

Abstract
PDGF-A is a key contributor to lung development in mice. Its expression is needed for secondary septation of the alveoli and deletion of the gene leads to abnormally enlarged alveolar air spaces in mice. In humans, the same phenotype is the hallmark of bronchopulmonary dysplasia (BPD), a disease that affects premature babies and may have long lasting consequences in adulthood. So far, the knowledge regarding adult effects of developmental arrest in the lung is limited. This is attributable to few follow-up studies of BPD survivors and lack of good experimental models that could help predict the outcomes of this early age disease for the adult individual. In this study, we used the constitutive lung-specific Pdgfa deletion mouse model to analyze the consequences of developmental lung defects in adult mice. We assessed lung morphology, physiology, cellular content, ECM composition and proteomics data in mature mice, that perinatally exhibited lungs with a BPD-like morphology. Histological and physiological analyses both revealed that enlarged alveolar air spaces remained until adulthood, resulting in higher lung compliance and higher respiratory volume in knockout mice. Still, no or only small differences were seen in cellular, ECM and protein content when comparing knockout and control mice. Taken together, our results indicate that Pdgfa deletion-induced lung developmental arrest has consequences for the adult lung at the morphological and functional level. In addition, these mice can reach adulthood with a BPD-like phenotype, which makes them a robust model to further investigate the pathophysiological progression of the disease and test putative regenerative therapies.
Keywords: lung, mouse model, PDGF-A
INTRODUCTION
The vital exchange of gases between the air and the bloodstream occurs in the distal compartment of the lung, where the numerous functional units named alveoli are located. Alveoli have a sac-like morphology surrounded by an extensive capillary bed. Mature alveoli exhibit a large surface area, essential for an efficient blood-gas exchange and good respiratory capacity. This large surface area is achieved during the last stage of lung development, through the process of secondary septation of pre-existing respiratory sacs. This complex process, named alveologenesis, requires coordination of different cell types such as the alveolar epithelial cells type 1 (AEC1s), alveolar epithelial cells type 2 (AEC2s), endothelial cells, myofibroblasts, components of the extracellular matrix (ECM), as well as many molecular mediators [reviewed in (65)].
Disruption of alveologenesis leads to alveolar simplification, less surface area, and consequently inefficient gas exchange. These are distinctive features of lung diseases such as chronic obstructive pulmonary disease (COPD) (56) and bronchopulmonary dysplasia (BDP) (55). BPD is a chronic lung disease mainly detected in premature babies (4). Two types of BPD (old and new) have been described and suggested to depend on the timing of prematurity and severity of the lung damage (5). Old BPD was common in slightly premature babies exposed to mechanical ventilation and high oxygen concentrations. The typical patterns of old BPD were structural damage, inflammation, and fibrosis. The new BPD is instead a consequence of developmental arrest in the lung, leading to immature lungs with reduced alveolar surface area. The new type of BPD emerged due to the increase in survival of more premature infants.
BPD can have negative consequences for a child’s development, both at the respiratory and neurological level (30, 35, 64). Some survivors exhibit long-term pulmonary defects (69, 71), and there are currently no therapeutic options to treat BPD, even though a few preventive approaches have shown some level of efficacy (44). Extensive efforts have been made to increase knowledge of the molecular mechanisms that regulate alveologenesis, with the ambition to discover new therapeutic approaches (57).
Platelet-derived growth factor A (PDGF-A) is a known key regulator of alveologenesis (9, 10, 38). PDGF-A is a secreted protein and one of a total of four ligands in the PDGF family, which can bind to its receptors as homo- or heterodimers. PDGF-A is together with PDGF-C the main ligands for the PDGF receptor-α (PDGFR-α), a tyrosine kinase receptor with important roles during developmental processes (1). The downstream signaling cascade of PDGFR-α induces cellular processes such as proliferation, differentiation, migration, and cytoskeleton rearrangements (67).
We recently reported on the generation of a mouse model with a lung-specific deletion of Pdgfa (28), focusing on lung development and the role of PDGF-A during alveologenesis. In the present study, we focus on adult mice. We have previously shown that adult AEC2s maintain expression of Pdgfa (27), but a possible role for PDGF-A in the adult lung remained unknown. In contrast to the global Pdgfa knockout (10), mice with a lung-specific deletion of Pdgfa survive until adulthood and were, therefore, used in this study. Here, we combine histological, physiological, and proteomics methods to analyze the consequences of deficient alveolar formation for the adult lung. We show that mice with a lung-specific constitutive Pdgfa deletion do not recover from alveolar developmental arrest. Their lungs exhibit enlarged distal air spaces, which results in lower lung capacity and higher lung compliance. Even so, the knockout mice were able to live as long as their littermate controls. Moreover, we provide compelling evidence that elastin deposition during alveologenesis is not dependent on PDGF-A signaling, contrary to what was previously thought (38).
Collectively, these results indicate a similarity between the phenotype of lung-specific Pdgfa knockout mice and human BPD survivors. Therefore, these mice can be considered a good model in which to study the pathophysiological process of this disease and are a powerful tool to test potential regenerative therapeutic approaches.
MATERIALS AND METHODS
Ethical permits.
Experiments were conducted both in Sweden and Germany, according to Swedish legislation, German animal welfare law, and European legislation for the protection of animals used for scientific purposes (2010/63/EU). Ethical permits in Sweden were approved by the Uppsala animal ethics committee (permit number C115/15 and C135/15) and in Germany by the regional board (RP Giessen, Hesse, Germany; Az: V 54-19 c 20 15 h 01 GI 20/10 Nr. G81/2017). All efforts were made to minimize suffering, and experimental procedures on mice were performed under anesthesia.
Mice.
Mice carrying different combinations of the following alleles were used: Pdgfafl/fl, conditional Pdgfa knockout [Pdgfatm1Vlcg (2)]; Pdgfa+/−, heterozygous global Pdgfa knockout (Pdgfatm1Cbet (10); PdgfraGFP reporter (Pdgfratm11(EGFP)Sor (31)]; and Spc-cre - constitutive Spc-cre expressing alleles (Tg(Sftpc-cre)tm1Blh (52)]. All mice were from the C57Bl/6J background. All analyses were performed on Pdgfafl/−; Spc-cre (PdgfaAEC2KO) and Pdgfafl/+ (Pdgfawt). DNA from toe biopsies was used for PCR genotyping, and the crossings to obtain knockout mice (PdgfaAEC2KO) were described previously (28).
Lung perfusion.
Mice were anesthetized using Ketamine (75 mg/kg) and Dexdomitor (0.5 mg/kg). All lungs were perfused with PBS through the heart’s right ventricle and inflated with 4% formaldehyde (VWR, 9713.5000) through the trachea. Lungs for vibratome sections were inflated with 2% low-melting agarose; samples were further immersion fixed in 4% formaldehyde for 4 h at 4°C and washed in PBS at 4°C.
Histology.
Perfusion fixed lungs for paraffin sections were dehydrated in a series of ethanol (70%, 95%, and 99%) and xylene, and paraffin embedded. Microtome sections (5 μm thick) were rehydrated in xylene and ethanol (99%, 95%, 80%) for 2 × 2 min. Sections were stained with hematoxylin and eosin and mounted in Neo-mount (109016, Merck).
Immunofluorescence.
Lungs inflated with 2% low-melting agarose were vibratome sectioned (70–100 μm). Sections were blocked overnight at 4°C in blocking buffer (1% BSA + 0.5% Triton X-100) and incubated with primary antibodies for 48 h at 4°C, washed in PBS, and incubated with secondary antibodies overnight at 4°C. Sections were stained with Hoechst (1:10,000; Invitrogen, 33342) for 10 min and mounted in Prolong Gold antifade reagent (Invitrogen, P36930). Primary antibodies used: goat anti-SPC (1:100, Santa Cruz, sc-7706), rabbit anti-Spc (1:100, Abcam, ab90716), rabbit anti-tropoelastin (1:100, Abcam, ab21600), goat anti-CD31 (1:500, R&D Systems, AF3628), rabbit anti-ERG (1:500, Abcam, ab92513), rat anti-Lama2 (1:100, Abcam, ab11576), goat anti-ColIV (1:200, Bio-Rad, 134001), rabbit anti-ColI (1:200, Abcam, ab21286), rabbit anti-Hopx (1:100, Santa Cruz, Sc-30216), rabbit anti-ADFP (1:200, Invitrogen, PA1-16972), rabbit anti-vWF (1:200, Dako, A0082), and mouse anti-human A-sma (alpha-smooth muscle actin) conjugated to Cy3 (1:200, Sigma-Aldrich, C6198). Secondary antibodies used: Cy3 AffiniPure Donkey Anti-Goat IgG (1:200, Jackson IR, 705-165-147), Alexa Fluor 647 AffiniPure Donkey Anti-Rabbit IgG (1:200, Jackson IR, 711-605-152) and Cy3 AffiniPure Donkey Anti-Rat IgG (1:200, Jackson IR, 712-165-150).
RNA extraction and quantitative PCR.
