEMKOV@UCDENVER.EDU.
Tiffany Thomas, Email: tt2254@cumc.columbia.edu.
Joseph Schwartz, Email: js2745@columbia.edu.
Chaitanya Divgi, Email: divgic@gmail.com.
Beth H. Shaz, Email: bshaz@nybloodcenter.org.
Yelena Ginzburg, Email: yelena.ginzburg@mssm.edu.
James C. Zimring, Email: jcz2k@virginia.edu.
Steven L. Spitalnik, Email: ss2479@cumc.columbia.edu.
Eldad A. Hod, Email: eh2217@cumc.columbia.edu.
References
- 1. Pfuntner A, Wier LM, Stocks C. Healthcare Cost and Utilization Project. Most frequent procedures performed in U.S. hospitals, 2011. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb165.jsp Published October 2013. Accessed March 20, 2020.
- 2.Yoshida T, Prudent M, D’alessandro A. Red blood cell storage lesion: causes and potential clinical consequences. Blood Transfus. 2019;17(1):27–52. doi: 10.2450/2019.0217-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hendrickson JE, Hod EA, Spitalnik SL, Hillyer CD, Zimring JC. Storage of murine red blood cells enhances alloantibody responses to an erythroid-specific model antigen. Transfusion. 2010;50(3):642–648. doi: 10.1111/j.1537-2995.2009.02481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prestia K, et al. Transfusion of stored blood impairs host defenses against Gram-negative pathogens in mice. Transfusion. 2014;54(11):2842–2851. doi: 10.1111/trf.12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barthel W, Markwardt F. Aggregation of blood platelets by adrenaline and its uptake. Biochem Pharmacol. 1975;24(20):1903–1904. doi: 10.1016/0006-2952(75)90415-3. [DOI] [PubMed] [Google Scholar]
- 6.Desai PC, et al. Alloimmunization is associated with older age of transfused red blood cells in sickle cell disease. Am J Hematol. 2015;90(8):691–695. doi: 10.1002/ajh.24051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dumont LJ, AuBuchon JP. Evaluation of proposed FDA criteria for the evaluation of radiolabeled red cell recovery trials. Transfusion. 2008;48(6):1053–1060. doi: 10.1111/j.1537-2995.2008.01642.x. [DOI] [PubMed] [Google Scholar]
- 8.Dern RJ, Gwinn RP, Wiorkowski JJ. Studies on the preservation of human blood. I. Variability in erythrocyte storage characteristics among healthy donors. J Lab Clin Med. 1966;67(6):955–965. [PubMed] [Google Scholar]
- 9.D’Alessandro A, et al. Heterogeneity of blood processing and storage additives in different centers impacts stored red blood cell metabolism as much as storage time: lessons from REDS-III-Omics. Transfusion. 2019;59(1):89–100. doi: 10.1111/trf.14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van‘t Erve TJ, et al. Heritability of glutathione and related metabolites in stored red blood cells. Free Radic Biol Med. 2014;76:107–113. doi: 10.1016/j.freeradbiomed.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van‘t Erve TJ, et al. The heritability of metabolite concentrations in stored human red blood cells. Transfusion. 2014;54(8):2055–2063. doi: 10.1111/trf.12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van‘t Erve TJ, et al. The heritability of hemolysis in stored human red blood cells. Transfusion. 2015;55(6):1178–1185. doi: 10.1111/trf.12992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis. 2009;42(3):267–278. doi: 10.1016/j.bcmd.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 14.Luzzatto L, Arese P. Favism and glucose-6-phosphate dehydrogenase deficiency. N Engl J Med. 2018;378(1):60–71. doi: 10.1056/NEJMra1708111. [DOI] [PubMed] [Google Scholar]
- 15.van Zwieten R, Verhoeven AJ, Roos D. Inborn defects in the antioxidant systems of human red blood cells. Free Radic Biol Med. 2014;67:377–386. doi: 10.1016/j.freeradbiomed.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 16.Scott MD, Zuo L, Lubin BH, Chiu DT. NADPH, not glutathione, status modulates oxidant sensitivity in normal and glucose-6-phosphate dehydrogenase-deficient erythrocytes. Blood. 1991;77(9):2059–2064. [PubMed] [Google Scholar]
