

Abstract
Poly (adenosine diphosphate ribose) polymerase (PARP) inhibitors benefit a small percentage of ovarian cancer patients with homologous recombination (HR) deficiency (HRD), which greatly limits the applications of PARP inhibitors. Given the function of CDK9 in homologous recombination repair (HRR), here, we show how to extend the utility of PARP inhibitors in BRCA1-proficient ovarian cancer by targeting CDK9. We found that high CDK9 expression is associated with a higher tumor stage in epithelial ovarian cancer patients, and CDK9 is co-expressed with BRCA1 by analyzing a public database. By using a CDK9 inhibitor CDKI-73, we found that its combination with the PARP inhibitor olaparib significantly suppressed cell viability and colony formation and induced apoptosis in BRCA1-proficient ovarian cancer cells. Consistently, the combination treatment remarkably reduced the tumor growth in mouse xenograft models. We demonstrated that CDKI-73 could downregulate BRCA1 expression, resulting in hypersensitivity to olaparib in BRCA1-proficient ovarian cancer. Taken together, our results show a synergetic effect of CDKI-73 combined with olaparib in BRCA1-proficient ovarian cancer, facilitating the clinical use of CDK9 as a predictive biomarker to exploit PARP inhibitors.
Keywords: PARP inhibitors, CDK9, homologous recombination deficiency, BRCA, ovarian cancer
Introduction
Currently, ovarian cancer remains one of the leading causes of gynaecologic malignancy-related death in the United States [1]. Due to the histologic and genetic heterogeneity of ovarian cancer, current treatment modalities, including cytoreductive surgery and chemotherapy, remain poorly effective, which highlights the necessity for new, more effective remedies [2,3].
The emergence of Poly (adenosine diphosphate ribose) polymerase (PARP) inhibitors undoubtedly brings a glimmer of light to the current state of treatment, especially for patients harboring germline or somatic BRCA1/2 mutations or other homologous recombination repair (HRR) gene deficiencies based on the concept of synthetic lethality [4]. Olaparib, a PARP inhibitor, initially received approval by the Food and Drug Administration (FDA) as the first monotherapy for the treatment of advanced-stage ovarian cancer patients with BRCA1/2-mutation [5]. Although the clinical response is favorable, the application of PARP inhibitors is still not extensive in ovarian cancers because only approximately 50% of ovarian cancers are HRR deficient [6]. Thus, maximizing their utilization in HRR-proficient ovarian cancer has become a crucial and urgent clinical problem. Defective HRR is an important therapeutic target to efficiently enhance the sensitivity of PARP inhibitors according to the synthetic lethality theory [6,7]. Relevant clinical trials on combined PARP inhibitors and HR deficiency (HRD) have been conducted on several human malignancies, including prostate cancer [8], ovarian cancer [6,9], and breast cancer [10]. To broaden the spectrum of HRD in ovarian cancer, recent studies have focused on pharmaceutically compromising HRR by suppressing the expression of HR genes, which might potentially benefit PARP inhibitor therapy [11].
Unlike most other CDKs functioning in cell cycling, cyclin-dependent kinase 9 (CDK9) plays a crucial role in RNA transcription elongation by phosphorylating the carboxyl terminus of RNA Pol II [12,13]. The deregulation of CDK9 has necessary implications for the development and maintenance of various human malignancies [14], and the overexpression and increased activation of CDK9 have been observed in melanoma and prostate cancer [15,16]. Recently, CDK9 was also shown to be highly expressed in human ovarian cancer cell lines, and the level of CDK9 expression was higher in metastatic and recurrent ovarian tumor tissue compared to that in the primary tumors; an elevated CDK9 protein level was significantly correlated with poor patient prognosis [17]. Consequently, CDK9 has been considered a novel prognostic biomarker and a promising therapeutic target for many cancer types [14,18-21]. However, the relationship between CDK9 expression and ovarian cancer progression still needs to be discussed.
CDK9 has also been shown to play a role in promoting genomic integrity and facilitating recovery from replication stress and DNA damage [22,23]. CDK9 participates in HR-mediated DNA double-strand breaks (DSBs) repair via the recruitment of BRCA1 to DNA damage sites, and depletion of CDK9 can sensitize cells to ionizing radiation (IR) treatment and PARP inhibitors in breast cancer [24], which encouraged an attempt to determine whether CDK9 inhibitors can act as sensitizers to PARP inhibitors in ovarian cancer.
CDKI-73, an effective CDK9 inhibitor, can specifically inhibit CDK9 kinase activity and further impede gene transcription [25]. In this study, we show that CDKI-73 can prevent BRCA1 expression, leading to increased spontaneous DNA damage and hypersensitivity to the PARP inhibitor olaparib in vitro and in vivo. Thus, we propose a new therapeutic strategy based on CDK9: CDK9 inhibitors can render BRCA1 wild-type ovarian cancer vulnerable to PARP inhibitors in a synthetic lethal manner.
Materials and methods
Cell lines and cell culture
The human ovarian cancer cell lines SKOV3, A2780, OVCAR-5, OVCAR-8, and OVCA433 were obtained from the American Type Culture Collection (ATCC) and stored in our laboratory. HO8910 cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). SKOV3 and A2780 cells were cultured in RPMI 1640 (HyClone, UT, USA) supplemented with 10% fetal bovine serum (FBS) (GIBCO BRL, BRA, USA) and 1 × 105 IU/L penicillin and streptomycin (Beyotime Biotechnology, SH, China). HO8910, OVCAR-5, OVCAR-8, and OVCA433 cells were maintained in DMEM (HyClone) containing 10% FBS and penicillin and streptomycin. All cells were incubated at 37°C with 5% CO2 and 95% humidity.
Drugs
CDKI-73 (MedChemExpress, NJ, USA) and olaparib (MedChemExpress) were dissolved in DMSO for all in vitro experiments. For in vivo studies, CDKI-73 and olaparib were dissolved in 0.5% sodium carboxymethyl cellulose. An equal amount of DMSO or sodium carboxymethyl cellulose was used as the vehicle for all experiments.
Clonogenic assay
After plating three thousand cells per well into six-well plates, cells were treated with the corresponding drugs. Every three days, the medium was replaced with a fresh medium containing the same concentration of inhibitors or vehicle. Ten days later, the cells were stained with 0.5% crystal violet stain solution (Yeasen, SH, China) after 4% polyformaldehyde fixation. Finally, the number of colonies (> 10 cells) was counted under a microscope, and cell viability was calculated relative to the vehicle.