RNA was extracted from the left lung lobe using RNeasy Mini Kit (Qiagen). The iScript cDNA synthesis kit (Bio-Rad, 1708891) was used to synthesize cDNA from 1 µg of total RNA. Analysis was performed using the CFX-96 Real Time System (Bio-Rad) and the following TaqMan probes (Thermo Fisher Scientific): Pdgfa (Mm00435540_m1), Pdgfra (Mm00440701_m1), Eln (Mm00514670_m1), Acta2 (Mm00725412_s1), Lama2 (Mm00550083_m1), Vim (Mm01333430_m1), Col1 (Mm00801666_s1), Vim (Mm01210125_m1), Sftpc (Mm00488144_m1), Pdpn (Mm01348912_g1), Plin2 (Mm00475794_m1), Vwf (Mm00550376_m1), Tagln (Mm00441661_g1). The Ct values were normalized to the 18s rRNA endogenous control (X03201.1, Applied Biosystems). Six biological replicates in each group (n = 6 knockout, n = 6 control) were run in triplicate, and each run was repeated three times (technical replicates). Relative gene expression was calculated using the Livak method (2-ddCt) (61).
Proteome analysis.
After lung perfusion, the left lobe was removed and snap-frozen on dry ice and isopentane. Next, the lobes were cut into smaller pieces, transferred to lysis buffer (4% SDS, 50 mM HEPES pH 7.6, 1 mM DTT), and disrupted using a tissue homogenizer. Samples were heated for 5 min at 95°C and sonicated afterwards. Samples were dissolved in 500 µl of lysis buffer, and the total protein amount was estimated (Bio-Rad DCC Assay). Preparation for mass spectrometry (MS) analysis was performed using a modified version of the SP3 protein clean-up and digestion protocol (45). Peptides were labeled with TMT10plex reagent according to the manufacturer’s protocol (Thermo Scientific). Labeled peptides were separated by immobilized pH gradient-isoelectric focusing (IPG-IEF) on 3–10 strips as described previously (13), and the extracted peptide fractions were separated using an online 3000 RSLCnano system coupled to a Thermo Scientific Q Exactive Mass Spectrometer. The MS spectra were matched to the Ensembl 82 mouse protein database (8) using MSGF + Percolator in the Galaxy platform. Protein false discovery rates (FDR) were calculated using the picked-FDR method using gene symbols as protein groups and limited to 1% FDR (60). The normalized data were loaded in R software (version 3.3.2) for downstream analysis. To compare the protein expression difference between the knockout samples and control samples, as well as between the female and male samples, two-way ANOVA statistics model was applied to the protein expression values. The proteins with a fold change >1.5-fold and P value <0.05 were considered to be differentially expressed.
Echocardiography.
Anesthesia was induced with 3% isoflurane and maintained with 1.5–2% isoflurane supplemented in 100% oxygen. Transthoracic 2D echocardiography, M-mode, and Doppler imaging were performed with a Vevo2100 high-resolution imaging system equipped with a 40 MHz transducer (VisualSonics, Toronto, Canada). Right ventricular cardiac output (CO), cardiac index (CI), left ventricular ejection fraction (EF), the ratio between pulmonary artery acceleration time (PAT) and pulmonary artery ejection time (PET), right ventricular internal diameter (RVID), right ventricular wall thickness (RVWT), and tricuspid annular plane systolic excursion (TAPSE) were measured as described previously (33, 66). Echocardiographic analyses were performed by an experienced observer who was blind to the genotype and hemodynamic measurements.
In vivo micro-CT imaging.
Images were acquired using a Quantum GX microCT scanner (PerkinElmer, Inc, Waltham, MA). Mice were placed into an induction chamber, anesthetized by inhalation of 3% isoflurane in oxygen, and afterwards placed on a scanner bed with a nose cone supplying isoflurane in oxygen adjusted to 1.5%. The scanner bed was translated longitudinally to align the animal chest within the center of the field of view. The scanner’s complementary metaloxide-semiconductor X-ray flat-panel detector was set to allow image acquisition with an X-ray tube voltage of 90 kV and current of 80 μA. For Hounsfield unit (HU) calibration, a phantom consisting of an air-filled 1.5 ml tube inside a water-filled 50 ml tube (water: 0 HU, air: −1,000 HU) was scanned.
Micro-CT data were collected in list-mode over a single complete gantry rotation with a total rotation time of 4 min (14,688 frames collected in total). Once the data were collected, the mouse was removed from the scanner and supervised during recovery from anesthesia. Raw projection images were processed using a proprietary algorithm for intrinsic retrospective respiratory gating and then reconstructed using a filtered back-projection algorithm on a dedicated graphics processing unit. Reconstructed volumes were loaded into Analyze Pro software (Analyze Direct, Mayo Clinic) and processed by a single observer blinded to the animal’s genotypes and invasive morphometric results. Lung segmentation and quantitative analysis of the CT density, functional residual capacity (FRC) were performed as described (24).
In vivo measurements of lung function and hemodynamic parameters.
Experiments were performed as described previously (62). Briefly, animals were inserted in a chamber and anesthetized with 3% isoflurane supplemented in 100% oxygen until surgical tolerance was achieved. After tracheotomy, mice were intubated using an 18G metal tube (SCIREQ Scientific Respiratory Equipment Inc.) and ventilated using the flexiVent system (SCIREQ Scientific Respiratory Equipment Inc.) at a frequency of 150 breaths/min and a tidal volume of 5 ml/kg.
Lung function tests were performed using the flexiVent predetermined script at a PEEP of 3 cmH2O, with consistent perturbation order, following the manufacturer’s recommendations. The results are presented as an average of multiple measurements with the coefficient of determination above 0.95. Pressure-volume curves were generated using a stepwise pressure-driven perturbation (PVs-P).
For right ventricular systolic pressure (RVSP) measurements, the jugular vein was catheterized by a micro-tip catheter (Millar Instruments, SPR 671, REF 8406719) that was placed into the right ventricle. Systemic arterial and left ventricular systolic pressures were measured by a catheter inserted into the carotid artery or left ventricle, respectively.
Heart ratio measurements.
After finishing the hemodynamic measurements, we injected heparin (Ratiopharm) solution at a concentration of 50,000 U/kg ip. The animals were euthanized by exsanguination, and the heart was removed and kept in PBS on ice until further analysis. Right ventricular (RV) hypertrophy was measured by separating the RV from the left ventricle plus septum (LV+S) and results are presented as a ratio of RV weight to LV+S weight.
Image acquisition and analysis.
For mean linear intercept (MLI) calculations, hematoxylin eosin-stained paraffin sections were imaged with a bright-field Axioimager microscope (Zeiss) at ×40 magnification. Thirty unbiased images were acquired from each sample, and MLI was calculated manually using imageJ software with a grid plugin (NIH). Eleven horizontal lines were superimposed on each image, and the intersections between the lines and the alveolar walls were counted. The number of lines was multiplied by the length of the lines and divided by the number of interception points.
All other image acquisition was performed using a TCS Sp8 Laser Scanning Confocal microscope (Leica Microsystems). For the quantification of immunofluorescence images, at least 10 ×40 or ×63 fields per sample were analyzed either manually or using the spots and surface modules in Imaris software (Bitplane, Oxford Instruments).
Statistical analysis.
Quantifications and mRNA gene expression statistical differences between knockout and control lungs were calculated using Student’s t test (unpaired and two-tailed). All analysis was performed with at least five different mice per group (knockout n = 5 vs. control n = 5), and P < 0.05 was considered statistically significant. The statistical analysis and data assembly was performed using GraphPad Prism version 6 for Mac (GraphPad Software, La Jolla, CA). All graphs are presented as means ± SE.
RESULTS
Constitutive deletion of Pdgfa leads to chronic alveolar defects.
To investigate the impact of constitutive Pdgfa deletion in the mature lung, we examined adult mice in which the deletion of Pdgfa was restricted to surfactant protein-C (Spc)-positive AEC2s (PdgfaAEC2KO) and compared them with littermate controls (PdgfaWT). Generation of PdgfaAEC2KO mice (Pdgfafl/−; Spc-cre) and PdgfaWT (Pdgfafl/+) was described previously (28), and the mice were euthanized for morphological analysis at postnatal day 60 (P60) or 1 yr (P365) of age.
At P60, the histological analysis of PdgfaAEC2KO lungs revealed enlarged distal air spaces and alveolar simplification (Fig. 1, A and B) with a significant increase in the mean linear intercept (Fig. 1C). Reduced expression of Pdgfa mRNA levels in left lung lobe homogenates was confirmed by quantitative (q)PCR and indicated a 40% reduction in expression (Fig. 1D). As comparison, in lungs from Pdgfa heterozygous mice, the mRNA levels were reduced to 75% compared with wild-type mice (data not shown). Despite the severe lung phenotype, the PdgfaAEC2KO mice survived through adulthood. At 1 yr of age, the histological analysis of the knockout lungs revealed the same enlarged distal air spaces (Fig. 1, E and F) in combination with a significantly higher mean linear intercept (Fig. 1G), when compared with the controls. Pdgfa mRNA expression levels were also significantly decreased (Fig. 1H). None of the phenotypes had changed from P60 until 1 yr, indicating a stabilization of the alveolar morphology.
Fig. 1.

Comparison of adult lung tissue between PdgfaAEC2KO and PdgfaWT mice. Hematoxylin eosin-stained lung paraffin sections of control (A) and knockout (B) mice at postnatal day 60 (P60). C: mean linear intercept (MLI) calculation results comparing control (blue) and knockout (red) alveolar area. D: relative levels of Pdgfa mRNA expression levels normalized to 18s rRNA endogenous control. Hematoxylin eosin-stained lung paraffin sections of control (E) and knockout (F) mice at 1 yr (P365) of age. G: MLI results comparing control (blue) and knockout (red) mice. H: relative levels of Pdgfa mRNA expression levels normalized to 18s rRNA endogenous control. Control mice n = 5–7; knockout mice n = 5–7. Graphs are represented as means ± SE. Statistical analysis was done using Student’s two-tailed unpaired t test and statistical significance is indicated as follows: *P < 0.05, **P < 0.01, ****P < 0.0001. Scale bars: 50 µm.
