

Abstract
Patients with recurrent glioblastoma achieving response to bevacizumab combined with chemotherapy have clinical improvement and prolonged survival. High gene expression of angiotensinogen (AGT) is associated with a poor bevacizumab response. Because AGT expression is epigenetically regulated, we aimed to investigate whether AGT promoter methylation in tumor tissue predicts response to bevacizumab combination therapy in patients with recurrent glioblastoma. The study included 159 patients with recurrent glioblastoma, treated with bevacizumab combination treatment (training cohort, n = 77; validation cohort, n = 82). All patients could be evaluated for treatment response and biomarkers. DNA methylation of 4 CpG sites in the AGT promoter was measured using pyrosequencing. A model for nonresponse was established using logistic regression analysis. In the training cohort, lower methylation of each of the four CpG sites in the AGT promoter was significantly associated with nonresponse (all P < 0.05). Moreover, the mean methylation level of all four CpG sites was associated with an increased likelihood of not achieving response to bevacizumab combination therapy (twofold decrease: odds ratio = 3.01; 95% confidence interval: 1.41–6.44; P = 0.004). We developed a model for nonresponse in the training cohort, where a threshold of mean AGT promoter methylation levels was set to below 12%. The model could predict bevacizumab nonresponse with 96% specificity. Importantly, this predictor was also significantly associated with nonresponse in the validation cohort (P = 0.037). Taken together, our findings suggest that low AGT promoter methylation in tumor tissue predicts nonresponse to bevacizumab combination treatment in patients with recurrent glioblastoma. We have, thus, established and successfully validated a predictor for nonresponse that can be used to identify patients who will not benefit from bevacizumab combination therapy.
Keywords: bevacizumab, DNA methylation, glioblastoma, local renin–angiotensin system, predictive biomarker
One of four recurrent glioblastoma patients responds to bevacizumab treatment. The remaining patients have no treatment benefit. In this study, we found lower angiotensinogen (AGT) promoter methylation in nonresponding compared with responding patients. A predictor for nonresponse was established and successfully validated. Applying this predictor in clinical practice, one of four patients can be spared an ineffective and toxic treatment.
Abbreviations
- 95% CI
95% confidence interval
- AGT
angiotensinogen
- AUC
area under the receiving operating characteristic curve
- CEBP
CCAAT/enhancer‐binding protein
- CR
complete response
- EIAED
enzyme‐inducing anti‐epileptic drug
- OR
odds ratio
- PD
progressive disease
- PR
partial response
- PS
ECOG performance status
- SD
stable disease
- STAT3
signal transducer and activator of transcription 3
- VEGF
vascular endothelial growth factor A
1. Introduction
Glioblastoma almost inevitable progress following standard treatment, comprising surgery, radiotherapy plus concomitant and adjuvant temozolomide (Stupp et al., 2005). At tumor progression, no standard treatment is available and most known agents have shown limited activity. Bevacizumab, an antibody targeting vascular endothelial growth factor A (VEGF), has been proposed as an active drug, given that glioblastoma is characterized by an abnormal tumor vasculature fueled by pro‐angiogenic stimulation from VEGF overexpression (Baumgarten et al., 2016). The inhibition of VEGF has shown to normalize the tumor vasculature and hereby improve tumor blood perfusion and drug delivery (Goel et al., 2011). This mechanism of action has been confirmed in recurrent glioblastoma patients responding to anti‐angiogenic therapy on the basis of molecular and imaging data (Batchelor et al., 2013; Goel et al., 2011; Urup et al., 2017).
Bevacizumab in combination with chemotherapy has been shown to produce high response rates of approximately 30% in recurrent glioblastoma patients (Friedman et al., 2009; Urup et al., 2016a; Wick et al., 2017). Although this treatment has not proven active in the total population of recurrent glioblastoma patients (Taal et al., 2014; Wick et al., 2017), patients whom achieve response to bevacizumab combination therapy obtain clinical improvement and have prolonged survival (Henriksson et al., 2011; Huang et al., 2016; Jakobsen et al., 2018). Especially, for the prognostic favorable group of recurrent glioblastoma patients (defined as baseline ECOG performance status ≤ 1, prednisolone ≤ 25 mg, and unifocal disease) we have based on retrospective data (Urup et al., 2016a) observed an impressive difference in median overall survival (OS) of 10 months between responding and nonresponding patients. This highlights the importance of identifying patients benefitting from bevacizumab combination treatment based on prognostic factors and predictive biomarkers for bevacizumab efficacy. However, to date no validated predictive tumor biomarkers for bevacizumab efficacy have been identified.