Cell viability assay
Cell viability was assessed using the Cell Counting Kit-8 (Beyotime Biotechnology). The IC50 values were analyzed with GraphPad Prism software, and the combined effect of CDKI-73 and olaparib was examined by the Chou-Talalay combination index (CI) method. CI values under 0.9 represent synergism, values 0.9 to 1.1 are additive, and values above 1.1 indicate antagonism. The fraction affected (FA) was calculated as follows: % reduction from the untreated control × 0.01.
Apoptosis analysis
After treatment with drugs or the vehicle, the cells were harvested and stained with annexin V-FITC and propidium iodide (PI) (BD Pharmingen, CA, USA). Then, cellular apoptosis was analyzed by flow cytometry on an ACEA NovoCyte flow cytometer (ACEA Biosciences, HZ, China).
Immunofluorescence staining
Cells were inoculated onto coverslips in 24-well plates with a culture medium containing different concentrations of drugs or the vehicle control for 48 hours. Then, immunofluorescence staining was performed as described previously [26]. After fixation, permeabilization, and blocking, coverslips were incubated with a mouse anti-γH2AX (Ser139) monoclonal antibody (ab26350, Abcam), followed by incubation with a secondary antibody and counterstaining with DAPI. γH2AX foci and the percentage of positive cells (> 10 foci per cell) were calculated under a fluorescence microscope.
Quantitative real-time PCR
Following total RNA extraction with an RNeasy Plus Mini Kit (Qiagen, Hilden, Germany), the reverse transcription synthesis of cDNA was carried out using ReverTra Ace qPCR RT Master Mix (TOYOBO, SH, China). Then, real-time PCR was performed using SYBR Green Master Mix (Yeasen). The human BRCA1 forward primer was 5’-GAAACCGTGCCAAAAGACTTC-3’, and the reverse primer was 5’-CCAAGGTTAGAGAGTTGGACAC-3’.
Western blot analysis
Cells were harvested, and proteins were isolated. Then, Western blotting and signal detection were performed as previously described [27]. The primary antibodies used were rabbit polyclonal anti-BRCA1 (ab9141, Abcam), rabbit monoclonal anti-CDK9 (ab76320, Abcam), rabbit polyclonal anti-CDK9 (phospho T186) (ab79178, Abcam), rabbit monoclonal anti-cleaved PARP1 (ab32064, Abcam), rabbit monoclonal anti-phospho-histone H2AX (Ser139) (#9718, Cell Signaling Technology), mouse monoclonal anti-Bcl-2 (#15071, Cell Signaling Technology) and rabbit monoclonal anti-cleaved caspase3 (#9664, Cell Signaling Technology).
Tumor Xenografts
Animal experiments were approved by the Ethics Committee of Experimental Research at Fudan University Shanghai Medical College. Six-week-old female nude mice (Shanghai JSJ Experimental Animal Center) were subcutaneously injected with 5 × 106 HO8910 cells in 100 μl PBS. One week later, tumors appeared, and mice were randomized into four treatment groups and administered intragastrically with drugs or the vehicle control daily for three weeks. Tumor volume and body weight were measured at 3-day and 5-day intervals, respectively. Tumor volume (V) was calculated as follows: maximal diameter × perpendicular diameter2/2. Finally, mice were sacrificed, and tumors were collected and fixed in formalin.
Immunohistochemical (IHC) Staining
Thirty-three paraffin-embedded ovarian high-grade serous carcinoma specimens and seven normal ovarian tissues were obtained from the Obstetrics & Gynecology Hospital of Fudan University. Our study was approved by the Ethics Committee. Tissue section slides (4 μm) were heated for 1 hour at 60°C, deparaffinized in xylene and rehydrated through a graded ethanol series (100, 95, 75, and 50%). Then, endogenous peroxidase activity was blocked by incubation with 3% hydrogen peroxide, and 1% Triton was used to permeabilize cells. Following antigen retrieval with Improved Antigen Retrieval Buffer (50x Citrate Sodium Buffer, pH 6.0) (Yeasen), slides were blocked with 5% donkey serum for 1 hour at room temperature. After incubation with the anti-human CDK9 primary antibody (ab76320; Abcam) in a humidified chamber at 4°C overnight and with the secondary antibody for 1 hour at 37°C, antibody binding was detected using a DAB Horseradish Peroxidase Color Development Kit (Beyotime Biotechnology). Then, slides were counterstained with hematoxylin (Beyotime Biotechnology) and mounted onto coverslips after dehydration. Paraffin-embedded xenograft sections (4 μm) were incubated with antibodies against Ki67 (#9027S, Cell Signaling Technology) and BRCA1 (ab213929, Abcam). The evaluation of protein expression was determined by the IHC score as described previously [28].
Statistical analysis
All data were analyzed with GraphPad Prism software and are presented as the mean ± S.D. of at least three independent experiments. One-way ANOVA was used to analyze the synergistic effect. Student’s t-test was used for the two-group comparison. P < 0.05 was considered statistically significant.
Results
CDK9 is highly expressed in ovarian cancer and is associated with an advanced pathologic stage
To explore whether the expression of CDK9 is associated with clinical progression, we analyzed the expression level of CDK9 protein in ovarian high-grade serous adenocarcinoma specimens and normal ovarian tissues through IHC staining. As shown in Figure 1A, CDK9 was mainly localized in the cell nuclei, consistent with the previous study [17]. CDK9 was weakly stained in normal ovarian tissues and increasingly stained in early-stage (stage I/II) and advanced-stage (stage III/IV) ovarian cancer tissues. CDK9 expression was significantly higher in specimens from advanced-stage patients (IHC score = 6.67±0.50) than early-stage (IHC score = 4.58±0.51) patients (Figure 1B). Furthermore, the protein expression of CDK9 in ovarian cancer tissues was higher than that in normal ovarian tissues by Western blot analysis (Figure 1C). Kaplan-Meier-plotter analysis revealed that 5-year progression-free survival (PFS) (Figure 1D, P=0.049) and overall survival (OS) (Figure 1E, P=0.41) were shorter in patients with higher CDK9 expression, although the difference was not statistically significant in the OS condition (Figure 1E). These results suggest that a higher level of CDK9 is associated with the invasive progression of human ovarian cancer.
Figure 1.
