

Abstract
This study sets out to establish the comparative contribution of PD-L1 expression by pulmonary endothelial cells (ECs) and/or epithelial cells (EpiCs) to the development of indirect acute lung injury (iALI) by taking advantage of the observation that treatment with naked siRNA by intratracheal delivery in mice primarily affects lung EpiCs, but not lung ECs, while intravenous delivery of liposomal-encapsulated siRNA largely targets vascular ECs including the lung, but not pulmonary EpiCs. We showed that using a mouse model of iALI [induced by hemorrhagic shock followed by septic challenge (Hem-CLP)], PD-L1 expression on pulmonary ECs or EpiCs was significantly upregulated in the iALI mice at 24 h post–septic insult. After documenting the selective ability of intratracheal versus intravenous delivery of PD-L1 siRNA to inhibit PD-L1 expression on EpiCs versus ECs, respectively, we observed that the iALI-induced elevation of cytokine/chemokine levels (in the bronchoalveolar lavage fluid, lung lysates, or plasma), lung myeloperoxidase and caspase-3 activities could largely only be inhibited by intravenous, but not intratracheal, delivery of PD-L1 siRNA. Moreover, intravenous, but not intratracheal, delivery led to a preservation of normal tissue architecture, lessened pulmonary edema, and reduced neutrophils influx induced by iALI. In addition, in vitro mouse endothelial cell line studies showed that PD-L1 gene knockdown by siRNA or knockout by CRISPR/Cas9-mediated gene manipulation, reduced monolayer permeability, and maintained tight junction protein levels upon recombinant IFN-γ stimulation. Together, these data imply a critical role for pulmonary vascular ECs in mediating PD-1:PD-L1–driven pathological changes resulting from systemic stimuli such as Hem-CLP.
Keywords: ARDS, co-inhibitory receptor, hemorrhage, permeability, siRNA
INTRODUCTION
Acute respiratory distress syndrome (ARDS), a rapidly progressive lung condition, is characterized by increased microvascular permeability, pulmonary edema, widespread inflammation, and severe hypoxemia (35). Risks of ARDS are normally divided into two categories: 1) direct lung injury, caused mainly by pneumonia, and 2) indirect lung injury (iALI) resulting from sepsis, trauma, hemorrhagic shock, pancreatitis, etc. Despite the significant advances in supportive care, e.g., lung protective strategies, proning, paralytics, extracorporeal membrane oxygenation, and development of antibiotics, the mortality remains unacceptably high (6, 26, 29, 34). Currently, protective ventilation with a low tidal volume is the only effective new therapeutic method developed recently, implying that the pathophysiology of ARDS needs to be better elucidated if we are to ever develop novel pharmacological targets for this condition (28).
It is thought that the pathogenesis of iALI involves the activation of neutrophils, native lung endothelial cells (ECs), and/or epithelial cells (EpiCs) and their respective interactions within the immune system (5, 30). Neutrophils are thought to be primed (e.g., exhibited an increased capacity to produce a respiratory burst and actively suppress their constitutive ability to undergo apoptosis) and recruited to the lung tissue through interaction with ECs, which then induce respiratory burst and oxidative tissue damage (2, 8, 15). Oxygen free radicals, mainly produced by primed neutrophils, trigger the local tissue injury/damage, inflammation, and dysfunction of native ECs/ EpiCs. The loss of endothelial cell integrity increases the vascular permeability, resulting in accumulating fluids and proteins within the lung tissue, which in turn facilitates the migration of neutrophils and lymphocytes out of circulation (13). However, the mechanism by which neutrophil infiltration or inflammatory mediator release leads to the loss of endothelial/epithelial barrier integrity is not fully understood.
Programmed cell death receptor-1 (PD-1 or CD279) is a co-inhibitory cell surface protein expressed on leukocytes; it is classically described as playing a central role in controlling the excessive activation of T cells. PD-L1, the primary ligand of PD-1, is widely expressed on immune cells as well as nonimmune cell lineages, such as lung/liver/intestinal endothelial and epithelial cells (12). Previous studies in our laboratory have shown that mice globally lacking the gene for PD-1 or PD-L1 were better able to survive experimental septic challenge (10, 11). Moreover, in a mouse model of iALI induced by hemorrhagic shock followed by cecal ligation and puncture (Hem-CLP), we have documented that mice globally deficient in either PD-1 or the ligand, PD-L1, not only reduced the indices of lung injury and survival (19, 23), but moreover, we found that PD-1 expression was associated with pulmonary inflammatory burden, neutrophil activation, and cell apoptosis, implying that PD-1:PD-L1 pathway may have a special role in the regulation of iALI development (24). While it is evident that PD-1:PD-L1 interaction inhibits the overactivation of lymphocytes, little is known about how the PD-L1 expression may influence the function of nonimmune cells, especially endothelial cells and epithelial cells. How PD-L1 expression on lung ECs/EpiCs may be regulated in iALI and/or if such changes effect EC/ EpiC monolayer integrity or permeability is poorly understood.
In this respect, our recent studies have shown that intratracheal (a local lung delivery format) delivery of naked (nonliposome encapsulated) small interfering (si)RNA primarily targets lung EpiCs, but does not target pulmonary macrophages (MØs) or ECs. On the other hand, intravenous (systemic delivery route) delivery of liposomal-encapsulated siRNA targets primarily vascular ECs and some blood phagocytes (but not lung EpiCs) (16, 21, 27, 31). Inasmuch, here we propose to test the hypothesis that by taking advantage of the unique nature of cell targeting produced by intratracheal as opposed to intravenous siRNA delivery, we should be able to establish at what cellular level the PD-L1 gene product contributes to the development of iALI resulting from the sequential insults of Hem-CLP in mice.
MATERIALS AND METHODS
Mice.
Male C57BL/6 mice from Jackson Laboratory (Bar Harbor, ME) were utilized at 8–12 wk old. This choice was made so as to maximize our ability to initially see an experimental difference in the iALI response based on previous reports that male mice did poorer in response to these experimental stressors of shock (hemorrhage) and/or septic (CLP) challenge than pro-estrus-stratified female mice (36, 38). Protocols were performed in accordance with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee of Rhode Island Hospital (AWC no. 0110-13 and 0040-16).
Mouse model of iALI (Hem-CLP).
Hemorrhagic shock (Hem; nonlethal, fixed pressure) followed by a septic challenge–cecal ligation and puncture (CLP) 24 h later was used to induce iALI, as previously described (2, 16, 18, 21, 25, 27, 31). Using this model we have shown induces arterial blood Po2/, (mmHg) of ~150 mmHg by 24 h post-Hem-CLP, along with evidence of protein leak, edema, morphological changes, increase lung tissue levels of pro-inflammatory cytokines/chemokines, and neutrophil influx into the lungs [as assessed by immune-histochemistry and tissue myeloperoxidase (MPO) levels] (2, 16, 18, 21, 25, 27, 31).
siRNA delivery.