In recurrent glioblastoma patients, a high RNA expression of the angiotensinogen gene (AGT) in tumor tissue has been found associated with nonresponse to bevacizumab combination therapy (Urup et al., 2016b). Angiotensinogen is the primary substrate of the renin–angiotensin system, a hormone system regulating blood pressure and fluid/electrolyte homeostasis. The existence of a local renin–angiotensin system in the brain as well as in glioblastoma has been confirmed (Juillerat‐Jeanneret et al., 2004; Paul et al., 2006). This paracrine system regulates cerebral blood flow, and increased activity of the main effector peptide angiotensin II has been shown to reduce cerebral blood perfusion and oxygenation (Kazama et al., 2004; Paul et al., 2006; Vallejo‐Ardila et al., 2018; Wei et al., 2007). Furthermore, angiotensin II signaling has been linked to resistance to bevacizumab‐induced vascular normalization (Johansen et al., 2018; Levin et al., 2017; Stylianopoulos and Jain, 2013).
The transcriptional activity of AGT has been found to be dependent on demethylation of the CEBP (CCAAT/enhancer‐binding protein) binding site of the AGT promoter region (Wang et al., 2014). Accordingly, the aim of this study was to investigate whether lower AGT promoter methylation in tumor tissue is predictive for glioblastoma patients not responding to bevacizumab combination therapy. This was initially investigated in a training cohort of 77 patients and subsequently studied in a validation cohort of 82 recurrent glioblastoma patients.
2. Materials and methods
2.1. Patients
All patients included in the study were identified using our clinical database of patients with histopathologically confirmed glioblastoma (WHO grade IV), whom at recurrence were consecutively treated with bevacizumab and irinotecan at Rigshospitalet, Copenhagen, Denmark. Eligibility criteria for the study were response evaluability and DNA methylation evaluable tumor tissue from the time of glioblastoma diagnosis. The selection of the two study cohorts is illustrated in REMARK diagrams (Figs S1 and S2).
2.1.1. Training cohort
All patients treated at recurrence with bevacizumab plus irinotecan between May 2005 and December 2011 were assessed for eligibility. These patients have also been included in a previous biomarker study (Urup et al., 2016b). During this period, bevacizumab (10 mg·kg−1) and irinotecan (125 mg·m−2, if EIAEDs 340 mg·m−2) were administered every 2 weeks and could be prescribed to recurrent glioblastoma patients in ECOG performance status 0‐2 according to published treatment protocols (Hasselbalch et al., 2010; Poulsen et al., 2009).
2.1.2. Validation cohort
All patients treated at recurrence with bevacizumab and irinotecan between January 2012 and February 2015 were assessed for eligibility. During this period, treatment was administered according to the same protocol as for the training cohort (Poulsen et al., 2009). Patients were prospectively included in the database which was regularly updated.
The study was conducted in accordance with the Helsinki Declaration, and the Danish Ethical Committee approved the study and granted exemption from the consent requirement (H‐2‐2012‐069).
2.2. Clinical follow‐up
Prior to administration of bevacizumab combination treatment, patients had to have measurable progressive disease (PD) by contrast‐enhanced MRI after standard therapy and be at least 4 weeks from prior chemotherapy and 3 months from completion of radiation therapy. In patients undergoing relapse surgery, bevacizumab combination therapy could be administered 4 weeks after surgery if the tumor was measurable at a postoperative MRI. Clinical follow‐up was performed every 4 weeks and MRI every 8 weeks. Treatment response was evaluated (investigator assessment by TU, VAL, and HSP) based on the Response Assessment in Neuro‐Oncology (RANO) criteria (Wen et al., 2010). Patients were categorized according to their best response; patients with complete response (CR) or partial response (PR) were classified as responders, while patients with stable disease (SD) or PD were classified as nonresponders. Patients not evaluable by MRI at first response evaluation (week 8) due to early toxicity, clinical progression, or death were classified as nonevaluable and excluded.
2.3. Tumor sample preparation
Archived formalin‐fixed, paraffin‐embedded brain tumor tissue samples from time of initial glioblastoma diagnosis were collected, and freshly cut sections (5 μm) were stored at 2–8 °C. A pathologist (HB) performed tissue review blinded to identifiers and clinical outcome. Areas containing representative tumor cells were marked on the hematoxylin and eosin‐stained slides. In the training cohort, tumors were microdissected to enrich for tumor cells. For the validation cohort, tumors were macrodissected to increase the tumor cell frequency to above 80%.