- 17.Francis RO, Jhang JS, Pham HP, Hod EA, Zimring JC, Spitalnik SL. Glucose-6-phosphate dehydrogenase deficiency in transfusion medicine: the unknown risks. Vox Sang. 2013;105(4):271–282. doi: 10.1111/vox.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roback JD, et al. Metabolomics of ADSOL (AS-1) red blood cell storage. Transfus Med Rev. 2014;28(2):41–55. doi: 10.1016/j.tmrv.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gevi F, D’Alessandro A, Rinalducci S, Zolla L. Alterations of red blood cell metabolome during cold liquid storage of erythrocyte concentrates in CPD-SAGM. J Proteomics. 2012;76(Spec No.):168–180. doi: 10.1016/j.jprot.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 20.Dumont LJ, Yoshida T, AuBuchon JP. Anaerobic storage of red blood cells in a novel additive solution improves in vivo recovery. Transfusion. 2009;49(3):458–464. doi: 10.1111/j.1537-2995.2008.02038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stowell SR, et al. Addition of ascorbic acid solution to stored murine red blood cells increases posttransfusion recovery and decreases microparticles and alloimmunization. Transfusion. 2013;53(10):2248–2257. doi: 10.1111/trf.12106. [DOI] [PubMed] [Google Scholar]
- 22.Reisz JA, et al. Oxidative modifications of glyceraldehyde 3-phosphate dehydrogenase regulate metabolic reprogramming of stored red blood cells. Blood. 2016;128(12):e32–e42. doi: 10.1182/blood-2016-05-714816. [DOI] [PubMed] [Google Scholar]
- 23.Tzounakas VL, et al. Glucose 6-phosphate dehydrogenase deficient subjects may be better “storers” than donors of red blood cells. Free Radic Biol Med. 2016;96:152–165. doi: 10.1016/j.freeradbiomed.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Tzounakas VL, et al. Data on how several physiological parameters of stored red blood cells are similar in glucose 6-phosphate dehydrogenase deficient and sufficient donors. Data Brief. 2016;8:618–627. doi: 10.1016/j.dib.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Francis RO, Jhang J, Hendrickson JE, Zimring JC, Hod EA, Spitalnik SL. Frequency of glucose-6-phosphate dehydrogenase-deficient red blood cell units in a metropolitan transfusion service. Transfusion. 2013;53(3):606–611. doi: 10.1111/j.1537-2995.2012.03765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orlina AR, Josephson AM, McDonald BJ. The poststorage viability of glucose-6-phosphate dehydrogenase-deficient erythrocytes. J Lab Clin Med. 1970;75(6):930–936. [PubMed] [Google Scholar]
- 27.Pertinhez TA, Casali E, Baroni F, Berni P, Baricchi R, Spisni A. A comparative study of the effect of leukoreduction and pre-storage leukodepletion on red blood cells during storage. Front Mol Biosci. 2016;3:13. doi: 10.3389/fmolb.2016.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sagiv E, et al. Glucose-6-phosphate-dehydrogenase deficient red blood cell units are associated with decreased posttransfusion red blood cell survival in children with sickle cell disease. Am J Hematol. 2018;93(5):630–634. doi: 10.1002/ajh.25051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shalev O, Manny N, Sharon R. Posttransfusional hemolysis in recipients of glucose-6-phosphate dehydrogenase-deficient erythrocytes. Vox Sang. 1993;64(2):94–98. doi: 10.1111/j.1423-0410.1993.tb02525.x. [DOI] [PubMed] [Google Scholar]
- 30.Kumar P, Sarkar S, Narang A. Acute intravascular haemolysis following exchange transfusion with G-6-PD deficient blood. Eur J Pediatr. 1994;153(2):98–99. doi: 10.1007/BF01959216. [DOI] [PubMed] [Google Scholar]
- 31.Gulati S, Singh S, Narang A, Bhakoo ON. Exchange transfusion with G-6-PD deficient donor blood causes exaggeration of neonatal hyperbilirubinemia. Indian Pediatr. 1989;26(5):499–501. [PubMed] [Google Scholar]
- 32.Renzaho AM, Husser E, Polonsky M. Should blood donors be routinely screened for glucose-6-phosphate dehydrogenase deficiency? A systematic review of clinical studies focusing on patients transfused with glucose-6-phosphate dehydrogenase-deficient red cells. Transfus Med Rev. 2014;28(1):7–17. doi: 10.1016/j.tmrv.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 33.Sirdah MM, Shubair ME, Al-Kahlout MS, Al-Tayeb JM, Prchal JT, Reading NS. Possible association of 3’ UTR +357 T>G, IVS11-nt 93 T>C, c.1311 C>T polymorphism with G6PD deficiency. Hematology. 2017;22(6):370–374. doi: 10.1080/10245332.2016.1276117. [DOI] [PubMed] [Google Scholar]
- 34.Arai T, Takahashi M, Araki K, Washizu T. Activities of enzymes related to the malate-aspartate shuttle in the blood cells of thoroughbred horses undergoing training exercise. Vet Res Commun. 2001;25(7):577–583. doi: 10.1023/A:1017977200420. [DOI] [PubMed] [Google Scholar]