Mice received siRNA at 2 h following shock resuscitation via intratracheal or intravenous delivery (16, 21, 27). Mouse PD-L1 siRNA-SMART pool sequences 1. 5′-UGAGCAUGAACUAAUAUGU-3′, 2. 5′- (19) GUAUCAGCUCUCAGAUUUC-3′, 3. 5′-UCUGUAGACACCAUUUAUA-3′, 4. 5′-CAUGGUGUUGGAUUGGUG-3′), and scrambled (control) sequence were from Dharmacon (Lafayette, CO).
Sample acquisition.
Mice were euthanized 24 h after CLP with an overdose of isoflurane. Blood/plasma, bronchoalveolar lavage fluids (BALF), and lung tissues were collected to assess cytokine/chemokine, protein concentration (as a measurement for pulmonary vascular leak), lung MPO, caspase-3, histology, and flow cytometry as described previously (2, 16, 21, 25, 27, 31).
Flow cytometry.
Cells were stained with anti-CD31 (EC), -CD326/Epcam (EpiC), and -CD274 (PD-L1), then examined by flow cytometry (Miltenyi Biotec, Auburn, CA) as previously described (20, 21). Data were analyzed using FlowJo software.
Quantification of cytokines and chemokines.
ELISA kits were employed to measure the levels of TNF-α, IL-6, IL-10, and MCP-1 (Biolegend, San Diego, CA); KC and MIP-2 (R&D Systems, Minneapolis, MN) in BALF, lung tissue homogenate supernatants, and plasma (10, 11).
MPO and caspase-3 activity assay.
MPO and caspase-3 in lung tissue homogenates were measured according to methods described previously (2, 16, 27).
Lung histology and immunohistochemical/immunofluorescent staining.
Lung sections were stained with H&E or naphthol AS-D chloroacetate esterase (Sigma-Aldrich, St. Louis, MO). To quantify the lung morphology as an index of tissue injury, lung morphometric analysis was performed using ImageJ software (NIH, Bethesda, MD) as previously described (2). The percentage of open alveolar space in each image was determined following thresholding out the bright alveolar space and the pixel number filled by the tissue (septae) using “flood-object measurements.” The % alveolar space/field was calculated from the total pixel number per field. Cell line monolayers were stained for tight junction protein, zona occludens-1 (ZO-1), or occludin (OCC). The images were collected with a Nikon Eclipse 80i microscope using a ×20 objective and a Spot RT3 camera. Slides were randomly screened and blindly evaluated with three to six images acquired per specimen (2, 16, 27, 37).
In vitro cell culture and PD-L1 gene silencing.
Mouse lung epithelial cell line-LA4 and endothelial cell line CRL-2167 (CRL) were obtained from ATCC (Manassas, VA). To confirm the efficacy of anti-PD-L1 siRNA, CRL cells were cultured, transfected with anti-PD-L1 or control siRNA, and stimulated with recombinant (r)IFN-γ to induce PD-L1 expression. PD-L1 mRNA and protein were determined following transfection (37). The optimal concentration of rIFN-γ needed to maximally stimulate the expression of PD-L1 by CRL cells was determined based on a dose-response study. We confirmed that CRL cells could not only constitutively express PD-L1 protein (by flow cytometry), but that maximal induction occurred at 20 ng/mL IFN-γ stimulation (Fig. 1). PD-L1 knockout (PD-L1−/−) CRL cells were generated by CRISPR/Cas9 system according to manufacturer’s instructions (Santa Cruz, Dallas, TX). The stable PD-L1−/− CRL cells were confirmed by analyzing gene and protein expression.
Fig. 1.
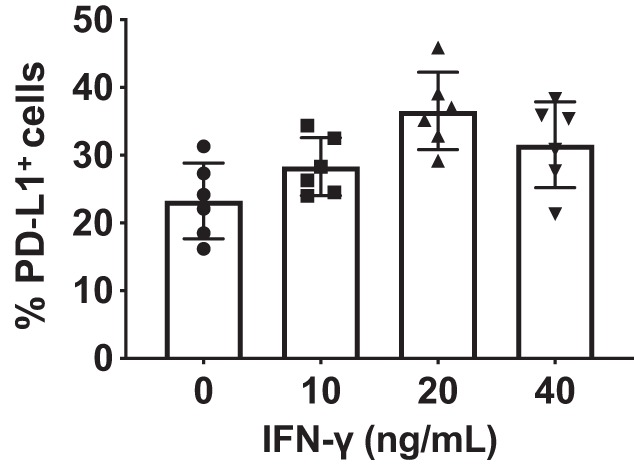
Dose response of mouse recombinant (r)IFN-γ induces an upregulation of PD-L1 expression on CRL-2167 (CRL) cells. Cells were plated and stimulated with mouse rIFN-γ in a dose-response manner to determine the optimum concentration for the expression of PD-L1 determined by flow cytometry. PD-L1 was constitutively expressed without simulation (0 IFN), however, after rIFN-γ stimulation at concentrations reached a plateau at 20–40 ng/mL. Data represent an average of three independent experiments.
Quantification of gene expression.
Total RNA was prepared for real-time qPCR following the manufacturer’s instructions (Life Technologies). The expression of PD-L1 was obtained by normalizing to 18S and relative to a control (nonstimulated cells) (37).
Permeability assay of CRL monolayer.
Paracellular permeability of CRL monolayer, stimulated with/without rIFN-γ, was accessed by flux of fluorescein isothiocyanate-dextran (FD4, Sigma) using the Transwell plates (37).
Western blot analysis.
Protein samples prepared for ZO-1 and OCC expression were determined by Western blot. Image densitometry was collected and analyzed with ImageJ software. GAPDH was used as a loading control (Life Technologies) (37).
Statistical analysis.
Data are expressed as means ± SE and analyzed using GraphPad Prism 5. Multigroup comparisons were performed using one-way analysis of variance (ANOVA) followed by Student–Newman–Keuls test. P < 0.05 was considered to be statistically significant.
RESULTS
PD-L1 expression on lung endothelial/epithelial cells was significantly elevated in iALI.