2.4. DNA methylation
Genomic DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany; 51304 or 51306) in the training cohort and Maxwell RSC DNA FFPE Kit (Promega; AS1450, Madison, WI, USA) in the validation cohort. DNA extracts were stored at −80 °C. DNA concentration was measured with a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), and genomic DNA was bisulfite‐converted using the EZ DNA Methylation‐Lightning Kit (Zymo Research, Irvine, CA, USA). DNA methylation of four selected CpG sites in the ATG promoter region was measured using pyrosequencing. The CpG sites analyzed were situated 282–229 base pairs upstream of the transcription start site in the AGT promoter (Fig. 1). The sites were selected based on a previous study where DNA methylation of these cytosines, which are situated in a CEBP binding region, has been associated with both chromatin accessibility in human adrenocortical cells and with lower AGT expression in both humans and rats (Wang et al., 2014). Primers for PCR amplification [forward: 5′‐GGTGGTTGGTTTTAGGTTGTTATATA‐TTTA‐3′, reverse (biotinylated): 5′‐ACTATTCCCAAACTACCTATACAC‐3′] and sequencing (5′‐TGTTATATATTTA‐GGGAGATGT‐3′) were designed using the pyromark assay design 2.0 software (Qiagen). Pyrosequencing was performed using the PyroMark Q24 instrument, and pyrograms were quality‐controlled using the pyromark q24 software version 2.0.7 (Qiagen). Samples that did not pass the quality control due to low DNA amount or poor DNA quality were considered nonevaluable by methylation analysis. In the training cohort and validation cohort, five and four patients were assessed nonevaluable by methylation analysis.
Fig. 1.
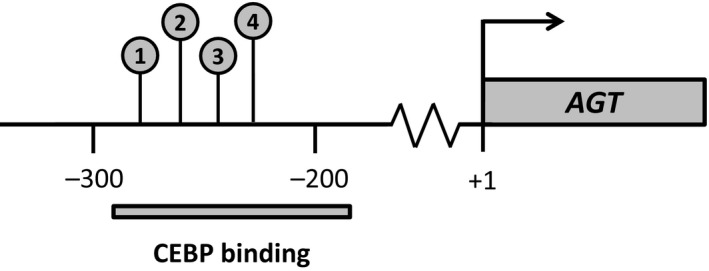
The four analyzed CpG sites in the angiotensinogen (AGT) promoter region. The four CpG sites analyzed by pyrosequencing were situated 282–229 base pairs (bp) upstream of the transcription start site of AGT in a CEBP binding region (CpG 1: −282; CpG 2: −261; CpG 3: −245; CpG 4: −229). Lower DNA methylation of these sites has been associated with a higher transcriptional activity of AGT (Wang et al., 2014).
2.5. Statistical analysis
Fisher’s exact tests and Mann–Whitney U‐tests were used for group comparison analyses. Survival was estimated with the Kaplan–Meier method. The probability of nonresponse was estimated by employing logistic regression, and the results are presented by odds ratios (ORs) with 95% confidence intervals (95% CIs) and the area under the receiver operating characteristic curve (AUC). Based on sample size, a fivefold cross‐validation was used for internal validation. The threshold estimated from the training set based on logistic regression analysis was used directly in the validation set in order to assess sensitivity and specificity. Continuous variables were log‐transformed (log base 2) prior to analysis. Assessment of the goodness of fit was done using the Hosmer–Lemeshow test (logistic regression). P‐values < 0.05 were considered significant. Calculations were performed using spss (v19.0; IBM Corp., Armonk, NY, USA), r version 3.5.0 (2018‐04‐23; R Development Core Team, Vienna, Austria, http://www.R-project.org), and sas (v9.4; SAS Institute, Cary, NC, USA).
3. Results
3.1. Patient characteristics
3.1.1. Training cohort
A total of 77 recurrent glioblastoma patients were included in the training cohort. Patients were aged 23–71 years, and 62% were men. All patients had progressed after radiation therapy and temozolomide. Baseline patient characteristics and clinical outcomes are summarized in Table 1. The majority of patients (69%) were categorized into a poor prognostic group defined as having at least one of three baseline factors: poor ECOG performance status (PS = 2), corticosteroid use (prednisolone > 25 mg), or multifocal disease. After progression on bevacizumab combination treatment, 11 patients underwent surgical resection and six patients received various types of experimental treatments. At the end of follow‐up, all patients had progressed and two patients were alive (median follow‐up: 8.2 months, range: 2–69 months).
Table 1.
Patient characteristics according to response and nonresponse to bevacizumab combination therapy in the training cohort. CI, confidence interval; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; PFS, progression free survival; OS, overall survival.