Adult PdgfaAEC2KO lungs reach normal proportions of PDGFR-α+ cells.
During development, in the alveolar stage, lungs from PdgfaAEC2KO mice have reduced numbers of PDGFR-α+ cells (28). To understand whether the low numbers of PDGFR-α+ cells were maintained in adult knockout lungs, we crossed PdgfaAEC2KO mice with the PdgfraH2B:GFP reporter mouse (hereafter referred to as PdgfraGFP). The total number of cells (Hoechst-positive) in PdgfaAEC2KO; PdgfraGFP lungs was significantly decreased compared with controls (Fig. 2A). However, there was no difference in the relative proportion of PDGFR-α+ cells versus total number of cells (Fig. 2, B, D–G). This proportion contrasted with earlier data observed at the alveolar stage (28). To confirm this result, we analyzed Pdgfra gene expression in whole left lobe lung homogenates by qPCR. No significant difference in Pdgfra mRNA expression levels was found between lung homogenates from PdgfaWT and PdgfaAEC2KO mice (Fig. 2J).
Fig. 2.
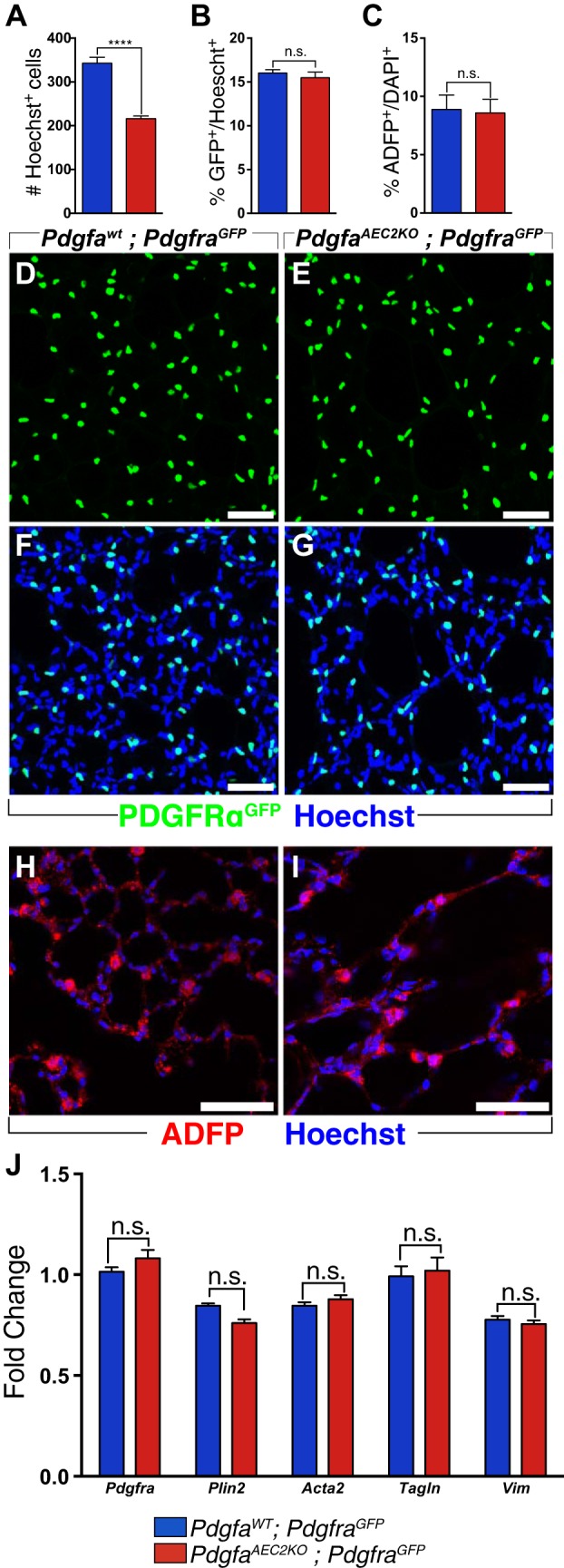
Comparable numbers of PDGFR-α+ cells between PdgfaAEC2KO and PdgfaWT lungs. A: total number of cells in control (blue) and knockout (red) lungs, quantified by counting Hoechst-positive nuclei. B: quantification of the number of Pdgfra-positive cells (GFP positive) as percentage of the total number of cells (Hoechst-positive) per field image. C: quantification of the number of lipofibroblasts by ADFP staining (red), as percentage of the total number of cells per field image (Hoechst-positive) Lung vibratome sections (70 µm thick) of control (D, F, H) and knockout (E, G, I) mice at postnatal day (P)60 showing PDGFR-αGFP expression (green), ADFP (red), and Hoechst (blue). J: relative levels of mRNA expression of different fibroblast markers normalized to 18s rRNA endogenous control in left lung lobe homogenates. Control mice n = 5–6; knockout mice n = 5–6. Graphs are represented as means ± SE. Statistical analysis was done using Student’s two-tailed unpaired t test and statistical significance is indicated as follows: ****P < 0.0001; n.s. nonsignificant. Scale bars: 50 µm.
Different populations of fibroblasts in the peripheral lung are PDGFR-α+. One of the most abundant PDGFR-α+ fibroblast subpopulation in the adult lung is the lipofibroblast (14). To understand if this cell subpopulation was affected in the knockout lungs, the relative number of lipofibroblasts was quantified as the number of ADFP+ cells per total number of cells. There was no difference between control and knockout lungs (Fig. 2, C, H, I). Neither were there any changes in the mRNA expression levels of the lipofibroblast marker Plin2 (Fig. 2J). Additionally, no differences were detected in mRNA expression levels of several other mesenchymal cell markers, Acta2, Tagln, and Vim (Fig. 2J). Collectively, these data indicate that adult control and knockout lungs exhibited the same relative numbers of PDGFRα+ cells and fibroblasts.
Normal cellular composition in adult PdgfaAEC2KO lungs.
The distal lung is composed of multiple cell types. In addition to the fibroblasts, we investigated the expression pattern and relative numbers of AEC2s, AEC1s, and endothelial cells in adult lungs from PdgfaAEC2KO mice. At P7, the proportion of AEC2s was increased, as previously shown by the expression of the AEC2 marker Spc (28). The adult PdgfaAEC2KO lungs maintained a higher proportion of Spc+ cells when compared with PdgfaWT lungs (14.34% ± 1.231 vs. 9.060% ± 0.9558, Fig. 3, A–C). However, no significant increase was seen in mRNA expression of Spc (Fig. 3D). No differences were seen for AEC1s. Hopx+ AEC1s covered the alveolar walls in a comparable pattern in both control and knockout lungs (Fig. 3, E and F). Furthermore, qPCR analysis of the AEC1 marker Pdpn confirmed that AEC1s were not affected in the knockout lungs (Fig. 3G). The vascular component of the lung was analyzed by the expression of the endothelial cell markers Erg (Fig. 3, H–J), Pecam1 (CD31) (Fig. 3, K and L), and vWF (Fig. 3M). We could see no differences in the expression pattern, the relative number of endothelial cells, or the mRNA expression levels, when comparing knockout and control lungs. In summary, the cellular composition in adult PdgfaAEC2KO lungs was comparable to control lung, with the exception of AEC2s.
Fig. 3.

Lung cellular composition of PdgfaAEC2KO is similar to PdgfaWT. A, B: lung paraffin sections (7 µm) stained with AEC2s marker Spc (red) and Hoechst (blue). C: quantification of the number of AEC2s (Spc-positive) as percentage of the total number of cells (Hoechst-positive). D: relative levels of mRNA expression of Sftpc normalized to 18s rRNA endogenous control. E, F: lung vibratome sections (70 µm) stained with AEC1s marker Hopx (red) and Hoechst (blue). G: relative levels of mRNA expression of Pdpn normalized to 18s rRNA endogenous control. H, I: lung vibratome sections (70 µm) stained with EC nuclear marker ERG (green) and Hoechst (blue). J: quantification of the number of EC (ERG-positive) as percentage of the total number of cells (Hoechst-positive). K, L: lung vibratome sections (70 µm) stained with EC marker Pecam1 (CD31, green) and Hoechst (blue). M: relative levels of mRNA expression of vWF normalized to 18s rRNA endogenous control. Control mice n = 5–6; knockout mice n = 5–6. Graphs are represented as means ± SE. Statistical analysis was done using Student’s two-tailed unpaired t test and statistical significance is indicated as follows: *P < 0.05; n.s., nonsignificant. Scale bars: 50 µm.
ECM deposition is not affected by Pdgfa deletion, not even elastin.
