

Abstract
Recent studies have shown that IRF-1 plays a significant role in various tumour-induced chemoresistance, but its role and mechanism in gastric cancer-associated chemoresistance are not clear. Our study showed that IRF-1 expression could reverse gastric cancer-related chemoresistance. Dysregulated DNA repair is an important cause of chemoresistance. We established a chemoresistant gastric cancer cell line and found that drug-resistant gastric cancer cells had increased DNA repair ability and that IRF-1 regulated DNA damage repair. Further studies showed that IRF-1 inhibited the expression of RAD51 directly by binding to the RAD51 promoter to affect DNA damage repair; this binding reversed resistance. However, restoring the expression of RAD51 halted the inhibitory effect of IRF-1 partially. Also, we revealed that the overexpression of IRF-1 in a mouse model synergized with chemotherapeutic drugs to inhibit tumour growth. Finally, IRF-1 expression correlated with RAD51 expression in gastric cancer specimens. The expression of IRF-1 and RAD51 are both related to the survival duration of patients with gastric cancer. These results suggest that targeting IRF-1-RAD51 could be an effective approach to reversing multidrug resistance in gastric cancer.
Keywords: IRF-1, RAD51, chemotherapy, multiple drug resistance, gastric cancer
Introduction
Gastric cancer (GC) is one of the most common malignancies and remains the third leading cause of cancer-related deaths worldwide, causing a considerable health burden [1]. Currently, the main treatment strategies for patients with GC include surgery, chemotherapy, and radiation therapy. Surgical resection is the only possible curative treatment for GC. However, due to the absence of obvious symptoms in the early stage, the initial examination of most GC cases occurs in the advanced stage [2]. Therefore, chemotherapy remains the primary treatment for patients with advanced GC.
Chemoresistance is one of the most intractable issues facing the successful treatment of cancer in current clinical practice [3]. The 5-year survival rate of patients with advanced GC is still less than 30% [4]. Chemotherapy resistance is a key factor affecting the efficacy of chemotherapy in GC. The mechanism of chemoresistance in GC is complicated. A recent study found that the overexpression of drug resistance-associated proteins, abnormal cell proliferation and apoptosis, DNA damage repair dysfunction, tumour microenvironment changes, and changes in cell drug metabolism are associated with chemoresistance in GC [3]. Among these changes, aberrant DNA damage repair is an important cause of chemotherapy resistance in GC [5,6].
Multiple drug resistance (MDR) is the primary reason why the chemotherapy of GC fails. MDR is defined as the resistance by cancer cells to multiple chemotherapeutic drugs with different structures and mechanisms of action [7]. Abnormal DNA repair capacity is one of the major causes of MDR in GC. An increase in the rate or level of DNA repair contributes to evading or counteracting the effects of cytotoxic drugs [8]. RAD51 is an important homologous recombination (HR) repair protein whose main function is to catalyse homologous DNA strand pairing and exchange [9]. After DNA damage stress-sensing molecules, such as ATM, ATR, etc., recognize DNA double-strand breaks, they remove the nucleotides from the double-strand breaks first and expose the 3’ ssDNA tail at the site of injury [10]. The single-stranded binding protein RPA is then appended to the end of the excised ssDNA. Subsequently, under the action of BRCA1 and RAD51 homologues, RPA dissociates from ssDNA, which then binds RAD51. BRCA2 can promote the binding of RAD51 and DNA to form the RAD51 ribosome [11]. RAD51 continuously aggregates to ssDNA and finally forms a nucleoprotein filament and catalyses an ATP-dependent reaction. This reaction mediates homologous pairing and the exchange of homologous sequences between ssDNA and homoduplex DNA [11].
At present, RAD51 overexpression, but not gene amplification or mutation, has been reported in human malignancies, such as breast cancer [12], pancreatic cancer [13], non-small-cell lung cancer [14], and prostate cancer [15]. High RAD51 levels are associated with increased HR, which can repair DNA damage caused by chemotherapy drugs, reducing the efficacy of these drugs, thereby causing chemotherapy resistance [6,16]. In addition to increasing the DNA repair capacity, RAD51 also causes MDR by participating in the regulation of cell cycle, apoptosis, and proliferation, and targeting RAD51 can reverse MDR effectively [17,18].
Interferon regulatory factor-1 (IRF-1) is a transcription factor marked by functional diversity. IRF-1 plays a crucial role in antiviral responses and autoimmunity and in the regulation of immune system development, differentiation, and function. In addition, IRF-1 is a tumour suppressor that plays an antitumour role by regulating the expression of various genes, such as P21, P53, caspases, cyclins, and survivin, and participating in the regulation of cell cycle, proliferation, apoptosis, and DNA damage response [19,20]. In recent years, the role of IRF-1 in the chemoresistance of tumour cells has attracted much attention, but there are few related studies. In high-grade serous ovarian cancer, IRF-1 enhances sensitivity to platinum-based chemotherapeutic drugs and is considered to be an independent predictor of both progression-free and overall survival [21]. On the other hand, in ovarian cancer cells, cisplatin induces IRF-1 expression, limiting the drug’s effectiveness [22], but the specific mechanism is still unclear. Hence, this project constituted more in-depth research in this field.
Our previous studies have demonstrated that IRF-1 can reverse chemotherapy resistance in GC by inhibiting the expression of P-gp. However, baseline levels of P-gp expression are lower in some GC cells, and exogenous expression of IRF-1 also affects the MDR of GC cells [23]. These phenomena suggest that in addition to reversing resistance by inhibiting P-gp expression, IRF-1 may reverse resistance via several other mechanisms. In this study, we found that the expression of IRF-1 is related to DNA damage repair capability. Furthermore, we showed that IRF-1 can inhibit the expression of the homologous recombinant protein RAD51 to sensitize cells to the therapeutic effect of chemotherapy drugs. Specifically, IRF-1 mediated the transcriptional inhibition of the expression of RAD51 in vitro and in vivo, and the re-expression of RAD51 in IRF-1-overexpressed GC cells partially rescued chemotherapy drug resistance. Additionally, we demonstrated that IRF-1 expression correlated highly with RAD51 expression in GC samples.
In conclusion, our study shows that IRF-1 reverses the MDR of GC by downregulating the expression of RAD51, which has potential clinical and surgical therapeutic significance. This may provide new insight into the role of IRF-1 in the chemotherapeutic resistance of tumour cells.
Materials and methods
Clinical sample collection
Paraffin-embedded specimens were prepared with tissue samples collected from 52 patients who had been diagnosed with GC at the Union Hospital (Wuhan, China) according to the original histopathological reports from August 2014 to August 2015. All participants provided written consent. The study and consent procedures were approved by the Institutional Ethics Committee at Huazhong University of Science and Technology.
Cell lines and cell culture
Human GC cell lines (MKN45, AGS, and SGC7901) were purchased from the BeNa Culture Collection (Beijing, China). Multi-drug resistant (MDR) GC cell lines were established as described previously (24). All cell lines were cultured in RPMI-1640 (Gibco, NY, USA) supplemented with 10% foetal bovine serum (FBS) (ScienCell, CA, USA) and penicillin/streptomycin (HyClone, UT, USA). Cells were maintained at 37°C in a humidified 5% CO2 atmosphere.
Drugs and antibodies
All chemotherapy drugs - cisplatin (CDDP), fluorouracil (5FU), and doxorubicin (ADR) were purchased from Selleck (Houston, TX, USA). Antibodies against IRF-1 (ab186384), RAD51 (ab88572), and γH2A (ab2893) were acquired from Abcam (Cambridge, UK). Antibodies against GAPDH (10494-1-AP) were obtained from Proteintech (Wuhan, China).
