

Abstract
The presence of immune cells in the tumor microenvironment has been associated with response to immunotherapies across several cancer types, including melanoma. Despite its therapeutic relevance, characterization of the melanoma immune microenvironments remains insufficiently explored. To distinguish the immune microenvironment in a cohort of 180 metastatic melanoma clinical specimens, we developed a method using promoter CpG methylation of immune cell type‐specific genes extracted from genome‐wide methylation arrays. Unsupervised clustering identified three immune methylation clusters with varying levels of immune CpG methylation that are related to patient survival. Matching protein and gene expression data further corroborated the identified epigenetic characterization. Exploration of the possible immune exclusion mechanisms at play revealed likely dependency on MITF protein level and PTEN loss‐of‐function events for melanomas unresponsive to immunotherapies (immune‐low). To understand whether melanoma tumors resemble other solid tumors in terms of immune methylation characteristics, we explored 15 different solid tumor cohorts from TCGA. Low‐dimensional projection based on immune cell type‐specific methylation revealed grouping of the solid tumors in line with melanoma immune methylation clusters rather than tumor types. Association of survival outcome with immune cell type‐specific methylation differed across tumor and cell types. However, in melanomas immune cell type‐specific methylation was associated with inferior patient survival. Exploration of the immune methylation patterns in a pan‐cancer context suggested that specific immune microenvironments might occur across the cancer spectrum. Together, our findings underscore the existence of diverse immune microenvironments, which may be informative for future immunotherapeutic applications.
Keywords: DNA methylation, immune cells, melanoma, pan‐cancer, tumor microenvironment
The importance of the tumor immune microenvironment has been highlighted in multiple cancers, but information regarding the contribution of individual immune cell types is lacking for melanoma. In this study, we have sought to fill the gap by identifying distinct DNA methylation patterns for specifc immune cells. These were corroborated by other molecular correlates and found to be shared across other types of solid tomors.
Abbreviations
- Bp
base pair
- CD
cluster of differentiation
- CNS
central nervous system
- CpG
cytosine phosphate guanine
- DHS
DNase hypersensitivity sites
- DMFS
distant metastasis‐free survival
- DSS
disease‐specific survival
- FDR
false discovery rate
- GDSC
genomics of drug sensitivity
- ICI
immune checkpoint inhibitor
- IHC
immunohistochemical
- MM
metastatic melanoma
- mRNA
messenger RNA
- NK cell
natural killer cell
- SCNA
somatic copy number alteration
- TCGA
The Cancer Genome Atlas
- TIL
tumor‐infiltrating lymphocyte
- TLS
tertiary lymphoid structures
- tSNE
t‐distributed stochastic neighbor embedding
- TSS
transcription start site
1. Introduction
Metastatic cutaneous melanoma is the most aggressive form of skin malignancy with poor patient survival (Surveillance, 2019). Based on advancement in targeted therapies and immunotherapies, survival outcome has, however, increased considerably (Chapman et al., 2011; McDermott et al., 2014; Robert et al., 2014). Immune checkpoint inhibitors (ICIs), such as CTLA4 and PD1/PDL1 blockade, that reactivate tumor‐associated effector T cells have demonstrated dramatic clinical efficacy and provide long‐term survival in a substantial fraction of patients (Hodi et al., 2010; Weber et al., 2015, 2017; Wolchok et al., 2017). Multiple studies have identified predictive biomarkers for therapy response that include activity of intratumor immunological effector cells (Riaz et al., 2017; Yuan et al., 2011), tumor mutational burden (Riaz et al., 2017; Van Allen et al., 2015), mismatch repair deficiency (Le et al., 2017b; Viale et al., 2017), tumor aneuploidy (Davoli et al., 2017), PDL1 levels (Long et al., 2016; Patel and Kurzrock, 2015), and intestinal microbiota (Gopalakrishnan et al., 2018; Matson et al., 2018). However, the still large fraction of nonresponding patients (Hodi et al., 2010; Wolchok et al., 2017) warrants further understanding of various factors determining primary resistance to ICIs, including the role of the tumor microenvironment.
The tumor microenvironment is a complex mixture of malignant and nonmalignant cells that includes immune cells. Clearly, the microenvironment of melanoma tumors, especially immune cells, plays an important role in determining progression (Passarelli et al., 2017; Tucci et al., 2018; Villanueva and Herlyn, 2008). Single‐cell RNA sequencing of melanoma tumors has confirmed that a complex mixture of immune T‐cell subtypes exists within a tumor (Jerby‐Arnon et al., 2018; Sade‐Feldman et al., 2018). In addition, bulk transcriptomic analyses have been utilized to explore the immune microenvironment from melanoma tumors, demonstrating that tumors with increased expression of genes involved in a diverse range of immune systems have increased survival (Angelova et al., 2015; Bindea et al., 2013). Current methods to decipher the immune landscape in tumors are based on the transcriptome (Angelova et al., 2015; Bindea et al., 2013). However, global DNA methylation patterns are largely associated with cellular lineage and show higher distinction between cellular lineages than mRNA expression in blood and skin lineages (Bock et al., 2012). Moreover, DNA methylation has been demonstrated to resolve cell of origin of peripheral blood cells (Houseman et al., 2012) and cell‐free DNA (Moss et al., 2018), and was introduced as a complementary approach to classify central nervous system (CNS) tumors (Capper et al., 2018). Nevertheless, DNA methylation has not been extensively exploited to determine the immune lineages occurring in the microenvironment of melanoma. Therefore, we created DNA methylation signatures that reflect a wide range of tumor‐associated immune cell subsets. These signatures grouped melanoma tumors into three clusters with distinct clinical and molecular properties that were re‐identified in independent data. Further molecular exploration revealed several immune exclusion mechanisms activated in tumors. Moreover, an immune‐rich microenvironment consisting of a broad range of immune cells was observed across several cancer types. In conclusion, DNA immune methylation signatures harbor significant clinical and biological information.
2. Materials and methods
2.1. Patient sample information
The study was approved by the Regional Ethics Committee at Lund University (Dnr. 191/2007 and 101/2013). The experiments were undertaken with the understanding and written consent of each subject. The study methodologies conformed to the standards set by the Declaration of Helsinki. The sample cohort consisted of 214 melanoma tumors and was obtained at the Department of Surgery at Skåne University Hospital. This is a historic cohort collected before the era of ICIs and targeted therapies.
2.2. Sample preparation and bisulfite conversion
DNA extraction from patient samples was done as described previously (Harbst et al., 2014) and bisulfite‐converted using EZ DNA Methylation Kit (Zymo Research Europe, Freiburg, Germany) according to the manufacturer's instructions and analyzed using Illumina Infinium MethylationEPIC BeadChip array. Methylation data are available in Gene Expression Omnibus with accession number http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144487.
2.3. Immune cell type‐specific CpG set for metastatic melanoma
We selected two immune cell type‐associated gene signatures (Angelova et al., 2015; Tirosh et al., 2016) to identify immune cell types in melanoma tumors. Next, we combined both gene signatures, and after removing conflicting genes (genes belonging to different cell types in each signature), we obtained a total of 920 genes associated with 30 immune cell subsets. Next, we selected corresponding promoter CpGs (< 1500 bp from TSS) of these genes, since their association with the transcriptional regulation of a gene is well known and thus more likely to be lineage‐specific. After mapping the gene signatures with the list of CpG probes shared between 450K and EPIC platforms, we found 4228 promoter CpGs belonging to 744 genes and associated with 30 immune cell subsets.
The CpGs were then further filtered using the following criteria:
Selecting the most differentially methylated CpGs across reference immune cells: We dichotomized the β‐values into robust methylation bins, as unmethylated (β < 0.3) and methylated (β ≥ 0.3). We then selected CpGs that have significantly different proportions of methylated and unmethylated signals among the reference immune cells using Fisher's exact test and an FDR < 0.01.
High methylation in nonimmune cells: To ensure that any methylation difference we observe is likely coming from immune cells and not from other cells present in the microenvironment, we further shortlisted CpGs with a high percentage and level of methylation (β > 0.7 in > 98% of samples) among nonimmune normal cells and melanoma cell lines.