Moving from our initial observation that the mouse endothelial cell line, CRL, not only constitutively expresses PD-L1 protein (by flow cytometry), but could be stimulated by a proinflammatory cytokine, such as IFN-γ, to express higher levels of PD-L1 in vitro (Fig. 1), we chose, subsequently, to use a mouse model of Hem-CLP-induced iALI (2, 16, 21, 25, 27, 31) to initially examine the PD-L1 expression in vivo on native lung EpiCs/ECs by flow cytometry. As shown in Fig. 2, A–D, at 24 h post–septic insult (48 h post-Hem), the PD-L1 expression on EpiCs (Fig. 2, A and B)/ECs (Fig. 2, C and D) was significantly upregulated compared with sham-operated mice. In addition, our recent studies have documented that PD-L1 gene deficiency is associated with a reduced mortality induced by iALI in adult mice (20). These data imply that PD-L1 expression might be involved in the development of iALI.
Fig. 2.

Hemorrhagic shock followed by cecal ligation and puncture (Hem-CLP) enhanced PD-L1 expression on lung epithelial and endothelial cells in vivo and PD-L1 siRNA suppresses PD-L1 gene/protein expression in CRL-2167 (CRL) and stimulated LA-4 cells in vitro. PD-L1 protein expression on Epcam+ or CD31+ lung cells was determined by flow cytometry. Typical PD-L1 expression/mean fluorescent intensity (MFI) histogram for those cells positive for either the epithelial cell (A), Epcam+, or the endothelial cell (C), CD31+, markers. The horizontal “line” in those histograms identifies cells determined to be positive for both PD-L1+Epcam+ or PD-L1+CD31+ (from which the %PD-L1+Epcam+ or %PD-L1+CD31+ was established) based on “isotype” antibody-chromophore control. The frequency of PD-L1+Epcam+ or PD-L1+CD31+ was significantly increased on lung epithelial (B) and endothelial (D) cells from Hem-CLP compared with sham-operated mice (means ± SE; n = 4–6 mice per group; *P < 0.05 vs. sham group, rank-sum test). To verify the efficacy of PD-L1 siRNA, the mouse lung epithelial cell line LA4 [E–F (F’)] and the endothelial cell line CRL (G and H) were used. PD-L1 mRNA and protein were determined following siRNA transfection. Subsequently, the expression of PD-L1 gene (E) and protein (F) [representative dot plots of LA4 cell side-scatter (SSC) profile vs. PD-L1 staining intensity for each condition summarized for multiple experiments is shown in F’] of LA4 cells were increased after rIFN-γ stimulation and reduced with anti-PD-L1 siRNA. Endothelial cell line CRL, which constitutively expresses PD-L1, when treated with anti-PD-L1, but not Control, siRNA treatment reduced PD-L1 gene (G) and protein (H) expression (means ± SE; n = 4–6 individual experiments/group; *P < 0.05 vs. PBS alone group; #P < 0.05 vs. PD-L1 siRNA-treated group, by one-way ANOVA and a Student–Newman–Keuls test).
PD-L1 siRNA treatment downregulated gene/protein expression in vitro.
To establish the pathological role of PD-L1 expression on pulmonary ECs and/or EpiCs in mouse iALI, we chose to treat the mice with anti-PD-L1 siRNA in vivo based on our previous experiences (16, 21, 27, 31). Before these experiments, however, we needed to broadly determine the efficiency of PD-L1 siRNA silencing. Using LA4 mouse lung epithelial cell line and CRL mouse endothelial cell line, we detected a significant increase in both the PD-L1 mRNA and protein expressions in LA4 cells stimulated with rIFN-γ. PD-L1 siRNA treatment markedly reduced PD-L1 gene expression (~75% and 70% reduction compared with PBS and control siRNA, respectively) (Fig. 2E), and protein levels reduced ~80% and 77% compared with PBS and control siRNA, respectively (Fig. 2F). This was further confirmed by flow cytometry (Fig. 2F’). Similarly, PD-L1 gene expression in CRL cells (without rIFN-γ treatment, as PD-L1 is constitutively expressed in CRL) was suppressed by treatment with PD-L1-specific siRNA (~50% and 20% reduction compared with PBS and control siRNA, respectively) (Fig. 2G). Suppression of CRL cell surface PD-L1 protein expression after siRNA treatment (~60% and 40% reduction compared with PBS and control siRNA, respectively) (Fig. 2H). Hence, these data support the use of PD-L1 siRNA in the subsequent experimental mouse model in vivo.
PD-L1 siRNA treatment selectively downregulated gene expression depending on the route/nature of siRNA administration in vivo.
To delineate the effects of PD-L1 on pulmonary ECs and/or EpiCs in mouse iALI, we took advantage of our previous observation that intratracheal delivery of naked siRNA primarily affects lung EpiCs (Epicam+ cells), but not lung ECs (CD31+ cells), while intravenous delivery of liposomal-encapsulated siRNA largely targets vascular ECs, including in the lung, but not pulmonary EpiCs (16, 21, 27, 31). Mice underwent hemorrhagic shock and were then treated with either intratracheal or intravenous delivery of PD-L1 or control siRNA 2 h after resuscitation. The dose we used in this study was selected according to previous research (16, 21, 27, 31). At 24 h following CLP, mice were euthanized and lung samples were examined. As shown in Fig. 3, a significantly lower level of PD-L1 gene expression in the whole lung tissue of mice that received either intravenous (Fig. 3A; ~50% and 55% reduction compared with PBS and control siRNA, respectively) delivery of liposomal-encapsulated or intratracheal (Fig. 3D; ~35% and 45% reduction compared with PBS and control siRNA, respectively) delivery of naked anti-PD-L1 siRNA suggests that the siRNA is effective in vivo as well. However, on closer flow cytometric phenotypic examination, we observed that the mice receiving intravenous delivery of siRNA had significantly lower PD-L1 expression on lung CD31+ cells (ECs) (Fig. 3B) when compared with mice that received control siRNA, and this was not seen on pulmonary Epcam+ cells (EpiCs) (Fig. 3E). In contrast, intratracheal delivery of naked PD-L1 siRNA inhibited the PD-L1 expression on pulmonary Epcam+ EpiCs (Fig. 3F), but not CD31+ ECs (Fig. 3E). These findings are also consistent with our prior research (16, 21, 27, 31), implying that the administration of siRNA can be cell-type specific when given via these different delivery methods.
Fig. 3.