- 35.D’Alessandro A, Hansen KC, Silliman CC, Moore EE, Kelher M, Banerjee A. Metabolomics of AS-5 RBC supernatants following routine storage. Vox Sang. 2015;108(2):131–140. doi: 10.1111/vox.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bordbar A, Yurkovich JT, Paglia G, Rolfsson O, Sigurjónsson ÓE, Palsson BO. Elucidating dynamic metabolic physiology through network integration of quantitative time-course metabolomics. Sci Rep. 2017;7:46249. doi: 10.1038/srep46249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stincone A, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. 2015;90(3):927–963. doi: 10.1111/brv.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simon TL, Marcus CS, Myhre BA, Nelson EJ. Effects of AS-3 nutrient-additive solution on 42 and 49 days of storage of red cells. Transfusion. 1987;27(2):178–182. doi: 10.1046/j.1537-2995.1987.27287150195.x. [DOI] [PubMed] [Google Scholar]
- 39.Maffi D, et al. Glucose-6-phosphate dehydrogenase deficiency in Italian blood donors: prevalence and molecular defect characterization. Vox Sang. 2014;106(3):227–233. doi: 10.1111/vox.12096. [DOI] [PubMed] [Google Scholar]
- 40.Piomelli S, Corash LM, Davenport DD, Miraglia J, Amorosi EL. In vivo lability of glucose-6-phosphate dehydrogenase in GdA- and GdMediterranean deficiency. J Clin Invest. 1968;47(4):940–948. doi: 10.1172/JCI105786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piomelli S, Siniscalco M. The haematological effects of glucose-6-phosphate dehydrogenase deficiency and thalassaemia trait: interaction between the two genes at the phenotype level. Br J Haematol. 1969;16(6):537–549. doi: 10.1111/j.1365-2141.1969.tb00435.x. [DOI] [PubMed] [Google Scholar]
- 42.Ragusa R, Di Cataldo A, Gangarossa S, Lo Nigro L, Schilirò G. Low-grade haemolysis and assessment of iron status during the steady state in G6PD-deficient subjects. Acta Haematol. 1993;90(1):25–28. doi: 10.1159/000204368. [DOI] [PubMed] [Google Scholar]
- 43.Agarwal P, Ray VL, Choudhury N, Agarwal S, Chaudhary RK. Effect of gamma irradiation on blood from glucose 6 phosphate dehydrogenase deficient blood donors. Hematology. 2007;12(3):267–270. doi: 10.1080/10245330701214137. [DOI] [PubMed] [Google Scholar]
- 44.Kanias T, et al. Ethnicity, sex, and age are determinants of red blood cell storage and stress hemolysis: results of the REDS-III RBC-Omics study. Blood Adv. 2017;1(15):1132–1141. doi: 10.1182/bloodadvances.2017004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang HY, et al. Inability to maintain GSH pool in G6PD-deficient red cells causes futile AMPK activation and irreversible metabolic disturbance. Antioxid Redox Signal. 2015;22(9):744–759. doi: 10.1089/ars.2014.6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lachant NA, Noble NA, Myrhe BA, Tanaka KR. Antioxidant metabolism during blood storage and its relationship to posttransfusion red cell survival. Am J Hematol. 1984;17(3):237–249. doi: 10.1002/ajh.2830170304. [DOI] [PubMed] [Google Scholar]
- 47.Prchal J, Srivastava SK, Beutler E. Active transport of GSSG from reconstituted erythrocyte ghosts. Blood. 1975;46(1):111–117. [PubMed] [Google Scholar]
- 48.Dudzinska W, Hlynczak AJ, Skotnicka E, Suska M. The purine metabolism of human erythrocytes. Biochemistry Mosc. 2006;71(5):467–475. doi: 10.1134/S0006297906050014. [DOI] [PubMed] [Google Scholar]
- 49.Nemkov T, et al. Hypoxia modulates the purine salvage pathway and decreases red blood cell and supernatant levels of hypoxanthine during refrigerated storage. Haematologica. 2018;103(2):361–372. doi: 10.3324/haematol.2017.178608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grek CL, Zhang J, Manevich Y, Townsend DM, Tew KD. Causes and consequences of cysteine S-glutathionylation. J Biol Chem. 2013;288(37):26497–26504. doi: 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laychock SG, Bauer AL. Epiandrosterone and dehydroepiandrosterone affect glucose oxidation and interleukin-1 beta effects in pancreatic islets. Endocrinology. 1996;137(8):3375–3385. doi: 10.1210/endo.137.8.8754764. [DOI] [PubMed] [Google Scholar]