| Training cohort |
Total n = 77 |
Response (CR + PR) n = 26 (34%) |
Nonresponse (SD + PD) n = 51 (66%) |
P‐value |
|---|---|---|---|---|
| Gender, n (%) | ||||
| Male | 48 (62) | 15 (31) | 33 (69) | 0.62 |
| Female | 29 (38) | 11 (38) | 18 (62) | |
| Age, years (range) | ||||
| Median | 56 (23–71) | 54 (23–65) | 57 (30–71) | 0.22 |
| ECOG performance status, n (%) | ||||
| 0 | 31 (40) | 12 (39) | 19 (61) | 0.46 |
| 1 | 35 (46) | 12 (34) | 23 (66) | |
| 2 | 11 (14) | 2 (18) | 9 (82) | |
| Prior lines of chemotherapy, n (%) | ||||
| 1 | 69 (90) | 24 (35) | 45 (65) | 0.71 |
| 2 | 8 (10) | 2 (25) | 6 (75) | |
| Glioblastoma diagnosis, n (%) | ||||
| Glioblastoma | 63 (82) | 22 (35) | 41 (65) | 0.76 |
| Secondary glioblastoma a | 14 (18) | 4 (29) | 10 (71) | |
| Multifocal disease, n (%) | ||||
| Yes | 21 (27) | 6 (29) | 15 (71) | 0.60 |
| No | 56 (73) | 20 (36) | 36 (64) | |
| Corticosteroid use, n (%) b | ||||
| Yes | 58 (75) | 18 (31) | 40 (69) | 0.41 |
| No | 19 (25) | 8 (42) | 11 (58) | |
| Neurocognitive deficit, n (%) | ||||
| Yes | 43 (56) | 13 (30) | 30 (70) | 0.48 |
| No | 34 (44) | 13 (36) | 21 (62) | |
| Prognostic group | ||||
| Favorable c | 24 (31) | 10 (42) | 14 (58) | 0.44 |
| Poor d | 53 (69) | 16 (30) | 37 (70) | |
| Survival outcome | ||||
| Median PFS, months (95% CI) | ||||
| Total cohort | 5.2 | 10.9 (9.6–12.3) | 3.9 (3.3–4.4) | < 0.01 |
| Median OS, months (95% CI) | ||||
| Total cohort | 8.2 | 13.5 (10.3–16.8) | 7.5 (6.3–8.6) | < 0.01 |
| Favorable prognostic group c | 13.3 | 20.3 (15.8–24.8) | 8.3 (7.0–9.7) | < 0.01 |
| Poor prognostic group d | 7.5 | 8.8 (7.2–10.4) | 6.5 (5.2–7.8) | < 0.01 |
Lower‐grade glioma progressing as grade IV glioma.
Prednisolone > 10 mg.
The favorable prognostic group was defined as ECOG performance status ≤ 1, prednisolone ≤ 25 mg, and unifocal disease prior to initiation of bevacizumab combination therapy.
The poor prognostic group was defined as having at least one of the following baseline factors: ECOG performance status = 2, prednisolone > 25 mg, or multifocal disease prior to initiation of bevacizumab combination therapy.
Twenty‐six patients (34%) achieved response to bevacizumab combination therapy. None of the clinical baseline characteristics differed significantly between the responding and nonresponding patients. Both progression‐free survival and OS were significantly longer in the responding versus nonresponding patients.
In comparison with patients belonging to a poor prognostic group presenting a median OS of 7.5 months, patients in the favorable prognostic group had a significantly better prognosis with a median OS of 13.3 months. Importantly, responding patients of the favorable prognostic group showed a better prognosis with a median OS of 20 months in comparison with 8 months in the nonresponding patients (P < 0.01; Table 1), resulting in a median survival difference of 12 months. In comparison, this difference in median OS was 2 months for the poor prognostic group.
3.1.2. Validation cohort
Eighty‐two patients were included in the validation cohort. There were no significant differences in clinical characteristics between the validation cohort and the training cohort except for those patients in the validation cohort were older (P = 0.04) and had a higher frequency of multifocal disease (P = 0.05) (Table S1). The response rate was 33%, and clinical characteristics were not significantly associated with response (data not shown). After progression on bevacizumab combination therapy, 9 patients had surgery and 19 patients were administered various experimental treatments. At the end of follow‐up, all patients had progressed and one patient was alive (median follow‐up: 7.3 months, range: 2–40 months).
3.2. AGT promoter methylation in responding versus nonresponding patients
AGT promoter methylation levels of the four CpG sites investigated in tumor tissue from the training cohort showed a significant and high degree of intercorrelation (P < 0.0001 for all possible combinations; Fig. S3). AGT promoter methylation levels showed high variability among patients on sites 1 (range: 4–58%), 2 (range: 10–67%), 3 (range 2–44%), and 4 (range: 3–47%). The mean methylation level of the four CpG sites (mean CpG sites 1–4) ranged from 5% to 50%. As shown in Fig. 2, AGT promoter methylation of all four CpG sites and their mean were significantly lower in nonresponding versus responding patients (all P ≤ 0.03).
Fig. 2.
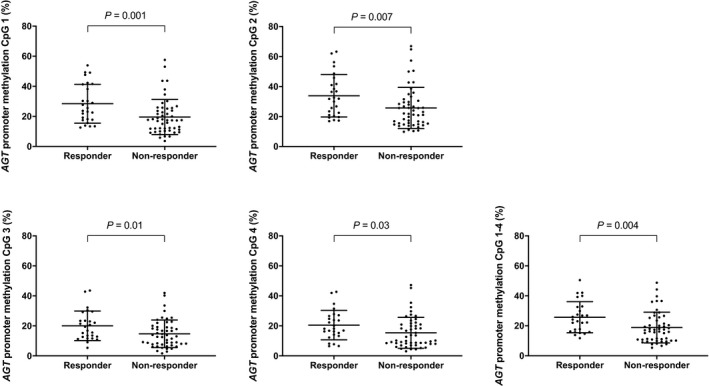
DNA methylation levels of the four analyzed CpG sites as well as the mean level of CpG sites 1–4 in the AGT promoter in responding and nonresponding patients of the training cohort. Mean values and standard deviations are shown by horizontal lines.
