

Abstract
Non-coding microRNAs (miRNAs) are powerful regulators of gene expression and critically involved in cardiovascular pathophysiology. The aim of the current study was to identify miRNAs regulating cardiac fibrosis. Cardiac samples of age-matched control subjects and sudden cardiac death (SCD) victims with primary myocardial fibrosis (PMF) were subjected to miRNA profiling. Old SCD victims with PMF and healthy aged human hearts showed increased expression of miR-1468-3p. In vitro studies in human cardiac fibroblasts showed that augmenting miR-1468-3p levels induces collagen deposition and cell metabolic activity and enhances collagen 1, connective tissue growth factor, and periostin expression. In addition, miR-1468-3p promotes cellular senescence with increased senescence-associated β-galactosidase activity and increased expression of p53 and p16. AntimiR-1468-3p antagonized transforming growth factor β1 (TGF-β1)-induced collagen deposition and metabolic activity. Mechanistically, mimic-1468-3p enhanced p38 phosphorylation, while antimiR-1468-3p decreased TGF-β1-induced p38 activation and abolished p38-induced collagen deposition. RNA sequencing analysis, a computational prediction model, and qPCR analysis identified dual-specificity phosphatases (DUSPs) as miR-1468-3p target genes, and regulation of DUSP1 by miR-1468-3p was confirmed with a dual-luciferase reporter assay. In conclusion, miR-1468-3p promotes cardiac fibrosis by enhancing TGF-β1-p38 signaling. Targeting miR-1468-3p in the older population may be of therapeutic interest to reduce cardiac fibrosis.
Keywords: extracellular matrix, microRNA, cardiac fibrosis, senescence, aging, p38, miR-1468-3p, dual-specificity phosphatases
Graphical Abstract
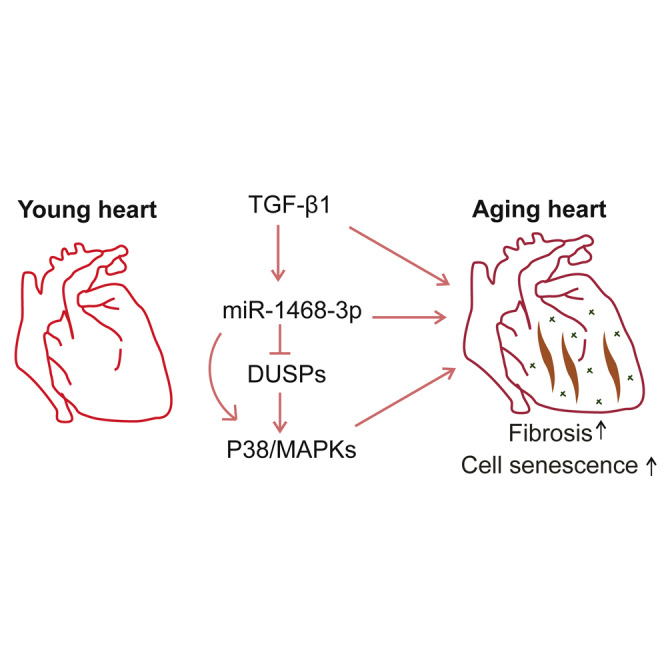
Cardiac expression of miR-1468-3p is increased in healthy aging subjects and in sudden cardiac death victims with primary myocardial fibrosis. miR-1468-3p promotes cell senescence and induces collagen production in fibroblasts by enhancing p38 signaling, and DUSP1 is a target gene of miR-1468-3p.
Introduction
Cardiovascular disease (CVD) remains the leading cause of death in the world.1 Various pathologies, such as myocardial ischemia, valve diseases, and metabolic disturbances, result in excessive deposition of extracellular matrix (ECM), which affects cardiac myocyte contraction/relaxation and disturbs cell-cell coupling, resulting in myocardial stiffness, diastolic dysfunction, and arrhythmogenesis.2,3 It has been reported that only a 3% expansion of the ECM volume elevates the risk of all-cause mortality by 50%.4 Moreover, in the FinGesture database of autopsy-verified sudden cardiac death (SCD) victims, we have observed primary myocardial fibrosis (PMF) without any known etiology to be a relatively common autopsy finding among the young SCD victims.5,6 Despite the adverse effects of cardiac fibrosis, the molecular mechanisms regulating the development of cardiac fibrosis are poorly understood.
Transforming growth factor β1 (TGF-β1) plays a key role in fibrosis-related pathologies, including cirrhosis, renal fibrosis, pancreatic fibrosis, and cardiac fibrosis.7,8 In the heart, cardiac fibroblasts (CFs) are the main cell type responsible for ECM production, and the majority of ECM is comprised of collagenous proteins (85%–90% collagen 1).2,7,9 TGF-β1 enhances CF proliferation and the ability of CFs to synthesize collagen, leading to the accumulation of ECM. Disruption of TGF-β1 signaling by knockdown of fibroblast SMAD3 results in incomplete scar formation along with a higher risk of tissue rupture and mortality.10 In contrast, late inhibition of TGF-β1 signaling after myocardial infarction is protective against cardiac fibrosis and adverse cardiac remodeling.11,12 More recently, both fibroblast-specific TGF-β1 receptor knockout and SMAD3 knockout were shown to attenuate pressure overload-induced cardiac fibrosis.13
There are two key pathways that mediate TGF-β1 signaling: the canonical SMAD pathway and the non-canonical signaling pathways. Canonical TGF-β1 signaling is transduced by phosphorylation of SMAD2/SMAD3, followed by their subsequent binding with SMAD4 and shuttling into the nucleus.14 Non-canonical pathways are comprised of several TGF-β1-activated signaling mechanisms, including the phosphatidylinositol 3-kinase (PI3K)/AKT and mitogen-activated protein kinase (MAPK) signaling pathways.14 The TGF-β1/MAPK pathways signal via a three-tiered cascade of MAPKKK/MAPKK/MAPK, which culminates in three major MAPKs: p38, c-Jun N-terminal kinase (JNK), or extracellular signal-regulated kinases (ERK1/2).15,16 MAPK activation requires phosphorylation by upstream MAPK kinases (MKKs), and p38 is preferentially activated by MKK3 and MKK6.15,16 Inactivation is largely achieved by dual-specificity phosphatases (DUSPs) that dephosphorylate MAPKs and serve as a key constraint for MAPK signals. Previously, dysregulation of DUSPs has been shown to result in sustained MAPK signaling and cardiac fibrosis.15
Aging increases the risks of CVD1,17 and aggravates CVD mortality in older individuals. During cardiac aging, accumulation of ECM in the myocardium as well as collagen deposition in the vascular media lead to a progressive decline in cardiac function.1,17 Cellular senescence is defined as the permanent cessation of cell proliferation.18 Instead of being removed from the body, senescent cells accumulate over a lifetime, which is considered both a hallmark as well as a contributor of aging.1,18,19 The indefinite cell cycle exit is mediated by p53/p21 (CDKN1A) or p16 (CDKN2A) through inhibition of cyclin-dependent kinases (CDKs) that are required for cell cycle progression.19,20 Senescent cells are characterized by several traits, including increased levels of p21, p16, and lysosome β-galactosidase activity.19,21 Inducible targeting of p16-positive cells in a genetic mouse model has been shown to prolong lifespan and to mitigate age-dependent deterioration of cardiac function.22
MircoRNAs (miRNAs) are small non-coding RNAs containing approximately 22 nt.23 They serve as post-translational regulators by binding to their complementary sequence within their target mRNA 3′ UTRs (untranslated regions), leading to mRNA degradation or blocked translation.23,24 The binding of miRNAs to their target mRNAs does not require full complementary, which allows each miRNA to regulate the expression of hundreds of genes.23 Numerous reports have identified the crucial role of miRNAs in fine-tuning cardiac function.23,25, 26, 27 The miR-29 family is one of the first miRNA families found to be associated with cardiac fibrosis. They are downregulated in the peri-infarct region and target collagens to modulate the fibrotic response.28 In the last few years, a number of other miRNAs have also been shown to modulate cardiac fibrosis, such as miR-378, miR-125b, and let-7i.29, 30, 31 miR-1468-3p was identified 13 years ago32 and, until now, it has been found solely in humans and goats. In this study, we identify a disease-associated and age-dependent increase in miR-1468-3p expression in human hearts and uncover the dual role of miR-1468-3p in promoting cardiac fibrosis and cell senescence. The dual function of miR-1468-3p exemplifies a signaling mechanism underpinning cardiac aging and development of CVD, and it provides a potential target for the treatment of aging-associated fibrosis.