Differential effects of intravenous (I.V.) versus intratracheal (I.T.) delivered PD-L1 siRNA on suppressing PD-L1 expression in the lungs of mice with indirect acute lung injury (iALI). The lungs were collected 24 h after hemorrhagic shock followed by cecal ligation and puncture (Hem-CLP) or sham operation. Lung tissue homogenate lysates were used for PD-L1 gene expression. PD-L1 gene expression was upregulated in all iALI mice compared with sham mice and was selectively suppressed by anti-PD-L1 siRNA when delivered either intravenously (A) or intratracheally (D). Enzymatic digested lung single-cell suspensions were examined for PD-L1 protein expression by flow cytometry. Intravenous delivery of siRNA suppressed PD-L1 expression by CD31+ lung endothelial cells (B and B′: a representative dot plot of CD31+ cells), but not by Epcam+ lung epithelial cells of Hem-CLP mice (C). Alternatively, intratracheal delivery of siRNA inhibited the PD-L1 expression on Epcam+ lung epithelial cells (F and F′: a representative dot plot of Epcam+ cells), but not CD31+ lung endothelial cells (E). Values are means ± SE; n = 4–6 mice per group; *P < 0.05 vs. Sham; #P < 0.05 vs. PD-L1 siRNA-treated Hem-CLP group, by one-way ANOVA and a Student–Newman–Keuls test.
Intravenous delivery of PD-L1-specific siRNA, but not intratracheal, attenuated the development of Hem-CLP-induced acute lung injury.
To assess the impact of upregulated PD-L1 on ECs in iALI, mice were intravenously injected with liposomal-encapsulated siRNA during resuscitation. We initially looked at the protein ratio of BALF/blood and found that intravenous delivery of anti-PD-L1-silencing siRNA significantly decreased the protein leak in the alveolus seen in Hem-CLP mice (Fig. 4A), whereas intratracheal delivery did not (Fig. 4B). Lung tissue histology further showed that PD-L1 silencing by intravenous delivery led to improved tissue architecture, lessened pulmonary edema, and reduced immune cell tissue influx (Fig. 4, C–F). To the extent these changes in morphology were quantifiable, when the percentage of open alveolar space of the lung sections was digitally determined (Fig. 4G), we observed that % alveolar space in all Hem-CLP groups was significantly lower compared with sham mice. Mouse lungs that had been treated with PD-L1 siRNA, via intravenous delivery, had higher % alveolar space than PBS and control siRNA-treated groups. Alternatively, intratracheal delivery did not produce such a change (Fig. 4, B, H–L).
Fig. 4.

Effects of intravenous (I.V.) versus intratracheal (I.T.) delivery of PD-L1 siRNA on pulmonary protein leak in mice with indirect acute lung injury (iALI). Intravenous delivery (A) of PD-L1 siRNA significantly decreased the protein leak in iALI mice compared with PBS or control siRNA-treated hemorrhagic shock followed by cecal ligation and puncture (Hem-CLP) groups. In contrast, intratracheal delivery (B) of PD-L1 siRNA did not alter the protein leak in all iALI groups. Values are means ± SE; n = 6 mice per group; *P < 0.05 vs. sham; #P < 0.05 vs. PD-L1 siRNA-treated group, by one-way ANOVA and a Student–Newman–Keuls test. Intravenous delivery of siRNA attenuated Hem-CLP-induced lung injury (H&E staining). Representative images of H&E-stained lung tissue sections from intravenous siRNA-treated mice: PBS+sham (C); PBS+Hem-CLP (D); control siRNA+Hem-CLP (E); and PD-L1 siRNA+Hem-CLP (F) (original magnification: ×200; scale bar: 100 μm), as well as semiquantitive digital determination of these morphological changes (G) [% of open alveolar space in each image as determined image analysis (means ± SE; n = 3 per group; *P < 0.05 vs. sham; #P < 0.05 vs. PD-L1 siRNA-treated group, by Mann–Whitney U test)]. Alternatively, no such protection was seen with intratracheal delivery of PD-L1 siRNA to Hem-CLP mice (H–L).
Subsequently, we examined changes in the cytokine/chemokine levels in plasma (Fig. 5), lung tissue lysates (Fig. 6), or BALF samples (Fig. 7). Cytokine analysis of plasma samples demonstrated the Hem-CLP induced a consistent increase in the levels of TNF-α, IL-6, KC, MCP-1, and MIP-2 in the vehicle and the control siRNA-treated Hem-CLP, as opposed to the sham animals. However, this Hem-CLP-induced increase was markedly suppressed (except for IL-10 and KC) by intravenously delivered anti-PD-L1 siRNA to the treatment group (Fig. 5A), but not when siRNA was delivered intratracheal (Fig. 5B). Although the lung tissue lysate cytokine/chemokine levels following Hem-CLP were again markedly increased in the vehicle and the control siRNA Hem-CLP groups, with respect to the elevated IL-6, MIP-2, and MCP-1 levels, these were all significantly decreased in samples from the intravenous (Fig. 6A), but not the intratracheal (Fig. 6B), delivered anti-PD-L1 siRNA treatment groups (while no difference was seen in TNF-α, IL-10, and KC). With respect to BALF cytokine levels, we observed that even though there were consistently increased cytokine/chemokine levels in the vehicle and the control siRNA-treated Hem-CLP groups, when compared with sham group, and trends toward suppression of this rise in a few cases when PD-L1 siRNA was administered, these changes did not reach the level of statistical significance in both delivery methods (Fig. 7).
Fig. 5.

PD-L1 gene suppression by intravenous (I.V.), but not intratracheal (I.T.), siRNA delivery markedly attenuated the increase in systemic plasma levels of inflammatory cytokine/chemokine produced in response to hemorrhagic shock followed by cecal ligation and puncture (Hem-CLP). The plasma levels of TNF-α, IL-6, KC, MCP-1, and MIP-2 in the vehicle and the control siRNA-treated Hem-CLP mice were increased compared with the sham animals. However, this Hem-CLP-induced increase was markedly suppressed (except for IL-10 and KC) by intravenous delivery of anti-PD-L1 siRNA (A), but not intratracheal delivery (B). Values are means ± SE; n = 6–10 mice per group; *P < 0.05 vs. sham; #P < 0.05 vs. PD-L1 siRNA-treated group, by one-way ANOVA and a Student–Newman–Keuls test.
Fig. 6.

PD-L1 gene suppression by intravenous (I.V.), but not intratracheal (I.T.), siRNA delivery markedly attenuated the increase in local lung tissue levels of inflammatory cytokine/chemokine produced in response to hemorrhagic shock followed by cecal ligation and puncture (Hem-CLP). TNF-α, IL-6, IL-10, KC, MCP-1, and MIP-2 levels in tissue homogenate samples were increased after Hem-CLP compared with sham mice. The levels of IL-6, MCP-1, and MIP-2 were significantly decreased in samples from the intravenous (A), but not the intratracheal (B), delivered anti-PD-L1 siRNA-treated indirect acute lung injury (iALI) groups. No changes were seen in TNF-α, IL-10, and KC levels from both intravenous and intratracheal delivery-treated iALI groups. Values are means ± SE; n = 6–10 mice per group; *P < 0.05 vs. sham; #P < 0.05 vs. PD-L1 siRNA-treated group, by one-way ANOVA and a Student–Newman–Keuls test.