The ECM network is important for proper formation of the alveoli in the distal lung (11, 14, 41, 48), and elastin is one of the key components of the lung ECM (32, 34, 68, 70, 72). Several years ago, we suggested that PDGF-A was needed for elastin production (10), a hypothesis that has not been confirmed so far. To test this, we examined elastin and several other ECM components expression in lung sections of PdgfaAEC2KO and PdgfaWT mice. Immunostaining revealed that elastin fibers were present in PdgfaAEC2KO lungs. At P7, both control and knockout lungs exhibited high presence of elastin fibers surrounding the alveolar air spaces (Fig. 4, A and B). The amount of elastin was further quantified by measuring the volume of the elastin network using 3D reconstruction images. These revealed that the amount of elastin per field image was similar in control and knockout lungs (Fig. 4C). Furthermore, there was no difference in mRNA expression levels of Eln (Fig. 4D). In adult mice, both control and knockout lungs showed a high number of elastin fibers, but the elastin fibers in the PdgfaAEC2KO mice were differently organized (Fig. 4, E and F). There was a higher density of elastin fibers in the interstitial spaces, as the knockout lungs lacked the small alveolar air spaces observed in the control mice. However, again no differences were detected in either the volume of the elastin network (Fig. 4G) or in the mRNA expression level (Fig. 4H). These results indicate that the emphysema-like phenotype in the PdgfaAEC2KO mice is not due to the lack of elastin and that production of elastin does not depend on PDGF-A signaling from AEC2s.
Fig. 4.

Elastin deposition is not dependent on PDGF-A expression from AEC2s. A, B: postnatal day 7 (P7) lung vibratome sections (100 µm) stained for Elastin (white). C: volume of Elastin in control and knockout P7 lungs. D: relative levels of mRNA expression of Eln normalized to 18s rRNA endogenous control. E, F: adult lung vibratome sections (100 µm) stained for Elastin (white). G: volume of Elastin in control and knockout adult lungs. H: relative levels of mRNA expression of Eln normalized to 18s rRNA endogenous control. Control mice n = 5–6; knockout mice n = 5–6. Graphs are represented as means ± SE. Statistical analysis was done using Student’s two-tailed unpaired t test. n.s. Nonsignificant. Scale bars: 50 µm.
Other examined ECM components, such as ColI, ColIV, and Lama2, did not show any differences between control and knockout mice. No changes were seen either through immunostaining (Supplemental Fig. S1; all supplemental material is available at https://doi.org/10.6084/m9.figshare.8870024) or in mRNA expression levels (Supplemental Fig. S1).
Proteomics analysis confirms stabilization of adult PdgfaAEC2KO lungs.
Besides the obvious differences in morphology, PdgfaAEC2KO lungs did not show any evidence of major alterations in ECM and cellular composition, when compared with PdgfaWT lungs. To investigate potential alterations in protein expression in the PdgfaAEC2KO mice, we employed mass spectrometry-based proteomics (13) on lungs from four knockout and four control mice at the age of 4 wk. We identified in total 10,314 proteins at 1% false discovery rate and 9,895 proteins were quantified in all eight lung samples. The differentially expressed proteins between knockout and control lungs were calculated with a two-way ANOVA, considering 1.5-fold change in expression and P < 0.05 as the cut-off values. To our surprise, only 10 proteins showed statistically significant differences between knockout and control lungs (Table 1), most of which are expressed by epithelial cells. Eight of the proteins were upregulated in knockout lungs. The most upregulated protein in knockout lungs was Resistin-like alpha (Retnla), a protein expressed by airway and alveolar type 2 epithelial cells. Retnla stimulates the production of α-smooth muscle actin and type I collagen in lung fibroblasts (19, 39, 40). In agreement with previously published data on lung proteomics (22), we observed a high number of differentially expressed proteins when comparing females versus males, regardless of the mouse genotype (Supplemental Table S1).
Table 1.
Differentially expressed proteins in PdgfaAEC2KO and PdgfaWT lungs
| Protein Accession | Gene | Protein | Log2 Fold (KO/control) | P Value |
|---|---|---|---|---|
| ENSMUSP00000023329.4 | ENSMUSG00000061100 | Retnla* | 2.23 | 0.0240 |
| ENSMUSP00000032089.2 | ENSMUSG00000030017 | Reg3g* | 1.28 | 0.03112 |
| ENSMUSP00000128276.1 | ENSMUSG00000066108 | Muc5b* | 1.07 | 0.01709 |
| ENSMUSP00000075945.7 | ENSMUSG00000047730 | Fcgbp* | 1.01 | 0.04027 |
| ENSMUSP00000028987.6 | ENSMUSG00000027485 | Bpifb1* | 1.01 | 0.04558 |
| ENSMUSP00000011178.2 | ENSMUSG00000011034 | Slc5a1* | 0.66 | 0.00696 |
| ENSMUSP00000113763.1 | ENSMUSG00000027937 | Jtb* | 0.61 | 0.04273 |
| ENSMUSP00000043672.9 | ENSMUSG00000033847 | Pla2g4c* | 0.61 | 0.04119 |
| ENSMUSP00000063549.4 | ENSMUSG00000089996 | Tmsb15b2† | −0.6 | 0.02133 |
| ENSMUSP00000005597.8 | ENSMUSG00000056592 | Zfp658† | −0.88 | 0.03305 |
Eight proteins were upregulated and
two proteins were downregulated >1.5-fold (P < 0.05) in knockout (KO) mice.
Physiological analysis confirms that PdgfaAEC2KO mice develop pulmonary emphysema.
Based on the morphological and histological phenotype we hypothesized that PdgfaAEC2KO mice suffered from emphysema. To evaluate the lung function of the mice, we studied important physiological parameters in knockout and control lungs. In vivo µCT measurements revealed lower lung density (Fig. 5A) and increased functional residual capacity (FRC) in the knockout mice (Fig. 5B). Additionally, µCT analysis of the lungs showed that the tissue volume was not altered (Fig. 5C) in mice, whereas the air-tissue volume ratio was elevated in PdgfaAEC2KO lungs (Fig. 5D), indicating an increase in air volume inside the knockout lungs. Additionally, there was an increase in both dynamic (Fig. 5E) and static (Fig. 5F) compliance in PdgfaAEC2KO lungs. The respiratory pressure-volume loop obtained in knockout mice had a characteristic shape, which suggests emphysema and atelectasis (Fig. 5G). The latter was further demonstrated by the higher hysteresis measured in knockout lungs (Fig. 5H). These findings were consistent with functional abnormalities associated with enlargement of the distal air spaces. Additional lung function data revealed increased inspiratory capacity, reduced tissue resistance, and unchanged respiratory system resistance in the knockout mice (Supplemental Fig. S2).
Fig. 5.
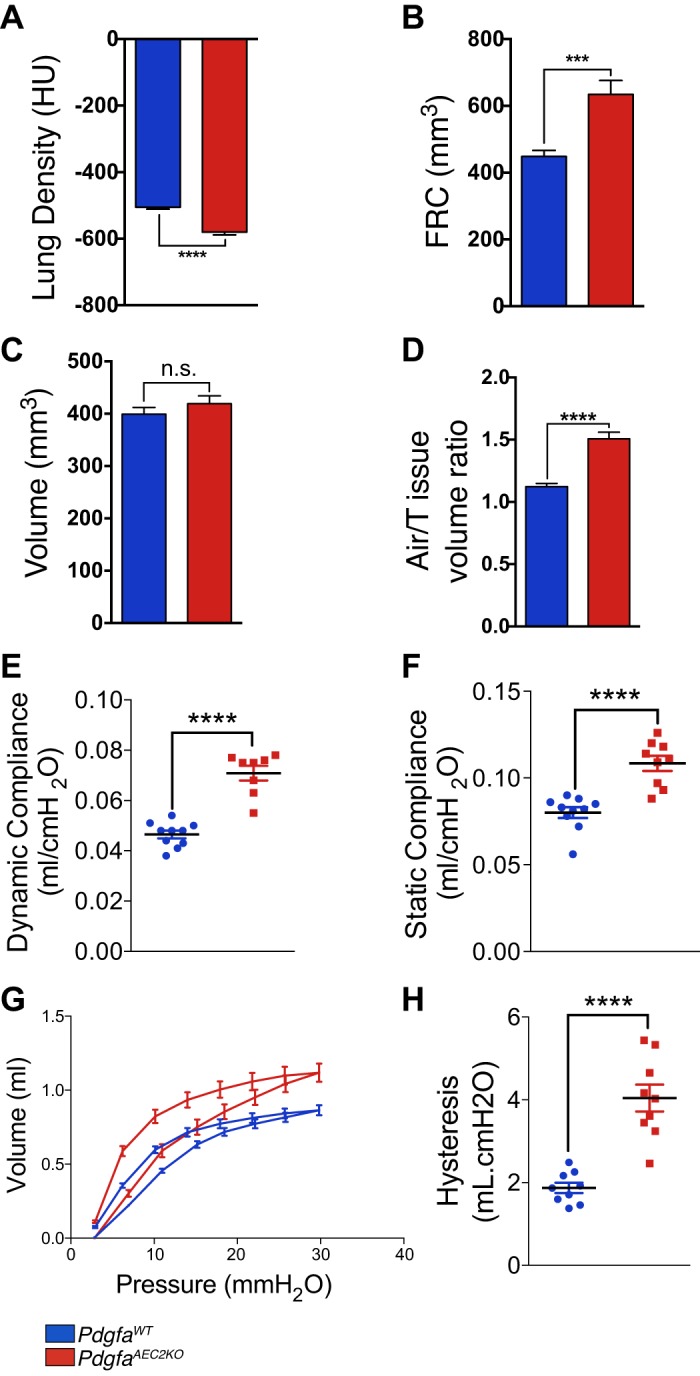
Mice lacking Pdgfa develop emphysema. In vivo µCT and lung function measurements of mouse lungs showing lung density in Hounsfield units (HU) (A) and functional residual capacity (FRC) volume (B), lung tissue compartment volume (C), lung air-to-tissue volume ratio (D), dynamic compliance (E), static compliance (F), pressure-volume loops (G), and Hysteresis (H). Hysteresis and pressure-volume loops were calculated using PV-sP. Control mice n = 9–10; knockout mice n = 8–9. Values are given as means ± SE. Statistical analysis was done using Student’s two-tailed unpaired t test and statistical significance is indicated as follows: ***P < 0.001 and ****P < 0.0001
Taken together, these data confirm that PdgfaAEC2KO mice suffer from lung emphysema.