Results
miR-1468-3p Expression Is Increased in Fibrotic Hearts and Promotes Senescence
We analyzed the miRNA profile in left ventricular samples taken at autopsy from SCD victims identified with PMF in the FinGesture study. Healthy myocardial samples (control) were collected from accidental death victims without evidence of cardiac diseases at autopsy. In the SCD victims with PMF, no other cardiac diseases such as cardiac hypertrophy and/or coronary artery disease were observed. From miRNA profiling data, we selected seven miRNA candidates (miR-1468-3p, miR-499a-5p, miR-30c-5p, miR-1285-3p, miR-3613-3p, miR-4286, and miR-874-3p) for individual qPCR validation. Among the candidate miRNAs analyzed, miR-1468-3p was upregulated in older SCD victims with PMF (age 40–55 years) when compared to age-matched controls, but it remained unchanged in diseased hearts of younger subjects (age 20–30 years) (Figure 1A). qPCR analysis of other candidate miRNAs showed a reduction in miR-1285-3p levels in young SCD victims with PMF, but a slight increase in older SCD victims with PMF (Figure S1). Cardiac expression of miR-499a-5p decreased in old SCD victims with PMF, but not in the young (Figure S1).
Figure 1.

Cardiac miR-1468-3p Expression Is Increased in Aged SCD Victims and It Promotes Senescence
(A and B) Analysis for cardiac miR-1468-3p (A and B) and miR-1468-5p (B) expression in control subjects (control) and victims of sudden cardiac death (SCD) with primary myocardial fibrosis (PMF case). All samples were paired with matched age. The data are shown as relative to respective age control of miR-1468-3p. (C and D) Human cardiac fibroblasts were treated with 20 nM of either mimic-1468-3p or control sequence (mimic-Ctrl) for 24 h. (C) Quantitative analysis of senescence-associated β-galactosidase (SA-β-GAL) activity and representative staining. Scale bar: 100 μm. N = 4. (D) Western blot (WB) analysis and densitometry analysis for expression of p16, p21, and p53. Vinculin was used as a loading control. (E) Analysis for miR-1468-3p and miR-1468-5p expression in healthy control hearts at different ages. (F) Analysis for miR-1468-3p expression in young healthy control hearts and in hearts of 45- to 65-year-old SCD victims with PMF (PMF case). Data are expressed as mean ± SD. Student’s t test was used. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To validate the increased miR-1468-3p expression in SCD victims with PMF, we collected additional cardiac samples from healthy control subjects and SCD victims with PMF. In agreement with the profiling data, cardiac miR-1468-3p levels were increased in older SCD victims with PMF compared to age-matched controls, but not in the young SCD victims with PMF (Figure 1B). Analysis for expression of miR-1468-5p produced from the same primary miRNA transcript showed no difference between healthy and diseased hearts (Figure 1B).
The increase in miR-1468-3p levels in aged human cardiac samples prompted us to test whether miR-1468-3p induces cell senescence. We overexpressed miR-1468-3p in cultured human CFs (hCFs) with a synthetic mimic (mimic-1468-3p) and compared them with a control sequence (scrambled nucleotides). Flow cytometry analysis showed a dose-dependent increase in the transfection efficiency of the fluorescence-labeled control sequence (Figure S2). Analysis for cell senescence by measuring for senescence-associated β-galactosidase (SA-β-GAL) activity showed induction in SA-β-GAL in hCFs overexpressing miR-1468-3p (Figure 1C). Western blot analysis showed that mimic-1468-3p induced p53 and p16 expression in hCFs, whereas no difference was observed in p21 levels (Figure 1D). To validate that miR-1468-3p-induced cell senescence is linked to human cardiac aging, we analyzed miR-1468-3p levels in healthy human heart samples between different age groups (20–30 versus 40–55 years old). As shown in Figure 1E, cardiac miR-1468-3p expression increases in healthy human hearts during aging. No difference was observed in miR-1468-5p expression between the age groups (Figure 1E). To assess for the effect of both aging and fibrosis (PMF) on miR-1468-3p levels, we further compared additional left ventricular samples from young healthy hearts to samples from aged SCD victims with PMF, which revealed a robust increase in miR-1468-3p levels in the diseased hearts (Figure 1F).
miR-1468-3p Promotes Collagen Deposition
Given that miR-1468-3p expression was increased in old SCD victims with cardiac fibrosis, we next explored its fibrotic potential. By definition, fibrosis is a pathological state with excess deposition of ECM. Analysis for total collagen deposition with Sirius Red staining showed that mimic-1468-3p significantly augmented collagen deposition in hCFs (Figure 2A). Consistent with the elevated total collagen deposition, western blot analysis showed increased collagen 1 (Col1a1), connective tissue growth factor (CTGF), and periostin (Postn) expression in mimic-1468-3p-treated hCFs (Figure 2B). We also measured Col1a1, CTGF, and Postn expression at the transcriptional level, all of which showed a tendency of increase without significance (Figure S3).
Figure 2.

miR-1468-3p Promotes Fibrosis, and Targeting miR-1468-3p Reduces TGF-β1-Induced Fibrosis
(A and B) Human cardiac fibroblasts (hCFs) were treated with 20 nM mimic-1468-3p or mimic-Ctrl. After 24 h, cells were fixed and stained for total collagen deposition, or collected for WB analysis. Shown are quantification of total collagen deposition (A) and WB and densitometry analysis for collagen 1, periostin (Postn), and CTGF expression (B). (C–E) hCFs were treated with 50 nM antagomir of miR-1468-3p (antimiR-1468-3p) or control sequence (antimiR-Ctrl). After 24 h, hCFs were treated TGF-β1 (5 ng/mL) where indicated. At the end of the experiment, cells were fixed and stained for total collagen deposition, collected for WB analysis, or collected for qPCR analysis. Shown are quantification of total collagen deposition (C); qPCR analysis for COL1A1, connective tissue growth factor (CTGF), and Postn (D); and WB and densitometry analysis (lower panel) for collagen 1, Postn, and CTGF expression (E). (F) qPCR analysis for miR-1468-3p expression. Vinculin was used as a loading control for WB. Collagen deposition, N = 5–6 per group; qPCR, N = 3–4 per group. Data are expressed as mean ± SD. One-way ANOVA followed by Tukey’s post hoc test and Student’s t test was used. $p < 0.05, $$p < 0.01 versus mimic-Ctrl; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus antimiR-Ctrl; ##p < 0.01, ###p < 0.001 versus antimiR-Ctrl+TGF-β1.
To investigate whether targeting miR-1468-3p regulates CF function, we employed an antisense oligonucleotide of miR-1468-3p (antimiR-1468-3p). Treatment of non-stimulated hCFs with antimiR-1468-3p had no effect on collagen deposition or fibrotic gene expression (Figure 2C; Figure S3). Given that, in a physiological setting, fibroblasts are quiescent and inactive with regard to collagen synthesis, we stimulated hCFs with TGF-β1. TGF-β1 substantially triggered collagen deposition in hCFs treated with the anti-miR control, and targeting miR-1468-3p with antimiR-1468-3p significantly decreased TGF-β1-induced collagen deposition (Figure 2C). qPCR analysis showed that antimiR-1468-3p blunted TGF-β1-induced Col1a1 and Postn expression, with a trend to decrease CTGF expression (Figure 2D). Similarly, western blot analysis showed that antimiR-1468-3p reduced TGF-β1-induced Col1a1 and Postn expression (Figure 2E). These data suggest that inhibition of miR-1468-3p does not counteract fibrosis at the basal level, but it attenuates TGF-β1-triggered fibrotic responses.
The lack of effect for antimiR-1468-3p in hCFs at baseline prompted us to investigate whether TGF-β1 regulates miR-1468-3p expression. In accordance with the collagen deposition data, qPCR analysis showed a 2-fold increase in miR-1468-3p levels in TGF-β1-treated hCFs, and antimiR-1468-3p treatment restored the TGF-β1-induced increase in miR-1468-3p expression to the basal level (Figure 2F).
To better understand the function of miR-1468-3p, we compared the expression level of miR-1468-3p to other miRNAs in hCFs. We found that the expression of miR-1468-3p is relatively low in quiescent hCFs and, similarly, the expression level of miR-1468-3p in healthy human hearts was among the lowest of those analyzed (Figure S4). We then compared the distribution of miR-1468-3p in fibroblasts and endothelial cells, the most abundant cell type in the heart.33 qPCR analysis showed that the expression of miR-1468-3p is equal in hCFs and human umbilical vein endothelial cells (HUVECs) (Figure S4).
miR-1468-3p Modulates Cell Proliferation and Metabolic Activity
To further characterize the role of miR-1468-3p in regulating biological functions in hCFs, we monitored cell proliferation and metabolic activity, both of which are key characteristics of hCFs that intensify overall collagen deposition and exacerbate fibrosis. Evaluation for hCF proliferation by analyzing for 5-ethynyl-2′-deoxyuridine (EdU) incorporation showed that augmenting miR-1468-3p levels did not affect cell proliferation (Figure 3A). Furthermore, antagonizing the function of miR-1468-3p did not affect EdU incorporation at baseline, but it attenuated TGF-β1-induced cell proliferation (Figure 3B). The metabolic activity of hCFs was markedly increased in mimic-1468-3p-treated hCFs (Figure 3C). Again, antimiR-1468-3p did not affect the metabolic activity of hCFs at baseline but attenuated the TGF-β1-induced cell metabolic activity (Figure 3D).