Forming gene–CpG pairs: We wanted to ensure that any single gene is not over‐represented through the presence of multiple CpGs. We therefore selected the most significant CpG for each gene from Fisher's exact test in step 1.
The selection processes resulted in 67 gene–CpG pairs belonging to 21 immune cell populations.
2.4. Immune cell type‐specific CpG set for nonmelanoma TCGA pan‐cancer cohorts
The CpG selection procedure was identical to the process we followed for metastatic melanoma (MM) tumor cohorts except step 2. At step 2, we filtered the immune CpGs against methylation profiles of matching tumor cell lines from Genomics of Drug Sensitivity (GDSC) database and selected CpGs for further analyses if they had shown high level and percentage of methylation in the tumor cell lines (β > 0.7 in > 90% of samples). Here, we had to relax the sample selection criteria since for a number of GDSC tumor types a minority of cell lines were displaying a methylation pattern that deviated from the majority of cell lines of the tumor type.
2.5. Immune methylation centroid‐based classification
Immune methylation centroid‐based classification was performed by correlating sample methylation profiles across centroid CpGs to each cluster centroid (Table S3) and then selecting the cluster that reported highest correlation (τKendall). If no cluster displayed correlation ≥ 0.3, then the sample was annotated unclassified.
2.6. Immune cell type‐specific methylation score calculation
Immune cell type‐specific methylation scores were calculated using the matching CpGs from the 67 CpG set for MM cohorts and by taking median methylation value of all CpGs belonging to a specific immune cell type. For non‐MM solid tumor cohorts, the process is almost identical to MM but here cohort‐specific immune CpG sets were used for score calculation.
2.7. PTEN promoter hypermethylation calculation
We selected promoter CpGs for the PTEN gene that are located at the DNase hypersensitivity sites (DHS) of the promoter, as the PTEN promoter contained a complex set of CpGs on the Illumina 450K array. Next, we called PTEN hypermethylation in tumors if more than 10% of the DHS promoter CpGs are hypermethylated (β > 0.7) and median β‐value for all PTEN promoter CpGs for the corresponding tumor is above 0.5.
2.8. Statistical analyses and calculations for immune cell type methylation and gene expression scores
All statistical and bioinformatics analyses were performed in R. For comparing numerical values, we used Spearman and Kendall correlation. Comparisons between two groups were performed using Mann–Whitney U‐test/Wilcoxon rank‐sum test, and for more than two groups, we used Kruskal–Wallis test. For survival analyses, we used univariate and multivariate Cox regression.
Both methylation and gene expression scores were calculated by median methylation/expression of CpGs/genes belonging to each immune cell type. We used median‐centered gene expression for calculating expression scores in Lund and TCGA cohort analyses; noncentered gene expression scores were used in pan‐cancer analysis of correlation between methylation and gene expression.
3. Results
3.1. DNA methylation‐based immune profiling of metastatic melanoma tumors
To explore the DNA methylation immune landscape in MM tumors, we first curated two previously described immune gene expression signatures (Angelova et al., 2015; Tirosh et al., 2016) (Fig. 1A) to identify corresponding gene promoter CpGs. Next, we identified those CpGs that were most differentially methylated across seven normal peripheral blood‐derived immune cell subsets (Reinius et al., 2012), while in parallel showing high methylation in nonimmune cell types in the tumor microenvironment (cancer cell lines and normal stromal cells—dermal fibroblasts, epidermal keratinocytes, and melanocytes) (Lauss et al., 2015). After subsequent selection of the best CpG (CpG showing lowest FDR in Fisher's exact test for differential methylation) for individual genes, we arrived at a final list of 67 CpGs (Table S1). These, we hypothesize, are DNA methylation signatures representative of different immunological cell types (Fig. 1B). Next, we used this CpG set to analyze Illumina EPIC methylation array‐based profiles from 180 MM tumors (the Lund cohort, Table S2).
Fig. 1.

Strategy to identify immune cell type‐specific CpGs. (A) Schematic outline of the strategy to select immune cell type‐associated CpGs by combining Angelova et al. (2015) and Tirosh et al. (2016) signatures. (B) Heatmap of the methylation profiles for the reference cell types (immune, normal, and melanoma cell lines) along the selected 67 CpGs. Color bars on the left indicate the corresponding cell type for the CpG. Color bar on the right indicates the corresponding range for methylation β. Row names indicate corresponding gene names for the CpGs.
3.2. Immune cell type‐associated CpGs reveal melanoma subgroups
Consensus clustering of the Lund cohort using the selected immune cell type‐specific CpGs identified three immune methylation clusters (Fig. S1A). Cluster 1 showed overall lower methylation in the majority of the immune CpGs, whereas Cluster 3 displayed the opposite, with Cluster 2 being intermediate in appearance (Fig. 2A). To explore the observed methylation differences further, we constructed a median methylation score for all CpGs associated with the described immune cell types for each tumor. We found that differences in methylation scores between Clusters 1 and 3 were most pronounced, with 19 out of 21 immune cell types being significant (Fig. 2B, Kruskal–Wallis and post hoc Dunn test; multiple testing corrections across cell types for each comparison individually, FDR < 0.001). The biggest difference in methylation values was observed for central memory CD4+ T cells (absolute Δβ: 1 vs 2: 0.25; 2 vs 3: 0.14; 1 vs 3: 0.39; FDR < 0.001) and effector memory CD8+ T cells (absolute Δβ: 1 vs 2: 0.3; 2 vs 3: 0.18; 1 vs 3: 0.48; FDR < 0.001). Significant differences between Clusters 1 and 2 were observed mostly in the lymphoid lineage and differences between Clusters 2 and 3 followed a similar pattern of significance to that of Clusters 1 and 3, albeit with lower level of methylation difference (absolute Δβ‐value).
Fig. 2.

Immune methylation clusters and association with survival. (A) Selected immune cell type‐specific CpGs for immune methylation clusters across Lund cohort. Color bar on the right indicates the corresponding range for methylation β. Color bars on the left indicate the corresponding cell type for the CpG. Row names indicate corresponding gene names for the CpGs. (B) Balloon plot of absolute methylation difference per immune cell type between clusters. Balloon colors are representative of absolute Δβ, and balloon sizes indicate level of significance for comparisons. Color bar on the bottom left indicates the corresponding range for methylation absolute Δβ. P‐values calculated using Kruskal–Wallis and post hoc Dunn test, and multiple testing corrections performed across cell types for each comparison individually. (C, D) Kaplan–Meier plots of the immune methylation clusters for DMFS (regional lymph node metastases only) and DSS (all metastases except local recurrences), respectively. Numbers at risk and percentages at risk (within brackets) are indicated below each plot. P‐values calculated using log‐rank test. Maximum time to display on x‐axis has been clipped to 10 years.
3.3. Immune methylation clusters are associated with patient survival
To determine whether the identified methylation patterns have prognostic value, we analyzed for differences in the survival outcome of the melanoma patients in Lund cohort. Immune methylation clusters showed significant difference in survival using both distant metastasis‐free survival (DMFS) for regional metastatic cases and melanoma‐specific survival for the entire metastatic cohort (DSS) using univariate Cox regression models (Fig. 2C,D, respectively). Cluster 2 showed significantly decreased DMFS [hazard ratio (HR): 2.45; 95% confidence interval (CI): 1.30–4.61; P = 0.005] compared to Cluster 1. Regarding DSS, we found both Cluster 2 (HR: 2.10; 95% CI: 1.22–3.64; P = 0.008) and Cluster 3 (HR: 2.18; 95% CI: 1.33–3.56; P = 0.002) to be of significantly higher risk when compared to Cluster 1. Furthermore, multivariable Cox regression models with adjustments for age and gender (DMFS) and metastasis type (DSS) reported analogous findings for both DMFS (Fig. S1B) and DSS (Fig. S1C). Overall, using DNA methylation‐based immune profiling we identified three melanoma subgroups with different patient survival.