3.3. AGT promoter methylation as a biomarker for response
To further test whether lower AGT promoter methylation levels were associated with an increased likelihood of not achieving response to bevacizumab combination therapy in the training cohort, we performed univariate analyses of the candidate CpG sites. As shown in Table 2, lower methylation levels of all four CpG sites and the mean methylation of CpG sites 1–4 were significantly associated with an improved likelihood of not achieving response (all P ≤ 0.02). The two most significant explanatory variables were CpG site 1 and mean CpG sites 1–4 with an area under the curve (AUC) of 0.72 and 0.70, respectively.
Table 2.
Univariate analysis of nonresponse to bevacizumab combination therapy based on AGT promoter methylation in the training cohort. OR, Odds ratio; CI, Confidence interval.
| AGT promoter methylation | Nonresponse | ||
|---|---|---|---|
| Twofold decrease | OR (95% CI) | P‐value | AUC |
| CpG site 1 | 2.93 (1.44–5.94) | 0.003 | 0.72 |
| CpG site 2 | 2.73 (1.28–5.83) | 0.01 | 0.69 |
| CpG site 3 | 2.18 (1.17–4.04) | 0.01 | 0.67 |
| CpG site 4 | 2.04 (1.14–3.66) | 0.02 | 0.66 |
| Mean CpG sites 1–4 | 3.01 (1.41–6.44) | 0.004 | 0.70 |
Based on the high degree of intercorrelation of the four CpG sites and to establish the most technical robust biomarker, the mean DNA methylation level of CpG sites 1–4 was chosen, in preference to CpG site 1, for the final predictive model for nonresponse. Using this predictor, a lower mean methylation level of CpG sites 1–4 was significantly associated with nonresponse to bevacizumab combination therapy (twofold decrease: OR = 3.01; 95% CI: 1.41–6.44; P = 0.004).
In multivariate analysis, clinical prognostic factors (performance status, neurocognitive deficit, corticosteroid use, and multifocal disease) were not associated with response (P > 0.30 for all factors). However, lower mean methylation of CpG sites 1–4 was significantly and independently associated with nonresponse (P = 0.005).
3.4. Establishment of a clinical useable predictor for nonresponse
To establish a model for clinical decision making, a biomarker cut point of AGT promoter methylation was determined in the training cohort. Considering the difficulties and limitations in response assessment and to increase the likelihood of identifying patients not responding to bevacizumab combination therapy, we prioritized a high specificity in preference to a high sensitivity for our model. Accordingly, our predictive model with a sensitivity of 37% at a specificity of 96% was established. Based on this model, the threshold for a positive test and being considered as a ‘nonresponder’ was set to below 12% of mean AGT promoter methylation of CpG sites 1–4. Table 3 shows the distribution of responders and nonresponders according to the predictor. Using this predictive model, 95% (19 of 20) of the patients that were predicted nonresponders according to our model were true nonresponders. The results of the internal cross‐validation confirmed the model. In clinical practice, this means that a patient with nonmethylated AGT promoter (methylation < 12%) will most likely not achieve response to bevacizumab combination therapy.
Table 3.
The predictor for nonresponse to bevacizumab combination therapy applied to the training cohort.
| Training cohort | Predictor for nonresponse | |
|---|---|---|
|
Negative test for nonresponse Methylated (≥ 12%) |
Positive test for nonresponse Nonmethylated (< 12%) |
|
| Nonresponse | 32 (56%) | 19 (95%) |
| Response | 25 (44%) | 1 (5%) |
| Total | 57 | 20 |
3.5. Validation of the predictor for nonresponse
To validate our predictive model for not responding to bevacizumab combination treatment, DNA methylation of the four CpG sites in the AGT promoter was investigated in the validation cohort of similar size. Importantly, application of the predictor with the cut point of 12% methylation established in the training cohort showed that AGT promoter demethylation was significantly associated with nonresponse in the validation cohort (P = 0.037). In the training cohort, 95% (19 of 20) of the patients with nonmethylated promoter were nonresponsive (Table 3). Similarly, in the validation cohort, 88% (15 of 17) of patients with nonmethylated promoter were nonresponsive (Table 4). Considering both cohorts of the study, 23% (37 of 159) of the patients were, according to our predictive model, predicted not to respond to treatment. Of these individuals, 92% (34 of 37) patients were correctly predicted nonresponders.