Fig. 7.

PD-L1 gene suppression, by intravenous (I.V.) or intratracheal (I.T.) siRNA delivery, had no effect on cytokine/chemokine levels detected in bronchoalveolar lavage fluid (BALF). BALF was collected 24 h after hemorrhagic shock followed by cecal ligation and puncture (Hem-CLP) or sham operation. TNF-α, IL-6, IL-10, KC, MCP-1, and MIP-2 levels were increased after indirect acute lung injury (iALI) compared with sham mice. There were no changes in the cytokine/chemokine levels between intravenous and intratracheal delivery of PD-L1 siRNA in all iALI groups. Values are means ± SE; n = 6–10 mice per group, by one-way ANOVA and a Student–Newman–Keuls test.
Consistent with the selective suppression of the elevated Hem-CLP-induced cytokine/chemokine levels seen in the intravenous PD-L1 siRNA-treated mice, the Hem-CLP-induced elevated MPO and caspase-3 activities were also reduced by PD-L1 gene suppression with intravenously delivered PD-L1 siRNA (Fig. 8, A and C). This was not seen when anti-PD-L1 siRNA was given intratracheally (Fig. 8, B and D). Again, there were fewer esterase-positive cells detected in the lungs of Hem-CLP mice after intravenously delivered PD-L1 siRNA, suggesting that the neutrophil influx was inhibited (Fig. 8, E–I).
Fig. 8.

PD-L1 gene suppression by intravenous (I.V.), but not intratracheal (I.T.), siRNA delivery markedly attenuated the development of pulmonary myeloperoxidase (MPO), caspase-3 activity, and esterase staining for neutrophil influx. Intravenous delivery of PD-L1 siRNA decreased the MPO (A) and caspase-3 activity (C) when compared with either PBS or control siRNA-treated Hem-CLP mice. In contrast, intratracheal delivery of PD-L1 siRNA did not alter either MPO (B) or caspase-3 activity (D) when compared with the other Hem-CLP groups. Values are means ± SE; n = 4–6 mice per group; *P < 0.05 vs. sham; #P < 0.05 vs. PD-L1 siRNA-treated group, by one-way ANOVA and a Student–Newman–Keuls test. Representative images of the typical immunohistological esterase (a marker for neutrophils)–stained lung tissue sections are shown in E–H, where E is from PBS+sham, F is from PBS+Hem-CLP, G is from control siRNA+ Hem-CLP, and H is from PD-L1 siRNA+Hem-CLP-treated mice. I: the summary data of esterase+ cell counts of 8 fields/slide/sample at ×400 magnification (scale bar: 25 μm). Values are means ± SE; n = 10 area per sample/group; *P < 0.05 vs. sham; #P < 0.05 vs. PD-L1 siRNA-treated group, by one-way ANOVA and a Student–Newman–Keuls test.
Knockdown or deletion of PD-L1 gene expression in CRL cells reduced rIFN-γ-induced monolayer permeability with restored tight junction protein levels.
To begin to get at a possible mechanistic explanation as to why intravenous delivery of PD-L1 siRNA reduces vascular leak in the lungs of Hem-CLP mice (Fig. 4, A, C–G), we set out to investigate the possible impact of PD-L1 on pulmonary vascular permeability using mouse CRL cell monolayer in vitro. CRL cells were treated with anti-PD-L1 siRNA to silence PD-L1 or a PD-L1 gene knockout (PD-L1−/−) CRL cell line was generated with CRISPR/Cas9-mediated system. As mentioned above, PD-L1 expression on CRL endothelial cells was significantly reduced in anti-PD-L1 siRNA-treated and PD-L1−/− CRL cells compared with their controls, respectively (Fig. 2, G–H). Consistent with the in vivo studies, after rIFNγ stimulation, CRL cell monolayers treated with anti-PD-L1 siRNA had a significantly reduced permeability as determined by FD4 influx through the Transwell membrane compared with rIFNγ with PBS only or control siRNA (Fig. 9A). This was associated with a significant restoration of tight junction protein ZO-1 (Fig. 9B) and less increase in OCC (Fig. 9C) levels. Immunofluorescent staining for ZO-1 (Fig. 9D) and OCC (Fig. 9E) in CRL monolayers showed a reduced intensity of ZO-1 and OCC staining, and disorganized/disrupted profiles of tight junctions were observed in the rIFNγ-stimulated control siRNA and PBS-treated CRL cell monolayers compared with nonstimulated (PBS alone) CRL cells. However, PD-L1 siRNA treatment restored tight junction protein levels. Additionally, PD-L1 gene knockout (PD-L1−/− CRL) reduced monolayer permeability (FD4 influx) (Fig. 9F) induced by rIFNγ and preserved the tight junction protein, ZO-1, levels when compared with rIFNγ-treated wild-type CRL (PD-L1+/+) cells (Fig. 9, G–H).
Fig. 9.

Knockdown or deletion of PD-L1 gene reduced recombinant (r)IFN-γ-induced monolayer permeability in CRL-2167 (CRL) cells. In vitro mouse CRL endothelial cell culture was set up to elucidate the role of PD-L1 on cell membrane integrity after rIFN-γ treatment. A: monolayer permeability was analyzed by FD4 influx through the Transwell membrane. Treatment with rIFN-γ induced an increase of the monolayer permeability (increase FD4 concentration in the basal chambers), while PD-L1 siRNA treatment preserved the barrier function with reduced FD4 influx. B: the increased permeability was associated with significantly reduced tight junction protein ZO-1 when compared with PD-L1 siRNA and no rIFN-γ-treated cells. C: there was slight restoration, but not statistically significant, observed in occludin (OCC) protein levels between PD-L1 siRNA-treated and other groups. Representative image of immunofluorescent staining for tight junction proteins ZO-1 (D) and OCC (E) in CRL monolayers. When compared with negative control (PBS alone), the monolayers treated with rIFN-γ (PBS or control sinRNA) showed obvious disruptions of the distribution and decreased intensity of ZO-1 (D) and OCC (E) protein staining. PD-L1 siRNA treatment preserved the integrity of monolayers closer to nontreated control compared with rIFN-γ treated plus PBS or control siRNA (original magnifications ×200). F: this was further confirmed by PD-L1 gene knockout in CRL cells using CRISPR-Cas9 system that rIFN-γ treatment induced an increase of the PD-L1+/+ CRL monolayer permeability with increased FD4 influx, while PD-L1−/− CRL monolayer reduced FD4 influx and restored the barrier function. This again was associated with reducing in ZO-1 (G) and less in OCC (H) protein level, in the PD-L1+/+ CRL monolayers when compared with PD-L1−/− CRL monolayers after stimulation with rIFN-γ. Values are means ± SE; data represent the average of 3 independent experiments with triplicate for each treatment; *P < 0.05 vs. PD-L1 siRNA-treated group or PD-L1−/− cells; #P < 0.05 vs. PBS with IFN-γ-treated group, by one-way ANOVA and a Student–Newman–Keuls test.