Transcriptome sequencing
Parental GC cells (MKN45 and AGS) and chemotherapy-resistant GC cells (MKN45/MDR and AGS/MDR) were assessed with de novo transcriptome analysis using the Illumina HiSeq 2000 system (GM, Shanghai, China). Three samples were used for the sequencing of each group of cells.
Cell transfection and virus infection
IRF-1 siRNA and negative siRNA controls were constructed by GenePharma (Shanghai, China) (Table S1). All transfections with siRNA and vectors were performed using Lipofectamine 3000 according to the manufacturer’s instructions (Invitrogen, Carlsbad, MA, USA). The fluid was changed 48 h after transfection. Lentiviruses overexpressing IRF-1 (Lv-IRF-1) and RAD51 (Lv-RAD51) and empty viruses (Lv-Null) were purchased from GeneChem (Shanghai, China). IRF-1 expression was induced by adding doxycycline (Dox) to the medium (5 μg/mL) as described previously [24].
CCK8 assay
The CCK8 assay was performed using Cell Counting Kit-8 (Dojindo, Kyushu, Japan) per the manufacturer’s protocol. Cell sensitivity to chemotherapy drugs was measured as described previously [24].
Apoptosis assay
Apoptosis was assessed using the Annexin V/FITC Apoptosis Detection Kit (AntGene, ant003, Wuhan, China) as described previously [23].
Quantitative real-time PCR
Total RNA from cultured cells or frozen tumour tissues was extracted using a Trizol reagent kit (Takara, Dalian, China). qRT-PCR was then performed as described previously [25]. All primers were synthesized by Sangon (Shanghai, China) (Table S2). GAPDH was used as an internal control.
Western blotting
Protein extraction was carried out on ice using a RIPA buffer (Sigma-Aldrich, Darmstadt, Germany) containing proteinase and phosphatase inhibitors. Western blotting was performed as described previously [25].
Comet assay
The Comet Assay Kit (Trevigen Inc, Gaithersburg, MD, USA) was used under alkalic conditions according to the manufacturer’s specifications. Cells were re-suspended in 10% low-melting-point agarose, and 50 μl was immediately pipetted onto two-well comet assay slides. After solidification of the agarose, the slides were placed in a cell lysate (Trevigen, Inc) bath at 4°C overnight. The following day, the samples on the slides were electrophoresed in an alkaline electrophoresis solution, stained with a gold solution, and observed under a fluorescence microscope. The tail moments (TMs) of the comets were scored using CASP software.
Immunofluorescent staining
Cells (1 × 105 per well in 12-well plates) were grown on coverslips (WHB, WHB-12-CS, Shanghai, China) and after drug exposure for 24 h, were fixed in 4% paraformaldehyde (Sigma-Aldrich, Darmstadt, Germany) for 15 min, permeabilized in 0.2% Triton X-100 for 10 min, washed with PBS three times, and blocked with normal goat serum for 1 h at room temperature. The cells were then incubated with IRF-1 (Abcam, 1:100 dilution) and RAD51 (Abcam, 1:100 dilution) antibodies overnight at 4°C, washed with PBS three times, and incubated with secondary antibodies (Bosterbio, BA1105, Wuhan, China) in the dark for 1 h. Cell nuclei were stained with DAPI (blue) for 10 min and observed with a fluorescence microscope.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using the SimpleChIP Enzymatic Chromatin IP Kit (CST, #9003, MA, USA). All procedures were performed according to the manufacturer’s instructions. A monoclonal anti-IRF-1 antibody (Santa, sc-74530, CA, USA) and the corresponding rabbit-IgG (CST, USA) were used as controls. Bound DNA fragments were amplified using PCR, and the resulting PCR products were analysed with gel electrophoresis on 2% agarose gels. PCR primers are listed in Table S3.
Luciferase assay
Wild-type RAD51 and mutant RAD51 promoter regions were inserted into pGL3-based vectors. MKN45 cells were co-transfected with the RAD51 luciferase reporter construct and the IRF-1 plasmid using the Lipofectamine 3000 reagent. In the double luciferase assay, the cells were transfected for 48 hours, and the luciferase reporter gene expression system was detected using the dual-luciferase reporter gene detection system (Promega, Madison, WI, USA). Luciferase readings were normalised to Renilla luciferase activity.
Immunohistochemistry
Immunohistochemical (IHC) analyses of mouse tumour tissues and clinical GC tissues were performed using anti-IRF-1 (Abcam, 1:100 dilution) or anti-RAD51 (Abcam, 1:200 dilution) antibodies and incubated overnight at 4°C. HRP-conjugated Affinipure Goat Anti-Rabbit IgG (Proteintech, SA00001-2, Wuhan, China, 1:5000 dilution) was then used for incubation for one hour. We quantitatively scored tissue sections according to the percentage of positive cells and staining intensity, as described previously [23].
Xenograft assay
Five-week-old BALB/c female nude mice were bought from HFK Bio-Technology Co. (Beijing, China) for this assay. Lentiviruses containing specific DNA sequences were transfected into SGC7901 cells. To assess tumour growth in vivo, a 200 µl RPMI 1640 medium (without FBS) containing 5 × 106 cells was resuspended and then injected subcutaneously into the nude mice; each group contained 5 mice. Two weeks later, the mice were injected intraperitoneally with PBS containing CDDP, 5FU, or ADR once per week. Throughout the experiment, mice were fed 2 mg/mL of Dox in drinking water. Tumour volumes were measured every 7 days according to the formula V = 0.5 × L (length) × W2 (width). Mice were sacrificed humanely 5 weeks after cell inoculation. The care and handling of mice were approved by the Institutional Animal Care and Use Committee of Tongji Medical College, Huazhong University of Science and Technology.
Statistical analyses
Statistical analyses were performed using SPSS 24.0. Comparisons between two groups were performed with Student’s t-test. IRF-1 expression and clinical characteristics were analysed using the chi-square test. Kaplan-Meier survival analysis was used to analyse GC patient survival. The median RAD51 expression value was set as the cut-off value between high and low expression levels. P < 0.05 was considered statistically significant.
Results
IRF-1 sensitizes GC cells with MDR to chemotherapy drugs and regulates DNA damage repair
To understand the MDR mechanism in GC chemotherapy, we cultured GC cells with MDR. We then treated GC cell lines with three chemotherapeutics; CDDP, 5FU, and ADR and screened for viable cells 4 hours later. The initial concentration of chemotherapeutic drugs was 5 μmol/l, but each dose was later increased by a concentration gradient of 10 μmol/l. Six months after drug administration, we performed CCK-8 experiments to confirm that MDR cell lines were less sensitive to chemotherapeutic drugs than parental cell lines (Figure S1A). Subsequent transcriptome sequencing results also showed increased expression of resistance-related proteins in drug-resistant cell lines (Figure S1B and S1C).
Next, we determined whether IRF-1 is involved in chemotherapy resistance in GC cells using stable GC cells with MDR (MKN45/MDR and AGS/MDR) overexpressing IRF-1 (Lv-IRF-1). After 24 h of chemotherapy, the survival rate of cells in the Dox-induced IRF-1 overexpression group was significantly lower than that in the control group, with the growth of cells inhibited dose-dependently (Figures 1A and S2A). When IRF-1 was stably knocked down, however, we observed the opposite outcome (Figure S2B). Also, the apoptosis rate increased significantly after the overexpression of IRF-1 with the same drug concentration (Figure S2C and S2D).
Figure 1.