Figure 3.

miR-1468-3p Enhances the Metabolic Activity of hCFs
(A and C) hCFs were treated with 20 nM mimic-Ctrl for 24 h and analyzed for cell proliferation (A) or cell metabolic activity (C). (B and D) hCFs were treated with 50 nM antimiR-Ctrl for 24 h and treated with TGF-β1 (5 ng/mL) for 24 h where indicated. Shown are analyses for cell proliferation (B) and cell metabolic activity (D). N = 5–7 per group. Data are shown as mean ± SD. One-way ANOVA followed by Tukey’s post hoc test and Student’s t test was used. $$p < 0.01 versus mimic-Ctrl; ∗∗p < 0.01, ∗∗∗p < 0.001 versus antimiR-Ctrl; -p < 0.05 versus antimiR-Ctrl+TGF-β1.
RNA Profiling for Profibrotic Role of miR-1468-3p
To identify the mechanisms involved in miR-1468-3p function, we performed RNA sequencing (RNA-seq) analysis of hCFs treated with either control antagomir (n = 2), TGF-β1+control antagomir (n = 3), or TGF-β1+antimiR-1468-3p (n = 3). We screened out differentially expressed (DE) genes by criteria as FC (fold change) >1.5 and p < 0.05. More than 2,000 transcripts were DE after TGF-β1 treatment in control cells. AntimiR-1468-3p significantly affected the expression of 353 transcripts in TGF-β1-treated cells, 131 of which were downregulated and 222 upregulated (Table S1). Gene Ontology (GO) enrichment analysis of DE genes identified 283 gene functions that were significantly affected with the most enriched processes related to cell cycle (Table S2).
GO enrichment analysis of the 131 downregulated DE transcripts identified four TGF-β/MAPK pathway signaling-related processes among the top 10 enriched processes: MAPK cascade, phosphorylation, SMAD phosphorylation, and SMAD signal transduction (Figure 4A). In addition, KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis of the 131 transcripts identified six pathways, including the TGF-β, insulin, and PI3K-Akt signaling pathways (Figure 4B). GO analysis of the genes upregulated by antimiR-1468-3p identified cell cycle and DNA repair as the major enriched biological processes.
Figure 4.

RNA Profiling for miR-1468-3p Target Genes in hCFs
hCFs were treated with antimiR-Ctrl for 24 h and treated with TGF-β1 (5 ng/mL) for 24 h where indicated. At the end of the experiment, RNA was collected for RNA sequencing analysis, qPCR analysis, or WB analysis. Shown are (A) Gene Ontology (GO) enrichment analysis and (B) KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis for the genes downregulated by antimiR-1468-3p. (C) Heatmap representation of genes that were significantly regulated by antimiR-1468-3p in TGF-β1-treated cells. (D) qPCR analysis of selected genes modulated by TGF-β1 and/or antimiR-1468-3p. (E) WB and densitometry analysis (right panel) for p16 and p21. GAPDH was used as a loading control. qPCR, N = 3 per group. Data are expressed as mean ± SD. One-way ANOVA followed by Tukey’s post hoc test was used. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus antimiR-Ctrl; #p < 0.05, ##p < 0.01, ###p < 0.001 versus antimiR-Ctrl+TGF-β1.
With the dominant role of cell cycle-related genes in GO analysis of full RNA-seq data, we further examined the data on TGF-β/SMAD, MAPK, phosphorylation, and fibrosis-related genes (Table S3). We found that antimiR-1468-3p significantly modulated 22 of the 75 fibrotic marker transcripts investigated, with significant downregulation of 20 transcripts, including COL1A1, COL3A1, matrix metalloproteinase-2 (MMP2), fibronectin (FN), and α-smooth muscle actin (ACTA2) (Table S3; Figure 4C). For all 47 MAPK cascade kinases analyzed, 10 were significantly regulated by antimiR-1468-3p with a decrease in six transcripts, including MAPK14 (p38α) (Table S3; Figure 4C). However, analysis for MAPK14 expression by qPCR did not show a significant reduction in MAPK14 levels in antimiR-1468-3p-treated cells (Figure 4D). To date, 25 human DUSP genes have been identified and approximately 11 of them have been shown to serve a major role in regulation of MAPK pathways.16 AntimiR-1468-3p treatment significantly altered expression of 11 out of 25 DUSP transcripts, 8 of which serve a major role in MAPK inactivation (Table S3; Figure 4C). AntimiR-1468-3p treatment also significantly altered expression of seven TGF-β/SMADs signaling components, including a decrease in TGF-β1-induced SMAD4 expression (Table S3; Figure 4C). In addition, we compared expression of all integrin subunits and found that antimiR-1468-3p treatment significantly decreased expression of five integrins (Table S3; Figure 4C).
Given the identified role of miR-1468-3p in promoting senescence, the expression of all CDKs and CDK inhibitors (CDKIs, encoded by CDKNs) were investigated. Upon antimiR-1468-3p delivery, expression of 11 CDKs and CDKIs changed significantly, including a decrease in CDKN1A, CDKN2A, CDKN1B, and CDKN2B expression and an increase in expression of all CDKs (Table S3; Figure 4C). Gene expression of selected miR-1468-3p-regulated transcripts was confirmed by qPCR, which validated the decrease in ACTA2, CDKN1A, and ITGB3 expression by antimiR-1468-3p in TGF-β1-treated cells (Figure 4D). Western blot analysis confirmed the decrease of p16 by antimiR-1468-3p in TGF-β1-treated cells whereas anti miR-1468-3p had no effect on p21 expression (Figure 4E).
miR-1468-3p Regulates TGF-β1 Signaling
Western blot analysis was then used to interrogate the effect of miR-1468-3p on TGF-β1/MAPK signaling. Overexpression of miR-1468-3p induced an increase in p38 phosphorylation and a modest increase in JNK phosphorylation (Figure 5A), whereas no apparent change was observed in phosphorylated (p-)Smad2, p-AKT, and p-ERK1/2 signals (Figure S5). In addition, mimic-1468-3p induced an increase in the expression of total p38, but not total JNK or total focal adhesion kinase (FAK) (Figure 5A). Despite the increase in total p38, mimic-1468-3p was still able to induce p38 phosphorylation when compared to total protein (Figure 5A). The increase in p38 phosphorylation thus is not only due to increased protein expression. Alternatively, antimiR-1468-3p attenuated TGF-β1-induced phosphorylation of p38, although without affecting the JNK pathway (Figure 5B), and there was also no change in p-Smad2, p-AKT, and p-ERK1/2 levels (Figure S6). In accordance with the qPCR data, antimiR-1468-3p had no effect on total p38 expression, total JNK, or total FAK (Figure 5B). In addition, mimic-1468-3p elevated FAK phosphorylation (Figure 5A), and antimiR-1468-3p reduced TGF-β1-activated FAK phosphorylation (Figure 5B).
Figure 5.

miR-1468-3p Modulates TGF-β1-Induced Signaling Pathways
(A) hCFs were treated with 20 nM mimic-Ctrl for 24 h. Shown are WB and densitometry analyses for phosphorylated p38, phosphorylated c-Jun N-terminal kinase (JNK), phosphorylated focal adhesion kinase (FAK), total p38, and total FAK. Vinculin was used as a loading control. Also, the ratios of phosphorylated p38 to total p38 as well as phosphorylated JNK to total JNK are shown. (B) hCFs were treated with 50 nM antimiR-Ctrl for 24 h and treated with TGF-β1 (5 ng/mL) for 24 h where indicated. Shown is WB and densitometry analysis for phosphorylated p38, phosphorylated JNK, p-FAK, total p38, and total FAK. The ratios of phosphorylated p38 to total p38 as well as phosphorylated JNK to total JNK are also shown. Vinculin was used as a loading control. Data are expressed as mean ± SD. Student’s t test and one-way ANOVA followed by Tukey’s post hoc test were used. $p < 0.05, $$p < 0.01 versus mimic-Ctrl; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus antimiR-Ctrl; #p < 0.05, ###p < 0.001 versus antimiR-Ctrl+TGF-β1.
miR-1468-3p Regulates DUSPs
Experimental data thus showed that the p38 pathway is regulated by miR-1468-3p, and RNA-seq data indicated dysregulation of DUSPs in antimiR-1468-3p-treated cells. Furthermore, computational prediction with a public database TargetScan indicated DUSPs as potential miR-1468-3p target genes (Figure 6A). Notably, among the eight predicted potential target DUSPs, six were also identified from RNA-seq data (Figure 6A). Based on the predicted list, we selected seven DUSPs for further analysis: DUSP1, DUSP5, DUSP6, DUSP7, and DUSP16, all of which are the predicted targets of miR-1468-3p, as well as DUSP8 and DUSP14, which have displayed preference for p38/JNK inactivation.16 Surprisingly, expression of four out of five predicted miR-1468-3p target DUSPs were decreased by miR-1468-3p treatment, with a decrease in DUSP1 and DUSP6 reaching significance (Figure 6B). miR-1468-3p also significantly reduced DUSP8 expression, whereas DUSP14 expression was increased. The reduction in DUSP1 expression was further confirmed with western blotting (Figure 6B).