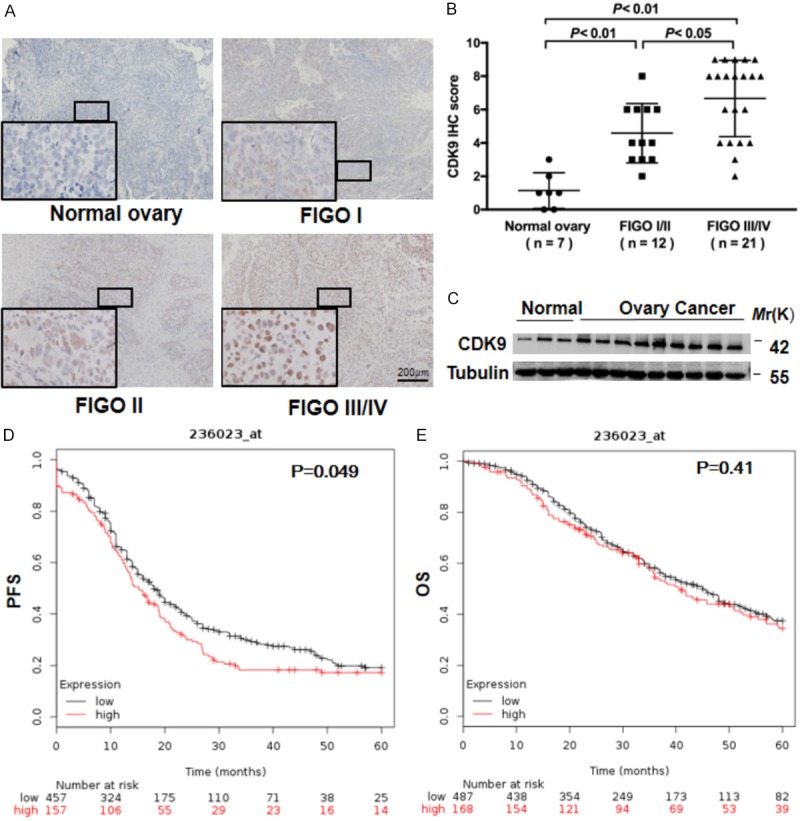
CDK9 is highly expressed in human ovarian cancer and is associated with invasive progression. A. Representative images of nuclear staining intensity for CDK9 in 7 normal ovary samples and 12 early-stage (stage I or II) and 21 advanced-stage (stage III or IV) ovarian high-grade serous adenocarcinoma samples. The stage of ovarian cancer was defined based on the International Federation of Gynecology and Obstetrics (FIGO) staging system. Scale bar, 200 μm. B. IHC scores for CDK9 in ovarian tumor tissues and normal ovarian tissues. ANOVA with the post hoc test. C. Comparison of the CDK9 protein levels in ovarian tumors and normal ovarian tissues by Western blot analysis. Tubulin was used as a loading control. D, E. The progression-free survival (PFS) and overall survival (OS) rates in the high-CDK9 and low-CDK9 groups of ovarian cancer patients by Kaplan-Meier plotter analysis (http://kmplot.com/analysis).
CDK9 is co-expressed with BRCA1 in ovarian cancer
Studies have shown that CDK9 facilitates HRR by recruiting BRCA1 to DNA damage sites [24]. Through GEPIA (gene expression profiling interactive analysis, http://gepia.cancer-pku.cn) analysis, we found that CDK9 was co-expressed with BRCA1 in ovarian cancer (Figure 2A), which was also confirmed in multiple ovarian cancer cell lines (Figure 2B). As shown in Figure 2B, CDK9 was highly expressed in HO8910, OVCAR-5, SKOV3, and A2780 (BRCA1 wild-type) cells, whereas in OVCAR-8 (BRCA1 methylated) and OVCA433 (BRCA2 deficient) cells [29], CDK9 expression was as low as that of BRCA1.
Figure 2.
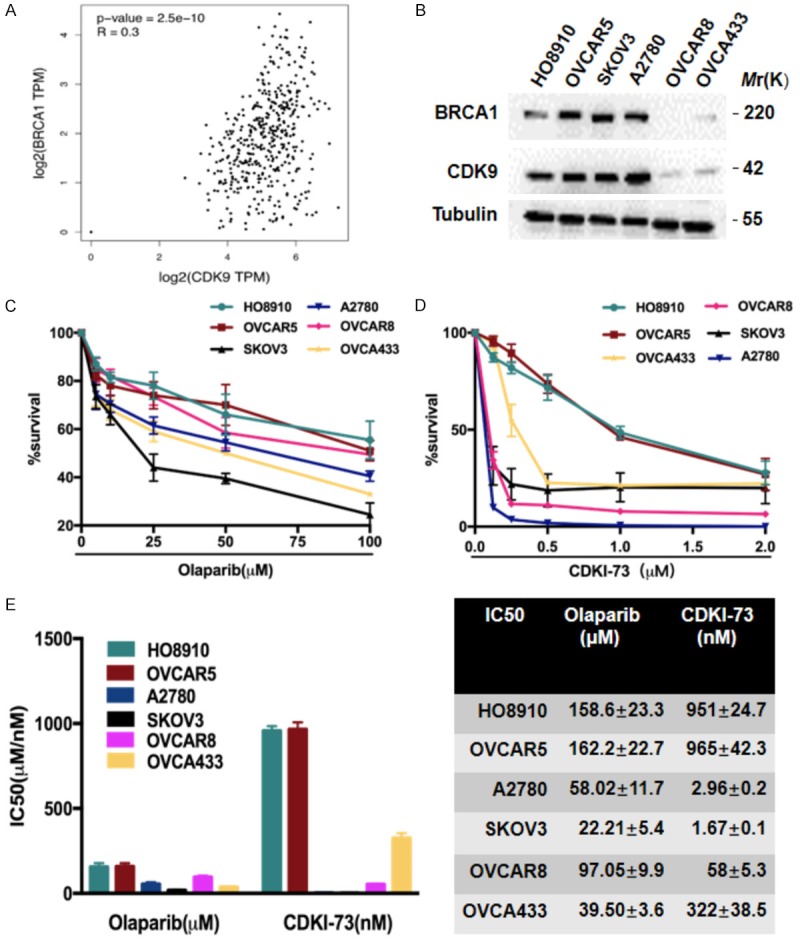
CDK9 is co-expressed with BRCA1 in ovarian cancer. A. The correlation between CDK9 and BRCA1 protein expression in ovarian cancer patients retrieved from the TCGA dataset was investigated by GEPIA (gene expression profiling interactive analysis, http://gepia.cancer-pku.cn). B. The expression of BRCA1 and CDK9 proteins in six ovarian cancer cell lines, HO8910, OVCAR-5, SKOV3, A2780, OVCAR-8, and OVCA433, as determined by Western blot analysis. C, D. The cytotoxic effects of olaparib (C) and CDKI-73 (D) at various concentrations on the six ovarian cancer cells for 48 hr were measured with the CCK-8 assay. E. IC50 values of the above cells were calculated using GraphPad Prism software.