Increased right heart ventricular pressure in PdgfaAEC2KO mice.
Lung function and cardiac performance are tightly regulated and may influence each other (25). We measured changes in right ventricular systolic pressure (RVSP) via right heart catheterization to reflect pressure changes in the pulmonary circulation (Fig. 6A). Increased RVSP in PdgfaAEC2KO mice went along with an increased Fulton index (Fig. 6B) that could indicate pulmonary hypertension in knockout mice when compared with control. However, at 2 mo of age, no increased muscularization of the vasculature was observed in the knockout lungs, assessed by measurement of α-SMA (Acta2) thickness around pulmonary arteries (Fig. 6, E–Q).
Fig. 6.

Effect of Pdgfa deletion on the development of pulmonary hypertension and right heart remodeling. A: hemodynamic measurements showing right ventricular systolic pressure (RVSP). B: right heart hypertrophy given as Fulton index (right ventricle weight/left ventricle + septum weight). Heart function as indicated by echocardiographically assessed tricuspid annular plane systolic excursion (TAPSE) (C) and cardiac index (CI) (D). Control mice n = 9–10, knockout mice n = 10–11. Immunostaining of control (E–J) and knockout lung vibratome sections (100 µm) (K–P) with α-smooth muscle actin (α-sma, red), von Willebrand factor (vWF, green), and Hoechst (blue). Q: quantification of α-sma thickness surrounding pulmonary arteries represented in pixels. Values are given as means ± SE. Statistical analysis was done using Student’s two-tailed unpaired t test, and statistical significance is indicated as follows: n.s. nonsignificant, *P < 0.05, **P < 0.01, and ***P < 0.001.
Both control and knockout mice showed a high variation within the group for the functional parameter TAPSE, and no differences were recorded (Fig. 6C). However, a slight increase in cardiac index (Fig. 6D) was observed in PdgfaAEC2KO animals as a response to the increased pulmonary resistance. No differences were seen in heart rate or systemic arterial pressure (including left ventricular systolic, arterial diastolic, and systolic pressure) between knockout and control mice (Supplemental Fig. S3). There was a reduction in the heart mass of the knockout mice, an impact that seemed to be restricted to the left ventricle (Supplemental Fig. S4). The overall weight of the knockout mice was, however, slightly reduced, but the length of the tibia indicated no general size differences between knockout and wild-type mice (Supplemental Fig. S4). No further alterations in heart parameters were detected by echocardiography (Supplemental Fig. S5).
DISCUSSION
The indispensable role of PDGF-A during lung development in mice is well known (9, 10, 28, 38), but its role in the adult lung has not previously been analyzed. The perinatal mortality of the global Pdgfa knockout mice has hampered the study of possible roles for PDGF-A during adulthood (10). In this study, we have used PdgfaAEC2KO mice and for the first time been able to investigate adult lungs lacking Pdgfa expression. PdgfaAEC2KO mice lack Pdgfa expression in Spc+ AEC2 cells but still survive until adulthood (28). Therefore, we focused on unraveling the consequences in the adult lung after Pdgfa deletion.
In the early studies of the global Pdgfa−/− mice, an emphysema-like lung phenotype with lack of elastin deposition in the alveolar septa was reported (10). Based on those data, we hypothesized that PDGF-A was needed for alveolar myofibroblasts to deposit elastin, an unchallenged hypothesis often referred to as dogma in subsequent publications. Using new methods and new mouse models we could finally dismiss that old hypothesis. Using thick vibratome lung sections, better imaging techniques, and 3D image reconstruction we detected elastin fibers in PdgfaAEC2KO lungs both during development and in adult mice. These data show that elastin is expressed even in the absence of PDGF-A from AEC2s, which was further proven by identical Eln mRNA expression levels in control and knockout mice. Thus, these results indicate that the emphysema-like phenotype in PdgfaAEC2KO mice is not due to the lack of elastin and that production of elastin does not depend on PDGF-A signaling from AEC2s. Recent reports have shown that elastin in the distal lung is present even before alveologenesis occurs (14, 32, 42). Additionally, mice lacking elastin do not survive more than 2 days after birth, as the lungs cannot withstand the mechanical stretch of breathing (70). These data strengthen our findings that the presence of elastin in the distal lung does not depend on PDGF-A signaling. We could nonetheless see that the 3D assembly pattern of elastin fibers was altered in PdgfaAEC2KO adult lungs. It is however difficult to assess if these alterations are caused by a downstream effect of the lack of Pdgfa expression in AEC2s or if they are consequences of the defective morphology in the knockout lungs. Interestingly, an altered elastin pattern was recently reported in mice lacking Pdgfra expression in Gli1+ myofibroblasts (36).
In the adult PdgfaAEC2KO lungs, Pdgfra+ fibroblasts were found in the same proportion as in PdgfaWT lungs. This was somehow surprising, since we previously observed that Pdgfra+ fibroblasts were both less proliferative and reduced in numbers in the knockout lungs at P7 (28). During the alveolar stage, we and others have observed a peak in the number of Pdgfra+ cells, reflecting the importance of these cells during alveologenesis (23, 27, 50). Our previously reported data (28), together with current quantifications, showed that the percentage of Pdgfra+ cells of the total number of cells in control lungs was 25% at P7 and reduced to 15% in the adult. Interestingly, in knockout lungs the proportion of Pdgfra+ cells was 15% both at P7 and in the adult. We suggest that PDGF-A signaling stimulates the proliferation of Pdgfra+ fibroblasts during alveologenesis, but that PDGF-A is not needed to maintain the Pdgfra+ fibroblast populations in the peripheral lung at later time points. Instead we hypothesize that a subpopulation of the Pdgfra+ fibroblasts that is present in the adult lung belongs to a separate subtype of fibroblasts that can be activated if needed. This was evidenced by other studies that showed that Pdgfra+ cells in the adult lung have a role during lung regeneration after pneumonectomy (16) and in bleomycin-induced fibrosis (37). In the alveolar stem cell niche, constituted by Pdgfra+ cells and AEC2s (6, 73), the Wnt and EGFR/KRAS signaling pathways have been suggested to function in parallel (46). It remains to be elucidated whether this interplay is further finetuned by the PDGF-A/PDGFR-α signaling pathway.
There were no major alterations in the cellular composition of the peripheral lung in the PdgfaAEC2KO mice, but there was a higher number of AEC2s. As we saw no increased proliferation of AEC2s after developmental stages (data not shown), we believe that the higher number of AEC2s in PdgfaAEC2KO adult lungs is a sequela from the increased proliferation and number of AEC2s that was observed during development at P7 (28). This assumption is justified by the same relative numbers of Spc+ observed both at P7 (28) and adulthood: ~10% in controls and 15% in knockout lungs. It is also motivated by the lack of differences in Spc mRNA levels observed in the adult lung (Fig. 3D). Nevertheless, an explanation for the increased number of AEC2s in knockout lungs remains to be elucidated. AEC2s function as stem cells, having the capacity to both self-renew and give rise to AEC1s in the adult lung (6, 18). This capacity is observed in only a small percentage of AEC2s (18), and it has been hypothesized that these AEC2s locally inhibit surrounding AEC2s to also behave as stem cells and self-renew. One possibility would be that this local inhibition also takes place during alveogenesis and it is lost in PdgfaAEC2KO lungs, leading to increased number of proliferating AEC2s. Lineage tracing studies would be required to find out if AEC2 self-renewing capacity is active during alveogenesis.
To our surprise, the proteomics analysis revealed very few differences between control and knockout lungs. However, this supports the lack of differences between knockout and control lungs in both ECM composition, as well as cellular composition based on endothelial cells, fibroblasts, and AEC1s markers.
Ten proteins were found to be altered in the knockout lungs, and five of those are normally expressed by AEC2s (Retnla, Reg3g, Muc5b, Bbpifb1, and Jtb), according to the LungGens database (20, 21). Several of the other proteins are also expressed by airway epithelial cells with distinct reported functions. The most upregulated protein in knockout lungs was Resistin-like alpha (Retnla), a protein expressed by airway and alveolar type 2 epithelial cells that stimulates the production of a-smooth muscle actin and type I collagen in lung fibroblasts (19, 39, 40). Reg3g is involved in antimicrobial activity (17), while Muc5b is important for maintaining immune homeostasis and mucociliary clearance (58). Upregulation of these proteins could reflect alterations in the airway epithelia of knockout mice, but we did not detect any effects either in the airway cell morphology or in mucus production. Interestingly, we found more differentially expressed proteins when comparing females and males than when comparing knockouts and controls. These results agreed with previous reports and growing evidence of a sex-related biological difference in the lung (15, 22, 26, 53). The PdgfaAEC2KO mice did not show any sex differences in any tested parameter, female and male knockout mice exhibited the same phenotype.