Figure 6.

miR-1468-3p Regulates Dual-Specificity Phosphatases (DUSPs) and p38 MAPK
Cultured hCFs were treated with 20 nM mimic-Ctrl for 24 h. At the end of the experiment, samples were collected for qPCR and WB analyses. (A) Computational analysis for potential miR-1468-3p target genes. (B) Shown is qPCR analysis for DUSPs 1, 5, 6, 7, 8, 14, and 16, as well as WB and densitometry analysis for DUSP1. Tubulin was used as a loading control. (C) Cultured human umbilical vein endothelial cells (HUVECs) were treated with 20 nM of mimic-Ctrl. Shown is qPCR analysis for DUSP1 (left panel), and WB (middle panel) and densitometry analysis (right panel) of DUSP1 and phosphorylated p38. GAPDH or tubulin were used as a loading control. (D) hCFs were treated with 20 nM mimic-1468-3p or 50 nM antimiR-1468-3p for 24 h, and then co-transfected with mutated (muDUSP1) or wild-type (WTDUSP1) DUSP1 luciferase reporter construct as indicated. Shown is normalized firefly luciferase activity (FLuc) to Renilla luciferase (RLuc) activity. N = 5–6 per group. (E) Analysis for DUSP1 expression in healthy control hearts at the age of 18–30 years or 40–65 years, and in hearts of SCD victims with PMF at the age of 40–65 years. Shown are blinded gradings for DUSP1 expression level and representative images. Scale bar: 100 μm. Data are expressed as mean ± SD. Student’s t test and Mann Whitney U test with Bonferroni correction were used. $p < 0.05, $$p < 0.01 versus mimic-Ctrl; ∗p < 0.05, ∗∗p < 0.01 versus healthy control hearts (age, 18–30 years)
In addition, given that the expression level of miR-1468-3p in HUVECs is comparable to that in hCFs, we tested whether a similar effect of miR-1468-3p on DUSP1 levels was also present in HUVECs. Again, overexpression of miR-1468-3p repressed DUSP1 expression at both the mRNA and protein levels, which was concomitant with increased p38 phosphorylation (Figure 6C). Phenotypically, mimic-1468-3p triggered an increase in p21 (CDKN1A), p53, and CTGF at the transcriptional level, whereas VEGFA expression decreased. Western blot analysis revealed upregulation of p16, p21, p53, and CTGF expression by mimic-1468-3p (Figure S7).
Prior studies have shown that DUSP6 has high specificity for ERK, but it does not dephosphorylate p38 or JNK.34, 35, 36, 37, 38 Given that augmenting miR-1468-3p had no effect on ERK, but modulated p38 phosphorylation (Figure 5A; Figure S5), we selected DUSP1 (which inactivates all three MAPKs) for further verification. We utilized a dual-luciferase assay in hCFs to confirm the direct regulation of DUSP1 by miR-1468-3p. A wild-type luciferase construct containing the miR-1468-3p seeding region in the 3′ UTR of DUSP1 (WTDUSP1), or a vector containing a mutated miR-1468-3p binding site (muDUSP1), was introduced into hCFs. Mimic-1468-3p reduced luciferase activity in WTDUSP1-transfected hCFs when compared to muDUSP1-transfected hCFs (Figure 6D). Consistent with the observed minimal effect of targeting miR-1468-3p in quiescent hCFs, no difference in luciferase activity was observed upon antimiR-1468-3p delivery (Figure 6D). Taken together, these data demonstrate that miR-1468-3p regulates DUSP1 expression by directly binding to its 3′ UTR region.
To translate our findings to human cardiac aging and fibrosis, immunostaining of DUSP1 in cardiac sections was performed, followed by blind grading of its expression in heart samples of SCD victims with PMF (age 40–65 years), healthy control individuals (age 40–65 years), and younger healthy individuals (age 18–30 years). Histological analysis showed that cardiac DUSP1 expression was increased with aging in healthy hearts, and the expression level was further increased in aged SCD victims with PMF (Figure 6E). Interestingly, histological analysis of total p38 and phosphorylated p38 also displayed an aging-associated increase in hearts of healthy human subjects, and the highest expression levels were observed in aged SCD victims with PMF (Figure S7). The noted induction of DUSP1 in human hearts may thus stem from induction of p38 signaling, and the enhanced miR-1468-3p expression possibly represents a compensatory response that only limits DUSP1 induction but is not sufficient to abolish it.
miR-1468-3p Functions through the p38 Pathway
To further evaluate the role of JNK and p38 pathways on the pro-fibrotic response of miR-1468-3p, we blocked each pathway with a respective inhibitor and validated the efficacy of each inhibitor using western blot (Figure S8). Analysis for the role of JNK and p38 on collagen deposition showed that the JNK inhibitor (JNK inhibitor I) abrogated the miR-1468-3p-induced increase in collagen deposition, but it also repressed collagen deposition in control-treated cells (Figure 7A). Importantly, miR-1468-3p was still able to induce collagen deposition in hCFs treated with the JNK inhibitor. Analysis for cell proliferation and cell metabolic activity showed that JNK inhibition severely blunted cell proliferation and reduced cell metabolic activity both in mimic-Ctrl (control) and mimic-1468-3p-treated cells (Figure 7A). These data thus suggest that JNK signaling is required to support and maintain basal activity of hCFs, and the decrease in collagen deposition by JNK inhibition is likely due to robust inhibition of cell proliferation. Differently from JNK inhibition, p38 inhibition by SB203580 slightly increased cell proliferation and abolished the miR-1468-3p-induced increase in cell metabolic activity and collagen deposition (Figure 7B). Importantly, p38 inhibition had no significant effect in control cells, indicating that the pro-fibrotic function of miR-1468-3p is p38-dependent. Consistent with the functional assays in hCFs, western blot analysis indicated that p38 inhibition attenuated miR-1468-3p-induced collagen 1 and CTGF expression, with a non-significant decrease in Postn expression (Figure 7C).
Figure 7.

miR-1468-3p Modulates Collagen Production via the p38 Pathway
(A and B) Cultured hCFs were treated with mimic-Ctrl, and co-treated with JNK inhibitor (JNKi, 2 μM) (A) or p38 inhibitor (SB203580, 5 μM) (B). Shown are analyses for collagen deposition, cell proliferation, and cell metabolic activity. (C) hCFs were treated with mimic-Ctrl, and co-treated with p38 inhibitor (SB203580, 5 μM) where indicated. Shown are WB and densitometry analysis (lower panel) for collagen 1, Postn, and CTGF. Vinculin was used as a loading control. (D) antimiR-Ctrl-treated hCFs were transduced with adenoviruses expressing LacZ or MKK3b (MKK3). Shown are analyses for collagen deposition (left panel) and cell metabolic activity (right panel). For collagen deposition, cell proliferation, and cell metabolic activity, N = 5–6 per group. Data are expressed as mean ± SD. One-way ANOVA followed by Tukey’s post hoc test was used. $p < 0.05, $$p < 0.01, $$$p < 0.001 versus mimic-Ctrl; †p < 0.05, ††p < 0.01, †††p < 0.001 versus mimic-1468-3p; ‡‡p < 0.01 versus mimic-Ctrl+JNKi; ∗∗∗p < 0.001 versus antimiR-Ctrl + LacZ; ##p < 0.01, ###p < 0.001 versus antimiR-Ctrl + MKK3.
To further validate the role of miR-1468-3p in regulating fibroblast function through the p38 pathway, we utilized adenoviral delivery of constitutively active MKK3b (upstream regulator of p38) to activate the p38 pathway. Forced activation of the p38 pathway was sufficient to induce collagen deposition and cell metabolic activity, both of which were significantly attenuated by targeting miR-1468-3p (Figure 7D).
Discussion
In the current study, we identified a substantial increase in cardiac miR-1468-3p levels in aged SCD victims with PMF and further showed that levels of miR-1468-3p are increased in healthy aged hearts. Our experimental data establish both a pro-senescent and a pro-fibrotic role for miR-1468-3p, the latter of which is at least partially mediated through induction of p38 signaling. The dual role of miR-1468-3p bridging fibrosis and senescence is an example how aging exacerbates CVD progression in older individuals.
miR-1468-3p in the Regulation of Fibroblast Function
The increase of miR-1468-3p expression in cardiac samples of SCD victims with PMF and with no evidence of cardiomyocyte pathology suggested a pro-fibrotic potential of miR-1468-3p. Studies in cultured fibroblasts showed that miR-1468-3p promotes collagen deposition and increases protein levels of collagen 1, CTGF, and Postn. Further assessment of cell proliferation and metabolic activity showed that the pro-fibrotic effect of miR-1468-3p is mainly attributed to potentiation on overall collagen-producing capacity instead of increasing CF proliferation.