DISCUSSION
In this study, we used a mouse model of hemorrhagic shock followed by septic insult to induce iALI. In line with our previous research, Hem-CLP produced a systemic inflammatory response and lung injury, which is commonly seen in ARDS patients (2, 16). To establish the role of PD-L1 expression in the development of iALI, we first examined its expression following Hem-CLP. Our data showed that PD-L1 expression was significantly elevated in iALI, especially on ECs and EpiCs. Then, we explored the therapeutic use of PD-L1 siRNA via cell-selective intratracheal as opposed to intravenous delivery in this experimental setting. We observed that shocked/septic mice exhibited a protection from developing iALI in response to intravenous delivery of siRNA, but this was not observed with intratracheal delivery in animals. Given the fact that intravenous delivery of siRNA goes largely into ECs while intratracheal delivery is taken up mainly by EpiCs (2, 16, 27), our study suggests that suppression of PD-L1 on/in ECs attenuates iALI, but not EpiCs. We further attempted to examine the mechanism by which PD-L1 might be contributing to the vascular permeability induced by inflammation, seen in iALI experimental mice, using an in vitro cell culture setting. Here we show that blocking PD-L1, by either siRNA knockdown or gene knockout, in EC cell line CRL was associated with reducing EC monolayer permeability by preserving tight junction integrity. Implying that ligation of PD-L1, in the absence of PD-1, is potentially sufficient to induce significant changes in EC function.
Acute respiratory distress syndrome is thought to represent a severe form of lung dysfunction characterized by an overwhelming inflammatory response (30). It is widely accepted that inflammatory mediators contribute to the development of ARDS. In patients, the persistent high levels of cytokines were associated with poor outcomes (23). IL-6 is believed to be an important cytokine in the pathogenesis of ALI. Plasma IL-6 levels are consistent and correlative with the development of ARDS (23). Goldman et al. (7) reported that IL-6 was able to increase the endothelial permeability, and IL-6−/− mice exhibited a decrease in BAL cellular inflammation. MIP-2, an important chemokine, is critically involved in the activation and recruitment of neutrophils (4). Blockade of CXCR-2 caused a reduction in lung neutrophil influx in response to Hem-CLP and additionally inhibited lung inflammation and protein leak (18). In this study, we observed higher expressions of cytokines/chemokines in the BALF, lung lysates, and plasma from mice subjected to Hem-CLP. Intravenous delivery of PD-L1 siRNA inhibited the inflammatory response to a certain extent. In plasma, IL-6, MIP-2, TNF-α, and MCP-1 were significantly reduced in iALI mice following PD-L1 silencing. However, the inhibitory effects seemed to be less efficient within the lung. In lung lysates, IL-6, MIP-2, and MCP-1 were decreased, while no significant difference was seen regarding TNF-α, IL-10, and KC. It appears that PD-L1 siRNA treatment has no effect on these cytokines/chemokines in the BALF after iALI. These data indicate that PD-L1 silencing may also have an influence on other cells, such as peripheral phagocytes, which produced substantial cytokines during Hem-CLP.
Furthermore, our previous study found that local pulmonary chemokine production of MIP-2, to a greater extent than KC, contributes to the development of neutrophil-associated ALI in this Hem-CLP mouse model (17). Therefore, the protective effects of PD-L1 silencing can be partially explained by the reduced inflammatory response, especially as mediated through the suppression of IL-6 and MIP-2. The observation that PD-L1 plays a role in monolayer permeability from the in vitro EC studies suggests that soluble factors in the lung may also have been affected. Consistent with the reduced MIP-2 levels seen in intravenous PD-L1 siRNA-treated mice, the neutrophil influx was largely inhibited following PD-L1 suppression. Previous studies have found that neutrophils accumulate in lung tissue as well as in BAL of ARDS patients (8). In our model of iALI, neutrophils were primed due to the predispositional effect of hemorrhage shock (2, 14). Inasmuch; upon the subsequent septic insult, these primed neutrophils are thought to be recruited to the lung, where they can produce substantial oxygen free radicals and inflammatory cytokines, while at the same time their constitutive apoptotic process is actively suppressed (1). Hence, it has been proposed that neutrophil activation represents the central part of the pathogenesis of lung injury. In line with the reduced neutrophil influx, we observed the MPO activity of the lungs was also significantly decreased after PD-L1 suppression. Therefore, the iALI mice were protected to some extend through the inhibition of neutrophil emigration. In particular, recent studies have reported that neutrophils also express certain levels of PD-L1 molecules, and, importantly, septic insult induced a significant upregulation of PD-L1 expression both in experimental mice and clinical patients (10). Wang et al. (32) showed that PD-L1+ neutrophils exhibited compromised migratory capacity. In our study, we observed upregulated PD-L1 expression on neutrophils following Hem-CLP, and intravenous delivery of siRNA suppressed its expression on neutrophils within the lung (data not shown). Thus, it is tempting to speculate as to whether PD-L1 silencing affects neutrophil migration in this study, and as such is a potential explanation for why the neutrophil influx into the lungs was decreased. Our suspicion is that neutrophil migration could be much more complicated, involving several aspects seen in iALI, such as chemokine alterations as indicated before, changes is adhesion molecule(s) expression, and/or endothelial cell function. Thus, the precise effect of PD-L1 silencing on neutrophils remains to be elucidated.
Importantly, the inability of intratracheally delivered PD-L1-specific siRNA to block the development of lung injury implies that the expression of PD-L1 on pulmonary EpiCs or its downstream signaling events are involved in mediating either the recruitment of neutrophils into the lung or altering local lung inflammation/injury here. Furthermore, since we have previously shown that intratracheal (but not intravenous) delivery of siRNA directed against key pro-apoptotic proteins, such as FasL, Fas, and caspase-3 (16, 31), have protective effects against the development of iALI, as modeled here, these cell death–mediated processes, in some aspects (the development of pulmonary EpiC death) would appear to be distal/downstream in the sequence of events initiated by circulatory and/or septic shock leading to iALI here. Conversely, the ineffectiveness of intratracheal versus intravenous delivery of both TNF-α, angiopoietin-2, and potentially MIP-2 places ECs and/or select leukocyte populations that express PD-L1 much more in a role as proximal conduits of signals (pathogenic or otherwise) produced by shock and/or septic insults seen in our iALI model (20). This does not necessarily mean that EpiCs are not important in this model or that the rise in PD-L1 expression on these cells is not related to a change in EpiC function. Destruction of epithelial integrity prompts a progressive influx of protein in alveoli and concurrently inhibits the reabsorption of alveolar edema (1, 32, 33). The extent of EpiC damage has been associated with the poorer clinical outcomes in ALI patients (3, 9, 22). However, since we did not examine the EpiCs following PD-L1 silencing in vivo, it remains to be determined whether PD-L1 expression would influence epithelial cell function. If any, the regulatory effect is of less importance in comparison with that in ECs under the scope of iALI.