The changes detected in the RSVP of PdgfaAEC2KO mice pointed toward functional alterations that are characteristic of pulmonary hypertension. A higher Fulton index was observed in PdgfaAEC2KO mice, caused by left ventricle hypotrophy (Supplemental Fig. S4A). Even though the increase in RSVP and CI indicated a pressure overload, it is fully compensated for at the age of analysis. The heart and vessels did not show any signs of typical pulmonary hypertension phenotype, as we detected neither right heart hypertrophy nor increased muscularization of the lung vasculature. Therefore, we conclude that at the age of analysis, PdgfaAEC2KO mice only display a very mild pulmonary hypertension.
The adult PdgfaAEC2KO mice presented chronic enlargement of alveolar air space area and reduced lung function, exhibiting similar symptoms to those of patients with emphysema and COPD (56). Despite the observed thin alveolar walls and enlargement of the alveolar area, PdgfaAEC2KO mice lived into adulthood without any obvious external phenotypes. Human patients suffering from emphysema and COPD can live for several years with the disease (63), so the long survival of PdgfaAEC2KO mice make them a potential model to study the long-term outcomes and possible therapies for chronic lung disease. However, further detailed analysis of the mouse model is necessary to make sure it can be considered a model suitable for chronic lung diseases. One approach to be considered would be the use of a design-based stereology approach. As it estimates the number of alveoli, the surface area for gas exchange could be obtained. If that could be performed also on human lung tissue, we would better know how well this model resembles human lung pathologies. Since the phenotype observed in the PdgfaAEC2KO mice was a consequence of developmental defects during the alveolar stage, it can also be considered a powerful tool to study treatment options for BPD. Human neonates that develop BPD express reduced levels of both Pdgfra (54) and Pdgfa (3), and mice that develop a BPD-like phenotype due to hyperoxia exposure display increased levels of the microRNA miR-34a (shown to interact with Pdgfra) (59). Together with our findings, these data strengthen the notion of the PDGF-A/PDGFR-α signaling pathway as a key regulator of late lung development and disturbances in the pathway being associated with a BPD phenotype.
When BPD was first described in 1967, it was defined as a disease in slightly premature infants exposed to mechanical ventilation and oxygen supplementation leading to lung inflammation and fibrosis (49). Data from BPD survivors indicated signs of lung structure abnormalities such as emphysema and irregular residual lung function (29, 30, 35, 64). From then until now, advances in neonatal care have resulted in higher survival rates of the very low birth weight and more premature infants, resulting in the appearance of a new form of BPD (5). The new BPD is the result of an arrest in lung development, resulting in immature lungs with fewer alveolar structures and reduced surface area for gas exchange (4, 5), similar to the PdgfaAEC2KO mice presented here. Since survivors of the new BPD are only now reaching adulthood, few longitudinal studies of long-term effects have been published, and studies in human tissue are limited. An obvious difference between human patients and PdgfaAEC2KO mice lies in the nature of lung development, as alveolarization starts in utero in humans but postnatally in mice. Whereas infants with BPD initially require hospital care, PdgfaAEC2KO mice grow up without external symptoms, and histological or functional studies are required to distinguish them from their wild-type littermate controls. Also, there are no signs of inflammation or disturbed pulmonary vasculature in the PdgfaAEC2KO mice. Molecular and cellular knowledge is to a large extent based on studies in other animal models (47). The most commonly used murine model to study BPD until now is based on neonatal hyperoxia-induced lung injury (7, 12, 14, 51), which has been useful to study the pathology of the old BDP. We believe that the PdgfaAEC2KO mouse model better resembles the new BPD. PdgfaAEC2KO mice could hence be used to study long-term effects of immature lungs with a reduced surface area for gas exchange and could also be used to test potential therapies to improve lung function in adulthood.
The focus of this paper is on the role of the PDGF-A ligand in the alveolar region. Homodimers of the PDGF-A ligand bind to and activate homodimers of the PDGFR-α. In cultured cells, the PDGFR-α show affinity for PDGF-A/B heterodimers and PDGF-BB homodimers as well. However, in vivo, there are no proofs for these interactions, even though the presence of PDGFR-α/β heterodimers was recently observed during palate formation (43).
Collectively, our data highlight that disruption of PDGF-A signaling from AEC2s during lung development has consequences on the adult lung at the morphological and physiological levels, without causing obvious deleterious effects on the deposition of structural proteins such as elastin.
GRANTS
This study was supported by research grants from the Swedish Research Council (Vetenskapsrådet; 2015-00550 to C. Betsholtz), the European Research Council (AdG294556 to C. Betsholtz), the Fondation Leducq (14CVD02 to C. Betsholtz), Swedish Cancer Society (Cancerfonden; 150735 to C. Betsholtz), Knut och Alice Wallenbergs Stiftelse (2015.0030 to C. Betsholtz), Magnus Bergvalls Stiftelse (2014-00174 to J. Andrae), and Åke Wiberg Stiftelse (362565719, 946216308 to J. Andrae). Funded by the Deutsche Forschungsgemeinschaft (German Research Foundation) project number 268555672 – SFB 1213, Project CP02, A07, and A08.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.G., S.K., S.H., N.W., C.B., and J.A. conceived and designed research; L.G., S.K., S.H., E.V.-L., B.K., C.-Y.W., C.V., G.M., and J.A. performed experiments; L.G., S.K., S.H., B.K., C.V., L.H., G.M., C.B., and J.A. analyzed data; L.G., S.K., S.H., B.K., C.B., and J.A. interpreted results of experiments; L.G., S.K., S.H., and B.K. prepared figures; L.G. and J.A. drafted manuscript; L.G., S.K., S.H., B.K., C.-Y.W., C.B., and J.A. edited and revised manuscript; L.G., S.K., S.H., E.V.-L., B.K., C.-Y.W., C.V., L.H., G.M., R.T.S., N.W., C.B., and J.A. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Jana Chmielniakova, Pia Peterson, Cecilia Olsson, and Helene Leksell for technical and administrative assistance with the mice, and the BioVis core facility at Uppsala University for microscopy technical support. We are grateful for support from Hillevi Andersson Sand at the Clinical Proteomics Mass Spectrometry facility and Karolinska Institutet, where the mass spectrometry analysis was performed. Dr. Mirja Fassbender is acknowledged for assistance with the animal proposal.
Present address of L. Gouveia: Vascular Biology and Signalling Laboratory, Institut d’Investigació Biomèdica de Bellvitge (IDIBELL), L’Hospitalet de Llobregat, Barcelona, Spain.
REFERENCES
- 1.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev 22: 1276–1312, 2008. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrae J, Gouveia L, He L, Betsholtz C. Characterization of platelet-derived growth factor-A expression in mouse tissues using a lacZ knock-in approach. PLoS One 9: e105477, 2014. doi: 10.1371/journal.pone.0105477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arjaans S, Wagner BD, Mourani PM, Mandell EW, Poindexter BB, Berger RMF, Abman SH. Early Angiogenic Proteins Associated with High Risk for Bronchopulmonary Dysplasia and Pulmonary Hypertension in Preterm Infants. Am J Physiol Lung Cell Mol Physiol 318: L644–L654, 2020. doi: 10.1152/ajplung.00131.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baraldi E, Carraro S, Filippone M. Bronchopulmonary dysplasia: definitions and long-term respiratory outcome. Early Hum Dev 85, Suppl: S1–S3, 2009. doi: 10.1016/j.earlhumdev.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med 357: 1946–1955, 2007. doi: 10.1056/NEJMra067279. [DOI] [PubMed] [Google Scholar]
- 6.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BLM. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123: 3025–3036, 2013. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benjamin JT, Smith RJ, Halloran BA, Day TJ, Kelly DR, Prince LS. FGF-10 is decreased in bronchopulmonary dysplasia and suppressed by Toll-like receptor activation. Am J Physiol Lung Cell Mol Physiol 292: L550–L558, 2007. doi: 10.1152/ajplung.00329.2006. [DOI] [PubMed] [Google Scholar]
- 8.Boekel J, Chilton JM, Cooke IR, Horvatovich PL, Jagtap PD, Käll L, Lehtiö J, Lukasse P, Moerland PD, Griffin TJ. Multi-omic data analysis using Galaxy. Nat Biotechnol 33: 137–139, 2015. doi: 10.1038/nbt.3134. [DOI] [PubMed] [Google Scholar]
- 9.Boström H, Gritli-Linde A, Betsholtz C. PDGF-A/PDGF alpha-receptor signaling is required for lung growth and the formation of alveoli but not for early lung branching morphogenesis. Dev Dyn 223: 155–162, 2002. doi: 10.1002/dvdy.1225. [DOI] [PubMed] [Google Scholar]