Analysis of miR-1468-3p expression showed that it is expressed in rather low abundance in human hearts when compared with other miRNAs analyzed. Similarly, we found that miR-1468-3p is expressed at low levels in quiescent hCFs. The relatively low expression of miR-1468-3p may account for the lack of effect of miR-1468-3p blockade on collagen production in quiescent hCFs. Alternatively, we found that TGF-β1 induces miR-1468-3p expression in vitro, which may also provide a rationale for the induction of miR-1468-3p levels in cardiac samples of SCD victims with PMF. Furthermore, blockade of miR-1468-3p function in TGF-β1-stimulated cells attenuates a TGF-β1-induced increase in total collagen deposition and increase in collagen 1, CTGF, and Postn expression.
miR-1468-3p Regulates the TGF-β1-p38 Pathway
Interrogation of RNA-seq data identified MAPK and SMAD (GO annotation)- and TGF-β (KEGG pathway)-related signaling pathways as key targets of miR-1468-3p. Our experimental data show that miR-1468-3p activates MAPKs as well as integrin/FAK signaling in the non-canonical TGF-β1 pathway. Conversely, blockade of miR-1468-3p reduces TGF-β1-induced p38 phosphorylation and FAK phosphorylation, which is consistent with the anti-fibrotic effect of antimiR-1468-3p.
Prior data indicate a central role for p38α in the regulation of cardiac fibrosis.39,40 In this study, we show that p38 inhibition abrogated miR-1468-3p-induced collagen deposition and cell metabolic activity, but it had no effect on CF function at the basal level. Alternatively, activation of p38 signaling by the adenovirus encoding for MKK3b (adv-MKK3) potentiated collagen deposition and cell metabolic activity, both of which were abolished by antimiR-1468-3p. Of note, differently from p38-induced fibrotic response, blockade of miR-1468-3p function did not fully abrogate the TGF-β1-induced pro-fibrotic response. Taken together, our data indicate a central role for p38 MAPK in mediating the pro-fibrotic effect of miR-1468-3p.
Integrin signaling mediates latent TGF-β activation and is thus intimately involved in regulating TGF-β-mediated fibrosis.8,41 FAK phosphorylation is the main step for integrin signaling conduction, and treatment mitigating FAK activation has been shown to attenuate cardiac fibrosis and improve cardiac function during post-ischemic cardiac remodeling.41 Analysis for integrin subunits in RNA-seq data revealed that expression levels of all integrin subunits significantly induced by TGF-β1 were reduced by antimiR-1468-3p, which is consistent with the decrease in TGF-β1-induced integrin/FAK signaling by antimiR-1468-3p. qPCR validation showed that ITGB3 is one of the integrin subunits that is reduced by antimiR-1468-3p. Interestingly, Rapisarda et al.42 proposed that ITGB3 accelerates the onset of senescence via activation of the TGF-β1 signal, and its expression could be used as a senescence marker. In addition, our data show that TGF-β1 induces miR-1468-3p expression, which in turn potentiates p38 and FAK signals, indicating that miR-1468-3p serves as a nodal point in the regulation of non-canonical TGF-β1 signaling.
miR-1468-3p Targets DUSPs
p38 MAPK, which belongs to the non-canonical TGF-β1-MAPK pathway, is regulated by phosphorylation and dephosphorylation by MKKs or DUSPs, respectively. Detailed analysis of MAPK and DUSP genes from RNA sequencing data indicates that DUSPs are key targets of miR-1468-3p. Eleven out of 25 human DUSP genes play a major role in MAPK-specific inactivation,16 and 8 out of those are significantly affected by miR-1468-3p blockade in TGF-β1-treated cells. Computational prediction of miR-1468-3p targets also identified eight DUSPs as potential targets for miR-1468-3p, five of which serve a role in MAPK inactivation. Based on computational analysis of predicted miR-1468-3p-targeted DUSPs, we performed qPCR analysis of selected DUSPs. Remarkably, we found that miR-1468-3p was sufficient to downregulate DUSP1 (inactivates all three major MAPKs, i.e., ERK, p38, and JNK), DUSP6 (primarily inactivates ERK), and DUSP8 (primarily inactivates p38 and JNK), and direct regulation of DUSP1 was further confirmed by analysis of luciferase construct driven by the 3′ UTR of DUSP1 containing the miR-1468-3p binding site.
miR-1468-3p in Cardiac Fibrosis and Aging
Analysis of human cardiac samples for miR-1468-3p levels showed an increase in older SCD victims with PMF and an age-dependent increase in hearts of healthy subjects. Experimental studies with cultured human fibroblasts showed that, in addition to promoting collagen production, miR-1468-3p promotes fibroblast senescence. Aging itself does not induce CVD; instead, it occurs as an independent risk factor for CVD and aggravates the cardiac pathologies in older individuals. Prior studies show that aging and fibrosis are tightly associated.19 Schafer et al.43 reported that in human idiopathic pulmonary fibrosis (IPF), fibroblasts acquire a senescent phenotype. They further demonstrated that the senescent fibroblasts are fibrogenic and promote IPF, and removal of senescent fibroblasts improved pulmonary function in a bleomycin-induced lung fibrosis model.43 In cardiac disease models, senescent CFs accumulate in the heart both after myocardial infarction44 and thoracic aortic constriction,45 and in the latter model genetic deletion of p53 and p16INK4 aggravated the stress-induced cardiac fibrosis. However, it is currently not clear whether aging-associated fibroblast senescence promotes or antagonizes collagen deposition in the myocardium.
Stimuli-induced senescence functions through either p53/p21 or p16, or through a combination of both. Interestingly, the p53/p21 pathway often responds to stimuli earlier to halt progression of the cell cycle, while the resulting senescence may be reversible and insufficient to maintain senescence, which suggests that p21 expression is also likely to be transient. Alternatively, the increase in p16 expression is considered to relate to natural aging. The p16 engaged senescence usually occurs secondary to the p53/p21 pathway and leads to an irreversible senescence.19,46,47 In agreement with a previously described pro-senescent role of TGF-β1,48,49 we found that TGF-β1 induced an increase in expression of p21 (CDKN1A), p16 (CDKN2A), and p15 (CDKN2B)19 in hCFs. In the current study, administration of antimiR-1468-3p reversed TGF-β1-triggered p21 and p16 expression in CFs, which supports the role of miR-1468-3p in regulating senescence.
miR-1468-3p increases both p53 and p16 levels, which suggests that miR-1468-3p-induced cell senescence resembles a full-stage natural senescence. This is also consistent with the increase in miR-1468-3p levels in hearts of healthy aging subjects. However, miR-1468-3p did not induce an increase in p21 levels in fibroblasts. This is consistent with the lack of effect of miR-1468-3p on the PI3K/Akt pathway, which plays a key role in promoting the accumulation of p21.50 Interestingly, in addition to the well-defined pro-fibrotic function of p38, it has been shown to serve a pivotal role in senescence; that is, p38 activation triggers senescence51 while inhibition of p38 antagonizes senescense.19,21 Previous data also indicate a key role for p38 in regulation of cell cycle inhibitors, including p16.52 Thus, the p38 pathway represents a potential target for miR-1468-3p in the regulation of senescence.
Overall, in the current study, we identify miR-1468-3p as a central regulator of CF function. miR-1468-3p facilitates TGF-β1-p38 signaling and serves a dual role, potentiating both cell senescence and collagen deposition. Cardiac expression of miR-1468-3p increases in aging healthy hearts and in SCD victims with PMF, bridging cardiac aging and fibrosis, and indicating the potential for miR-1468-3p as a therapeutic target in aging-associated cardiac fibrosis.
Materials and Methods
Human Cardiac Samples
The human samples were obtained from the FinGesture study, which has systematically collected both clinical and autopsy data from SCD victims between 1998 and 2018 in northern Finland (n = 5,869). The study complies with the Declaration of Helsinki and was approved by the Ethics Committee of the Northern Ostrobothnia Hospital District, Finland. The National Authority for Medicolegal Affairs (Valvira) approved the review of postmortem data by the investigators. The autopsies were performed by experienced forensic pathologists (each performing more than 100 autopsies per year) using contemporary guidelines for diagnosis of the cause of death. Sudden death was defined as a witnessed death within 6 h of the onset of symptoms or an unwitnessed death within 24 h of the time victim was last seen in a good state of health. The precise definition of PMF has been previously described.53 Samples of healthy cardiac tissue were collected from autopsies of traffic accident victims with no history or evidence of CVD at autopsy.