It is well established that the downregulation of BRCA1 may represent a potential biomarker for the sensitivity to PARP inhibitors [30]. Therefore, we speculate that a CDK9 inhibitor may promote sensitivity to PARP inhibitors. To choose eligible ovarian cancer cell lines, we first performed a Cell Counting Kit-8 (CCK-8) cell viability assay to test the IC50s of the above-mentioned cell lines to the PARP inhibitor olaparib and the CDK9 inhibitor CDKI-73 (Figure 2C and 2D). The results showed that HO8910 and OVCAR-5 cells (BRCA1 wild-type ovarian cancer cells) were more insensitive to both inhibitors than OVCAR-8 and OVCA433 cells (BRCA1-deficient ovarian cancer cells) (Figure 2E), therefore, we used the two cells for our following study. Notably, the BRCA1 wild-type ovarian cancer cell lines SKOV3 and A2780 were more sensitive to both inhibitors than the OVCAR-8 and OVCA433 cell lines, which demonstrated that the efficiency of the PARP inhibitors was not totally dependent on BRCA1 status [6].
CDKI-73 synergizes with olaparib in BRCA1-proficient ovarian cancer cells
Based on the role of CDK9 in HRR and the theory of synthetic lethality [24], we then evaluated the synergistic efficiency of CDKI-73 combined with olaparib in HO8910, OVCAR-5 and OVCAR-8 cells. As shown in Figure 3A-C, olaparib (at a concentration of 4 μM) had nearly no impact on the viability of HO8910 and OVCAR-5 cells, but CDKI-73 markedly reduced cell viability in a concentration-dependent manner. Compared to single-agent treatments, the drug combination treatment resulted in a synergistic inhibition of HO8910 and OVCAR-5 cell viability. The synergism indicated by combination index (CI) values < 0.7 was observed in Figure 3A, 3B, 3D. The synergistic effect was also observed but less pronounced in OVCAR-8 cells (Figure 3C, 3D), suggesting that the two inhibitors showed a better synergistic effect on BRCA1-proficient ovarian cancer cells.
Figure 3.
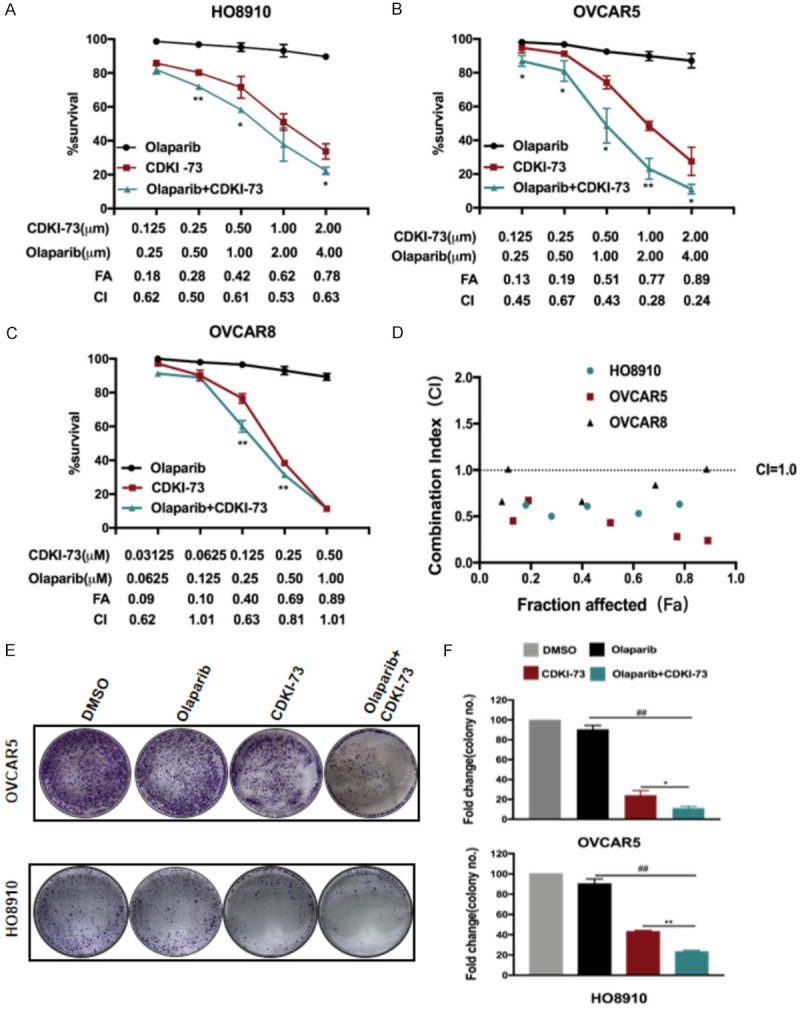
Synergistic effect of CDKI-73 combined with olaparib on the survival and growth of ovarian cancer cells. A-D. Cytotoxic effect of CDKI-73 and olaparib, either as monotherapy or in combination, on ovarian cancer cells (HO8910, OVCAR-5, and OVCAR-8). The synergistic effect of the combination was judged according to the combination index (CI) and fraction affected (FA). DMSO was used as a vehicle. Values are shown as the mean ± S.D. representative of three independent experiments. *P < 0.05 vs CDK9i; **P < 0.01 vs CDK9i. E, F. The proliferation inhibitory effect of CDKI-73 (0.25 μM) and olaparib (0.5 μM) on HO8910 and OVCAR-5 cells was determined with a clonogenic assay. DMSO was used as a vehicle. Values are shown as the mean ± S.D. representative of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 (Student’s t-test).
To detect the long-term inhibitory effects in cell proliferation, we then conducted a cell colony formation assay. Olaparib monotherapy (0.50 μM) and CDKI-73 monotherapy (0.25 μM) exhibited only weak and moderate proliferation inhibitory effects, respectively, whereas the combination of CDKI-73 and olaparib markedly reduced the growth of both cells (Figure 3E and 3F).
We proceeded to investigate the effect of the combination of CDKI-73 and olaparib on cell death by performing an apoptosis assay. Drug combination treatment yielded a higher apoptotic cell population in both cell lines than the vehicle and single-agent treatment (Figure 4A and 4B). Moreover, CDKI-73 combined with olaparib also induced a remarkable decrease in the expression of antiapoptotic protein Bcl-2 in both ovarian cancer cells, while the established apoptotic marker cleaved PARP showed a sharp increase in OVCAR-5 cells but a slight increase in HO8910 cells (Figure 4C).