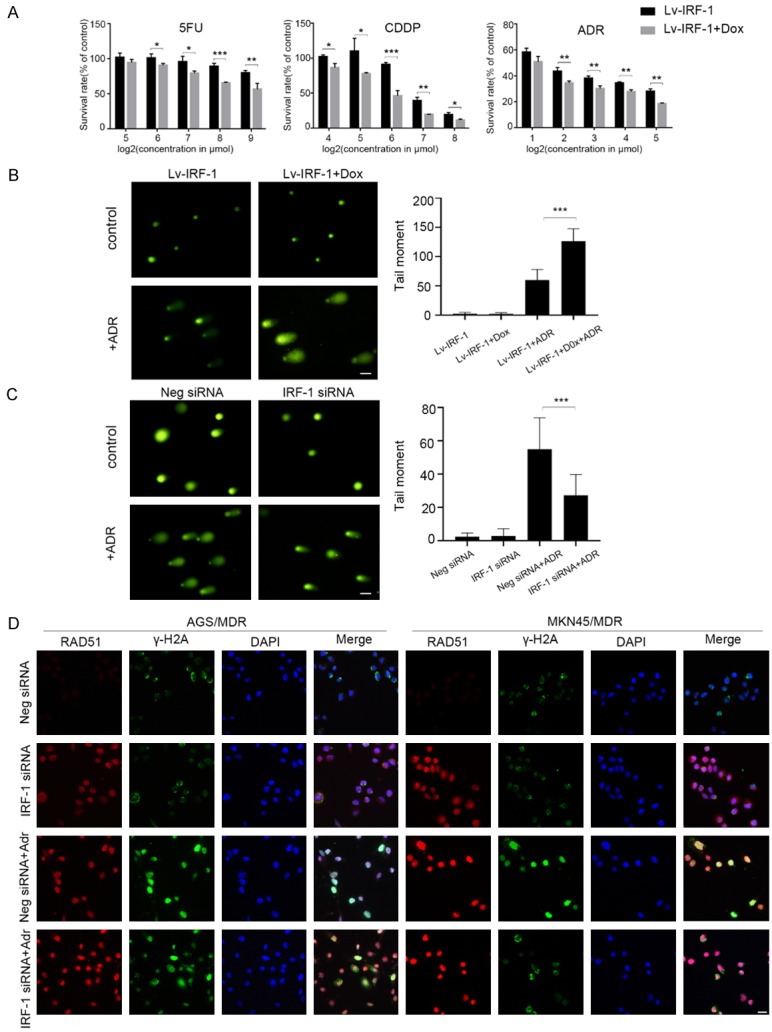
IRF-1 regulates DNA damage repair. (A) Cell viability assay showing the cell survival rates of AGS MDR/Lv-IRF-1 cells in the presence or absence of 2 μg/ml Dox 24 h after treatment with various concentrations of 5FU, CDDP, and ADR. (B) Representative images of the alkaline comet assay of AGS MDR/Lv-Null and AGS MDR/Lv-IRF-1 cells after exposure to 5 μmol/l ADR for 18 h. The tail moment is equal to the tail length multiplied by the tail DNA content. Scale bars, 50 μm. (C) Representative images of the alkaline comet assay after transfecting AGS MDR cells with Neg-siRNA or IRF-1 siRNA and exposing them to 5 μmol/l ADR for 18 h. Scale bars, 50 μm. (D) Immunofluorescent detection of RAD51 and γH2A expression levels after transfecting AGS MDR cells with Neg-siRNA or IRF-1 siRNA and exposing them to 5 μmol/l ADR for 18 h. Scale bars, 20 μm. In (A), the data are represented as the mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
To verify further that IRF-1 is involved in the DNA damage repair process, we used chemotherapeutic drugs to treat GC cells and examined the effect of IRF-1 on DNA repair ability. DNA double-strand breaks are relatively severe and can result in cell death. We used a comet assay to study the effect of IRF-1 on DNA double-strand damage repair in GC cells. The results suggest that the overexpression of IRF-1 causes the reduction in DNA double-strand damage repair ability in GC cell lines (AGS MDR and SGC7901) (Figures 1B and S3A). On the other hand, downregulating IRF-1 increases the double-strand repair ability (Figures 1C and S3B).
The presence of a γH2A-positive focus positive in a cell is a marker of DNA double-strand damage [26]. We found that the proportion of γH2A-positive cells in AGS cells with MDR and transfected with IRF-1 siRNA decreased significantly after 24 h of Dox treatment, indicating that the DNA double-strand damage repair ability was enhanced (Figure 1D). At the same time, decreasing IRF-1 expression levels increased the expression of the key protein RAD51 in the DNA double-strand break repair pathway (Figure 1D). These findings suggest that IRF-1 could inhibit DNA double-strand damage caused by chemotherapy drugs in GC cells.
RAD51 expression is elevated in MDR GC cells
To understand the difference between chemotherapy-resistant GC cells and parental cells, we performed transcriptome sequencing of chemotherapy-resistant GC cells (MKN45 MDR and AGS MDR) and parental cells (MKN45 and AGS). There was a difference in the expression of mRNA between the two groups of cells. In the AGS cell line with MDR, the expression of six genes associated with DNA damage repair was elevated, and that of three genes was diminished compared to the parental cell line (Figure 2A). In the MKN45 cell line with MDR, three genes associated with DNA damage repair were upregulated, and three genes were downregulated (Figure 2B). In particular, the RAD51 family gene was elevated in both drug-resistant cell lines compared to the parental cell lines. Additionally, we also found that the level of IRF-1 transcription was significantly lower in the chemotherapy-resistant cell lines than in the parental cell lines (Figure 2C). Subsequently, we verified in the cells, as shown in Figure 2E and 2F, that RAD51 was upregulated in GC chemotherapy-resistant cells, both at the mRNA and protein levels. Compared with the parental cells, the expression level of IRF-1 protein in chemotherapy-resistant GC cells decreased (Figure 2D).
Figure 2.
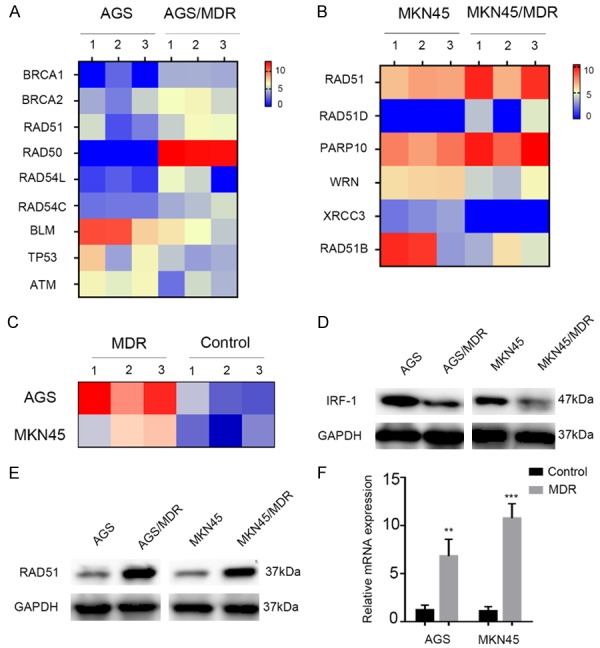
Gene expression profile analyses and validation. (A) Heatmap of the DNA repair gene transcript expression in the AGS cell line with or without MDR. (B) Heatmap of the DNA repair gene transcript expression in the MKN45 cell line with or without MDR. (C) Heatmap of the IRF-1 transcripts in cell lines with MDR versus parental gastric cancer cell lines. (D) Western blot analysis of the expression levels of IRF-1 in cell lines with MDR and parental gastric cancer cell lines. (E) Western blot analysis of the expression levels of RAD51 in cell lines with MDR and parental gastric cancer cell lines. (F) RAD51 mRNA expression level in cell lines with MDR and parental gastric cancer cell lines, as revealed by qRT-PCR. In (F), the data are represented as the mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
IRF-1 inhibits RAD51 expression in GC cells
The process of DNA damage repair is complex, and there are many factors involved in its regulation. When performing functional enrichment analysis of differential genes in AGS and AGS MDR cell lines, we found that proteins involved in the DNA damage repair of gastric cancer included RAD51, ATM, BRCA1, and more (Figure S4A). We also uncovered, through the protein interaction network analysis (https://string-db.org), that the expression level of RAD51, a protein repaired by DNA damage, may be associated with the expression of IRF-1 (Figure S4B).