3.4. Molecular characterization of immune methylation clusters
Next, we set out to explore whether the identified immune methylation clusters were associated with transcriptional differences of the immune cell‐specific genes in matched tumors (n = 179). Overall, transcriptional differences followed the observations on DNA methylation levels (Fig. 3A). Indeed, for most selected immune cell CpGs, moderate‐to‐high negative correlations (−0.2 to −0.5 and below, τKendall) were found between methylation and expression of the corresponding genes, as expected for the promoter CpGs involved in the regulation of gene expression (Fig. S1D). Next, we investigated whether the identified methylation clusters were accompanied by differences in immune cell infiltration using hematoxylin and eosin staining for tumor‐infiltrating lymphocytes (TILs) along with immunohistochemical staining (IHC) for CD3 and CD8 for T cells, and CD68 and CD163 for myeloid cells. Using matched staining data from 127 of 180 MM tumors, we analyzed association between clusters and staining categories (strong infiltration, localized infiltration, and absent for TILs and T cells; absent/low infiltration, nontumor infiltration, and tumor infiltration for myeloid cells). In line with the DNA methylation data, Cluster 1 had the highest percentage of samples with strong and localized infiltration of TILs, CD3+, and CD8+ T cells (P < 0.001; FDR < 0.001) (Fig. 3B). The presence of TILs showed significant association with DSS in the Lund cohort (Fig. S1E). Regarding CD68+ and CD163+ myeloid cells, Cluster 1 had fewer absent/low‐infiltrating samples compared to Cluster 3. Together, these results support that tumor infiltration of lymphocytes and myeloid cells is accurately reflected by DNA methylation.
Fig. 3.
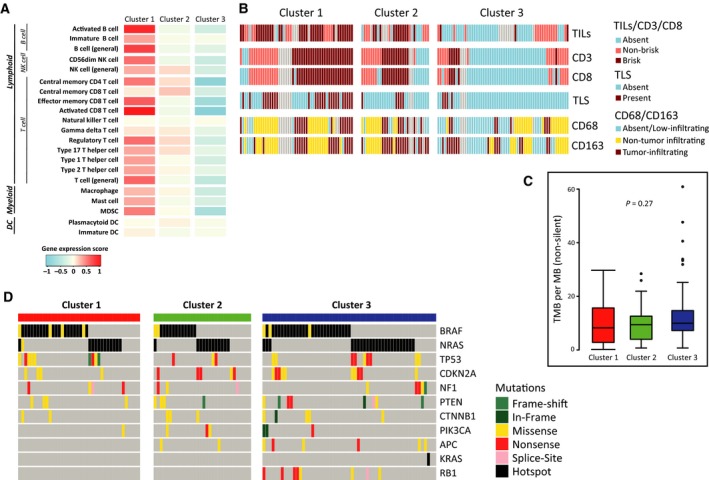
Molecular characteristics of immune methylation clusters. (A) Microarray‐based gene expression score for the selected immune cell types across clusters. Color bar on the bottom indicates the corresponding range for expression score. (B) Distribution of staining categories across clusters for TILs, and CD3+ and CD8+ T cells along with the information on the presence of TLS in the tumor and similar IHC staining for CD68+ and CD163+ myeloid cells. Staining categories are represented as follows: absent (absence of infiltration), nonbrisk (localized infiltration), and brisk (Strong infiltration) for TILs, CD3, and CD8; absent/low‐infiltrating (absence or low infiltration), non‐tumor‐infiltrating (infiltration outside tumor boundary), and tumor‐infiltrating (infiltration inside tumor boundary) for CD68 and CD163. (C) Distribution of nonsilent tumor mutational burden (per MB) across clusters. P‐value calculated using Kruskal–Wallis test. (D) Oncoplot of important melanoma‐associated mutations across clusters. Color bar on the right indicates the mutation category. Hotspot mutations are defined to involve V600, K601 for BRAF, and Q61, G12, and G13 for RAS.
Recently, the existence and importance of tertiary lymphoid structures (TLS) in the melanoma tumors have been reported along with their close relationship with prognosis and response to immunotherapeutic treatments (Cabrita et al., 2020; Helmink et al., 2020). Here, using the matched TLS information for the tumors (Cabrita et al., 2020), we observed significant enrichment of TLSs in Cluster 1 tumors (P < 0.001; FDR < 0.001) (Fig. 3B), indicating the possibility of a successful antitumor immunity in these tumors.
Tumor mutational burden has emerged as an important biomarker for predicting response to ICI treatments (reviewed in Chan et al., 2019). Using mutation data from 1368 cancer genes in 129 of 180 MM tumors from the Lund cohort, we did not observe a relationship between immune methylation clusters and tumor mutational burden or specific melanoma hotspot mutations (Fig. 3C,D, respectively). Additionally, we analyzed association between immune methylation clusters and our previously reported gene expression phenotypes (Cirenajwis et al., 2015) and observed strong association between high‐immune class and low methylation Cluster 1 (Fisher's exact P < 0.001, Table S3).
Overall, the immune methylation clusters are independent of mutational patterns and tumor mutational burden but are confirmed by the presence or absence of immune cells on the mRNA and protein level.
3.5. Analysis of immune exclusion mechanism associated with immune methylation clusters
Several molecular mechanisms have been proposed to explain why certain tumors are able to escape the immune system (Casey et al., 2016; Gettinger et al., 2017; Le et al., 2017a; Peng et al., 2016; Spranger et al., 2015; further reviewed in Spranger and Gajewski, 2018; Trujillo et al. 2018). Clearly, immune methylation Cluster 3 was characterized by an immune‐poor DNA methylation pattern reflected by increased methylation levels of immune cell‐specific CpGs (Fig. 2A). Further, we hypothesized that the differentiation state of the melanoma tumor may determine the ability of Cluster 3 tumors to evade the immune system. Thus, we used matched staining information for 127 of 180 MM tumors for the MITF protein, which is a melanocyte‐specific transcription factor important in melanocyte differentiation and melanoma cell survival (Carreira et al., 2005; Garraway et al., 2005; McGill et al., 2002; reviewed in Levy et al., 2006; Steingrímsson et al., 2004). Overall, 25% of the matched stained tumors were negative for MITF protein. Intriguingly, we found fewer MITF‐negative tumors in the immune‐rich methylation Cluster 1 (Fig. 4A; chi‐square P = 0.06). As MITF‐positive and MITF‐negative tumors are known to harbor different transcriptional programs and presumably represent two distinct melanoma states (Hoek et al., 2006), we thus performed subsequent analyses in MITF‐positive and MITF‐negative tumors separately. In pathway analysis of gene expression data, we found that MITF‐positive tumors showed enrichment for c‐MYC (MYC) target genes and DNA repair‐associated genes in Cluster 3 (FDR < 0.05); however, such observations were not found for the MITF‐negative group. Upon stratification by MITF status, we observed a significant difference in MYC expression across clusters for MITF‐positive tumors (Fig. 4B, P = 0.001), which was not observed for the combined data (Fig. S2A). Furthermore, we explored the molecular profiles of known immune exclusion‐associated genes (beta‐catenin pathway, PI3K pathway, TP53, reviewed in Spranger and Gajewski (2018)) across clusters. CTNNB1 showed a significant mRNA expression difference across clusters (Fig. 4C, P < 0.001, FDR < 0.05); however, when stratified by MITF status, characteristics similar to MYC were revealed (Fig. S2B, P < 0.001). Together, these observations delineate that MYC and CTNNB1 may act as immune exclusion molecules in MITF‐positive melanoma tumors. Among PI3K‐Akt pathway genes, we observed significant gene expression differences across clusters for PTEN and PIK3R1 (Fig. 4D and Fig. S2C, P = 0.005; FDR < 0.05 and P = 0.03; FDR < 0.05, respectively) irrespective of MITF status. Upon further investigation, we found that PTEN somatic copy number alterations (SCNAs) showed good overlap with PTEN promoter hypermethylation and nonsynonymous mutations for the same tumors. However, PTEN hypermethylation and mutations were found to be mostly exclusive (Fig. 4E, Fig. S2D). Next, we summed up all molecular alteration events of PTEN for each tumor and found significant concordance between PTEN alteration events and corresponding mRNA expression (Fig. S2E). Also, the distribution of tumors harboring any PTEN alteration event varied significantly across clusters (chi‐square P < 0.001) with Cluster 3 showing highest enrichment (Fig. 4F). We further explored the association between PTEN mRNA expression and TILs and observed a trend in difference in terms of PTEN expression between tumors with absent and brisk TILs (Fig. S2F). The association of TILs with molecular events in PTEN was more prominent (Fig. 4E), and a significant difference in terms of TILs was observed between tumors with and without PTEN molecular events (chi‐square P = 0.001). Association between immune methylation clusters and molecular event status in PTEN was also validated in the TCGA MM cohort (Fig. S2G). Together, we observed a known immune exclusion mechanism in the immune‐poor MM tumors displaying a complex pattern with upregulation of PI3K‐Akt pathway by blocking PTEN functioning through molecular alterations and upregulation of MYC and CTNNB1 in MITF‐positive tumors only.