Figure 4.

CDKI-73 combined with olaparib synergistically induces cell apoptosis. A, B. Apoptosis was detected by flow cytometry. HO8910 and OVCAR-5 cells were exposed to CDKI-73 (0.25 μM) and olaparib (0.50 μM) or their combination for 48 hours, and apoptosis (annexin V+/PI- and annexin V+/PI+) was analyzed by flow cytometry. DMSO was used as a vehicle. Values are shown as the mean ± S.D. of three independent experiments. **P < 0.01; ***P < 0.001 (Student’s t-test). C. Western blot analysis of Bcl-2 and cleaved PARP in the two ovarian cancer cell lines treated with monotherapy or the combination of CDKI-73 and olaparib. Tubulin was used as a loading control.
Coadministration of CDKI-73 and olaparib demonstrates a strong therapeutic effect in vivo
To assess the synergy of CDKI-73 and olaparib in vivo, we further explored their therapeutic effect in ovarian xenograft models. Nude mice were injected subcutaneously with BRCA1-proficient HO8910 cells and were allowed to form tumors for one week. Following tumor formation, mice were randomly divided into four treatment groups: the vehicle control (ctrl), CDKI-73 (0.5 mg/kg), olaparib (0.25 mg/kg), and the combination of CDKI-73 and olaparib groups. During administration, all groups showed a steady body weight gains (Figure 5E) and no significant differences in the activity status, feeding, reaction to stimulation, or psychosis (no specific data), indicating all treatments were well tolerated in mice. Consistent with the effect of the combination of olaparib and CDKI-73 in vitro, the co-administration of olaparib and CDKI-73 resulted in a greater inhibition of tumor growth (i.e., tumor volumes and weights) than other groups (Figure 5A-C, P < 0.01 at day 21). Similarly, the combination treatment caused a marked decrease in the percent change in tumor burden compared to other treatments (Figure 5D). BRCA1 and Ki67 (a proliferation marker) staining in harvest xenograft tumor samples was shown in Figure 5F. A reduction in BRCA1 and Ki67 protein expression was detected in the co-administration group, suggesting impaired HR and proliferative ability.
Figure 5.
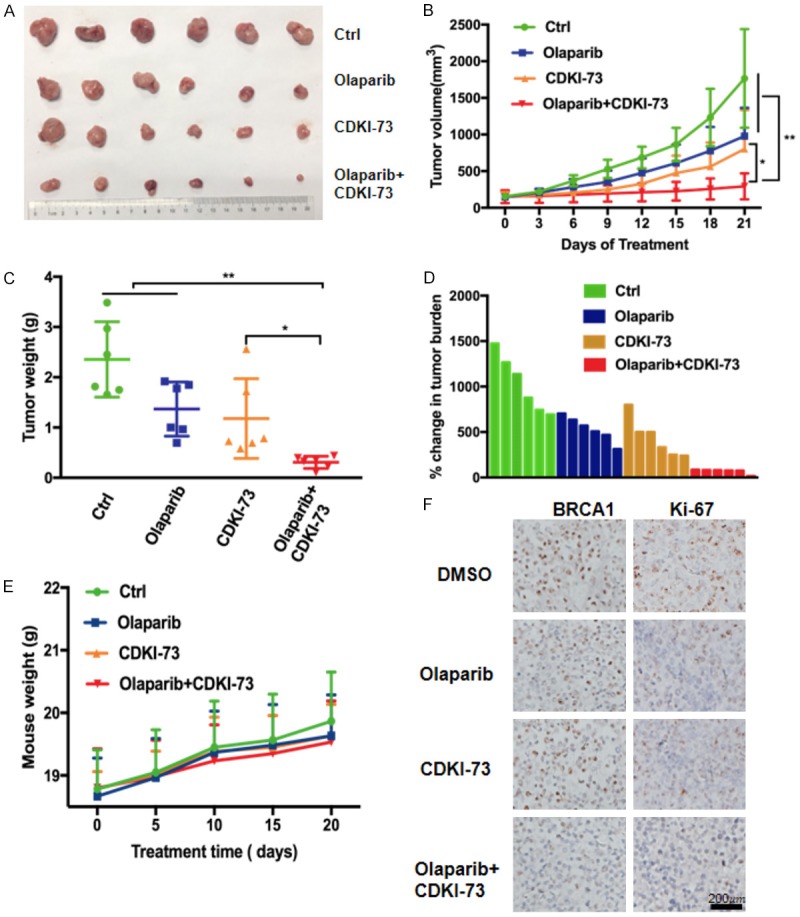
Coadministration of CDKI-73 and olaparib demonstrates a strong therapeutic effect on ovarian cancer cells in vivo. Tumor-bearing nude mice were orally administered vehicle control (Ctrl), CDKI-73 (25 mg/kg), olaparib (50 mg/kg), or the combination of the two drugs daily for three weeks (n = 6). The coadministration of CDKI-73 and olaparib remarkably suppressed tumor growth. A. Images of harvested tumors from each treatment group. B. Tumor growth curves measured during treatment. C. Box and whiskers plot of tumor weight at sacrifice (minimum to maximum, the line represents the mean). *P < 0.05; **P < 0.01. D. Tumor volume changes (from the start to the end of treatment) in each treatment group are shown in a waterfall plot. E. Mouse weight was measured every 5 days during treatments. F. Representative IHC images of Ki67 and BRCA1 in formalin-fixed paraffin-embedded tumors. Scale bar, 200 μm.
Interestingly, in this BRCA1-proficient model, olaparib monotherapy distinctly suppressed tumor growth (P < 0.05, day 21), manifested by a reduction in tumor volume and tumor weight (Figure 5A-C). The more sensitivity of HO8910 to olaparib in vivo may be due to the high doses used in vivo (0.25 mg/kg), which may also explain why the synergetic effect in vivo was not obvious.
CDKI-73 downregulates BRCA1 expression
Next, we explored the specific mechanism of CDKI-73 sensitizing ovarian cancer cells to olaparib. We evaluated DNA damage by measuring H2AX phosphorylation status (γ-H2AX), which is a biomarker for DSBs [31]. As shown in Figure 6A, the formation of γ-H2AX foci was increased following treatment with CDKI-73 at different concentration (0.125 μM, 0.25 μM). And the expression of γ-H2AX protein, cleaved PARP and caspase-3 were significantly enhanced, while Bcl2 expression was inhibited after CDKI-73 treatment (Figure 6C). Above results indicated that more DNA damage and more DSBs were generated in CDKI-73-treated cells. As presumed, aggregated DSBs eventually increased cell apoptosis due to the inability to repair DNA damage (Figure 6B).