Table 4.
The predictor for nonresponse to bevacizumab combination therapy applied to the validation cohort.
| Validation cohort | Predictor for nonresponse | |
|---|---|---|
|
Negative test for nonresponse Methylated (≥ 12%) |
Positive test for nonresponse Nonmethylated (< 12%) |
|
| Nonresponse | 40 (62%) | 15 (88%) |
| Response | 25 (38%) | 2 (12%) |
| Total | 65 | 17 |
4. Discussion
In this study of 77 recurrent glioblastoma patients, a low methylation of the CEBP binding region in the AGT promoter was found significantly associated with a lack of response to bevacizumab combination treatment. A clinically useful model able to predict whether a patient is likely or not to achieve response to bevacizumab combination therapy was established and validated successfully in a cohort of 82 recurrent glioblastoma patients.
Our results suggest that the methylation status of the AGT promoter can be used to identify recurrent glioblastoma patients, with a specificity of 96%, who will not respond to bevacizumab combination therapy. Applying this predictor in clinical practice, our data suggest that nearly one of four recurrent glioblastoma patients can be spared from an ineffective and potentially toxic treatment.
In the present study, response to bevacizumab combination treatment was found significantly associated with an improved survival. This difference in survival between responding and nonresponding patients was relatively modest for patients who were categorized as having a poor prognosis, while the difference was pronounced for patients with a favorable prognostic profile. This suggests that patients of the poor prognostic group, irrespective of response, obtain limited survival benefit of bevacizumab combination therapy.
The relatively low sensitivity of our predictive model suggests that AGT promoter methylation at the time of glioblastoma diagnosis is not the only factor influencing lack of response to bevacizumab combination treatment. One explanatory factor is that DNA demethylation of the CEBP region in the AGT promoter may occur from the time of glioblastoma diagnosis to the time of glioblastoma recurrence as a result of continues AGT gene activation (Wang et al., 2014). Such stimulatory signals of AGT activation include the pro‐inflammatory cytokine interleukin‐6 and glucocorticosteroids (Wang et al., 2014), which both have been reported to predict poor outcome in bevacizumab‐treated cancer patients (Duerinck et al., 2015; Noonan et al., 2018; Urup et al., 2016a). In addition, the most prominent transcription factors known to induce angiotensinogen transcription are CEBP and STAT3 (Jain et al., 2007; Wang et al., 2014). In glioblastoma, these two transcriptions factors have been linked to hypoxia, necrosis, and induction of the mesenchymal subtype (Carro et al., 2010; Cooper et al., 2012). Consequently, DNA methylation levels of the AGT promoter could be reduced during tumor progression as a result of hypoxia/necrosis, inflammation, or long‐lasting corticosteroid dependency. If this is the case, angiotensinogen promoter methylation status at time of recurrence may more reliably predict treatment response. The change in AGT promoter methylation over time should be investigated further in longitudinal studies.
In support of our results, high protein expression of angiotensinogen in tumors from patients with metastatic colorectal cancer has been found associated with a poor response to bevacizumab combination therapy (Martin et al., 2014). Accordingly, our findings may be applicable to other solid tumors. In addition, preclinical studies have shown that angiotensin II‐induced remodeling of the tumor microenvironment promotes resistance to anti‐VEGF‐induced vascular normalization (Stylianopoulos and Jain, 2013). Furthermore, increased activity of the local brain renin–angiotensin system through angiotensin II signaling has shown to cause dysregulation of cerebral blood flow by promoting cerebrovascular remodeling, vascular inflammation, and oxidative stress (Kazama et al., 2004; Paul et al., 2006; Wei et al., 2007). Collectively, increased activity of local brain renin–angiotensin system may promote a dysregulated tumor vasculature resistant to bevacizumab‐induced vascular normalization. This could explain the findings of retrospective clinical studies, suggesting an improved benefit in terms of survival when bevacizumab is combined with an angiotensin II inhibitor (Johansen et al., 2018; Levin et al., 2017). Prospective clinical trials are needed to evaluate the efficacy of this combination regimen and to what extent AGT promoter methylation is associated with efficacy in recurrent glioblastoma patients.
This study was limited by its retrospective design, including the lack of isocitrate dehydrogenase 1 (IDH1) mutation status and O6‐methylguanine‐DNA methyltransferase (MGMT) gene promoter methylation status. However, IDH1 status and MGMT status have previously not been associated with bevacizumab efficacy (Taal et al., 2014; Wick et al., 2017), suggesting that these biomarkers would not have had an impact on the results of this study.