Fig. 4.
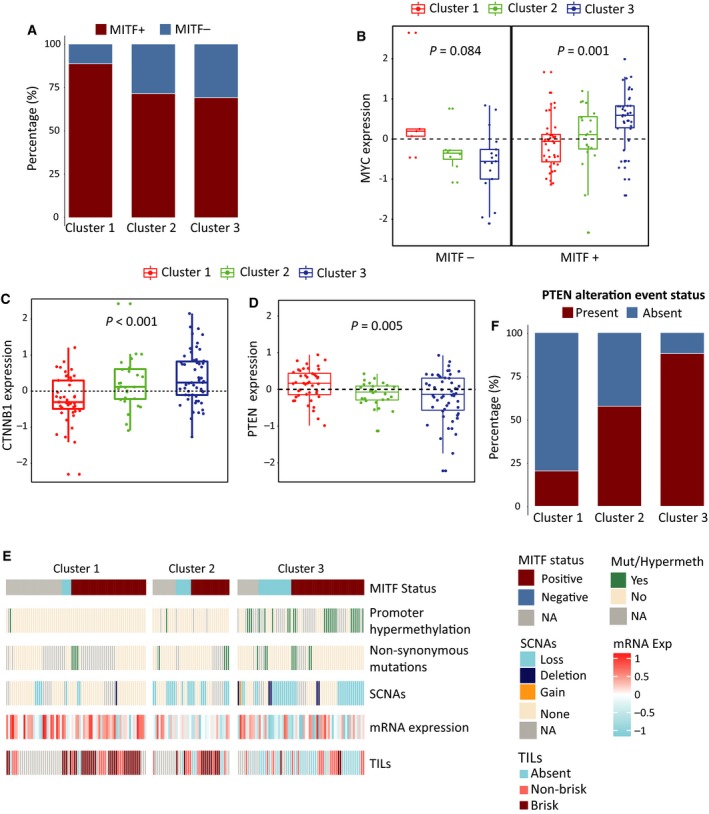
Exploration of immune exclusion mechanisms in the Lund clusters. (A) Barplot showing the distribution of MITF+ and MITF− samples across immune methylation clusters. (B) Boxplot showing distribution of c‐MYC (MYC) mRNA expression for MITF− samples across clusters (left) and same for MITF+ samples (right). P‐value from Kruskal–Wallis test. (C, D) Boxplots showing distribution of β‐catenin (CTNNB1) and PTEN mRNA expression across clusters, respectively. P‐value from Kruskal–Wallis test. (E) Heatmap showing distribution of different molecular alteration events in PTEN along with the corresponding gene expression for samples across clusters and staining information for TILs, further stratified by sample MITF status. Staining categories are represented as follows: absent (absence of infiltration), nonbrisk (localized infiltration), and brisk (Strong infiltration) for TILs. (F) Barplot showing distribution of overall PTEN alteration event status across clusters. A PTEN alteration event is defined as either promoter hypermethylation, nonsynonymous mutation, or copy number loss/deletion.
3.6. Validation of immune methylation clusters in external melanoma cohorts
To confirm the immune methylation clusters, we built methylation‐based centroids to classify samples belonging to the TCGA MM cohort. For this, we selected CpGs from the immune CpG set (Table S1) which showed a significant methylation difference across Lund clusters (Kruskal–Wallis test, FDR < 0.0001). Median methylation values for these significant CpGs (n = 51) were computed for each cluster to form methylation centroids (Table S4). A minor fraction of samples (2.37%) could not be classified (τKendall < 0.3). Immune methylation clusters in TCGA cohort displayed methylation profiles similar to corresponding clusters in the Lund cohort (Fig. S3A). When we grouped both the Lund and TCGA cohorts together using common centroid CpGs and tSNE (van der Maaten and Hinton, 2008) (perplexity = 30; θ = 0.1), we noticed clear segregation between the immune methylation clusters in the first two dimensions, as expected (Fig. 5A, left). Such segregation was not observed when samples were annotated on the basis of the site of metastasis (metastasis type) or cohort (Fig. 5A middle and right, respectively). Additionally, gene expression scores, tumor mutational burden, and mutational patterns displayed identical characteristics for clusters as observed in the Lund cohort (Fig. 5B,C, Fig. S3B, respectively). Importantly, Cluster 3 displayed a significant inferior overall survival (OS, HR: 2.29; 95% CI: 1.54–3.40; P < 0.001) compared to Cluster 1 in TCGA data (Fig. 5D) and remained significant in multivariate analyses with adjustment for metastasis type (P < 0.001, Fig. S3C). Additionally, we analyzed the association between immune methylation clusters in TCGA cohorts and previously reported gene expression classes for the same cohort (Akbani et al., 2015) and observed strong association between Immune class and low methylation Cluster 1 (Fisher's exact P < 0.001, Table S5).
Fig. 5.
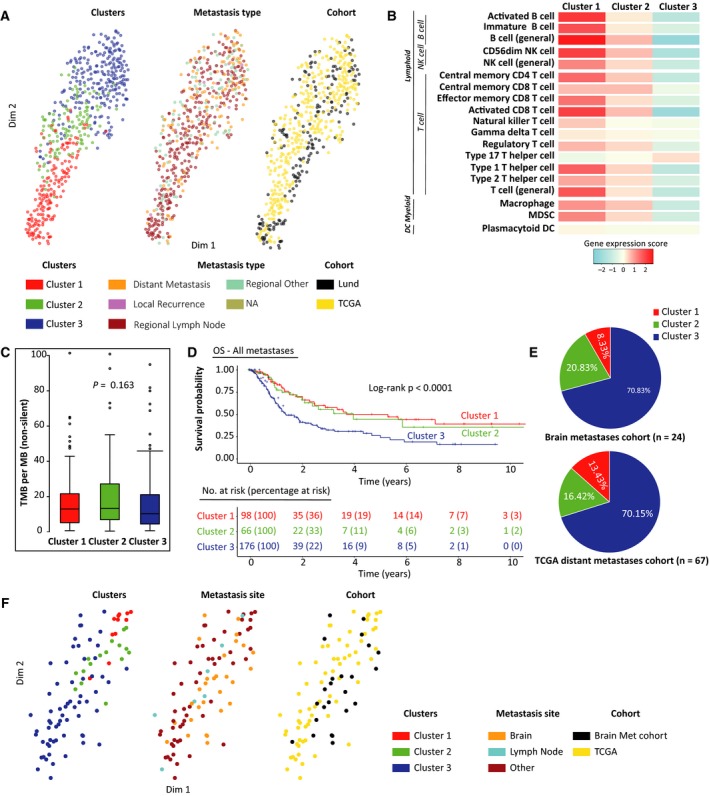
Validation of immune methylation clusters in external melanoma cohorts. (A) tSNE plot of methylation profiles for Lund and TCGA tumors using common centroid CpGs, annotated by clusters (left), metastasis type (site) (middle), and cohort membership (right). (B) RNA‐Seq‐based gene expression scores for the selected immune cell types across TCGA clusters. Color bar on the bottom indicates the corresponding range for expression score. (C) Distribution of nonsilent tumor mutational burden (per MB) across TCGA clusters. P‐value calculated using Kruskal–Wallis test. (D) Kaplan–Meier plot for the TCGA clusters for OS. Numbers at risk and percentages at risk (within brackets) are indicated below the plot. P‐value calculated using log‐rank test. Maximum time to display on x‐axis has been clipped to 10 years. (E) Pie charts of cluster membership distribution for brain metastases cohort (top) and TCGA distant metastases (bottom). (F) tSNE plot of methylation profiles for TCGA distant metastatic tumors and melanoma brain metastases from the brain metastasis cohort using common centroid CpGs, annotated by cluster membership (left), metastasis site (middle), and cohort membership (right).