Cell Cultures Experiments
hCFs and culture medium were purchased from ScienceCell (#6300), plated on poly-l-lysine (2 μg/cm2)-coated plates (ScienceCell, #0413), and cultured in fibroblast medium-2 (FM-2, #2331) supplemented with 5% fetal bovine serum (FBS, ScienceCell, #0025), 1% fibroblast growth supplement-2 (FGS-2, ScienceCell, #2382), and penicillin/streptomycin solution (P/S, ScienceCell, #0503). HUVECs were purchased from Cell Applications (#200p-05n), cultured according to the manufacturer’s instructions. Human endothelial cell basal medium (#210-500), human endothelial cell growth supplement (#211-GS), and attachment factor solution (contains bovine gelatin) (#123-100) were all from Cell Applications.
For mimic and anti-miR experiments, oligonucleotides were transfected with Lipofectamine 2000 (Thermo Fisher Scientific, #11668-019) in culture medium according to the manufacturer’s protocol. 50 nM concentration was used for the antisense oligonucleotide of miR-1468-3p (antimiR-1468-3p) (QIAGEN, #339131) and the control sequence (antimiR-Ctrl) (QIAGEN, #339136). Mimic-1468-3p and mimic-Ctrl (QIAGEN, #339173) were transfected at 20 nM concentration.
For TGF-β1 treatment, recombinant human TGF-β1 (Peprotech, #100-21) was used at 5 ng/mL concentration. Cultured hCFs were first transfected with 50 nM antimiR-Ctrl or antimiR-1468-3p for 24 h and treated with TGF-β1 for 24 h, after which cells were collected or fixed with 4% paraformaldehyde for analysis.
Collagen Deposition, Cell Proliferation, and Cell Metabolic Assay
Cultured hCFs, at 70% confluence, were treated with mimic-1468-3p or antimiR-1468-3p, with or without TGF-β1. Collagen deposition evaluation was conducted with Sirius Red/fast green staining (Chondrex/Amsbio, #9046). The cell proliferation assay was performed with a Click-iT EdU Alexa Fluor 488 HCS (high-content screening) assay (Thermo Fisher Scientific, #C10350) after a 2-h incubation with 10 μM EdU. Cell metabolic activity was measured after a 2-h incubation with 10 μM resazurin (Sigma, #R7017) measured with excitation at 544 nm and emission at 590 nm. All assays were performed according to the manufacturers’ instructions and measured by using a multiplate reader (Varioskan Flash 4.00.53).
SA-β-GAL Activity
Cultured hCFs were transfected with 20 nM mimic-1468-3p or mimic-Ctrl. After 24 h, cells were fixed and stained with a cell senescence detection kit (Abcam, #ab65351). Five randomly chosen fields in each well were imaged and analyzed using ImageJ with color deconvolution and the same threshold.
Dual-Luciferase Reporter Assay
WT dual-luciferase reporter plasmid containing human DUSP1 3′ UTR was purchased from GeneCopoeia (HmiT117930-MT06; GenBank: NM_004417.4). Five nucleotides from the binding site were mutated (5′-TTTTGC-3′→5′-TACCTA-3′) by using 5′ phosphorylated primers (forward, 5′-ACCTATCGCCCCTGTTTTTTGTAGAATC-3’; reverse, 5′-AAGAAACCGGATCACACACTGAG-3′). The amplification reaction was performed by using Phusion Hot Start II high-fidelity DNA polymerase (Thermo Fisher Scientific) according to the protocols of the manufacturer. The PCR product was DpnI (New England BioLabs) digested and ligated by T4 ligase (New England BioLabs) according to the manufacturer’s instructions. Clones were sequenced to confirm the correct mutation in the binding site. Cultured hCFs were delivered with 20 nM mimic-1468-3p or 50 nM antimiR-1468-3p for 24 h, then transfected with Lipofectamine 2000 (Thermo Fisher Scientific, #11668-019) WT or mutant luciferase construct for 24 h. Luciferase activity was measured with a Luc-Pair duo-luciferase assay kit 2.0 (GeneCopoeia) according to the manufacturer’s instructions. Firefly luciferase activity was normalized with Renilla luciferase activity.
Adenoviral Vectors and Plasmids
For adenoviral transfections, hCFs at 70% confluence were transduced with adenoviruses encoding for MKK3 or LacZ. After 24 h, cells were fixed for further analysis. adv-MKK3b has been previously described.54
qPCR Analysis for miRNA and mRNA Expression in Human Heart Samples and Cultured hCFs
RNA was extracted from formalin-fixed, paraffin-embedded (FFPE) tissues sections using a RecoverAll total nucleic acid isolation kit for FFPE (Thermo Fisher Scientific, #AM1975). miRNAs were isolated from cultured hCFs with an E.Z.N.A. miRNA kit (Omega Bio-tek, #R7034-01). Reverse transcription of miRNAs was done by using a TaqMan advanced miRNA cDNA synthesis kit (Thermo Fisher Scientific, #A28007) according to the manufacturer’s instructions. qPCR analysis for miRNA expression was carried out on QuantStudio 5 system (Thermo Fisher Scientific) with TaqMan advanced miRNA assays (Applied Biosystems), TaqMan fast advanced master mix (#4444557), and miRNA primers (#A25576). For data analysis, comparative CT method was used to calculate the results. All samples were determined in duplicate. hsa-miR-186-5p was used for the internal control of miRNA expression.
Total RNA was isolated with the E.Z.N.A. total RNA kit I (OMEGA Bio-tek, #OMEGR6834-02) and reverse-transcribed with a Transcriptor First Strand cDNA Synthesis Kit (Roche, #04896866001). qPCR was performed with SYBR Green PCR master mix (Thermo Fisher Scientific, #4309155) on QuantStudio 5 System (Thermo Fisher Scientific). For data analysis, comparative CT method was used to calculate the results. Primers used in qPCR are presented in Table 1. 18S ribosomal RNA was used as the internal reference gene.
Table 1.
Primers Used in qPCR
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) | |
|---|---|---|---|
| Sigma | ACTA2 | CTATGAGGGCTATGCCTTGCC | GCTCAGCAGTAGTAACGAAGGA |
| Sigma | POSTN | GACCGTGTGCTTACACAAATTG | AAGTGACCGTCTCTTCCAAGG |
| Sigma | CTGF | AAAAGTGCATCCGTACTCCCA | CCGTCGGTACATACTCCACAG |
| Sigma | COL1A1 | GAGGGCCAAGACGAAGACATC | CAGATCACGTCATCGCACAAC |
| Sigma | CDKN1A | AGGTGGACCTGGAGACTCTCAG | TCCTCTTGGAGAAGATCAGCCG |
| Sigma | ITGB3 | CCGGCCAGATGATTCGAAGA | CAGGGTAATCCTCCACCTGC |
| Sigma | DUSP1 | CTGCCTTGATCAACGTCTCA | ACCCTTCCTCCAGCATTCTT |
| Sigma | DUSP5 | GCCAGCTTATGACCAGGGTG | GTCCGTCGGGAGACATTCAG |
| Sigma | DUSP6 | CGAGACCCCAATAGTGC | AATGGCCTCAGGGAAA |
| Sigma | DUSP7 | GACGTGCTCGGCAAGTATG | GGATCTGCTTGTAGGTGAACTC |
| Sigma | DUSP8 | TCAGCTCCGTCAACATCTGC | CGCGTGCTCTGGTCATAGA |
| Sigma | DUSP14 | CTGCTCACTTAGGACTTTCT | CCTTGGTAGCGTGCTG |
| Sigma | DUSP16 | GCCCATGAGATGATTGGAACTC | CGGCTATCAATTAGCAGCACTTT |
| Sigma | MAPK14 | CTGGTACAGACCATATTAACCAGC | GGTGTTCCTGTCAGACGCAT |
| Sigma | 18S | CTCAACACGGGAAACCTCAC | CGCTCCACCAACTAAGAACG |
Western Blotting
For western blot, lysis buffer containing 20 mM Tris-HCl, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, and 1 mM dithiothreitol (pH 7.5) supplemented with protease and phosphatase inhibitors (Sigma) was used to extract the total protein content from hCFs. The lysate was centrifuged at 12,500 rpm at 4°C for 20 min. The concentration was determined by colorimetric protein assay (Bio-Rad). The protein samples (40 μg) were denatured for 5 min at 97°C, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto 0.45-μm nitrocellulose membranes by Trans-Blot turbo transfer system (Bio-Rad). The membranes were blocked for 1 h at room temperature (RT) by blocking buffer (Odyssey, LI-COR Biosciences) diluted into Tris-buffered saline (TBS, 50 mM Tris, 200 mM NaCl [pH 7.4]).