- 10.Boström H, Willetts K, Pekny M, Levéen P, Lindahl P, Hedstrand H, Pekna M, Hellström M, Gebre-Medhin S, Schalling M, Nilsson M, Kurland S, Törnell J, Heath JK, Betsholtz C. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85: 863–873, 1996. doi: 10.1016/S0092-8674(00)81270-2. [DOI] [PubMed] [Google Scholar]
- 11.Bourbon J, Boucherat O, Chailley-Heu B, Delacourt C. Control mechanisms of lung alveolar development and their disorders in bronchopulmonary dysplasia. Pediatr Res 57: 38R–46R, 2005. doi: 10.1203/01.PDR.0000159630.35883.BE. [DOI] [PubMed] [Google Scholar]
- 12.Bozyk PD, Bentley JK, Popova AP, Anyanwu AC, Linn MD, Goldsmith AM, Pryhuber GS, Moore BB, Hershenson MB. Neonatal periostin knockout mice are protected from hyperoxia-induced alveolar simplication. PLoS One 7: e31336, 2012. [Erratum in PLoS One 10: e0130369, 2015] doi: 10.1371/journal.pone.0031336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Branca RMM, Orre LM, Johansson HJ, Granholm V, Huss M, Pérez-Bercoff Å, Forshed J, Käll L, Lehtiö J. HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat Methods 11: 59–62, 2014. doi: 10.1038/nmeth.2732. [DOI] [PubMed] [Google Scholar]
- 14.Branchfield K, Li R, Lungova V, Verheyden JM, McCulley D, Sun X. A three-dimensional study of alveologenesis in mouse lung. Dev Biol 409: 429–441, 2016. doi: 10.1016/j.ydbio.2015.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carey MA, Card JW, Voltz JW, Arbes SJ Jr, Germolec DR, Korach KS, Zeldin DC. It’s all about sex: gender, lung development and lung disease. Trends Endocrinol Metab 18: 308–313, 2007. doi: 10.1016/j.tem.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen L, Acciani T, Le Cras T, Lutzko C, Perl A-KT. Dynamic regulation of platelet-derived growth factor receptor α expression in alveolar fibroblasts during realveolarization. Am J Respir Cell Mol Biol 47: 517–527, 2012. doi: 10.1165/rcmb.2012-0030OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi S-M, McAleer JP, Zheng M, Pociask DA, Kaplan MH, Qin S, Reinhart TA, Kolls JK. Innate Stat3-mediated induction of the antimicrobial protein Reg3γ is required for host defense against MRSA pneumonia. J Exp Med 210: 551–561, 2013. doi: 10.1084/jem.20120260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507: 190–194, 2014. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong L, Wang S-J, Camoretti-Mercado B, Li H-J, Chen M, Bi W-X. FIZZ1 plays a crucial role in early stage airway remodeling of OVA-induced asthma. J Asthma 45: 648–653, 2008. doi: 10.1080/02770900802126941. [DOI] [PubMed] [Google Scholar]
- 20.Du Y, Guo M, Whitsett JA, Xu Y. ‘LungGENS’: a web-based tool for mapping single-cell gene expression in the developing lung. Thorax 70: 1092–1094, 2015. doi: 10.1136/thoraxjnl-2015-207035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du Y, Kitzmiller JA, Sridharan A, Perl AK, Bridges JP, Misra RS, Pryhuber GS, Mariani TJ, Bhattacharya S, Guo M, Potter SS, Dexheimer P, Aronow B, Jobe AH, Whitsett JA, Xu Y. Lung Gene Expression Analysis (LGEA): an integrative web portal for comprehensive gene expression data analysis in lung development. Thorax 72: 481–484, 2017. doi: 10.1136/thoraxjnl-2016-209598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egaña I, Kaito H, Nitzsche A, Becker L, Ballester-Lopez C, Niaudet C, Petkova M, Liu W, Vanlandewijck M, Vernaleken A, Klopstock T, Fuchs H, Gailus-Durner V, Hrabe de Angelis M, Rask-Andersen H, Johansson HJ, Lehtiö J, He L, Yildirim AÖ, Hellström M; German Mouse Clinic Consortium . Female mice lacking Pald1 exhibit endothelial cell apoptosis and emphysema. Sci Rep 7: 15453, 2017. doi: 10.1038/s41598-017-14894-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Endale M, Ahlfeld S, Bao E, Chen X, Green J, Bess Z, Weirauch MT, Xu Y, Perl A-K. Temporal, spatial, and phenotypical changes of PDGFRα expressing fibroblasts during late lung development. Dev Biol 425: 161–175, 2017. doi: 10.1016/j.ydbio.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ford NL, Martin EL, Lewis JF, Veldhuizen RAW, Drangova M, Holdsworth DW. In vivo characterization of lung morphology and function in anesthetized free-breathing mice using micro-computed tomography. J Appl Physiol (1985) 102: 2046–2055, 2007. doi: 10.1152/japplphysiol.00629.2006. [DOI] [PubMed] [Google Scholar]
- 25.Forfia PR, Vaidya A, Wiegers SE. Pulmonary heart disease: The heart-lung interaction and its impact on patient phenotypes. Pulm Circ 3: 5–19, 2013. doi: 10.4103/2045-8932.109910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fulton CT, Cui TX, Goldsmith AM, Bermick J, Popova AP. Gene Expression Signatures Point to a Male Sex-Specific Lung Mesenchymal Cell PDGF Receptor Signaling Defect in Infants Developing Bronchopulmonary Dysplasia. Sci Rep 8: 17070, 2018. doi: 10.1038/s41598-018-35256-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gouveia L, Betsholtz C, Andrae J. Expression analysis of platelet-derived growth factor receptor alpha and its ligands in the developing mouse lung. Physiol Rep 5: e13092, 2017. doi: 10.14814/phy2.13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gouveia L, Betsholtz C, Andrae J. PDGF-A signaling is required for secondary alveolar septation and controls epithelial proliferation in the developing lung. Development 145: dev161976, 2018. doi: 10.1242/dev.161976. [DOI] [PubMed] [Google Scholar]
- 29.Hakulinen AL, Heinonen K, Länsimies E, Kiekara O. Pulmonary function and respiratory morbidity in school-age children born prematurely and ventilated for neonatal respiratory insufficiency. Pediatr Pulmonol 8: 226–232, 1990. doi: 10.1002/ppul.1950080404. [DOI] [PubMed] [Google Scholar]
- 30.Halvorsen T, Skadberg BT, Eide GE, Røksund OD, Carlsen KH, Bakke P. Pulmonary outcome in adolescents of extreme preterm birth: a regional cohort study. Acta Paediatr 93: 1294–1300, 2004. doi: 10.1111/j.1651-2227.2004.tb02926.x. [DOI] [PubMed] [Google Scholar]
- 31.Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol Cell Biol 23: 4013–4025, 2003. doi: 10.1128/MCB.23.11.4013-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hrycaj SM, Marty-Santos L, Cebrian C, Rasky AJ, Ptaschinski C, Lukacs NW, Wellik DM. Hox5 genes direct elastin network formation during alveologenesis by regulating myofibroblast adhesion. Proc Natl Acad Sci USA 115: E10605–E10614, 2018. doi: 10.1073/pnas.1807067115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovacevic I, Müller M, Kojonazarov B, Ehrke A, Randriamboavonjy V, Kohlstedt K, Hindemith T, Schermuly RT, Fleming I, Hoffmeister M, Oess S. The F-BAR Protein NOSTRIN Dictates the Localization of the Muscarinic M3 Receptor and Regulates Cardiovascular Function. Circ Res 117: 460–469, 2015. doi: 10.1161/CIRCRESAHA.115.306187. [DOI] [PubMed] [Google Scholar]
- 34.Kroon AA, Wang J, Post M. Alterations in expression of elastogenic and angiogenic genes by different conditions of mechanical ventilation in newborn rat lung. Am J Physiol Lung Cell Mol Physiol 308: L639–L649, 2015. doi: 10.1152/ajplung.00293.2014. [DOI] [PubMed] [Google Scholar]
- 35.Lemons JA, Bauer CR, Oh W, Korones SB, Papile L-A, Stoll BJ, Verter J, Temprosa M, Wright LL, Ehrenkranz RA, Fanaroff AA, Stark A, Carlo W, Tyson JE, Donovan EF, Shankaran S, Stevenson DK. Very low birth weight outcomes of the National Institute of Child health and human development neonatal research network, January 1995 through December 1996. NICHD Neonatal Research Network. Pediatrics 107: E1, 2001. doi: 10.1542/peds.107.1.e1. [DOI] [PubMed] [Google Scholar]
- 36.Li C, Lee MK, Gao F, Webster S, Di H, Duan J, Yang C-Y, Bhopal N, Peinado N, Pryhuber G, Smith SM, Borok Z, Bellusci S, Minoo P. Secondary crest myofibroblast PDGFRα controls the elastogenesis pathway via a secondary tier of signaling networks during alveologenesis. Development 146: dev176354, 2019. doi: 10.1242/dev.176354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li R, Bernau K, Sandbo N, Gu J, Preissl S, Sun X. Pdgfra marks a cellular lineage with distinct contributions to myofibroblasts in lung maturation and injury response. eLife 7: e36865, 2018. doi: 10.7554/eLife.36865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindahl P, Karlsson L, Hellström M, Gebre-Medhin S, Willetts K, Heath JK, Betsholtz C. Alveogenesis failure in PDGF-A-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development 124: 3943–3953, 1997. [DOI] [PubMed] [Google Scholar]