To further explore immune gene methylation in distant metastases, we classified brain, lymph node, and other metastases from distant sites from TCGA skin cutaneous melanoma (SKCM) cohort (n = 68) and brain metastases (n = 30) from another cohort (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108576) (Orozco et al., 2018) into the immune methylation clusters using methylation‐based centroids. We found that samples in both cohorts were enriched for Cluster 3 irrespective of the metastasis location (Fig. 5E). In TCGA cohort, we did not find any significant association between metastasis location and cluster membership. When both cohorts were analyzed together across the common centroid CpGs and tSNE (perplexity 30; θ = 0.1), observations similar to earlier Lund and TCGA joint analysis were made (Fig. 5F). Together, the immune cell methylation characteristics of the Lund cohort were re‐identified in external data, with similar prognostic implications, and the immune methylation profiles were largely independent of the site of metastasis.
3.7. Implications of the epigenetic immune clusters in a pan‐cancer context
Next, we wanted to investigate whether immune methylation signatures have the same prognostic implications in other cancer types as in melanoma, and to which extent immune environments display shared characteristics among solid tumors. At first, we wanted to dissect the role of specific immune cell lineages across cancer types. To this purpose, we created immune cell methylation scores for MM tumors (both Lund and TCGA cohorts) using previously derived melanoma‐specific immune CpG set. Similar scores for non‐MM TCGA solid tumor cohorts were constructed by identifying immune cell type‐specific CpGs individually for each cohort in a process analogous to MM. Here, the immune CpGs were filtered against cancer type‐specific CpGs using cell lines from the Genomics of Drug Sensitivity (GDSC) database. The resulting methylation scores were highly anticorrelated to the corresponding gene expression scores (computed in the similar manner as methylation scores, using genes instead of the corresponding CpGs), for most immune cell types and tumor cohorts (Fig. 6A). Next, to explore the relations among the immune microenvironments across tumor types, we constructed a tSNE plot using aforementioned methylation scores for 17 immune cell types. When tumor types were annotated on the basis of their tissue of origin, we observed some tumor cohorts were more localized than others (Fig. 6B, Left). Known immune‐deprived tumor types like CNS tumors (lower grade glioma, LGG; and glioblastoma multiforme, GBM) colocalized with a group of similarly immune‐poor tumor types from the developmental gastrointestinal tumors (pancreatic adenocarcinoma, PAAD; and liver hepatocellular carcinoma, LIHC), thus indicating that immune microenvironments of these tumor types bear resemblance despite their diverse tissue of origin. Moreover, the immune methylation Clusters 1 and 3 from MM separated well in this immune microenvironment plot (Fig. 6B, Right). Cluster 1 tumors were found to be colocalizing with a group of diverse tumor types, indicating existence of similar type of immunologically active tumors across the cancer spectrum. Conversely, we found Cluster 3 tumors colocalizing with most CNS and endocrine tumors, indicating that the immune microenvironment of these immune‐poor melanomas likely resembles other immune‐poor tumor types. Next, we analyzed the prognostic implications of immune cell type methylation scores across the cohorts using overall patient survival (OS). For this, we dichotomized the methylation scores into two categories, β > 0.7 as hypermethylated and β ≤ 0.7 as not hypermethylated. We found methylation scores for CD56+ dim NK cells, and activated and effector memory CD8+ T cells, to be significantly prognostic across multiple cancer cohorts including melanoma, with low methylation levels inferring good prognosis in most cancer types (Fig. 6C). Overall, we found low methylation scores of immune cell types to be associated with good outcome for the majority of cell types, particularly for immune cells from the lymphoid lineage, which might reflect the role of the adaptive immune system for an effective response to tumor neoantigens. However, the majority of significant associations with good outcome (adjusted P < 0.1) occurred in only two tumor types, that is, MM and head and neck squamous cell carcinoma (HNSC). Collectively, methylation‐based immune cell signatures inform on prognosis in several solid tumor cohorts, particularly in melanoma.
Fig. 6.
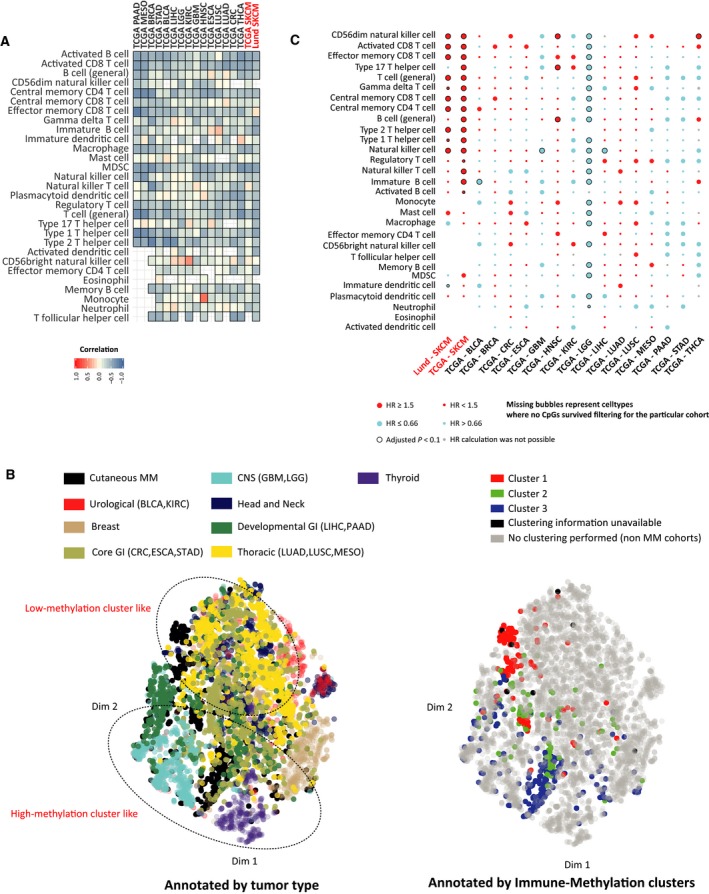
Immune‐specific methylation in pan‐cancer context. (A) Correlation between tissue‐specific immune cell type methylation and corresponding gene expression scores for MM cohorts (Lund and TCGA SKCM) and 15 other TCGA primary cancer cohorts (BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CRC, colorectal carcinoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KIRC, kidney renal clear cell carcinoma; LGG, lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma of lung; PAAD, pancreatic adenocarcinoma; STAD, stomach adenocarcinoma; THCA, thyroid carcinoma). MM cohorts are highlighted in red. Methylation scores for MM cohorts were calculated from the melanoma‐specific 67 immune CpG set, whereas for other tumor types, they were calculated using individual tissue type‐specific immune CpG sets in the similar fashion to MM. (B) tSNE plot of pan‐cancer cohorts based on the previously mentioned methylation scores of common 17 immune cell types; plot is annotated by tissue of origin (left) and immune methylation cluster membership for MM (right). (C) Balloon plot of hazard ratios (HR) for dichotomized immune methylation scores (β > 0.7: hypermethylated, β ≤ 0.7: not hypermethylated) from univariate Cox regression models for each immune cell type and cancer cohorts. MM cohorts are highlighted in red. Balloon colors are indicative of direction of hazard ratios, and sizes indicate value category of HR (large: HR ≥ 1.5 or HR ≤ 0.66; small: HR < 1.5 or HR > 0.66). Additional black circles indicate significance after correction for multiple testing (Benjamini–Hochberg corrected P‐value < 0.1). Gray circles indicate situations where HR calculations were not possible, and missing circles indicate cell types where no CpG survived filtering.