In summary, we found promoter methylation of the gene encoding angiotensinogen as being associated with response to bevacizumab combination therapy in recurrent glioblastoma patients. Based on these findings, we established and validated a model which in clinical practice can predict patients who will not achieve response to bevacizumab combination treatment. We hypothesize that this model, in a favorable prognostic group of recurrent glioblastoma patients, has the potential to identify patients who will benefit from bevacizumab combination therapy in terms of durable response and improved survival. This hypothesis will be tested in a prospective clinical study.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
TU, LG, UL, and HSP designed the study, and HSP coordinated it. VAL, HSP, and TU performed the RANO response evaluation. MJSL and TU analyzed the data with assistance from IJC. HB performed the histopathological evaluations. LG, KK, and KG performed the pyrosequencing analysis. TU, SRM, and LG prepared the figures and tables and wrote the manuscript with input from HSP and UL. All authors revised and approved the final version of the manuscript.
Supporting information
Fig. S1. REMARK diagram for the training cohort.
Fig. S2. REMARK diagram for the validation cohort.
Fig. S3. Correlation analyses for all combinations of DNA methylation levels of the four CpG sites analyzed in the AGT promoter and mean CpG sites 1‐4.
Table S1. Patient characteristics and outcome to bevacizumab combination therapy in the training cohort and the validation cohort.
Acknowledgements
The study was supported by Rigshospitalet, the Danish Cancer Society, Doctor Sophus Friis and Wife Olga Friis’ grant, and I.M. Daehnfeldt Foundation. Genentech funded the microdissection and DNA extraction of material from the training cohort. Genentech was not involved in the interpretation of the results or presentation of the data of this study.
References
- Batchelor TT, Gerstner ER, Emblem KE, Duda DG, Kalpathy‐Cramer J, Snuderl M, Ancukiewicz M, Polaskova P, Pinho MC, Jennings D et al (2013) Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc Natl Acad Sci USA 110, 19059–19064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgarten P, Blank AE, Franz K, Hattingen E, Dunst M, Zeiner P, Hoffmann K, Bahr O, Mader L, Goeppert B et al (2016) Differential expression of vascular endothelial growth factor A, its receptors VEGFR‐1, ‐2, and ‐3 and co‐receptors neuropilin‐1 and ‐2 does not predict bevacizumab response in human astrocytomas. Neuro Oncol 18, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H et al (2010) The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper LA, Gutman DA, Chisolm C, Appin C, Kong J, Rong Y, Kurc T, Van Meir EG, Saltz JH, Moreno CS et al (2012) The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. Am J Pathol 180, 2108–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerinck J, Clement PM, Bouttens F, Andre C, Neyns B, Staelens Y, Van Fraeyenhove F, Baurain JF, Luce S, D'Hondt L et al (2015) Patient outcome in the Belgian medical need program on bevacizumab for recurrent glioblastoma. J Neurol 262, 742–751. [DOI] [PubMed] [Google Scholar]
- Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R et al (2009) Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27, 4733–4740. [DOI] [PubMed] [Google Scholar]
- Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D and Jain RK (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91, 1071–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselbalch B, Lassen U, Hansen S, Holmberg M, Sorensen M, Kosteljanetz M, Broholm H, Stockhausen MT and Poulsen HS (2010) Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: a phase II trial. Neuro Oncol 12, 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson R, Asklund T and Poulsen HS (2011) Impact of therapy on quality of life, neurocognitive function and their correlates in glioblastoma multiforme: a review. J Neurooncol 104, 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RY, Rahman R, Ballman KV, Felten SJ, Anderson SK, Ellingson BM, Nayak L, Lee EQ, Abrey LE, Galanis E et al (2016) The impact of T2/FLAIR evaluation per RANO criteria on response assessment of recurrent glioblastoma patients treated with bevacizumab. Clin Cancer Res 22, 575–581. [DOI] [PubMed] [Google Scholar]
- Jain S, Li Y, Patil S and Kumar A (2007) HNF‐1alpha plays an important role in IL‐6‐induced expression of the human angiotensinogen gene. Am J Physiol Cell Physiol 293, C401–C410. [DOI] [PubMed] [Google Scholar]
- Jakobsen JN, Urup T, Grunnet K, Toft A, Johansen MD, Poulsen SH, Christensen IJ, Muhic A and Poulsen HS (2018) Toxicity and efficacy of lomustine and bevacizumab in recurrent glioblastoma patients. J Neurooncol 137, 439–446. [DOI] [PubMed] [Google Scholar]
- Johansen MD, Urup T, Holst CB, Christensen IJ, Grunnet K, Lassen U, Friis S and Poulsen HS (2018) Outcome of bevacizumab therapy in patients with recurrent glioblastoma treated with angiotensin system inhibitors. Cancer Invest 36, 512–519. [DOI] [PubMed] [Google Scholar]