- 39.Liu T, Dhanasekaran SM, Jin H, Hu B, Tomlins SA, Chinnaiyan AM, Phan SH. FIZZ1 stimulation of myofibroblast differentiation. Am J Pathol 164: 1315–1326, 2004. doi: 10.1016/S0002-9440(10)63218-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu T, Yu H, Ullenbruch M, Jin H, Ito T, Wu Z, Liu J, Phan SH. The in vivo fibrotic role of FIZZ1 in pulmonary fibrosis. PLoS One 9: e88362, 2014. doi: 10.1371/journal.pone.0088362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loscertales M, Nicolaou F, Jeanne M, Longoni M, Gould DB, Sun Y, Maalouf FI, Nagy N, Donahoe PK. Type IV collagen drives alveolar epithelial-endothelial association and the morphogenetic movements of septation. BMC Biol 14: 59, 2016. [Erratum in BMC Biol 14: 73, 2016] doi: 10.1186/s12915-016-0281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo Y, Li N, Chen H, Fernandez GE, Warburton D, Moats R, Mecham RP, Krenitsky D, Pryhuber GS, Shi W. Spatial and temporal changes in extracellular elastin and laminin distribution during lung alveolar development. Sci Rep 8: 8334, 2018. doi: 10.1038/s41598-018-26673-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCarthy N, Liu JS, Richarte AM, Eskiocak B, Lovely CB, Tallquist MD, Eberhart JK. Pdgfra and Pdgfrb genetically interact during craniofacial development. Dev Dyn 245: 641–652, 2016. doi: 10.1002/dvdy.24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McEvoy CT, Jain L, Schmidt B, Abman S, Bancalari E, Aschner JL. Bronchopulmonary dysplasia: NHLBI Workshop on the Primary Prevention of Chronic Lung Diseases. Ann Am Thorac Soc 11, Suppl 3: S146–S153, 2014. doi: 10.1513/AnnalsATS.201312-424LD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moggridge S, Sorensen PH, Morin GB, Hughes CS. Extending the Compatibility of the SP3 Paramagnetic Bead Processing Approach for Proteomics. J Proteome Res 17: 1730–1740, 2018. doi: 10.1021/acs.jproteome.7b00913. [DOI] [PubMed] [Google Scholar]
- 46.Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359: 1118–1123, 2018. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nardiello C, Mižíková I, Morty RE. Looking ahead: where to next for animal models of bronchopulmonary dysplasia? Cell Tissue Res 367: 457–468, 2017. doi: 10.1007/s00441-016-2534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nguyen NM, Kelley DG, Schlueter JA, Meyer MJ, Senior RM, Miner JH. Epithelial laminin α5 is necessary for distal epithelial cell maturation, VEGF production, and alveolization in the developing murine lung. Dev Biol 282: 111–125, 2005. doi: 10.1016/j.ydbio.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 49.Northway WH Jr, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl J Med 276: 357–368, 1967. doi: 10.1056/NEJM196702162760701. [DOI] [PubMed] [Google Scholar]
- 50.Ntokou A, Klein F, Dontireddy D, Becker S, Bellusci S, Richardson WD, Szibor M, Braun T, Morty RE, Seeger W, Voswinckel R, Ahlbrecht K. Characterization of the platelet-derived growth factor receptor-α-positive cell lineage during murine late lung development. Am J Physiol Lung Cell Mol Physiol 309: L942–L958, 2015. doi: 10.1152/ajplung.00272.2014. [DOI] [PubMed] [Google Scholar]
- 51.Oak P, Pritzke T, Thiel I, Koschlig M, Mous DS, Windhorst A, Jain N, Eickelberg O, Foerster K, Schulze A, Goepel W, Reicherzer T, Ehrhardt H, Rottier RJ, Ahnert P, Gortner L, Desai TJ, Hilgendorff A. Attenuated PDGF signaling drives alveolar and microvascular defects in neonatal chronic lung disease. EMBO Mol Med 9: 1504–1520, 2017. doi: 10.15252/emmm.201607308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okubo T, Hogan BLM. Hyperactive Wnt signaling changes the developmental potential of embryonic lung endoderm. J Biol 3: 11, 2004. doi: 10.1186/jbiol3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pinkerton KE, Harbaugh M, Han MK, Jourdan Le Saux C, Van Winkle LS, Martin WJ II, Kosgei RJ, Carter EJ, Sitkin N, Smiley-Jewell SM, George M. Women and Lung Disease. Sex Differences and Global Health Disparities. Am J Respir Crit Care Med 192: 11–16, 2015. doi: 10.1164/rccm.201409-1740PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Popova AP, Bentley JK, Cui TX, Richardson MN, Linn MJ, Lei J, Chen Q, Goldsmith AM, Pryhuber GS, Hershenson MB. Reduced platelet-derived growth factor receptor expression is a primary feature of human bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 307: L231–L239, 2014. doi: 10.1152/ajplung.00342.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Popova AP. Mechanisms of bronchopulmonary dysplasia. J Cell Commun Signal 7: 119–127, 2013. doi: 10.1007/s12079-013-0190-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Randell SH. Airway epithelial stem cells and the pathophysiology of chronic obstructive pulmonary disease. Proc Am Thorac Soc 3: 718–725, 2006. doi: 10.1513/pats.200605-117SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodríguez-Castillo JA, Pérez DB, Ntokou A, Seeger W, Morty RE, Ahlbrecht K. Understanding alveolarization to induce lung regeneration. Respir Res 19: 148, 2018. doi: 10.1186/s12931-018-0837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, Alexander SN, Bellinghausen LK, Song AS, Petrova YM, Tuvim MJ, Adachi R, Romo I, Bordt AS, Bowden MG, Sisson JH, Woodruff PG, Thornton DJ, Rousseau K, De la Garza MM, Moghaddam SJ, Karmouty-Quintana H, Blackburn MR, Drouin SM, Davis CW, Terrell KA, Grubb BR, O’Neal WK, Flores SC, Cota-Gomez A, Lozupone CA, Donnelly JM, Watson AM, Hennessy CE, Keith RC, Yang IV, Barthel L, Henson PM, Janssen WJ, Schwartz DA, Boucher RC, Dickey BF, Evans CM. Muc5b is required for airway defence. Nature 505: 412–416, 2014. doi: 10.1038/nature12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruiz-Camp J, Quantius J, Lignelli E, Arndt PF, Palumbo F, Nardiello C, Surate Solaligue DE, Sakkas E, Mižíková I, Rodríguez-Castillo JA, Vadász I, Richardson WD, Ahlbrecht K, Herold S, Seeger W, Morty RE. Targeting miR-34a/Pdgfra interactions partially corrects alveologenesis in experimental bronchopulmonary dysplasia. EMBO Mol Med 11: e9448, 2019. doi: 10.15252/emmm.201809448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Savitski MM, Wilhelm M, Hahne H, Kuster B, Bantscheff M. A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol Cell Proteomics 14: 2394–2404, 2015. doi: 10.1074/mcp.M114.046995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108, 2008. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 62.Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, Milger K, Egemnazarov B, Turowska A, Fuchs B, Nikam S, Roth M, Sydykov A, Medebach T, Klepetko W, Jaksch P, Dumitrascu R, Garn H, Voswinckel R, Kostin S, Seeger W, Schermuly RT, Grimminger F, Ghofrani HA, Weissmann N. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 147: 293–305, 2011. doi: 10.1016/j.cell.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 63.Shavelle RM, Paculdo DR, Kush SJ, Mannino DM, Strauss DJ. Life expectancy and years of life lost in chronic obstructive pulmonary disease: findings from the NHANES III Follow-up Study. Int J Chron Obstruct Pulmon Dis 4: 137–148, 2009. doi: 10.2147/COPD.S5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singer L, Yamashita T, Lilien L, Collin M, Baley J. A longitudinal study of developmental outcome of infants with bronchopulmonary dysplasia and very low birth weight. Pediatrics 100: 987–993, 1997. doi: 10.1542/peds.100.6.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Surate Solaligue DE, Rodríguez-Castillo JA, Ahlbrecht K, Morty RE. Recent advances in our understanding of the mechanisms of late lung development and bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 313: L1101–L1153, 2017. doi: 10.1152/ajplung.00343.2017. [DOI] [PubMed] [Google Scholar]
- 66.Szardien S, Nef HM, Voss S, Troidl C, Liebetrau C, Hoffmann J, Rauch M, Mayer K, Kimmich K, Rolf A, Rixe J, Troidl K, Kojonazarov B, Schermuly RT, Kostin S, Elsässer A, Hamm CW, Möllmann H. Regression of cardiac hypertrophy by granulocyte colony-stimulating factor-stimulated interleukin-1β synthesis. Eur Heart J 33: 595–605, 2012. doi: 10.1093/eurheartj/ehr434. [DOI] [PubMed] [Google Scholar]
- 67.Tallquist M, Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev 15: 205–213, 2004. doi: 10.1016/j.cytogfr.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 68.Wagenseil JE, Mecham RP. New insights into elastic fiber assembly. Birth Defects Res C Embryo Today 81: 229–240, 2007. doi: 10.1002/bdrc.20111. [DOI] [PubMed] [Google Scholar]
- 69.Walter EC, Ehlenbach WJ, Hotchkin DL, Chien JW, Koepsell TD. Low birth weight and respiratory disease in adulthood: a population-based case-control study. Am J Respir Crit Care Med 180: 176–180, 2009. doi: 10.1164/rccm.200901-0046OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wendel DP, Taylor DG, Albertine KH, Keating MT, Li DY. Impaired distal airway development in mice lacking elastin. Am J Respir Cell Mol Biol 23: 320–326, 2000. doi: 10.1165/ajrcmb.23.3.3906. [DOI] [PubMed] [Google Scholar]
- 71.Wong PM, Lees AN, Louw J, Lee FY, French N, Gain K, Murray CP, Wilson A, Chambers DC. Emphysema in young adult survivors of moderate-to-severe bronchopulmonary dysplasia. Eur Respir J 32: 321–328, 2008. doi: 10.1183/09031936.00127107. [DOI] [PubMed] [Google Scholar]
- 72.Yeo GC, Keeley FW, Weiss AS. Coacervation of tropoelastin. Adv Colloid Interface Sci 167: 94–103, 2011. doi: 10.1016/j.cis.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 73.Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, Morrisey EE. Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell 170: 1134–1148.e10, 2017. doi: 10.1016/j.cell.2017.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]