The membranes were probed with antibodies against p21 (sc-6246, Santa Cruz, 1:250), human p16INK4a/ CDKN2A (AF5779, R&D Systems, 1:1,000), p53 (sc-47698, Santa Cruz, DO-7, 1:200), collagen 1 (ab34710, Abcam, 1:500), CTGF (sc-14939, Santa Cruz, 1:200), Postn (sc-398631, Santa Cruz, 1:500), p-p38 MAPK (Thr180/Tyr182) (#9211, Cell Signaling Technology, 1:1,000), active JNK antibody (V7932, Promega, 1:3,000), FAK (p-Y397) (ab81298, Abcam, 1:1,000), DUSP1 (sc-373841, Santa Cruz, 1:200), p38 MAPK (#9212, Cell Signaling Technology, 1:1,000), p-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (#9106, Cell Signaling Technology, 1:1,000), p-Akt (Ser473) (#9271, Cell Signaling Technology, 1:1,000), p-SMAD2 (Ser465/Ser467) (#3108, Cell Signaling Technology, 1:1,000), p-HSP27 (Ser82) (Ser465/Ser467) (#2401, Cell Signaling Technology, 1:1,000), SAPK/JNK antibody (#9252, Cell Signaling Technology, 1:1,000), and purified mouse anti-FAK (#610088, BD Transduction Laboratories, 1:500). Vinculin (ab18058, Abcam, 1:1,000), α-tubulin (#2144, Cell Signaling Technology, 1:1,000), or glyceraldehyde-3-phosphate dehydrogenase (MAB374, Merck, 1:100,000) was used as a loading control. Secondary antibodies purchased from Life Technologies (Alexa Fluor A11371, A21058, and A21076) were used with a dilution of 1:5,000. All antibodies were diluted in Odyssey blocking buffer from LI-COR Biosciences. Odyssey infrared imaging (LI-COR Biosciences) was used to detect the antibody binding.
Flow Cytometry
Transfection efficiency was determined after delivery of a fluorescence-labeled (5′6-FAM [fluorescein]) control sequence (QIAGEN, #339136) to cells with Lipofectamine 2000 (Thermo Fisher Scientific, #11668-019). Cells without delivery were used as a negative control for setting the gating region. The percentage of cells that pass the gating threshold was counted for each sample. 10,000 events were acquired per sample on a FACSCalibur flow cytometer equipped with a 488-nm laser (BD Biosciences), and data analysis was processed using CellQuest Pro software (BD Biosciences).
Histology
For assessment of DUSP1, total p38, and phosphorylated p38 expression, FFPE sections of human cardiac tissue were used. Paraffin was removed in xylene and the sections were dehydrated in a graded ethanol series. Primary antibodies were diluted in blocking buffer and incubated on tissue sections overnight at 4°C. Primary antibodies used were as follows: 1:50 DUSP1 (sc-373841, Santa Cruz), 1:50 p-p38 MAPK (Thr180/Tyr182) (#9211, Cell Signaling Technology), and 1:100 p38 MAPK (#9212, Cell Signaling Technology). The sections were viewed with a Nikon Eclipse 80i microscope (Nikon, Melville, NY, USA), and captured with an ORCA-Flash4.0 LT digital camera (Hamamatsu, Hamamatsu City, Japan) and NIS-Elements AR v4.30.01 software (Nikon, Melville, NY, USA).
RNA-Seq Analysis
Total RNA was isolated from hCFs by using an E.Z.N.A. miRNA kit (Omega Bio-tek, #R7034-01). RNA sequencing was carried out with an Illumina TruSeq stranded mRNA kit on a HiSeq 3000 (50 bp, Illumina). Homo sapiens hg38 was the reference genome used, and the reference gene annotation was RefSeq-based annotation, which was downloaded from the Illumina iGenomes website.
Functional enrichment analyses of the gene profile (GO biological process and KEGG pathway analysis) were performed with the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 or the PANTHER (Protein Analysis through Evolutionary Relationships) classification system.
Further investigation of TGF-β/SMAD, p38/MAPK, phosphorylation, and fibrosis-related genes was performed using normalized RNA-seq data. A Student’s t test with equal variances was performed between antimiR-Ctrl+TGF-β1- versus antimiR-1468-3p+TGF-β1-treated cells, and all genes with significance (p < 0.05) were defined as DE.
Statistical Analysis
IBM SPSS Statistics software (IBM, Armonk, NY, USA) was used for determining statistical significance. Normally distributed data were analyzed with one-way ANOVA followed by Tukey’s post hoc test. When two groups were compared, a Student’s t test was used. p <0.05 was defined as statistically significant.
Author Contributions
R.L., L.R.-K., J.U., H.H., and R.K. designed the study. R.L., L.R,-K., J.M., J.U., A.K., and L.P. performed acquisition of data. R.L., L.R.-K., J.M., J.U., J.S., A.K., L.P., K.P., H.H., J.J., and R.K. analyzed and interpreted the data. R.L. and R.K. drafted the manuscript. L.R.-K., J.M., and J.J. made critical revisions to the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Marja Arbelius and Kirsi Salo for technical assistance. We thank the Medical Bioinformatics Centre of the Turku Centre for Biotechnology for the NGS (next-generation sequencing) data analysis. The Medical Bioinformatics Centre is supported by the University of Turku, Åbo Akademi University, and Biocenter Finland. This work was supported by the Academy of Finland (grants 131020 and 297094 to R.K. and 268505 to J.M.), the Finnish Foundation for Cardiovascular Research (to L.R.-K., J.M., H.H., and R.K.), and by the Jane and Aatos Erkko Foundation (to R.K., H.H., and J.J.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.04.001.
Supplemental Information
References
- 1.Gude N.A., Broughton K.M., Firouzi F., Sussman M.A. Cardiac ageing: extrinsic and intrinsic factors in cellular renewal and senescence. Nat. Rev. Cardiol. 2018;15:523–542. doi: 10.1038/s41569-018-0061-5. [DOI] [PubMed] [Google Scholar]
- 2.Weber K.T., Sun Y., Bhattacharya S.K., Ahokas R.A., Gerling I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2013;10:15–26. doi: 10.1038/nrcardio.2012.158. [DOI] [PubMed] [Google Scholar]
- 3.Zeisberg E.M., Tarnavski O., Zeisberg M., Dorfman A.L., McMullen J.R., Gustafsson E., Chandraker A., Yuan X., Pu W.T., Roberts A.B. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 4.Wong T.C., Piehler K., Meier C.G., Testa S.M., Klock A.M., Aneizi A.A., Shakesprere J., Kellman P., Shroff S.G., Schwartzman D.S. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short-term mortality. Circulation. 2012;126:1206–1216. doi: 10.1161/CIRCULATIONAHA.111.089409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hookana E., Junttila M.J., Puurunen V.P., Tikkanen J.T., Kaikkonen K.S., Kortelainen M.L., Myerburg R.J., Huikuri H.V. Causes of nonischemic sudden cardiac death in the current era. Heart Rhythm. 2011;8:1570–1575. doi: 10.1016/j.hrthm.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 6.Junttila M.J., Holmström L., Pylkäs K., Mantere T., Kaikkonen K., Porvari K., Kortelainen M.L., Pakanen L., Kerkelä R., Myerburg R.J., Huikuri H.V. Primary myocardial fibrosis as an alternative phenotype pathway of inherited cardiac structural disorders. Circulation. 2018;137:2716–2726. doi: 10.1161/CIRCULATIONAHA.117.032175. [DOI] [PubMed] [Google Scholar]
- 7.Rockey D.C., Bell P.D., Hill J.A. Fibrosis—a common pathway to organ injury and failure. N. Engl. J. Med. 2015;372:1138–1149. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 8.Munger J.S., Sheppard D. Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb. Perspect. Biol. 2011;3:a005017. doi: 10.1101/cshperspect.a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frangogiannis N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Invest. 2017;127:1600–1612. doi: 10.1172/JCI87491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong P., Shinde A.V., Su Y., Russo I., Chen B., Saxena A., Conway S.J., Graff J.M., Frangogiannis N.G. Opposing actions of fibroblast and cardiomyocyte Smad3 signaling in the infarcted myocardium. Circulation. 2018;137:707–724. doi: 10.1161/CIRCULATIONAHA.117.029622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikeuchi M., Tsutsui H., Shiomi T., Matsusaka H., Matsushima S., Wen J., Kubota T., Takeshita A. Inhibition of TGF-β signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc. Res. 2004;64:526–535. doi: 10.1016/j.cardiores.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 12.Okada H., Takemura G., Kosai K., Li Y., Takahashi T., Esaki M., Yuge K., Miyata S., Maruyama R., Mikami A. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111:2430–2437. doi: 10.1161/01.CIR.0000165066.71481.8E. [DOI] [PubMed] [Google Scholar]
- 13.Khalil H., Kanisicak O., Prasad V., Correll R.N., Fu X., Schips T., Vagnozzi R.J., Liu R., Huynh T., Lee S.J. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Invest. 2017;127:3770–3783. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Massagué J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auger-Messier M., Accornero F., Goonasekera S.A., Bueno O.F., Lorenz J.N., van Berlo J.H., Willette R.N., Molkentin J.D. Unrestrained p38 MAPK activation in Dusp1/4 double-null mice induces cardiomyopathy. Circ. Res. 2013;112:48–56. doi: 10.1161/CIRCRESAHA.112.272963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu R., Molkentin J.D. Regulation of cardiac hypertrophy and remodeling through the dual-specificity MAPK phosphatases (DUSPs) J. Mol. Cell. Cardiol. 2016;101:44–49. doi: 10.1016/j.yjmcc.2016.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fontana L. Interventions to promote cardiometabolic health and slow cardiovascular ageing. Nat. Rev. Cardiol. 2018;15:566–577. doi: 10.1038/s41569-018-0026-8. [DOI] [PubMed] [Google Scholar]