4. Discussion
In this study, we report that immune methylation‐specific signatures harbor important prognostic information and simultaneously inform us about the different tumor immune microenvironments across cancer landscape with a particular focus on MM. DNA methylation has established its role as a major epigenetic driver in cancer progression and development. However, its contribution to define the characteristics of tumor microenvironment remains poorly understood. It has been pointed out recently that DNA hypomethylation promotes immune evasion in the corresponding tumors (Jung et al., 2019). Also, DNA methylation pattern predicting response to ICI treatment for non‐small‐cell lung cancer has been unveiled (Duruisseaux et al., 2018). Thus, further emphasis on DNA methylation for the distinction of different tumor microenvironments is warranted. Our analyses with an immune cell type‐specific CpG set have unraveled diversity in the immune landscape of MM tumors in terms of DNA methylation. We identified three immune methylation clusters that were significantly associated with patient survival and further supported by transcriptomic and immunostaining data. Together, these analyses have hinted at the inverse relationship between immune cell type‐specific methylation and enrichment of the corresponding immune cell types in the tumor microenvironment.
Tumor mutational burden has often been associated with positive clinical response to ICI treatments for several tumor types including melanoma (Goodman et al., 2017; Van Allen et al., 2015, reviewed in Chan et al., 2019). However, we did not observe a clear association of immune methylation clusters with the tumor mutational burden, in line with a previous report (Spranger et al., 2016). This might be due to a universally high immunogenicity of melanoma cells, where immune evasion, as observed in Cluster 3, that is, immune methylation high cluster, can rather be achieved by transcriptional changes (Jerby‐Arnon et al., 2018). Therefore, in this study we additionally explored the possible immune exclusion mechanisms (reviewed in Spranger and Gajewski, 2018) at play for the Cluster 3 tumors in our cohort. Upregulation of PI3K‐Akt pathway and subsequent downregulation of PTEN are a major immune evasion mechanism that has been observed across cancers including melanoma (reviewed in Dituri et al., 2011; Spranger and Gajewski, 2018). However, somatic mutations in PTEN gene have found to be infrequent in melanomas (reviewed in Aguissa‐Touré and Li, 2012); hence, alternative molecular mechanisms such as SCNAs and promoter hypermethylation have been proposed as an alternative mechanism of blocking PTEN functions (Roh et al., 2016; Stahl et al., 2003). Our observations for Cluster 3 tumors further supported the hypothesis that immune‐poor melanomas likely achieve immune evasion through blocking of PTEN functioning through promoter hypermethylation and SCNAs and often in conjunction with one another. The role of MITF as a major transcription factor regulating melanoma progression and development is well established (Carreira et al., 2005; Garraway et al., 2005; McGill et al., 2002; reviewed in Levy et al., 2006; Steingrímsson et al., 2004). MITF‐low melanomas have been shown to be resistant to multiple targeted treatments (Müller et al., 2014). Studies have also shown that decreased expression of melanocyte differentiation antigens through downregulation of MITF could possibly trigger immune evasion in melanomas (Kono et al., 2006; Woods et al., 2014). Nonetheless, exact role of MITF in the immune exclusion mechanism is still unclear. Our analyses with immune‐poor melanomas stratified by MITF immunostaining suggested transcriptional upregulation of β‐catenin‐dependent canonical Wnt signaling pathway along with the upregulation of c‐Myc in the MITF‐positive group. Immune evasion through upregulation of canonical Wnt signaling pathway is common in melanomas (Spranger et al., 2015), and the role of c‐Myc in the immune exclusion process has previously been described (reviewed in Casey et al., 2018). Since β‐catenin‐induced melanomas require functional MITF (Schepsky et al., 2006; Widlund et al., 2002) and loss of MITF expression affects the corresponding expression of c‐Myc (Seoane et al., 2019), thus the role of MITF seems important in distinction of the immune evasion mechanisms in melanoma. As immune‐low tumors respond poorly to immunotherapeutic treatments (Cristescu et al., 2018; Van Allen et al., 2015), hence these results suggest further studies to clarify the role of PTEN and MITF levels in immune exclusion.
Notably, our analyses on distant MMs did not reveal a major difference in the immune microenvironment characterization among tumors from different locations, most notably brain. This is further supported by clinical trials showing efficacy of ICI for intracranial metastases concordant with the extracranial ones and systemic response (Kluger et al., 2019; Margolin et al., 2012; Tawbi et al., 2018). A recent study using mouse models suggested such efficacy is likely due to infiltration of T cells especially CD44+CD62L− effector memory cells from the extracranial sites (Taggart et al., 2018).
Our analyses on a pan‐cancer cohort revealed similarities in the immune microenvironments of tumors from diverse tumor types in terms of immune methylation. Generally, our findings are compatible with the concept of inflammatory and noninflammatory tumor environments (Chakravarthy et al., 2018); however, our pan‐cancer analyses suggest that immune environments carry additional complexity beyond this concept, as has been demonstrated before (Thorsson et al., 2018). Using cohort‐specific immune methylation scores, we observed grouping of MM tumors belonging to the low immune methylation cluster along with tumors from lung and gastrointestinal (GI) tract. Such observation indicates that immune‐rich microenvironments from melanoma have similarities to microenvironment of lung and GI tumors. In this regard, it is interesting that ICI treatments have garnered significant attention for non‐small‐cell lung cancer (NSCLC), which is among the cancer types where such an immune‐rich microenvironment likely occurs (reviewed in Moon et al., 2017). Therefore, in addition to a high mutational burden, such as induced by microsatellite instability (MSI) (Le et al., 2015), an immune‐rich environment may confer improved outcome to immunotherapy agents. Among the different immune cell types, the prognostic value of effector memory T cells could also be established in several tumor types of the pan‐cancer cohort. Memory T cells have received attention recently, as their presence in the tumor microenvironment was associated with clinical response to ICI treatment in melanoma (Gide et al., 2019; Sade‐Feldman et al., 2018).
5. Conclusion
In summary, we explored the immune microenvironment in MM tumors from a DNA methylation perspective. DNA methylation offers several advantages over transcriptomic characterization of mixed cellular environments, primarily due to its higher frequency in terms of differences between cellular lineages compared to gene expression (Bock et al., 2012). Additionally, the biologically limited range of methylation β‐values, being restricted from 0 (unmethylated) to 1 (methylated), facilitates the comparison across experiments and laboratories, whereas transcriptomic data naturally have a higher dynamic range. Also, greater stability of DNA compared to RNA in formalin‐fixed, paraffin‐embedded (FFPE) samples (Okello et al., 2010) makes it easier to work with DNA methylation in archival, historical cohorts.
Our use of a largely untreated historic cohort ensured that the immune cell type methylation patterns are not biased by treatment. Also, the extension of our study to other solid tumors revealed an immune‐rich environment in several other cancer types. The prognostic implications of some innate and adaptive immune cell types were re‐identified in other cancer types. These results point toward a more tailored approach to immunotherapy based on tumor immune microenvironment.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
GJ and KMB conceived the project and received appropriate funding; GJ, ML, and SM designed the study with inputs from JS and MR; KMB, JC, and TZ performed Illumina EPIC array hybridization and initial preprocessing of the data; KI, HO, CI, AC, and KN provided patient tumor materials; SM performed bioinformatic analyses with inputs from ML, GJ, RC, JS, and MR; RC performed IHC analyses; SM, ML, and GJ contributed to the manuscript writing with inputs from KMB, JS, and MR. All authors approved the final draft of the manuscript.