Figure 6.

CDKI-73 downregulates BRCA1. A. Representative immunofluorescence images of γ-H2AX staining in HO8910 cells treated with CDKI-73 at various concentrations (0, 0.125, and 0.25 μM) for 48 hours. DMSO was used as a vehicle. Scale bar, 20 μm. The percentage of γ-H2AX-positive cells (> 10 foci per nucleus) in HO8910 cells treated as described above. B. The percentage of apoptotic cells in HO8910 cells treated as described above was measured by flow cytometry. C. The expression of BRCA1, p-CDK9, CDK9, Bcl-2 proteins, γ-H2AX, cleaved PARP, and cleaved caspase-3 in HO8910 cells treated with CDKI-73. D. The expression of BRCA1 protein in the above cells. Values are shown as the mean ± S.D. and are representative of three independent experiments. *P < 0.05; **P < 0.01 (Student’s t-test). E. Downregulation of BRCA1 mRNA expression in HO8910 cells treated with CDKI-73 at various concentrations (0, 0.125, 0.25, and 0.50 μM) for 48 hours. Values are shown as the mean ± S.D. and are representative of three independent experiments. *P < 0.05; ***P < 0.001 (Student’s t-test). F. Time-dependent reduction in the expression of BRCA1 in HO8910 cells after treatment with 0.25 μM CDKI-73. G. Western blot analysis of BRCA1 in the two ovarian cancer cell lines treated with vehicle control (DMSO), CDKI-73 (0.25 μM), olaparib (0.5 μM) or the combination of the two drugs. Tubulin was used as a loading control. H. (Left) Representative immunofluorescence images of γ-H2AX staining in ovarian cancer cells treated with vehicle control (DMSO), CDKI-73 (0.25 μM), olaparib (0.5 μM), or the combination treatment. Scale bar, 20 μm. (Right) The percentage of positive cells (> 10 foci per nucleus) with γH2AX foci relative to the vehicle control. All data are shown as the mean ± S.D. representative of three independent experiments. *P < 0.05; ***P < 0.001 (Student’s t-test).
Due to the role of CDK9 in RNA transcription [13] and its co-expression relationship with BRCA1, we further investigated whether CDKI-73 regulates the expression of BRCA1 in ovarian cancer. As shown in Figure 6C-E, CDKI-73 dose-dependently downregulated the expression of BRCA1 at the mRNA and protein level in HO8910 cells. Furthermore, Figure 6F shows a time-dependent reduction in the protein levels of BRCA1 in HO8910 cells treated with 0.25 μM CDKI-73 for 0, 12, 24, 48, and 72 hours. Thus, our data indicated that CDKI-73 might compromise the HR function by down-regulating BRCA1 expression in ovarian cancer.
To explore whether CDKI-73 combined with olaparib can lead to further increased DSBs, we conducted an immunofluorescence staining assay. As shown in Figure 6H, markedly increased γ-H2AX foci formation was observed in HO8910 cell nuclei following treatment with the drug combination. Moreover, the drug combination group exhibited a marked concomitant reduction in the expression of BRCA1 protein in HO8910 and OVCAR-5 cells compared to the other groups (Figure 6G).
Discussion
PARP, a key enzyme that repairs DNA single-strand breaks (SSBs), binds to the stalled replication forks and facilitates the recruitment of HRR enzymes to DNA damage sites [32,33]. PARP inhibitors can cause the generation and accumulation of DSBs by blocking PARP from repairing SSBs [34]. DSBs are mainly repaired by HRR; therefore, accumulated DSBs would result in HR deficient cell death [35]. BRCA1/2 participates in the process of HRR, and mutations in the BRCA1/2 genes lead to HRD [36]. In theory, mutations in other genes or the abnormal expression of proteins involved in the HRR pathway will also lead to HRD and increase the sensitivity to PARP inhibitors [37]. Previous studies have found that the BRCA wild-type population may also benefit from PARP inhibitors treatment, and the probable cause may be that these BRCA wild-type ovarian cancer patients possess HRD [6]. For this reason, Turner proposed the concept of the “BRCAness” phenotype, which describes HRD without a BRCA mutation but with tumor characteristics similar to those with a BRCA mutation [37-39]. Since CDK9-knockdown cells have been demonstrated to show compromised HRR and sensitization to DNA damaging agents [24], CDK9 inhibitors would have a strong rationale to generate a “BRCAness” phenotype. Thus, in this study, we explored the synergistic effect of the CDK9 inhibitor CDKI-73 combined with the PARP inhibitor olaparib in BRCA1 wild-type ovarian cancer.
Here, we reported for the first time that the CDK9 protein was highly expressed in ovarian cancer specimens compared to that in normal ovarian tissues. Patients with higher CDK9 expression had higher tumor stage, showing that a higher CDK9 may predict ovarian cancer progression. Consistent with the former report [40], we verified that CDKI-73 significantly inhibited the survival and proliferation of ovarian cancer cells and led to increased cell apoptosis. These results suggest that CDK9 is associated with the invasive progression of human ovarian cancer and may be a potential therapeutic biomarker for ovarian cancer.
HRR mainly acts on the G2 and S phases of the cell cycle [41]. When DSBs occur, the MRN complex composed of MRE11A-NBS1-RAD50 detects the damage and recruits ATM to phosphorylate H2AX and further binds to 53BP1 and NBS1 [42]. Simultaneously, BRCA1 interacts with 53BP1 at DSBs, and subsequent DSB repair initiates [43]. Here, we examined the γ-H2AX and BRCA1 expression following CDKI-73 treatment. Results showed that CDKI-73 induced more γ-H2AX foci formation and more γ-H2AX expression accompanied by reduced BRCA1 expression in ovarian cancer cells. All of these results suggest that CDKI-73 might have compromised HRD in BRCA1 wild-type ovarian cancer cells, naturally more sensitive to olaparib. Compared to the vehicle control and monotherapy treatment, the combination of CDKI-73 and olaparib indeed blocked cell survival and proliferation effectively in vitro and in vivo. The body weight, behavior, and psychosis of all mice were not significantly different during the administration, indicating that the overall toxicity of the treatment to the mice was minimal. However, the toxicity in humans remains unknown and needs more clinical study.