- Juillerat‐Jeanneret L, Celerier J, Chapuis Bernasconi C, Nguyen G, Wostl W, Maerki HP, Janzer RC, Corvol P and Gasc JM (2004) Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br J Cancer 90, 1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA and Iadecola C (2004) Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase‐derived radicals. Circ Res 95, 1019–1026. [DOI] [PubMed] [Google Scholar]
- Levin VA, Chan J, Datta M, Yee JL and Jain RK (2017) Effect of angiotensin system inhibitors on survival in newly diagnosed glioma patients and recurrent glioblastoma patients receiving chemotherapy and/or bevacizumab. J Neurooncol 134, 325–330. [DOI] [PubMed] [Google Scholar]
- Martin P, Noonan S, Mullen MP, Scaife C, Tosetto M, Nolan B, Wynne K, Hyland J, Sheahan K, Elia G et al (2014) Predicting response to vascular endothelial growth factor inhibitor and chemotherapy in metastatic colorectal cancer. BMC Cancer 14, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan SA, Morrissey ME, Martin P, Biniecka M, O'Meachair S, Maguire A, Tosetto M, Nolan B, Hyland J, Sheahan K et al (2018) Tumour vasculature immaturity, oxidative damage and systemic inflammation stratify survival of colorectal cancer patients on bevacizumab treatment. Oncotarget 9, 10536–10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul M, Poyan Mehr A and Kreutz R (2006) Physiology of local renin‐angiotensin systems. Physiol Rev 86, 747–803. [DOI] [PubMed] [Google Scholar]
- Poulsen HS, Grunnet K, Sorensen M, Olsen P, Hasselbalch B, Nelausen K, Kosteljanetz M and Lassen U (2009) Bevacizumab plus irinotecan in the treatment patients with progressive recurrent malignant brain tumours. Acta Oncol 48, 52–58. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352, 987–996. [DOI] [PubMed] [Google Scholar]
- Stylianopoulos T and Jain RK (2013) Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci USA 110, 18632–18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taal W, Oosterkamp HM, Walenkamp AM, Dubbink HJ, Beerepoot LV, Hanse MC, Buter J, Honkoop AH, Boerman D, de Vos FY et al (2014) Single‐agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol 15, 943–953. [DOI] [PubMed] [Google Scholar]
- Urup T, Dahlrot RH, Grunnet K, Christensen IJ, Michaelsen SR, Toft A, Larsen VA, Broholm H, Kosteljanetz M, Hansen S et al (2016a) Development and validation of a prognostic model for recurrent glioblastoma patients treated with bevacizumab and irinotecan. Acta Oncol 55, 418–422. [DOI] [PubMed] [Google Scholar]
- Urup T, Michaelsen SR, Olsen LR, Toft A, Christensen IJ, Grunnet K, Winther O, Broholm H, Kosteljanetz M, Issazadeh‐Navikas S et al (2016b) Angiotensinogen and HLA class II predict bevacizumab response in recurrent glioblastoma patients. Mol Oncol 10, 1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urup T, Staunstrup LM, Michaelsen SR, Vitting‐Seerup K, Bennedbaek M, Toft A, Olsen LR, Jonson L, Issazadeh‐Navikas S, Broholm H et al (2017) Transcriptional changes induced by bevacizumab combination therapy in responding and non‐responding recurrent glioblastoma patients. BMC Cancer 17, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo‐Ardila DL, Fifis T, Burrell LM, Walsh K and Christophi C (2018) Renin‐angiotensin inhibitors reprogram tumor immune microenvironment: a comprehensive view of the influences on anti‐tumor immunity. Oncotarget 9, 35500–35511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Demura M, Cheng Y, Zhu A, Karashima S, Yoneda T, Demura Y, Maeda Y, Namiki M, Ono K et al (2014) Dynamic CCAAT/enhancer binding protein‐associated changes of DNA methylation in the angiotensinogen gene. Hypertension 63, 281–288. [DOI] [PubMed] [Google Scholar]
- Wei Y, Whaley‐Connell AT, Chen K, Habibi J, Uptergrove GM, Clark SE, Stump CS, Ferrario CM and Sowers JR (2007) NADPH oxidase contributes to vascular inflammation, insulin resistance, and remodeling in the transgenic (mRen2) rat. Hypertension 50, 384–391. [DOI] [PubMed] [Google Scholar]
- Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, Degroot J, Wick W, Gilbert MR, Lassman AB et al (2010) Updated response assessment criteria for high‐grade gliomas: Response Assessment in Neuro‐Oncology working group. J Clin Oncol 28, 1963–1972. [DOI] [PubMed] [Google Scholar]
- Wick W, Gorlia T, Bendszus M, Taphoorn M, Sahm F, Harting I, Brandes AA, Taal W, Domont J, Idbaih A et al (2017) Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med 377, 1954–1963. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. REMARK diagram for the training cohort.
Fig. S2. REMARK diagram for the validation cohort.
Fig. S3. Correlation analyses for all combinations of DNA methylation levels of the four CpG sites analyzed in the AGT promoter and mean CpG sites 1‐4.
Table S1. Patient characteristics and outcome to bevacizumab combination therapy in the training cohort and the validation cohort.