Supporting information
Fig S1. Additional characteristics of Lund immune‐methylation clusters.
Fig S2. Additional characteristics of immune exclusion in the immune‐methylation clusters.
Fig S3. Additional characteristics of TCGA immune‐methylation clusters.
Table S1. List of immune cell type associated CpGs with corresponding gene and associated cell type information.
Table S2. Clinical characteristics for immune‐methylation clusters (Lund cohort).
Table S3. Overlap between Lund gene expression phenotypes and Lund immune‐methylation clusters.
Table S4. List of centroid CpGs with methylation β values for each cluster.
Table S5. Overlap between TCGA gene expression classes and TCGA immune‐methylation clusters.
Appendix S1. Supplementary methods.
Acknowledgements
The study was supported by the Swedish Cancer Society, the Swedish Research Council, BioCARE, the Berta Kamprad Foundation, the King Gustaf V Jubilee Foundation, Mats Paulsson's Foundation, Stefan Paulsson's Foundation, and the governmental funding for healthcare research (ALF). This study was also supported by the Intramural Research Program (IRP) of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, US National Institutes of Health (NIH); the contents of the publication do not necessarily reflect the views of policies of the US Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government. This work utilized the computational resources of the NIH high‐performance computational capabilities Biowulf cluster (http://hpc.nih.gov). We acknowledge contributions to measurement of methylation from The National Cancer Institute Cancer Genomics Research Laboratory (CGR). This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No 641458. MR is financially supported by the Knut and Alice Wallenberg Foundation as part of the National Bioinformatics Infrastructure Sweden at SciLifeLab. We also wish to thank the members of the Swedish Bioinformatics Advisory Program and MELGEN consortium for their valuable suggestions for this study.
References
- Aguissa‐Touré A‐H and Li G (2012) Genetic alterations of PTEN in human melanoma. Cell Mol Life Sci 69, 1475–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, Arachchi H, Arora A, Auman JT, Ayala B et al (2015) Genomic classification of cutaneous melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, Waldner MJ, Bindea G, Mlecnik B, Galon J et al (2015) Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol 16, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf Anna C, Angell H, Fredriksen T, Lafontaine L, Berger A et al (2013) Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. [DOI] [PubMed] [Google Scholar]
- Bock C, Beerman I, Lien W‐H, Smith ZD, Gu H, Boyle P, Gnirke A, Fuchs E, Rossi DJ and Meissner A (2012) DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell 47, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon‐Christersson J et al (2020) Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565. [DOI] [PubMed] [Google Scholar]
- Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE et al (2018) DNA methylation‐based classification of central nervous system tumours. Nature 555, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S, Goodall J, Aksan I, La Rocca SA, Galibert M‐D, Denat L, Larue L and Goding CR (2005) Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature 433, 764–769. [DOI] [PubMed] [Google Scholar]
- Casey SC, Baylot V and Felsher DW (2018) The MYC oncogene is a global regulator of the immune response. Blood 131, 2007–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M et al (2016) MYC regulates the antitumor immune response through CD47 and PD‐L1. Science 352, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy A, Khan L, Bensler NP, Bose P and De Carvalho DD (2018) TGF‐β‐associated extracellular matrix genes link cancer‐associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun 9, 4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A and Peters S (2019) Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol 30, 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirenajwis H, Ekedahl H, Lauss M, Harbst K, Carneiro A, Enoksson J, Rosengren F, Werner‐Hartman L, Törngren T, Kvist A et al (2015) Molecular stratification of metastatic melanoma using gene expression profiling: prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget 6, 12297–12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M et al (2018) Pan‐tumor genomic biomarkers for PD‐1 checkpoint blockade–based immunotherapy. Science 362, eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Uno H, Wooten EC and Elledge SJ (2017) Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 355, eaaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dituri F, Mazzocca A, Giannelli G and Antonaci S (2011) PI3K functions in cancer progression, anticancer immunity and immune evasion by tumors. Clin Dev Immunol 2011, 947858–947858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duruisseaux M, Martínez‐Cardús A, Calleja‐Cervantes ME, Moran S, Castro de Moura M, Davalos V, Piñeyro D, Sanchez‐Cespedes M, Girard N, Brevet M et al (2018) Epigenetic prediction of response to anti‐PD‐1 treatment in non‐small‐cell lung cancer: a multicentre, retrospective analysis. Lancet Respir Med 6, 771–781. [DOI] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J et al (2005) Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117–122. [DOI] [PubMed] [Google Scholar]
- Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, Wurtz A, Dong W, Cai G, Melnick MA et al (2017) Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov 7, 1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, Madore J, Lim SY, Velickovic R, Wongchenko M et al (2019) Distinct immune cell populations define response to anti‐PD‐1 monotherapy and anti‐PD‐1/anti‐CTLA‐4 combined therapy. Cancer Cell 35, 238–255.e236. [DOI] [PubMed] [Google Scholar]
- Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA and Kurzrock R (2017) Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 16, 2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC et al (2018) Gut microbiome modulates response to anti–PD‐1 immunotherapy in melanoma patients. Science 359, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbst K, Lauss M, Cirenajwis H, Winter C, Howlin J, Törngren T, Kvist A, Nodin B, Olsson E, Häkkinen J et al (2014) Molecular and genetic diversity in the metastatic process of melanoma. J Pathol 233, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade‐Feldman M, Blando J, Han G et al (2020) B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek KS, Schlegel NC, Brafford P, Sucker A, Ugurel S, Kumar R, Weber BL, Nathanson KL, Phillips DJ, Herlyn M et al (2006) Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res 19, 290–302. [DOI] [PubMed] [Google Scholar]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK and Kelsey KT (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby‐Arnon L, Shah P, Cuoco MS, Rodman C, Su M‐J, Melms JC, Leeson R, Kanodia A, Mei S, Lin J‐R et al (2018) A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997.e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Kim HS, Kim JY, Sun J‐M, Ahn JS, Ahn M‐J, Park K, Esteller M, Lee S‐H and Choi JK (2019) DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat Commun 10, 4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluger HM, Chiang V, Mahajan A, Zito CR, Sznol M, Tran T, Weiss SA, Cohen JV, Yu J, Hegde U et al (2019) Long‐term survival of patients with melanoma with active brain metastases treated with pembrolizumab on a phase II trial. J Clin Oncol 37, 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono M, Dunn IS, Durda PJ, Butera D, Rose LB, Haggerty TJ, Benson EM and Kurnick JT (2006) Role of the mitogen‐activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res 4, 779. [DOI] [PubMed] [Google Scholar]
- Lauss M, Haq R, Cirenajwis H, Phung B, Harbst K, Staaf J, Rosengren F, Holm K, Aine M, Jirström K et al (2015) Genome‐wide DNA methylation analysis in melanoma reveals the importance of CpG methylation in MITF regulation. J Invest Dermatol 135, 1820–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS et al (2017a) Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 357, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D et al (2015) PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 372, 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy C, Khaled M and Fisher DE (2006) MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med 12, 406–414. [DOI] [PubMed] [Google Scholar]
- Long GV, Larkin J, Ascierto PA, Hodi FS, Rutkowski P, Sileni V, Hassel J, Lebbe C, Pavlick AC, Wagstaff J et al (2016) PD‐L1 expression as a biomarker for nivolumab (NIVO) plus ipilimumab (IPI) and NIVO alone in advanced melanoma (MEL): a pooled analysis. Ann Oncol 27(Suppl_6), 1112PD. [Google Scholar]
- Margolin K, Ernstoff MS, Hamid O, Lawrence D, McDermott D, Puzanov I, Wolchok JD, Clark JI, Sznol M, Logan TF et al (2012) Ipilimumab in patients with melanoma and brain metastases: an open‐label, phase 2 trial. Lancet Oncol 13, 459–465. [DOI] [PubMed] [Google Scholar]
- Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M‐L, Luke JJ and Gajewski TF (2018) The commensal microbiome is associated with anti–PD‐1 efficacy in metastatic melanoma patients. Science 359, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott D, Lebbé C, Hodi FS, Maio M, Weber JS, Wolchok JD, Thompson JA and Balch CM (2014) Durable benefit and the potential for long‐term survival with immunotherapy in advanced melanoma. Cancer Treat Rev 40, 1056–1064. [DOI] [PubMed] [Google Scholar]
- McGill GG, Horstmann M, Widlund HR, Du J, Motyckova G, Nishimura EK, Lin Y‐L, Ramaswamy S, Avery W, Ding H‐F et al (2002) Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 109, 707–718. [DOI] [PubMed] [Google Scholar]
- Moon EK, Langer CJ and Albelda SM (2017) The era of checkpoint blockade in lung cancer: taking the brakes off the immune system. Ann Am Thorac Soc 14, 1248–1260. [DOI] [PubMed] [Google Scholar]
- Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, Samet Y, Maoz M, Druid H, Arner P et al (2018) Comprehensive human cell‐type methylation atlas reveals origins of circulating cell‐free DNA in health and disease. Nat Commun 9, 5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, Kong X, Possik PA, Cornelissen‐Steijger PDM, Foppen MHG et al (2014) Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 5, 5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okello JBA, Zurek J, Devault AM, Kuch M, Okwi AL, Sewankambo NK, Bimenya GS, Poinar D and Poinar HN (2010) Comparison of methods in the recovery of nucleic acids from archival formalin‐fixed paraffin‐embedded autopsy tissues. Anal Biochem 400, 110–117. [DOI] [PubMed] [Google Scholar]
- Orozco JIJ, Knijnenburg TA, Manughian‐Peter AO, Salomon MP, Barkhoudarian G, Jalas JR, Wilmott JS, Hothi P, Wang X, Takasumi Y et al (2018) Epigenetic profiling for the molecular classification of metastatic brain tumors. Nat Commun 9, 4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passarelli A, Mannavola F, Stucci LS, Tucci M and Silvestris F (2017) Immune system and melanoma biology: a balance between immunosurveillance and immune escape. Oncotarget 8, 106132–106142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP and Kurzrock R (2015) PD‐L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 14, 847. [DOI] [PubMed] [Google Scholar]
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X et al (2016) Loss of PTEN promotes resistance to T cell‐mediated immunotherapy. Cancer Discov 6, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén S‐E, Greco D, Söderhäll C, Scheynius A and Kere J (2012) Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS ONE 7, e41361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martín‐Algarra S, Mandal R, Sharfman WH et al (2017) Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171, 934–949.e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L et al (2014) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372, 30–39. [DOI] [PubMed] [Google Scholar]
- Roh MR, Gupta S, Park K‐H, Chung KY, Lauss M, Flaherty KT, Jönsson G, Rha SY and Tsao H (2016) Promoter methylation of PTEN is a significant prognostic factor in melanoma survival. J Invest Dermatol 136, 1002–1011. [DOI] [PubMed] [Google Scholar]
- Sade‐Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily‐Rokni M et al (2018) Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepsky A, Bruser K, Gunnarsson GJ, Goodall J, Hallsson JH, Goding CR, Steingrimsson E and Hecht A (2006) The microphthalmia‐associated transcription factor Mitf interacts with beta‐catenin to determine target gene expression. Mol Cell Biol 26, 8914–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane M, Buhs S, Iglesias P, Strauss J, Puller A‐C, Müller J, Gerull H, Feldhaus S, Alawi M, Brandner JM et al (2019) Lineage‐specific control of TFIIH by MITF determines transcriptional homeostasis and DNA repair. Oncogene 38, 3616–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R and Gajewski TF (2015) Melanoma‐intrinsic β‐catenin signalling prevents anti‐tumour immunity. Nature 523, 231. [DOI] [PubMed] [Google Scholar]
- Spranger S and Gajewski TF (2018) Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer 18, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, Gajewski AP, Andrade J and Gajewski TF (2016). Density of immunogenic antigens does not explain the presence or absence of the T‐cell–inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci USA 113, E7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S and Robertson GP (2003) Loss of PTEN promotes tumor development in malignant melanoma. Can Res 63, 2881. [PubMed] [Google Scholar]
- Steingrímsson E, Copeland NG and Jenkins NA (2004) Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet 38, 365–411. [DOI] [PubMed] [Google Scholar]
- Surveillance, E., and End Results (SEER) Program (2019) SEER*Stat Database: Incidence – SEER 18 Regs Research Data + Hurricane Katrina Impacted Louisiana Cases, Nov 2018 Sub (2000–2016).
- Taggart D, Andreou T, Scott KJ, Williams J, Rippaus N, Brownlie RJ, Ilett EJ, Salmond RJ, Melcher A and Lorger M (2018). Anti–PD‐1/anti–CTLA‐4 efficacy in melanoma brain metastases depends on extracranial disease and augmentation of CD8+ T cell trafficking. Proc Natl Acad Sci USA 115, E1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, Khushalani NI, Lewis K, Lao CD, Postow MA et al (2018) Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med 379, 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T‐H, Porta‐Pardo E, Gao GF, Plaisier CL, Eddy JA et al (2018) The immune landscape of cancer. Immunity 48, 812–830.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G et al (2016) Dissecting the multicellular ecosystem of metastatic melanoma by single‐cell RNA‐seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo JA, Sweis RF, Bao R and Luke JJ (2018) T cell‐inflamed versus non‐T cell‐inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res 6, 990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucci M, Mannavola F, Passarelli A, Stucci LS, Cives M and Silvestris F (2018) Exosomes in melanoma: a role in tumor progression, metastasis and impaired immune system activity. Oncotarget 9, 20826–20837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM et al (2015) Genomic correlates of response to CTLA‐4 blockade in metastatic melanoma. Science 350, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L and Hinton G (2008) Visualizing data using t‐SNE. J Mach Learn Res 9, 2579–2605. [Google Scholar]
- Viale G, Trapani D and Curigliano G (2017) Mismatch repair deficiency as a predictive biomarker for immunotherapy efficacy. Biomed Res Int 2017, 4719194–4719194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J and Herlyn M (2008) Melanoma and the tumor microenvironment. Curr Oncol Rep 10, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr and Lao CD (2015) Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti‐CTLA‐4 treatment (CheckMate 037): a randomised, controlled, open‐label, phase 3 trial. Lancet Oncol 16, 375–384. [DOI] [PubMed] [Google Scholar]
- Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, Dalle S, Schenker M, Chiarion‐Sileni V and Marquez‐Rodas I (2017) Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med 377, 1824–1835. [DOI] [PubMed] [Google Scholar]
- Widlund HR, Horstmann MA, Price ER, Cui J, Lessnick SL, Wu M, He X and Fisher DE (2002) Beta‐catenin‐induced melanoma growth requires the downstream target microphthalmia‐associated transcription factor. J Cell Biol 158, 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok JD, Chiarion‐Sileni V, Gonzalez R, Rutkowski P, Grob J‐J, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF et al (2017) Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 377, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods K, Pasam A, Jayachandran A, Andrews MC and Cebon J (2014) Effects of epithelial to mesenchymal transition on T cell targeting of melanoma cells. Front Oncol 4, 367–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Adamow M, Ginsberg BA, Rasalan TS, Ritter E, Gallardo HF, Xu Y, Pogoriler E, Terzulli SL and Kuk D (2011). Integrated NY‐ESO‐1 antibody and CD8+ T‐cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci USA 108, 16723–16728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Additional characteristics of Lund immune‐methylation clusters.
Fig S2. Additional characteristics of immune exclusion in the immune‐methylation clusters.
Fig S3. Additional characteristics of TCGA immune‐methylation clusters.
Table S1. List of immune cell type associated CpGs with corresponding gene and associated cell type information.
Table S2. Clinical characteristics for immune‐methylation clusters (Lund cohort).
Table S3. Overlap between Lund gene expression phenotypes and Lund immune‐methylation clusters.
Table S4. List of centroid CpGs with methylation β values for each cluster.
Table S5. Overlap between TCGA gene expression classes and TCGA immune‐methylation clusters.
Appendix S1. Supplementary methods.