It has been reported that CDK9 plays a crucial role in RNA translational elongation. In our study, CDKI-73 significantly reduced the expression of BRCA1 at the mRNA and protein level, which was also accompanied by an increase in γ-H2AX foci formation. Surprisingly, GEPIA showed a positive correlation between CDK9 and BRCA1, which was also demonstrated in various ovarian cancer cell lines. Moreover, the lowest expression of BRCA1 protein and the highest levels of γ-H2AX protein and γ-H2AX foci formation were observed in drug combination compared to the vehicle control and monotherapy treatment, suggesting that the most severe DNA damage occurred in the combination treatment. Therefore the synthetic lethality between CDKI-73 and olaparib might be clarified by the BRCA1 downregulation induced by CDKI-73 in ovarian cancer cells. However, the mechanism by which the CDK9 protein directly regulates the transcription of the BRCA1 gene is still unknown and should be further studied.
In conclusion, our study revealed that in addition to having a direct antitumor effect, the CDK9 inhibitor CDKI-73 also led to HRD by suppressing BRCA1 expression and sensitized BRCA1-proficient ovarian cancer cells to olaparib. As CDK9 inhibitors and olaparib advance clinically, we believe this treatment strategy has value for further research.
Acknowledgements
We thank Dr. Yiqun Zhang (Obstetrics & Gynecology Hospital, Fudan University), Dr. Xiangxiang Wei, and Dr. Mengping Jia (Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fudan University) for their kind support and assistance. Thanks to Mr. Alex Kang (Boston College) and Mr. Matthew Horwedel (Worcester Polytechnic Institute) for language editing. Meanwhile, we are also grateful to the Obstetrics & Gynecology Hospital of Fudan University for tissue provision. This work was supported by the General Programs (81571401 to L. Yao) of the National Natural Science Foundation of China, the Research Fund of Shanghai Municipal Commission of Health and Family Planning (18039 to M. Chen).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384:1376–88. doi: 10.1016/S0140-6736(13)62146-7. [DOI] [PubMed] [Google Scholar]
- 3.Allemani C, Weir HK, Carreira H, Harewood R, Spika D, Wang XS, Bannon F, Ahn JV, Johnson CJ, Bonaventure A, Marcos-Gragera R, Stiller C, Azevedo e Silva G, Chen WQ, Ogunbiyi OJ, Rachet B, Soeberg MJ, You H, Matsuda T, Bielska-Lasota M, Storm H, Tucker TC, Coleman MP CONCORD Working Group. Global surveillance of cancer survival 1995-2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2) Lancet. 2015;385:977–1010. doi: 10.1016/S0140-6736(14)62038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mateo J, Lord CJ, Serra V, Tutt A, Balmana J, Castroviejo-Bermejo M, Cruz C, Oaknin A, Kaye SB, de Bono JS. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol. 2019;30:1437–47. doi: 10.1093/annonc/mdz192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, Chiu HJ, Ricks TK, Palmby T, Russell AM, Ladouceur G, Pfuma E, Li H, Zhao L, Liu Q, Venugopal R, Ibrahim A, Pazdur R. FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res. 2015;21:4257–61. doi: 10.1158/1078-0432.CCR-15-0887. [DOI] [PubMed] [Google Scholar]
- 6.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5:1137–54. doi: 10.1158/2159-8290.CD-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–8. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, Gillman A, Figueiredo I, Paulding C, Seed G, Jain S, Ralph C, Protheroe A, Hussain S, Jones R, Elliott T, McGovern U, Bianchini D, Goodall J, Zafeiriou Z, Williamson CT, Ferraldeschi R, Riisnaes R, Ebbs B, Fowler G, Roda D, Yuan W, Wu YM, Cao X, Brough R, Pemberton H, A’Hern R, Swain A, Kunju LP, Eeles R, Attard G, Lord CJ, Ashworth A, Rubin MA, Knudsen KE, Feng FY, Chinnaiyan AM, Hall E, de Bono JS. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, Konecny GE, Coleman RL, Tinker AV, O’Malley DM, Kristeleit RS, Ma L, Bell-McGuinn KM, Brenton JD, Cragun JM, Oaknin A, Ray-Coquard I, Harrell MI, Mann E, Kaufmann SH, Floquet A, Leary A, Harding TC, Goble S, Maloney L, Isaacson J, Allen AR, Rolfe L, Yelensky R, Raponi M, McNeish IA. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87. doi: 10.1016/S1470-2045(16)30559-9. [DOI] [PubMed] [Google Scholar]
- 10.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J. Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–44. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 11.Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, Primack B, Cao S, Bernhardy AJ, Coulson R, Lazaro JB, Kochupurakkal B, Sun H, Unitt C, Moreau LA, Sarosiek KA, Scaltriti M, Juric D, Baselga J, Richardson AL, Rodig SJ, D’Andrea AD, Balmaña J, Johnson N, Geyer M, Serra V, Lim E, Shapiro GI. CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 2016;17:2367–81. doi: 10.1016/j.celrep.2016.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang S, Fischer PM. Cyclin-dependent kinase 9: a key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol Sci. 2008;29:302–13. doi: 10.1016/j.tips.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 13.Laitem C, Zaborowska J, Isa NF, Kufs J, Dienstbier M, Murphy S. CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II-transcribed genes. Nat Struct Mol Biol. 2015;22:396–403. doi: 10.1038/nsmb.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morales F, Giordano A. Overview of CDK9 as a target in cancer research. Cell Cycle. 2016;15:519–27. doi: 10.1080/15384101.2016.1138186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdullah C, Wang X, Becker D. Expression analysis and molecular targeting of cyclin-dependent kinases in advanced melanoma. Cell Cycle. 2011;10:977–88. doi: 10.4161/cc.10.6.15079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rahaman MH, Kumarasiri M, Mekonnen LB, Yu M, Diab S, Albrecht H, Milne RW, Wang S. Targeting CDK9: a promising therapeutic opportunity in prostate cancer. Endocr Relat Cancer. 2016;23:T211–26. doi: 10.1530/ERC-16-0299. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Dean DC, Hornicek FJ, Shi H, Duan Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in ovarian cancer. FASEB J. 2019;33:5990–6000. doi: 10.1096/fj.201801789RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma H, Seebacher NA, Hornicek FJ, Duan Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in osteosarcoma. Ebiomedicine. 