Our study has limitations. While we observed protective effects of intravenous delivery with siRNA in mice subjected to Hem-CLP, it is not known whether this is mainly attributed to the interruption of PD-1:PD-L1 or PD-L1:CD80 ligation, as we have not examined the possible changes in intracellular signaling within ECs following PD-L1 suppression. In addition, as we assessed the therapeutic effects of PD-L1 silencing at a single time point, the long-term outcomes were not determined.
In conclusion, in this study, we demonstrated that silencing of the expression of PD-L1, primarily on ECs, but not EpiCs, protects mice against Hem-CLP-induced iALI. These data suggest that PD-L1 may have a novel role to play in the development of iALI, making it an interesting therapeutic target in ARDS patients.
GRANTS
This work was supported by NIH National Institute of General Medical Sciences Grants R01-GM107149 and R35-GM118097 (A.A.), National Natural Science Foundation of China Grant No. 81800081 (S.X.), as well as a fellowship from the China Scholarship Council No. 201306580023 (T.T.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.X., Q.Y., J.B., and A.A. conceived and designed research; S.X., Q.Y., Y.C., and C.-S.C. performed experiments; S.X., Q.Y., Y.C., and C.-S.C. analyzed data; S.X., Q.Y., J.B., T.T., L.T., and C.-S.C. interpreted results of experiments; S.X., Q.Y., C.-S.C., and A.A. prepared figures; S.X., Q.Y., and C.-S.C. drafted manuscript; J.B., L.T., C.-S.C., E.A.F., and A.A. edited and revised manuscript; S.X., Q.Y., J.B., T.T., L.T., Y.C., C.-S.C., E.A.F., and A.A. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Virginia Hovanesian, Core Research Laboratories, Rhode Island Hospital, for assistance with microscopy and imaging analysis of these specimens.
REFERENCES
- 1.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 279: L1137–L1145, 2000. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 2.Ayala A, Chung CS, Lomas JL, Song GY, Doughty LA, Gregory SH, Cioffi WG, LeBlanc BW, Reichner J, Simms HH, Grutkoski PS. Shock-induced neutrophil mediated priming for acute lung injury in mice: divergent effects of TLR-4 and TLR-4/FasL deficiency. Am J Pathol 161: 2283–2294, 2002. doi: 10.1016/S0002-9440(10)64504-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker CS, Evans TW, Randle BJ, Haslam PL. Damage to surfactant-specific protein in acute respiratory distress syndrome. Lancet 353: 1232–1237, 1999. doi: 10.1016/S0140-6736(98)09449-5. [DOI] [PubMed] [Google Scholar]
- 4.Burdon PC, Martin C, Rankin SM. The CXC chemokine MIP-2 stimulates neutrophil mobilization from the rat bone marrow in a CD49d-dependent manner. Blood 105: 2543–2548, 2005. doi: 10.1182/blood-2004-08-3193. [DOI] [PubMed] [Google Scholar]
- 5.Calfee CS, Eisner MD, Ware LB, Thompson BT, Parsons PE, Wheeler AP, Korpak A, Matthay MA; Acute Respiratory Distress Syndrome Network, National Heart, Lung, and Blood Institute . Trauma-associated lung injury differs clinically and biologically from acute lung injury due to other clinical disorders. Crit Care Med 35: 2243–2250, 2007. doi: 10.1097/01.CCM.0000280434.33451.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Combes A, Hajage D, Capellier G, Demoule A, Lavoué S, Guervilly C, Da Silva D, Zafrani L, Tirot P, Veber B, Maury E, Levy B, Cohen Y, Richard C, Kalfon P, Bouadma L, Mehdaoui H, Beduneau G, Lebreton G, Brochard L, Ferguson ND, Fan E, Slutsky AS, Brodie D, Mercat A; EOLIA Trial Group, REVA, and ECMONet . Extracorporeal membrane oxygenation for severe acute respiratory distress syndrome. N Engl J Med 378: 1965–1975, 2018. doi: 10.1056/NEJMoa1800385. [DOI] [PubMed] [Google Scholar]
- 7.Goldman JL, Sammani S, Kempf C, Saadat L, Letsiou E, Wang T, Moreno-Vinasco L, Rizzo AN, Fortman JD, Garcia JG. Pleiotropic effects of interleukin-6 in a “two-hit” murine model of acute respiratory distress syndrome. Pulm Circ 4: 280–288, 2014. doi: 10.1086/675991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med 17: 293–307, 2011. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gropper MA, Wiener-Kronish J. The epithelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care 14: 11–15, 2008. doi: 10.1097/MCC.0b013e3282f417a0. [DOI] [PubMed] [Google Scholar]
- 10.Huang X, Chen Y, Chung CS, Yuan Z, Monaghan SF, Wang F, Ayala A. Identification of B7-H1 as a novel mediator of the innate immune/proinflammatory response as well as a possible myeloid cell prognostic biomarker in sepsis. J Immunol 192: 1091–1099, 2014. doi: 10.4049/jimmunol.1302252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, Swan R, Kherouf H, Monneret G, Chung CS, Ayala A. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci USA 106: 6303–6308, 2009. doi: 10.1073/pnas.0809422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26: 677–704, 2008. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med 11: e19, 2009. doi: 10.1017/S1462399409001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lomas JL, Chung CS, Grutkoski PS, LeBlanc BW, Lavigne L, Reichner J, Gregory SH, Doughty LA, Cioffi WG, Ayala A. Differential effects of macrophage inflammatory chemokine-2 and keratinocyte-derived chemokine on hemorrhage-induced neutrophil priming for lung inflammation: assessment by adoptive cells transfer in mice. Shock 19: 358–365, 2003. doi: 10.1097/00024382-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 15.Lomas-Neira J, Chung C-S, Perl M, Gregory S, Biffl W, Ayala A. Role of alveolar macrophage and migrating neutrophils in hemorrhage-induced priming for ALI subsequent to septic challenge. Am J Physiol Lung Cell Mol Physiol 290: L51–L58, 2006. doi: 10.1152/ajplung.00028.2005. [DOI] [PubMed] [Google Scholar]