We further investigated whether RAD51 is a functional target of IRF-1 and found that the overexpression of IRF-1 decreased RAD51 expression both at the mRNA and protein levels in AGS, MKN45, and SGC7901 cells (Figure 3A and 3B). In addition, knocking down IRF-1 with siRNA increased RAD51 protein expression in both AGS and MKN45 cells (Figure 3C). Immunofluorescence analysis also confirmed that the increased expression of IRF-1 reverses the mechanism of GC MDR associated with the decreased expression of RAD51 and increased DNA damage (Figure 3D).
Figure 3.

IRF-1 inhibits RAD51 expression. Western blotting detection of IRF-1 and RAD51 protein expression levels after transfection of AGS MDR, MKN45 MDR, and SGC7901 cells with Lv-IRF-1 or Lv-Null and treatment with the indicated amounts of Dox (A) and qRT-PCR (B). (C) Western blot analysis of RAD51 protein levels after knocking down IRF-1 in AGS MDR and MKN45 MDR cells. Neg siRNA, with a non-targeting sequence, was used as a negative control. (D) Immunofluorescent detection of RAD51 and γH2A expression levels after transfection of AGS MDR and MKN45 MDR cells with Lv-IRF-1 or Lv-Null and treatment with ADR for 18 h. Scale bars, 20 μm. (E) Western blotting detection of IRF-1 and RAD51 protein expression levels after transfecting MKN45/Lv-IRF-1 and SGC7901/Lv-IRF-1 cells with an empty vector virus or RAD51 overexpression virus, respectively, in the presence or absence of the indicated doses of Dox. (F) Cell viability assay of the cell survival rates of AGS MDR cells expressing Lv-IRF-1 and Lv-IRF-1 plus RAD51. All experiments were performed in triplicates. In (B and F), the data are represented as the mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
To determine whether the IRF-1-mediated reversal of GC cell chemoresistance depends on RAD51, we designed a rescue experiment to verify the role of RAD51. We transfected a RAD51 overexpression lentivirus and a negative control virus into MKN45 MDR/Lv-IRF-1 and SGC7901/Lv-IRF-1 cells. As shown in the Western blot, the expression level of RAD51 was not affected by the empty RAD51 vector, but the RAD51 lentivirus reversed the inhibitory effect of IRF-1 on RAD51 (Figure 3E).
Next, we analysed cell responses to chemotherapeutic drugs after RAD51 re-expression in IRF-1-overexpressing cells. CCK-8 assays demonstrated that RAD51 overexpression partially rescued the growth inhibition of AGS, MDR/Lv-IRF-1, and SGC7901/Lv-IRF-1 cells (Figures 3F and S3C). These data suggest that RAD51 plays a critical role in the IRF-1-mediated reversal of the MDR in GC.
IRF-1 regulates RAD51 promoter activity in GC cells
To determine whether IRF-1 directly regulates RAD51 expression at the transcriptional level, we determined two potential IRF-1 transcriptional binding sites in the promoter region of the RAD51 gene by sequence analysis, -1924 to -1904 and -1445 to -1442 (Figure 4A). ChIP experiments were then performed in MKN45 cells using two pairs of primers covering the two identified regions of the RAD51 promoter. As expected, the immunoprecipitation of chromatin-fractionated IRF-1 showed increased PCR products around putative binding sites 1 and 2 in MKN45 cells (Figure 4B). After Dox treatment, we found that the overexpression of IRF-1 resulted in significant RAD51 promoter enrichment compared to untreated samples (Figure 4C). Similarly, the electrophoretic analysis of DNA fragments amplified by PCR produced results consistent with earlier findings (Figure 4D).
Figure 4.

IRF-1 suppresses RAD51 promoter activity. (A) Schematic structure of the putative IRF-1 binding sites in the RAD51 promoter. (B) The characterisation of the recruitment of IRF-1 to the RAD51 promoter using Chromatin immunoprecipitation (ChIP) experiments on MKN45/Lv-IRF-1 cells. IgG was used as a control. (C) Assessment of the binding of IRF-1 to the RAD51 promoter using the ChIP assay after treatment of MKN45/Lv-IRF-1 cells with 2 μg/ml Dox for 48 h. (D) Agarose gel electrophoretic analysis of the DNA fragment obtained using ChIP. (E) IRF-1-binding site sequences in both wild-type (WT) and mutant (Mut) forms. (F) RAD51 promoter activities of the promoter, with or without mutations, in the predicted IRF-1 binding sites. In (B, C, and F), the data are represented as the mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
To confirm further that the binding of IRF-1 to the RAD51 promoter is effective and to determine which binding site is functional, we performed a luciferase reporter assay. We constructed mutant reporter genes for different mutations, including mutation 1, mutation 2, and mutations 1 plus 2 (Figure 4E), in the predicted IRF-1 binding sites using the wild-type RAD51 promoter. Compared with the negative control group, the IRF-1 overexpression group had significantly reduced luciferase activity of the wild-type RAD51 promoter, but this reduction was reversed by the mutation in target sequence 2 and the mutation in sequences 1 plus 2 of RAD51 (Figure 4F). Surprisingly, the luciferase activity of the promoter with mutation site 1 was lower than that of the wild type promoter. Therefore, we inferred that binding site 1 was not the site of IRF-1’s inhibition of the activity of the RAD51 promoter, and this site may even increase the activity of the RAD51 promoter. The reversal effect observed with the promoter mutation of site 2 and with the simultaneous mutation of site 1 plus site 2 revealed that IRF-1 exerted an inhibitory effect on RAD51 promoter activity through binding site 2.
IRF-1 expression reverses chemotherapy resistance, and RAD51 rescues chemotherapy resistance in vivo
To further establish whether elevated IRF-1 expression reversed MDR in vivo, MKN45 cells expressing Lv-NC, Lv-IRF-1, or Lv-IRF-1 with the re-expression of RAD51 were injected intracranially into nude mice (Figure 5A). After feeding mice with 2 mg/ml Dox water, IRF-1 overexpression had no significant effect on the growth of xenogenic tumours compared with the control group. However, after treatment with different chemotherapeutic drugs, the volumes and weights of tumours in the overexpressing IRF-1 group diminished significantly (Figure 5B and 5C). Additionally, the IRF-1 gene overexpression plus RAD51 gene re-expression group had partial restoration of tumour volumes and weights.
Figure 5.