- 52.Fang F, et al. Sex hormone intake in female blood donors: impact on haemolysis during cold storage and regulation of erythrocyte calcium influx by progesterone. Blood Transfus. 2019;17(4):263–273. doi: 10.2450/2019.0053-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanias T, et al. Testosterone-dependent sex differences in red blood cell hemolysis in storage, stress, and disease. Transfusion. 2016;56(10):2571–2583. doi: 10.1111/trf.13745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rapido F, et al. Prolonged red cell storage before transfusion increases extravascular hemolysis. J Clin Invest. 2017;127(1):375–382. doi: 10.1172/JCI90837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tzounakas VL, et al. Redox status, procoagulant activity, and metabolome of fresh frozen plasma in glucose 6-phosphate dehydrogenase deficiency. Front Med (Lausanne) 2018;5:16. doi: 10.3389/fmed.2018.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peters AM, Osman S, Reavy HJ, Chambers B, Deenmamode M, Lewis SM. Erythrocyte radiolabelling: in vitro comparison of chromium, technetium, and indium in undamaged and heat damaged cells. J Clin Pathol. 1986;39(7):717–721. doi: 10.1136/jcp.39.7.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cline MJ, Berlin NI. The red cell chromium elution rate in patients with some hematologic diseases. Blood. 1963;21:63–69. doi: 10.1182/blood.V21.1.63.63. [DOI] [PubMed] [Google Scholar]
- 58.Hermann PB, Pianovski MA, Henneberg R, Nascimento AJ, Leonart MS. Erythrocyte oxidative stress markers in children with sickle cell disease. J Pediatr (Rio J) 2016;92(4):394–399. doi: 10.1016/j.jped.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 59.Lanteri MC, et al. Intradonor reproducibility and changes in hemolytic variables during red blood cell storage: results of recall phase of the REDS-III RBC-Omics study. Transfusion. 2019;59(1):79–88. doi: 10.1111/trf.14987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanias T, Busch MP, National Heart, Lung, Blood Institute Recipient Epidemiology Donor Evaluation Study III (REDS-III) Programme Diversity in a blood bag: application of omics technologies to inform precision transfusion medicine. Blood Transfus. 2019;17(4):258–262. doi: 10.2450/2019.0056-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.D’Alessandro A. From omics technologies to personalized transfusion medicine. Expert Rev Proteomics. 2019;16(3):215–225. doi: 10.1080/14789450.2019.1571917. [DOI] [PubMed] [Google Scholar]
- 62.D’Alessandro A, Liumbruno G. Personalised transfusion medicine. Blood Transfus. 2019;17(4):255–257. doi: 10.2450/2018.0142-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luzzatto L, Nannelli C, Notaro R. Glucose-6-phosphate dehydrogenase deficiency. Hematol Oncol Clin North Am. 2016;30(2):373–393. doi: 10.1016/j.hoc.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 64.Xu JZ, et al. G6PD Deficiency in an HIV clinic setting in the Dominican Republic. Am J Trop Med Hyg. 2015;93(4):722–729. doi: 10.4269/ajtmh.14-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Acker JP, M Croteau I, Yi QL. An analysis of the bias in red blood cell hemolysis measurement using several analytical approaches. Clin Chim Acta. 2012;413(21–22):1746–1752. doi: 10.1016/j.cca.2012.06.028. [DOI] [PubMed] [Google Scholar]
- 66.Nemkov T, Reisz JA, Gehrke S, Hansen KC, D’Alessandro A. High-throughput metabolomics: isocratic and gradient mass spectrometry-based methods. Methods Mol Biol. 2019;1978:13–26. doi: 10.1007/978-1-4939-9236-2_2. [DOI] [PubMed] [Google Scholar]
- 67.Nemkov T, D’Alessandro A, Hansen KC. Three-minute method for amino acid analysis by UHPLC and high-resolution quadrupole orbitrap mass spectrometry. Amino Acids. 2015;47(11):2345–2357. doi: 10.1007/s00726-015-2019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nemkov T, Hansen KC, D’Alessandro A. A three-minute method for high-throughput quantitative metabolomics and quantitative tracing experiments of central carbon and nitrogen pathways. Rapid Commun Mass Spectrom. 2017;31(8):663–673. doi: 10.1002/rcm.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moroff G, Sohmer PR, Button LN. Proposed standardization of methods for determining the 24-hour survival of stored red cells. Transfusion. 1984;24(2):109–114. doi: 10.1046/j.1537-2995.1984.24284173339.x. [DOI] [PubMed] [Google Scholar]
- 70.Francis RO, et al. Reexamination of the chromium-51-labeled posttransfusion red blood cell recovery method. Transfusion. 2019;59(7):2264–2275. doi: 10.1111/trf.15310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.