2019;39:182–93. doi: 10.1016/j.ebiom.2018.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narita T, Ishida T, Ito A, Masaki A, Kinoshita S, Suzuki S, Takino H, Yoshida T, Ri M, Kusumoto S, Komatsu H, Imada K, Tanaka Y, Takaori-Kondo A, Inagaki H, Scholz A, Lienau P, Kuroda T, Ueda R, Iida S. Cyclin-dependent kinase 9 is a novel specific molecular target in adult T-cell leukemia/lymphoma. Blood. 2017;130:1114–24. doi: 10.1182/blood-2016-09-741983. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Pandey S, Travers M, Sun H, Morton G, Madzo J, Chung W, Khowsathit J, Perez-Leal O, Barrero CA, Merali C, Okamoto Y, Sato T, Pan J, Garriga J, Bhanu NV, Simithy J, Patel B, Huang J, Raynal NJ, Garcia BA, Jacobson MA, Kadoch C, Merali S, Zhang Y, Childers W, Abou-Gharbia M, Karanicolas J, Baylin SB, Zahnow CA, Jelinek J, Graña X, Issa JJ. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell. 2018;175:1244–58. doi: 10.1016/j.cell.2018.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang CH, Lujambio A, Zuber J, Tschaharganeh DF, Doran MG, Evans MJ, Kitzing T, Zhu N, de Stanchina E, Sawyers CL, Armstrong SA, Lewis JS, Sherr CJ, Lowe SW. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014;28:1800–14. doi: 10.1101/gad.244368.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu DS, Zhao R, Hsu EL, Cayer J, Ye F, Guo Y, Shyr Y, Cortez D. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. Embo Rep. 2010;11:876–82. doi: 10.1038/embor.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Park SH, Pantazides BG, Karpiuk O, Warren MD, Hardy CW, Duong DM, Park SJ, Kim HS, Vassilopoulos A, Seyfried NT, Johnsen SA, Gius D, Yu DS. SIRT2 directs the replication stress response through CDK9 deacetylation. Proc Natl Acad Sci U S A. 2013;110:13546–51. doi: 10.1073/pnas.1301463110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nepomuceno TC, Fernandes VC, Gomes TT, Carvalho RS, Suarez-Kurtz G, Monteiro AN, Carvalho MA. BRCA1 recruitment to damaged DNA sites is dependent on CDK9. Cell Cycle. 2017;16:665–72. doi: 10.1080/15384101.2017.1295177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rahaman MH, Yu Y, Zhong L, Adams J, Lam F, Li P, Noll B, Milne R, Peng J, Wang S. CDKI-73: an orally bioavailable and highly efficacious CDK9 inhibitor against acute myeloid leukemia. Invest New Drugs. 2019;37:625–635. doi: 10.1007/s10637-018-0661-2. [DOI] [PubMed] [Google Scholar]
- 26.Wang T, Fu X, Jin T, Zhang L, Liu B, Wu Y, Xu F, Wang X, Ye K, Zhang W, Ye L. Aspirin targets P4HA2 through inhibiting NF-kappaB and LMCD1-AS1/let-7g to inhibit tumor growth and collagen deposition in hepatocellular carcinoma. Ebiomedicine. 2019;45:168–80. doi: 10.1016/j.ebiom.2019.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blachere NE, Parveen S, Frank MO, Dill BD, Molina H, Orange DE. High-titer rheumatoid arthritis antibodies preferentially bind fibrinogen citrullinated by peptidylarginine deiminase 4. Arthritis Rheumatol. 2017;69:986–95. doi: 10.1002/art.40035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naidoo K, Wai PT, Maguire SL, Daley F, Haider S, Kriplani D, Campbell J, Mirza H, Grigoriadis A, Tutt A, Moseley PM, Abdel-Fatah TMA, Chan SYT, Madhusudan S, Rhaka EA, Ellis IO, Lord CJ, Yuan Y, Green AR, Natrajan R. Evaluation of CDK12 protein expression as a potential novel biomarker for DNA damage response-targeted therapies in breast cancer. Mol Cancer Ther. 2018;17:306–15. doi: 10.1158/1535-7163.MCT-17-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stordal B, Timms K, Farrelly A, Gallagher D, Busschots S, Renaud M, Thery J, Williams D, Potter J, Tran T, Korpanty G, Cremona M, Carey M, Li J, Li Y, Aslan O, O’Leary JJ, Mills GB, Hennessy BT. BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol Oncol. 2013;7:567–79. doi: 10.1016/j.molonc.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst) 2018;71:172–6. doi: 10.1016/j.dnarep.2018.08.021. [DOI] [PubMed] [Google Scholar]
- 31.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–94. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Do K, Chen AP. Molecular pathways: targeting PARP in cancer treatment. Clin Cancer Res. 2013;19:977–84. doi: 10.1158/1078-0432.CCR-12-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai P. Biology of Poly (ADP-Ribose) polymerases: the factotums of cell maintenance. Mol Cell. 2015;58:947–58. doi: 10.1016/j.molcel.2015.01.034. [DOI] [PubMed] [Google Scholar]
- 34.Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J. Clin. Oncol. 2015;33:1397–406. doi: 10.1200/JCO.2014.58.8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tewari KS, Eskander RN, Monk BJ. Development of olaparib for BRCA-deficient recurrent epithelial ovarian cancer. Clin Cancer Res. 2015;21:3829–35. doi: 10.1158/1078-0432.CCR-15-0088. [DOI] [PubMed] [Google Scholar]
- 37.Byrum AK, Vindigni A, Mosammaparast N. Defining and modulating ‘BRCAness’. Trends Cell Biol. 2019;29:740–51. doi: 10.1016/j.tcb.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 38.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110–20. doi: 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- 39.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 40.Lam F, Abbas AY, Shao H, Teo T, Adams J, Li P, Bradshaw TD, Fischer PM, Walsby E, Pepper C, Chen Y, Ding J, Wang S. Targeting RNA transcription and translation in ovarian cancer cells with pharmacological inhibitor CDKI-73. Oncotarget. 2014;5:7691–704. doi: 10.18632/oncotarget.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathiasen DP, Lisby M. Cell cycle regulation of homologous recombination in Saccharomyces cerevisiae. Fems Microbiol Rev. 2014;38:172–84. doi: 10.1111/1574-6976.12066. [DOI] [PubMed] [Google Scholar]
- 42.Szilard RK, Jacques PE, Laramee L, Cheng B, Galicia S, Bataille AR, Yeung M, Mendez M, Bergeron M, Robert F, Durocher D. Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nat Struct Mol Biol. 2010;17:299–305. doi: 10.1038/nsmb.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801–17. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]