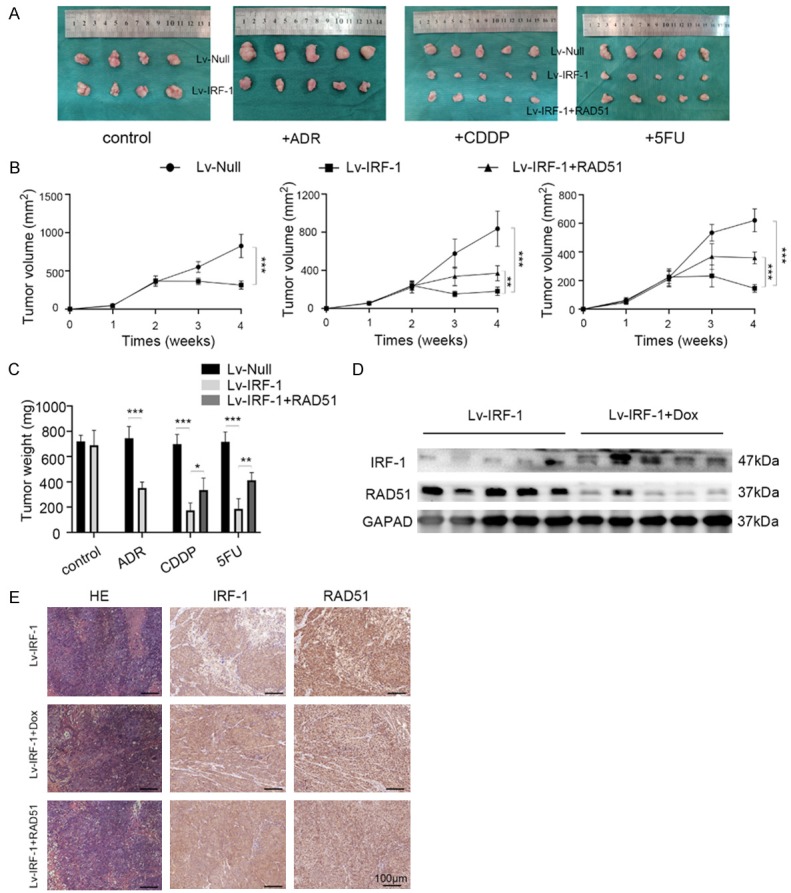
IRF-1 expression reverses chemotherapy resistance, while RAD51 rescues chemotherapy resistance in vivo. (A) Images showing tumours in different groups after chemotherapy and graphs depicting tumour volumes (B) and weights (C) of the different groups after injecting MKN45/Lv-null, MKN45/Lv-IRF-1, and MKN45/Lv-IRF-1 plus RAD51 cells into nude mice. Tumour volumes were measured every 7 days (tumour volume = 1/2 × length × width2). Western blotting (D) and immunohistochemical (E) detection of the expression levels of IRF-1 and RAD51. Scale bars, 100 μm. *P < 0.05, **P < 0.01, and ***P < 0.001.
Twenty-eight days after the inoculation of tumour cells, the nude mice were sacrificed humanely, and tissue proteins were extracted. Western blotting analyses showed that the Dox-induced IRF-1 overexpression group had lower RAD51 expression than the control group (Figure 5D). Immunohistochemistry findings confirmed the Western blotting results (Figure 5E). Consistent with the in vitro cell experimental outcomes, the overexpression of IRF-1 in vivo could reverse multidrug resistance in GC by inhibiting the expression of RAD51.
IRF-1 and RAD51 expression levels are correlated in GC and are independently predictive of poor prognosis
To understand the relationship between IRF-1 and RAD51 in GC tissues, we performed an IHC analysis of IRF-1 and RAD51 proteins in 52 GC samples (Figure 6A). The results revealed that IRF-1 and RAD51 correlated negatively with GC expression (Figure 6B). Previously published research showed that the low expression of IRF-1 correlated significantly with poor prognosis, as indicated by the Kaplan-Meier analysis. The 1-year and 3-year OS rates of patients in the high IRF-1 expression group were significantly higher than those of patients in the low IRF-1 expression group.
Figure 6.
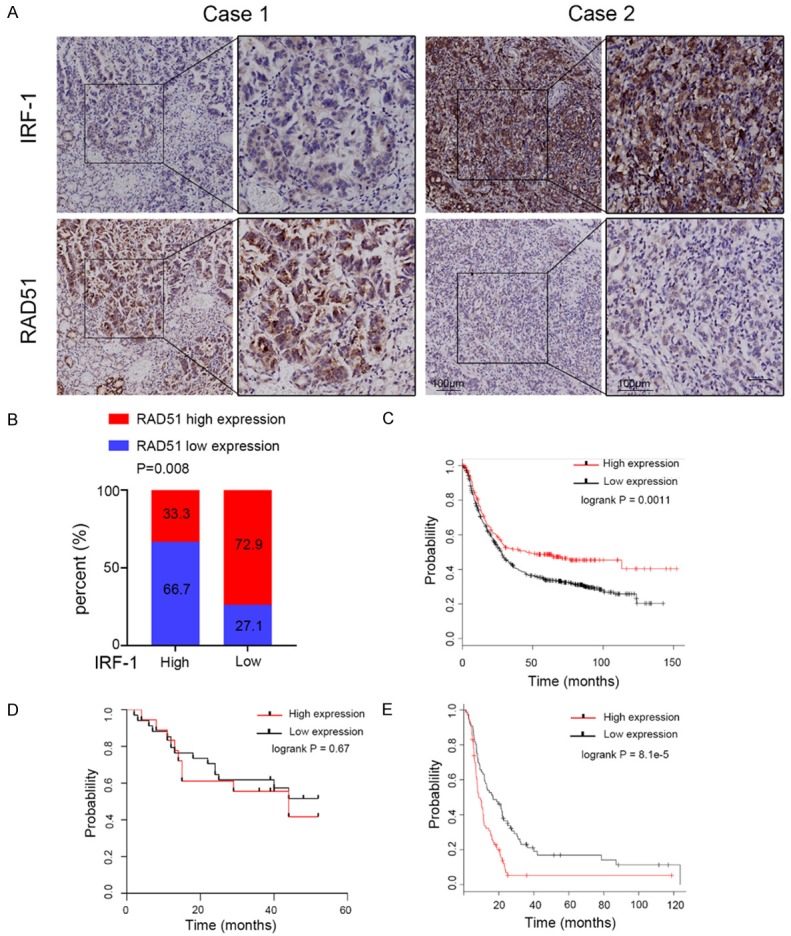
IRF-1 and RAD51 expression levels correlated highly with each other and are predictive of poor prognosis. A. Representative image of the immunohistochemical staining of IRF-1 and RAD51 in 52 GC tissues. Case 1 is the representative image from the IRF-1 low expression group. Case 2 is the representative image from the IRF-1 high expression group. Scale bars, 100 μm. The right panel represents a magnified view of the area of the box in the corresponding left panel. B. Statistical analysis of the correlation between IRF-1 and RAD51 expression in GC tissues. C. Survival curves of GC patients with high and low IRF-1 expression levels based on datasets. D. Survival curves of 52 GC patients with high and low RAD51 expression levels. E. Survival curves of GC patients with high and low RAD51 expression levels based on datasets.
To further confirm the clinical value of IRF-1, we used database analysis to determine that high IRF-1 expression was significantly associated with survival duration in GC patients [27] (Figure 6C). However, we found no significant correlation between RAD51 expression levels and survival duration in all 52 GC samples (Figure 6D), perhaps due to the small number of patients we selected and the short duration of follow-up. Nevertheless, we used the Kaplan-Meier Plotter online analysis tool to determine that high RAD51 expression was significantly associated with poor prognosis in GC patients receiving 5FU adjuvant chemotherapy (Figure 6E) [27].
Discussion
Our previous studies have shown that the overexpression of IRF-1 enhances the sensitivity of GC cells to chemotherapeutic drugs [24]. To further clarify the mechanism by which IRF-1 reverses GC resistance, we examined the expression of various DNA repair-related proteins and found that IRF-1 downregulated the expression of RAD51. We used bioinformatics to analyse the promoter region of the RAD51 gene and found that there was an IRF-1 binding site. ChIP and luciferase activity assays also demonstrated that IRF-1 directly interacted with the promoter through the IRF-1 binding site, regulating the expression of RAD51. Immunohistochemical analysis revealed that IRF-1 expression correlated negatively with RAD51 expression in GC specimens. Also, IRF-1-overexpressing GC cells with overexpressed RAD51 were resistant to chemotherapeutic drugs, indicating that the IRF-1-RAD51 axis plays a key role in chemoresistance.