- 18.Hernandez-Segura A., Nehme J., Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–453. doi: 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Childs B.G., Durik M., Baker D.J., van Deursen J.M. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat. Med. 2015;21:1424–1435. doi: 10.1038/nm.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rayess H., Wang M.B., Srivatsan E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer. 2012;130:1715–1725. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muñoz-Espín D., Serrano M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 22.Baker D.J., Childs B.G., Durik M., Wijers M.E., Sieben C.J., Zhong J., Saltness R.A., Jeganathan K.B., Verzosa G.C., Pezeshki A. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gebert L.F., MacRae I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019;20:21–37. doi: 10.1038/s41580-018-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berezikov E. Evolution of microRNA diversity and regulation in animals. Nat. Rev. Genet. 2011;12:846–860. doi: 10.1038/nrg3079. [DOI] [PubMed] [Google Scholar]
- 25.Kim J.H., Lee B.R., Choi E.S., Lee K.M., Choi S.K., Cho J.H., Jeon W.B., Kim E. Reverse expression of aging-associated molecules through transfection of miRNAs to aged mice. Mol. Ther. Nucleic Acids. 2017;6:106–115. doi: 10.1016/j.omtn.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D’Alessandra Y., Devanna P., Limana F., Straino S., Di Carlo A., Brambilla P.G., Rubino M., Carena M.C., Spazzafumo L., De Simone M. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur. Heart J. 2010;31:2765–2773. doi: 10.1093/eurheartj/ehq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anfossi S., Babayan A., Pantel K., Calin G.A. Clinical utility of circulating non-coding RNAs—an update. Nat. Rev. Clin. Oncol. 2018;15:541–563. doi: 10.1038/s41571-018-0035-x. [DOI] [PubMed] [Google Scholar]
- 28.van Rooij E., Sutherland L.B., Thatcher J.E., DiMaio J.M., Naseem R.H., Marshall W.S., Hill J.A., Olson E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagpal V., Rai R., Place A.T., Murphy S.B., Verma S.K., Ghosh A.K., Vaughan D.E. miR-125b is critical for fibroblast-to-myofibroblast transition and cardiac fibrosis. Circulation. 2016;133:291–301. doi: 10.1161/CIRCULATIONAHA.115.018174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X., Wang H.X., Li Y.L., Zhang C.C., Zhou C.Y., Wang L., Xia Y.L., Du J., Li H.H. MicroRNA Let-7i negatively regulates cardiac inflammation and fibrosis. Hypertension. 2015;66:776–785. doi: 10.1161/HYPERTENSIONAHA.115.05548. [DOI] [PubMed] [Google Scholar]
- 31.Yuan J., Liu H., Gao W., Zhang L., Ye Y., Yuan L., Ding Z., Wu J., Kang L., Zhang X. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress. Theranostics. 2018;8:2565–2582. doi: 10.7150/thno.22878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berezikov E., van Tetering G., Verheul M., van de Belt J., van Laake L., Vos J., Verloop R., van de Wetering M., Guryev V., Takada S. Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res. 2006;16:1289–1298. doi: 10.1101/gr.5159906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinto A.R., Ilinykh A., Ivey M.J., Kuwabara J.T., D’Antoni M.L., Debuque R., Chandran A., Wang L., Arora K., Rosenthal N.A., Tallquist M.D. Revisiting cardiac cellular composition. Circ. Res. 2016;118:400–409. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Y., Zhang Z.Y. The mechanism of dephosphorylation of extracellular signal-regulated kinase 2 by mitogen-activated protein kinase phosphatase 3. J. Biol. Chem. 2001;276:32382–32391. doi: 10.1074/jbc.M103369200. [DOI] [PubMed] [Google Scholar]
- 35.Zhou B., Wang Z.X., Zhao Y., Brautigan D.L., Zhang Z.Y. The specificity of extracellular signal-regulated kinase 2 dephosphorylation by protein phosphatases. J. Biol. Chem. 2002;277:31818–31825. doi: 10.1074/jbc.M203969200. [DOI] [PubMed] [Google Scholar]
- 36.Maillet M., Purcell N.H., Sargent M.A., York A.J., Bueno O.F., Molkentin J.D. DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J. Biol. Chem. 2008;283:31246–31255. doi: 10.1074/jbc.M806085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purcell N.H., Wilkins B.J., York A., Saba-El-Leil M.K., Meloche S., Robbins J., Molkentin J.D. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. USA. 2007;104:14074–14079. doi: 10.1073/pnas.0610906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y., Shepherd E.G., Nelin L.D. MAPK phosphatases—regulating the immune response. Nat. Rev. Immunol. 2007;7:202–212. doi: 10.1038/nri2035. [DOI] [PubMed] [Google Scholar]
- 39.Molkentin J.D., Bugg D., Ghearing N., Dorn L.E., Kim P., Sargent M.A., Gunaje J., Otsu K., Davis J. Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation. 2017;136:549–561. doi: 10.1161/CIRCULATIONAHA.116.026238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yokota T., Wang Y. p38 MAP kinases in the heart. Gene. 2016;575:369–376. doi: 10.1016/j.gene.2015.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valiente-Alandi I., Potter S.J., Salvador A.M., Schafer A.E., Schips T., Carrillo-Salinas F., Gibson A.M., Nieman M.L., Perkins C., Sargent M.A. Inhibiting fibronectin attenuates fibrosis and improves cardiac function in a model of heart failure. Circulation. 2018;138:1236–1252. doi: 10.1161/CIRCULATIONAHA.118.034609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rapisarda V., Borghesan M., Miguela V., Encheva V., Snijders A.P., Lujambio A., O’Loghlen A. Integrin beta 3 regulates cellular senescence by activating the TGF-β pathway. Cell Rep. 2017;18:2480–2493. doi: 10.1016/j.celrep.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schafer M.J., White T.A., Iijima K., Haak A.J., Ligresti G., Atkinson E.J., Oberg A.L., Birch J., Salmonowicz H., Zhu Y. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu F., Li Y., Zhang J., Piao C., Liu T., Li H.H., Du J. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS ONE. 2013;8:e74535. doi: 10.1371/journal.pone.0074535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer K., Hodwin B., Ramanujam D., Engelhardt S., Sarikas A. Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J. Am. Coll. Cardiol. 2016;67:2018–2028. doi: 10.1016/j.jacc.2016.02.047. [DOI] [PubMed] [Google Scholar]
- 46.Campisi J., d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 47.de Keizer P.L. The Fountain of Youth by targeting senescent cells? Trends Mol. Med. 2017;23:6–17. doi: 10.1016/j.molmed.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 48.Lyu G., Guan Y., Zhang C., Zong L., Sun L., Huang X., Huang L., Zhang L., Tian X.L., Zhou Z. TGF-β signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging. Nat. Commun. 2018;9:2560. doi: 10.1038/s41467-018-04994-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferreira-Gonzalez S., Lu W.Y., Raven A., Dwyer B., Man T.Y., O’Duibhir E., Lewis P.J.S., Campana L., Kendall T.J., Bird T.G. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat. Commun. 2018;9:1020. doi: 10.1038/s41467-018-03299-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Astle M.V., Hannan K.M., Ng P.Y., Lee R.S., George A.J., Hsu A.K., Haupt Y., Hannan R.D., Pearson R.B. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene. 2012;31:1949–1962. doi: 10.1038/onc.2011.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wagner E.F., Nebreda A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 52.Wong E.S.M., Le Guezennec X., Demidov O.N., Marshall N.T., Wang S.T., Krishnamurthy J., Sharpless N.E., Dunn N.R., Bulavin D.V. p38MAPK controls expression of multiple cell cycle inhibitors and islet proliferation with advancing age. Dev. Cell. 2009;17:142–149. doi: 10.1016/j.devcel.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 53.Haukilahti M.A.E., Holmström L., Vähätalo J., Kenttä T., Tikkanen J., Pakanen L., Kortelainen M.L., Perkiömäki J., Huikuri H., Myerburg R.J., Junttila M.J. Sudden cardiac death in women. Circulation. 2019;139:1012–1021. doi: 10.1161/CIRCULATIONAHA.118.037702. [DOI] [PubMed] [Google Scholar]
- 54.Koivisto E., Kaikkonen L., Tokola H., Pikkarainen S., Aro J., Pennanen H., Karvonen T., Rysä J., Kerkelä R., Ruskoaho H. Distinct regulation of B-type natriuretic peptide transcription by p38 MAPK isoforms. Mol. Cell. Endocrinol. 2011;338:18–27. doi: 10.1016/j.mce.2011.02.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.