- 16.Lomas-Neira J, Venet F, Chung C-S, Thakkar RK, Heffernan DS, Ayala A. Neutrophil-endothelial interactions mediate angiopoietin-2 associated pulmonary cell dysfunction in idirect ALI in mice. Am J Respir Cell Mol Biol 50: 193–200, 2014. doi: 10.1165/rcmb.2013-0148OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lomas-Neira JL, Chung C-S, Wesche DE, Perl M, Ayala A. In vivo gene silencing (with siRNA) of pulmonary expression of MIP-2 versus KC results in divergent effects on hemorrhage-induced, neutrophil-mediated septic acute lung injury. J Leukoc Biol 77: 846–853, 2005. doi: 10.1189/jlb.1004617. [DOI] [PubMed] [Google Scholar]
- 18.Lomas-Neira JL, Chung CS, Grutkoski PS, Miller EJ, Ayala A. CXCR2 inhibition suppresses hemorrhage-induced priming for acute lung injury in mice. J Leukoc Biol 76: 58–64, 2004. doi: 10.1189/jlb.1103541. [DOI] [PubMed] [Google Scholar]
- 19.Lomas-Neira JL, Heffernan DS, Ayala A, Monaghan SF. Blockade of endothelial growth factor, angiopoietin-2, reduces indices of ARDS and mortality in mice resulting from the dual-insults of hemorrhagic shock and sepsis. Shock 45: 157–165, 2016. doi: 10.1097/SHK.0000000000000499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lomas-Neira J, Monaghan SF, Huang X, Fallon EA, Chung CS, Ayala A. Novel role for PD-1:PD-L1 as a mediator of pulmonary vascular endothelial cell functions in the pathogenesis of indirect ARDS in mice. Front Immunol 9: 3030, 2018. doi: 10.3389/fimmu.2018.03030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lomas-Neira JL, Perl M, Venet F, Chung CS, Ayala A. The role and source of TNF-α in hemorrhage induced priming for septic lung injury. Shock 37: 611–620, 2012. doi: 10.1097/SHK.0b013e318254fa6a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, Martin TR. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 163: 2217–2225, 1999. [PubMed] [Google Scholar]
- 23.Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, Leeper K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 107: 1062–1073, 1995. doi: 10.1378/chest.107.4.1062. [DOI] [PubMed] [Google Scholar]
- 24.Monaghan SF, Thakkar RK, Heffernan DS, Huang X, Chung CS, Lomas-Neira J, Cioffi WG, Ayala A. Mechanisms of indirect acute lung injury: a novel role for the coinhibitory receptor, programmed death-1. Ann Surg 255: 158–164, 2012. doi: 10.1097/SLA.0b013e31823433ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monaghan SF, Thakkar RK, Tran ML, Huang X, Cioffi WG, Ayala A, Heffernan DS. Programmed death 1 expression as a marker for immune and physiological dysfunction in the critically ill surgical patient. Shock 38: 117–122, 2012. doi: 10.1097/SHK.0b013e31825de6a3. [DOI] [PubMed] [Google Scholar]
- 26.Papazian L, Forel JM, Gacouin A, Penot-Ragon C, Perrin G, Loundou A, Jaber S, Arnal JM, Perez D, Seghboyan JM, Constantin JM, Courant P, Lefrant JY, Guérin C, Prat G, Morange S, Roch A; ACURASYS Study Investigators . Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med 363: 1107–1116, 2010. doi: 10.1056/NEJMoa1005372. [DOI] [PubMed] [Google Scholar]
- 27.Perl M, Chung CS, Perl U, Thakkar R, Lomas-Neira J, Ayala A. Therapeutic accessibility of caspase-mediated cell death as a key pathomechanism in indirect acute lung injury. Crit Care Med 38: 1179–1186, 2010. doi: 10.1097/CCM.0b013e3181d4563f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petrucci N, De Feo C. Lung protective ventilation strategy for the acute respiratory distress syndrome. Cochrane Database Syst Rev 18: CD003844, 2007. [Erratum in Cochrane Database Syst Rev 2013: CD003844, 2013.] doi: 10.1002/14651858.CD003844.pub4. [DOI] [PubMed] [Google Scholar]
- 29.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 30.Sharp C, Millar AB, Medford AR. Advances in understanding of the pathogenesis of acute respiratory distress syndrome. Respiration 89: 420–434, 2015. doi: 10.1159/000381102. [DOI] [PubMed] [Google Scholar]
- 31.Thakkar RK, Chung CS, Chen Y, Monaghan SF, Lomas-Neira J, Heffernan DS, Cioffi WG, Ayala A. Local tissue expression of the cell death ligand, fas ligand, plays a central role in the development of extrapulmonary acute lung injury. Shock 36: 138–143, 2011. doi: 10.1097/SHK.0b013e31821c236d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang JF, Li JB, Zhao YJ, Yi WJ, Bian JJ, Wan XJ, Zhu KM, Deng XM. Up-regulation of programmed cell death 1 ligand 1 on neutrophils may be involved in sepsis-induced immunosuppression: an animal study and a prospective case-control study. Anesthesiology 122: 852–863, 2015. doi: 10.1097/ALN.0000000000000525. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Adler KB, Erjefalt J, Bai C. Airway epithelial dysfunction in the development of acute lung injury and acute respiratory distress syndrome. Expert Rev Respir Med 1: 149–155, 2007. doi: 10.1586/17476348.1.1.149. [DOI] [PubMed] [Google Scholar]
- 34.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 35.Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet 369: 1553–1564, 2007. doi: 10.1016/S0140-6736(07)60604-7. [DOI] [PubMed] [Google Scholar]
- 36.Wichmann MW, Ayala A, Chaudry IH. Male sex steroids are responsible for depressing macrophage immune function after trauma-hemorrhage. Am J Physiol Cell Physiol 273: C1335–C1340, 1997. doi: 10.1152/ajpcell.1997.273.4.C1335. [DOI] [PubMed] [Google Scholar]
- 37.Wu Y, Chung CS, Chen Y, Monaghan SF, Patel S, Huang X, Heffernan DS, Ayala A. A novel role for programmed cell death receptor ligand-1 in sepsis-induced intestinal dysfunction. Mol Med 22: 830–840, 2016. doi: 10.2119/molmed.2016.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zellweger R, Wichmann MW, Ayala A, Stein S, DeMaso CM, Chaudry IH. Females in proestrus state maintain splenic immune functions and tolerate sepsis better than males. Crit Care Med 25: 106–110, 1997. doi: 10.1097/00003246-199701000-00021. [DOI] [PubMed] [Google Scholar]