MDR is the most important cause of tumour chemotherapy failure, and it plays a vital role in tumour metastasis and recovery. Once MDR is acquired, the anticancer effect of chemotherapeutic drugs diminishes significantly [28]. Although the MDR mechanism is complex, it is currently believed that MDR is related primarily to the ABC transporter family expression, DNA damage repair, apoptosis induction, autophagy induction, and cancer stem cell regulation [29]. Our previous studies have also shown that IRF-1 can reverse drug resistance by regulating the expression of drug-resistant proteins and reducing drug retention [23]. Here, we reveal that IRF-1 reversed DNA resistance by inhibiting RAD51 expression to reduce DNA damage repair. Increased expression of drug-resistant proteins and enhanced DNA damage repair capacity are the main causes of multidrug resistance in tumours [8]. Our results demonstrate that IRF-1 reversed tumour MDR in a variety of ways. In the study of patient samples, we performed a survival analysis of 52 patients with GC and found that the expression level of IRF-1 correlated significantly with the survival time of patients. We did not, however, find a correlation between RAD51 expression and patient survival, possibly because of the limited number of cases or short follow-up time. Nevertheless, in the TCGA database, we found, in patients with gastric cancer receiving 5-fluorouracil, that the survival time of the group with high expression of RAD51 was significantly lower than that of the low expression group. This outcome is consistent with the association between the high expression of RAD51 and poor prognosis in ovarian cancer [30]. These findings suggest that IRF-1 is a potential predictor of survival and chemotherapy response and that the overexpression of IRF-1 could be an effective strategy to overcome chemotherapy resistance.
Many studies have shown a strong link between IRF-1 and DNA damage repair. Prost et al. observed that IRF-1 dysregulation in cells may reduce the ability of cells to repair DNA damage and alter their sensitivity to the damage, but that the specific mechanism is less clear [31]. Mattia et al. performed a functional analysis of the IRF-1 target gene identified by ChIP-chip and found that IRF-1 is closely related to DNA damage response [32]. Our studies have shown that IRF-1 decreases the expression of RAD51 and inhibits DNA damage responses. Compared with chemotherapy alone, the expression of γH2A and the length of tails in the comet assay were greater when IRF-1 was combined with chemotherapy. Contrasting results were observed in the AGS and SGC7901 cell lines with MDR when IRF-1 was knocked down. However, we found that the re-expression of RAD51 did not fully restore the effect of IRF-1 on DNA damage response. These phenomena suggest that IRF-1 may also regulate the expression of other genes involved in DNA damage repair. In SGC7901 cells, we found that IRF-1 also affected the expression of the DNA damage repair gene PCNA (data not shown). These results indicate that IRF-1 overexpression reduces DNA damage response and increases the toxicity of chemotherapeutic drugs.
HR is the primary repair pathway for DSBs, using intact sister chromatids as a template for RAD51-catalysed DNA strand exchange and guiding error-free repair. HR is a very delicate and complex DNA repair process. Undoubtedly, RAD51, the core protein in the process of HR, is also regulated by several mechanisms. RAD51 has been reported previously to be regulated by wild-type p53 [33-35]. Carman et al. found that wild-type p53 binds to p53 response elements on the RAD51 promoter in vivo and downregulates RAD51 messenger RNA and protein expression. Moreover, wild-type p53 also inhibits the formation of Rad51 foci during double-strand breaks [33]. In another study, the oncogenic transcription factor Foxm1 activated RAD51 expression and caused chemoresistance in glioblastoma [36]. This study was the first to implicate RAD51 as an IRF-1-regulated gene that can mediate chemoresistance in cancer cells. Luciferase assay results confirmed that the transcription factor IRF-1 is directly involved in the regulation of RAD51. Surprisingly, the mutation of binding site 1 alone increased the activity of the RAD51 promoter. Mutations in the second binding site, alone and combined with other mutations, significantly reduced RAD51 promoter activity. This may suggest differential regulation of RAD51 by IRF-1 and appears to provide a reasonable explanation for the absence of a significant increase in the baseline expression of RAD51 when knocking down IRF-1 expression (Figure 3C). The reversal of chemoresistance in gastric cancer by using IRF-1 to mediate RAD51 expression seems to provide a new direction for chemotherapy.
Many anticancer drugs achieve the purpose of treatment by damaging the DNA of tumour cells, but tumour cells can activate the DNA damage repair mechanism and, thus, become resistant to DNA damage drugs. Hypermethylation of the mismatch repair gene, MLH1, can lead to resistance to cisplatin and carboplatin [37]. ERCC1 is a major protein in nucleotide excision repair systems. High ERCC1 expression is associated with increased removal of DNA-platinum adducts and results in increased tumour resistance to cisplatin [38]. In contrast, cancers with DNA repair deficiencies are more sensitive to chemotherapy drugs. Defects in HR repair caused by germline or somatic mutations in BRCA1 or BRCA2 make tumours susceptible to PARPi treatment [39]. Similarly, the enhancement of DNA repair capacity plays a vital role in tumour radiotherapy tolerance. In non-small cell lung cancer, highly expressed RAD50 increases radiation tolerance [40]. Ku80 is a key protein in the repair of non-homologous end junctions. Ku80 plays an important role not only in tumorigenesis but also in radiation resistance in esophageal cancer cells [41]. In contrast, ATM kinase inhibitors significantly increase the radiation sensitivity of glioma cells [42].
There is increasing evidence that overactive HR repair mechanisms are associated with chemoresistance [43]. Our experiments demonstrated that RAD51 expression was higher in drug-resistant cells than in parental cells, both at the mRNA and protein levels. These results are consistent with previously published findings showing that RAD51 overexpression is associated with chemoresistance [44-46]. Although we did not find that RAD51 overexpression correlated significantly with the clinical outcome in 52 patients with GC, many studies have revealed that RAD51 expression levels correlate significantly with poor prognosis [47,48]. RAD51 expression is also involved in a variety of non-DNA repair pathways, such as tumour metastasis [49], tumour growth, and differentiation [50]. For that reason, we suggest that RAD51 be researched further as a therapeutic target for malignant tumours; its role in chemotherapy has become the focus of recent research. Liu et al. demonstrated that the overexpression of RAD51 compensates for BRCA1 deficiency and mediates breast cancer stem cell resistance to PARPi [45]. Per Sampath et al., HDAC inhibition induces miR-182 to target RAD51 and HR, making acute myeloid leukaemia cells sensitive to DNA-damaging agents [51]. Although RAD51 has shown synergy in multiple-drug screenings of multiple cancer types, and there are no clinical trials directly targeting RAD51, we recommend additional targeting of RAD51 to improve clinical outcomes.
Conclusion
Our study reveals a key role for the RAD51-dependent IRF-1 pathway in chemical sensitization. This is unexpected because studies on the role of IRF-1 in chemoresistance are few. In summary, we found that IRF-1 expression levels were lower in chemotherapy-resistant GC cells than in parental cells and that overexpressing IRF-1 reversed MDR in GC. Mechanistically, IRF-1 directly regulated the DNA damage repair gene, RAD51, at the transcriptional level. Overexpressing IRF-1 inhibited RAD51 expression, and the re-expression of RAD51 partially rescued IRF-1 overexpression’s inhibitory effect on chemotherapy resistance. These results suggest that IRF-1 plays a key role in tumour cell resistance and that targeting IRF-1 is an effective approach to increasing the chemosensitivity of tumour cells.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 81572411). All authors have read and approved the submission of the manuscript. The care and handling of animals were approved by the Institutional Animal Care and Use Committee of Tongji Medical College, Huazhong University of Science and Technology.
Disclosure of conflict of interest
None.
Abbreviations
- GC
gastric cancer
- MDR
multiple drug resistance
- HR
homologous recombination
- RAD51
recombinant RAD51 homolog
- IRF-1
Interferon regulatory factor-1
- CDDP
cisplatin
- 5FU
fluorouracil
- ADR
doxorubicin
- Dox
doxycycline
- TMs
tail moments
- P-gp
P glycoprotein
- IHC
immunohistochemical
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Yamamoto H, Watanabe Y, Maehata T, Morita R, Yoshida Y, Oikawa R, Ishigooka S, Ozawa S, Matsuo Y, Hosoya K, Yamashita M, Taniguchi H, Nosho K, Suzuki H, Yasuda H, Shinomura Y, Itoh F. An updated review of gastric cancer in the next-generation sequencing era: insights from bench to bedside and vice versa. World J Gastroenterol. 2014;20:3927–3937. doi: 10.3748/wjg.v20.i14.3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marin JJ, Al-Abdulla R, Lozano E, Briz O, Bujanda L, Banales JM, Macias RI. Mechanisms of resistance to chemotherapy in gastric cancer. Anticancer Agents Med Chem. 2016;16:318–334. doi: 10.2174/1871520615666150803125121. [DOI] [PubMed] [Google Scholar]
- 4.In H, Solsky I, Palis B, Langdon-Embry M, Ajani J, Sano T. Validation of the 8th edition of the AJCC TNM staging system for Gastric Cancer using the National Cancer Database. Ann Surg Oncol. 2017;24:3683–3691. doi: 10.1245/s10434-017-6078-x. [DOI] [PubMed] [Google Scholar]
- 5.Xie X, Huang N, Zhang Y, Wei X, Gao M, Li M, Ning J, Liu W, Zhao Q, Wang H, Gu K. MiR-192-5p reverses cisplatin resistance by targeting ERCC3 and ERCC4 in SGC7901/DDP cells. J Cancer. 2019;10:1039–1051. doi: 10.7150/jca.25814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ronchetti L, Melucci E, De Nicola F, Goeman F, Casini B, Sperati F, Pallocca M, Terrenato I, Pizzuti L, Vici P, Sergi D, Di Lauro L, Amoreo CA, Gallo E, Diodoro MG, Pescarmona E, Vitale I, Barba M, Buglioni S, Mottolese M, Fanciulli M, De Maria R, Maugeri-Sacca M. DNA damage repair and survival outcomes in advanced gastric cancer patients treated with first-line chemotherapy. Int J Cancer. 2017;140:2587–2595. doi: 10.1002/ijc.30668. [DOI] [PubMed] [Google Scholar]
- 7.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 8.Zhang D, Fan D. Multidrug resistance in gastric cancer: recent research advances and ongoing therapeutic challenges. Expert Rev Anticancer Ther. 2007;7:1369–1378. doi: 10.1586/14737140.7.10.1369. [DOI] [PubMed] [Google Scholar]
- 9.Symington LS. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. In: Richardson CC, editor. Annual Review of Biochemistry. 1997. pp. 61–92. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Doty T, Gibson B, Heyer WD. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol. 2010;17:1260–1262. doi: 10.1038/nsmb.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maacke H, Opitz S, Jost K, Hamdorf W, Henning W, Kruger S, Feller AC, Lopens A, Diedrich K, Schwinger E, Sturzbecher HW. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int J Cancer. 2000;88:907–913. doi: 10.1002/1097-0215(20001215)88:6<907::aid-ijc11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 13.Maacke H, Jost K, Opitz S, Miska S, Yuan Y, Hasselbach L, Luttges J, Kalthoff H, Sturzbecher HW. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000;19:2791–2795. doi: 10.1038/sj.onc.1203578. [DOI] [PubMed] [Google Scholar]
- 14.Takenaka T, Yoshino I, Kouso H, Ohba T, Yohena T, Osoegawa A, Shoji F, Maehara Y. Combined evaluation of Rad51 and ERCC1 expressions for sensitivity to platinum agents in non-small cell lung cancer. Int J Cancer. 2007;121:895–900. doi: 10.1002/ijc.22738. [DOI] [PubMed] [Google Scholar]
- 15.Mitra A, Jameson C, Barbachano Y, Sanchez L, Kote-Jarai Z, Peock S, Sodha N, Bancroft E, Fletcher A, Cooper C, Easton D IMPACT Steering Committee and IMPACT and EMBRACE Collaborators. Eeles R, Foster CS. Overexpression of RAD51 occurs in aggressive prostatic cancer. Histopathology. 2009;55:696–704. doi: 10.1111/j.1365-2559.2009.03448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu ZY, Loignon M, Han FY, Panasci L, Aloyz R. Xrcc3 induces cisplatin resistance by stimulation of Rad51-related recombinational repair, S-phase checkpoint activation, and reduced apoptosis. J Pharmacol Exp Ther. 2005;314:495–505. doi: 10.1124/jpet.105.084053. [DOI] [PubMed] [Google Scholar]
- 17.Henning W, Sturzbecher HW. Homologous recombination and cell cycle checkpoints: Rad51 in tumour progression and therapy resistance. Toxicology. 2003;193:91–109. doi: 10.1016/s0300-483x(03)00291-9. [DOI] [PubMed] [Google Scholar]
- 18.He WL, Li YH, Hou WJ, Ke ZF, Chen XL, Lu LY, Cai SR, Song W, Zhang CH, He YL. RAD51 potentiates synergistic effects of chemotherapy with PCI-24781 and cis-diamminedichloroplatinum on gastric cancer. World J Gastroenterol. 2014;20:10094–10107. doi: 10.3748/wjg.v20.i29.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 20.Chen FF, Jiang G, Xu K, Zheng JN. Function and mechanism by which interferon regulatory factor-1 inhibits oncogenesis (Review) Oncol Lett. 2013;5:417–423. doi: 10.3892/ol.2012.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohen S, Mosig R, Moshier E, Pereira E, Rahaman J, Prasad-Hayes M, Halpert R, Billaud JN, Dottino P, Martignetti JA. Interferon regulatory factor 1 is an independent predictor of platinum resistance and survival in high-grade serous ovarian carcinoma. Gynecol Oncol. 2014;134:591–598. doi: 10.1016/j.ygyno.2014.06.025. [DOI] [PubMed] [Google Scholar]
- 22.Pavan S, Olivero M, Cora D, Di Renzo MF. IRF-1 expression is induced by cisplatin in ovarian cancer cells and limits drug effectiveness. Eur J Cancer. 2013;49:964–973. doi: 10.1016/j.ejca.2012.09.024. [DOI] [PubMed] [Google Scholar]
- 23.Yuan J, Yin Z, Tan L, Zhu W, Tao K, Wang G, Shi W, Gao J. Interferon regulatory factor-1 reverses chemoresistance by downregulating the expression of P-glycoprotein in gastric cancer. Cancer Lett. 2019;457:28–39. doi: 10.1016/j.canlet.2019.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Gao J, Tian Y, Zhang J. Overexpression of interferon regulatory factor 1 enhances chemosensitivity to 5-fluorouracil in gastric cancer cells. J Cancer Res Ther. 2012;8:57–61. doi: 10.4103/0973-1482.95175. [DOI] [PubMed] [Google Scholar]
- 25.Gao J, Senthil M, Ren B, Yan J, Xing Q, Yu J, Zhang L, Yim JH. IRF-1 transcriptionally upregulates PUMA, which mediates the mitochondrial apoptotic pathway in IRF-1-induced apoptosis in cancer cells. Cell Death Differ. 2010;17:699–709. doi: 10.1038/cdd.2009.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W. Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev. 2002;12:162–169. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- 27.Szasz AM, Lanczky A, Nagy A, Foerster S, Hark K, Green JE, Boussioutas A, Busuttil R, Szabo A, Gyorffy B. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget. 2016;7:49322–49333. doi: 10.18632/oncotarget.10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fruci D, Cho WC, Romania P, Nobili V, Locatelli F, Alisi A. Drug transporters and multiple drug resistance in the most common pediatric solid tumors. Curr Drug Metab. 2016;17:308–316. doi: 10.2174/1567205010666131212110948. [DOI] [PubMed] [Google Scholar]
- 29.Wu Q, Yang Z, Nie Y, Shi Y, Fan D. Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches. Cancer Lett. 2014;347:159–166. doi: 10.1016/j.canlet.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Xiao M, Cai J, Cai L, Jia J, Xie L, Zhu Y, Huang B, Jin D, Wang Z. Let-7e sensitizes epithelial ovarian cancer to cisplatin through repressing DNA double strand break repair. J Ovarian Res. 2017;10:24. doi: 10.1186/s13048-017-0321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prost S, Bellamy CO, Cunningham DS, Harrison DJ. Altered DNA repair and dysregulation of p53 in IRF-1 null hepatocytes. FASEB J. 1998;12:181–188. doi: 10.1096/fasebj.12.2.181. [DOI] [PubMed] [Google Scholar]
- 32.Frontini M, Vijayakumar M, Garvin A, Clarke N. A ChIPchip approach reveals a novel role for transcription factor IRF1 in the DNA damage response. Nucleic Acids Res. 2009;37:1073–1085. doi: 10.1093/nar/gkn1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arias-Lopez C, Lazaro-Trueba I, Kerr P, Lord CJ, Dexter T, Iravani M, Ashworth A, Silva A. P53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006;7:219–224. doi: 10.1038/sj.embor.7400587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazaro-Trueba I, Arias C, Silva A. Double bolt regulation of Rad51 by p53: a role for transcriptional repression. Cell Cycle. 2006;5:1062–1065. doi: 10.4161/cc.5.10.2764. [DOI] [PubMed] [Google Scholar]
- 35.Gatz SA, Wiesmueller L. p53 in recombination and repair. Cell Death Differ. 2006;13:1003–1016. doi: 10.1038/sj.cdd.4401903. [DOI] [PubMed] [Google Scholar]
- 36.Zhang N, Wu X, Yang L, Xiao F, Zhang H, Zhou A, Huang Z, Huang S. FoxM1 inhibition sensitizes resistant glioblastoma cells to temozolomide by downregulating the expression of DNA-repair gene Rad51. Clin Cancer Res. 2012;18:5961–5971. doi: 10.1158/1078-0432.CCR-12-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. 1998;4:1–6. [PubMed] [Google Scholar]
- 38.Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res. 2016;106:27–36. doi: 10.1016/j.phrs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 39.Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Gudikote J, Giri U, Yan J, Deng W, Ye R, Jiang W, Li N, Hobbs BP, Wang J, Swisher SG, Fujimoto J, Wistuba II, Komaki R, Heymach JV, Lin SH. RAD50 expression is associated with poor clinical outcomes after radiotherapy for resected non-small cell lung cancer. Clin Cancer Res. 2018;24:341–350. doi: 10.1158/1078-0432.CCR-17-1455. [DOI] [PubMed] [Google Scholar]
- 41.Yang QS, Gu JL, Du LQ, Jia LL, Qin LL, Wang Y, Fan FY. ShRNA-mediated Ku80 gene silencing inhibits cell proliferation and sensitizes to gamma-radiation and mitomycin C-induced apoptosis in esophageal squamous cell carcinoma lines. J Radiat Res. 2008;49:399–407. doi: 10.1269/jrr.07096. [DOI] [PubMed] [Google Scholar]
- 42.Biddlestone-Thorpe L, Sajjad M, Rosenberg E, Beckta JM, Valerie NC, Tokarz M, Adams BR, Wagner AF, Khalil A, Gilfor D, Golding SE, Deb S, Temesi DG, Lau A, O’Connor MJ, Choe KS, Parada LF, Lim SK, Mukhopadhyay ND, Valerie K. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin Cancer Res. 2013;19:3189–3200. doi: 10.1158/1078-0432.CCR-12-3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ward A, Khanna KK, Wiegmans AP. Targeting homologous recombination, new pre-clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treat Rev. 2015;41:35–45. doi: 10.1016/j.ctrv.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 44.Gachechiladze M, Skarda J, Soltermann A, Joerger M. RAD51 as a potential surrogate marker for DNA repair capacity in solid malignancies. Int J Cancer. 2017;141:1286–1294. doi: 10.1002/ijc.30764. [DOI] [PubMed] [Google Scholar]
- 45.Liu Y, Burness ML, Martin-Trevino R, Guy J, Bai S, Harouaka R, Brooks MD, Shang L, Fox A, Luther TK, Davis A, Baker TL, Colacino J, Clouthier SG, Shao ZM, Wicha MS, Liu S. RAD51 mediates resistance of cancer stem cells to PARP inhibition in triple-negative breast cancer. Clin Cancer Res. 2017;23:514–522. doi: 10.1158/1078-0432.CCR-15-1348. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, Peri S, van der Gulden H, van der Heijden I, O’Brien SW, Zhang Y, Harrell MI, Johnson SF, Candido Dos Reis FJ, Pharoah PD, Karlan B, Gourley C, Lambrechts D, Chenevix-Trench G, Olsson H, Benitez JJ, Greene MH, Gore M, Nussbaum R, Sadetzki S, Gayther SA, Kjaer SK kConFab Investigators. D’Andrea AD, Shapiro GI, Wiest DL, Connolly DC, Daly MB, Swisher EM, Bouwman P, Jonkers J, Balmana J, Serra V, Johnson N. The BRCA1-delta 11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016;76:2778–2790. doi: 10.1158/0008-5472.CAN-16-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Wang JC, Li YH, Wang RX, Fan XM. Expression of PH domain leucine-rich repeat protein phosphatase, forkhead homeobox type O 3a and RAD51, and their relationships with clinicopathologic features and prognosis in ovarian serous adenocarcinoma. Chin Med J (Engl) 2017;130:280–287. doi: 10.4103/0366-6999.198932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y, Wang WY, Xiao JH, Xu F, Liao DY, Xie L, Wang J, Luo F. Overexpression of Rad51 predicts poor prognosis in colorectal cancer: our experience with 54 patients. PLoS One. 2017;12:e0167868. doi: 10.1371/journal.pone.0167868. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Alshareeda AT, Negm OH, Aleskandarany MA, Green AR, Nolan C, TigHhe PJ, Madhusudan S, Ellis IO, Rakha EA. Clinical and biological significance of RAD51 expression in breast cancer: a key DNA damage response protein. Breast Cancer Res Treat. 2016;159:41–53. doi: 10.1007/s10549-016-3915-8. [DOI] [PubMed] [Google Scholar]
- 50.Sarwar R, Sheikh AK, Mahjabeen I, Bashir K, Saeed S, Kayani MA. Upregulation of RAD51 expression is associated with progression of thyroid carcinoma. Exp Mol Pathol. 2017;102:446–454. doi: 10.1016/j.yexmp.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 51.Lai TH, Ewald B, Zecevic A, Liu C, Sulda M, Papaioannou D, Garzon R, Blachly JS, Plunkett W, Sampath D. HDAC inhibition induces microRNA-182, which targets RAD51 and impairs HR repair to sensitize cells to sapacitabine in acute myelogenous leukemia. Clin Cancer Res. 2016;22:3537–3549. doi: 10.1158/1078-0432.CCR-15-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.