

Abstract
Inhaled granulocyte/macrophage colony-stimulating factor (GM-CSF) shows promise as a therapeutic to treat viral and bacterial pneumonia, but no mouse model of inhaled GM-CSF has been described. We sought to 1) develop a mouse model of aerosolized recombinant mouse GM-CSF administration and 2) investigate the protection conferred by inhaled GM-CSF during influenza A virus (IAV) infection against secondary bacterial infection with pneumococcus. To assess lower respiratory tract delivery of aerosolized therapeutics, mice were exposed to aerosolized fluorescein (FITC)-labeled dextran noninvasively via an aerosolization tower or invasively using a rodent ventilator. The efficiency of delivery to the lower respiratory tracts of mice was 0.01% noninvasively compared with 0.3% invasively. The airway pharmacokinetics of inhaled GM-CSF fit a two-compartment model with a terminal phase half-life of 1.3 h. To test if lower respiratory tract levels were sufficient for biological effect, mice were infected intranasally with IAV, treated with aerosolized recombinant mouse GM-CSF, and then secondarily infected with Streptococcus pneumoniae. Inhaled GM-CSF conferred a significant survival benefit to mice against secondary challenge with S. pneumoniae (P < 0.05). Inhaled GM-CSF did not reduce airway or lung parenchymal bacterial growth but significantly reduced the incidence of S. pneumoniae bacteremia (P < 0.01). However, GM-CSF overexpression during influenza virus infection did not affect lung epithelial permeability to FITC-dextran ingress into the bloodstream. Therefore, the mechanism of protection conferred by inhaled GM-CSF appears to be locally mediated improved lung antibacterial resistance to systemic bacteremia during IAV infection.
Keywords: aerosol, GM-CSF, inhaled, pharmacokinetics, pneumonia
INTRODUCTION
Respiratory viruses such as influenza A virus (IAV) continue to be a leading cause of death due to infectious diseases in the United States. Unfortunately, even with appropriate antiviral and antibiotic therapies, the rate of mortality is 7–8% (14, 30), with hospital mortality being significantly higher in bacterially coinfected patients (26). New therapeutic strategies are required to prevent and treat respiratory virus-associated secondary bacterial pneumonia (SBP). Granulocyte/macrophage colony-stimulating factor (GM-CSF), a member of the IL-3/IL-5/GM-CSF family of cytokines, may play an important role in influenza pathogenesis. The absence of GM-CSF signaling in GM-CSF cytokine knockout (Csf2−/−) (28, 29) and GM-CSF common β-chain receptor knockout (Csf2rb−/−) (28) mice increases their susceptibility to IAV, which can be partially alleviated by reconstitution of alveolar macrophages (AMs) that can respond to GM-CSF (28). Conversely, multiple preclinical mouse models have shown that increased airway levels of GM-CSF confer protection against IAV infection in mice. Two studies (12, 40) have demonstrated that constitutive airway GM-CSF overexpression using the surfactant protein C (SPC)-GM+/+ model confers protection from IAV, with this effect possibly being dependent on dendritic cells (40), but more likely from AMs (12, 28). Using an inducible airway GM-CSF overexpression model on the GM-CSF-deficient background, Sever-Chroneos et al. showed that GM-CSF must be induced ≥3 days before IAV infection for maximum protection (29). GM-CSF signaling and AMs are also critical for protection against SBP, as depletion of AMs increases susceptibility to SBP with Streptococcus pneumoniae (4) and administration of GM-CSF may improve resistance to SBP by recruitment of phagocytes (32). Despite the plethora of data from these preclinical models, the precise mechanisms of protection of GM-CSF have not truly been elucidated.
We recently reported that airway GM-CSF overexpression after IAV infection markedly decreased mortality but that GM-CSF did not affect the initial weight loss associated with virus infection and, instead, rescued mice from mortality during the slow recovery phase (9). This fact, along with the knowledge that influenza infection mortality is frequently associated with SBP (20, 21, 24), led us to hypothesize that the primary effect of GM-CSF overexpression was prevention of SBP. To investigate this possibility, we decided to move away from the doxycycline-induced Tet-On double-transgenic GM-CSF (DTGM) mouse model (9) and, instead, developed a model wherein known quantities of exogenous recombinant mouse GM-CSF (rmGM-CSF) can be administered repeatedly to the lower airways of mice without the use of sedatives.
For several years, inhaled GM-CSF has been used off-label for the condition known as a pulmonary alveolar proteinosis (PAP) (25), where impaired GM-CSF signaling in the lung causes surfactant accumulation. Interestingly, subcutaneous administration of GM-CSF is not very effective in improving the symptoms of PAP. However, when aerosolized and administered by inhalation, GM-CSF has shown benefit in PAP patients (38). A phase I clinical trial of inhaled GM-CSF for PAP enrolled 35 patients and demonstrated efficacy and no serious adverse effects (35). Inhaled GM-CSF shows promise as a therapeutic to treat viral and bacterial pneumonia (11). A multicenter, double-blind, placebo-controlled, randomized clinical trial of inhaled GM-CSF with pneumonia-associated acute respiratory distress syndrome (ARDS) is underway within the nationwide German Center for Lung Research (NCT02601365). However, to better design future clinical trials, a clearer understanding of the mechanism of action of airway GM-CSF is required to determine who can benefit, what lung disease processes can be influenced, and when GM-CSF should be administered.
In this study we created and characterized a mouse model in which we could exogenously administer GM-CSF, repeatedly, to the airways of mice. We then tested if inhaled GM-CSF could provide any survival benefit to mice during IAV infection and subsequent S. pneumoniae SBP challenge (3, 15, 33).
MATERIALS AND METHODS
Mice.
All animal experiments were approved by the Pennsylvania State University College of Medicine Institutional Animal Care and Use Committee (protocol no. 47450). Wild-type C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Adult (10- to 14-wk-old) mice were used for all experiments except when stated otherwise. Male mice were used for all experiments that examined aerosol delivery and recovery of FITC-dextran. Male and female mice were used for studies examining GM-CSF delivery and pharmacokinetics and for infectious in vivo experiments.
Noninvasive aerosolization exposures.
For noninvasive aerosolization experiments, we used the inExpose aerosolization tower (Scireq, Montreal, PQ, Canada), which generates aerosols via an Aerogen Aeroneb [standard (4- to 6-μm droplet size) mist] vibrating mesh nebulizer, with a bias flow of 1.5 L/min. Mice were held in soft restraints during exposure to the aerosol via a nose cone.
Invasive aerosolization exposures.
For invasive aerosolization experiments, mice were anesthetized with ketamine (100 mg/kg)-xylazine (10 mg/kg), tracheotomized, and paralyzed (vecuronium, 1 mg/kg). Mice were ventilated with the flexiVent rodent ventilator (Scireq) using a respiratory rate of 150 breaths/min, 3 cmH2O positive end-expiratory pressure, and 10 mL/kg tidal volume, and anesthesia was maintained with sevoflurane (1%). Aerosols were generated with an Aerogen Aeroneb [fine (2- to 4-μm droplet size) mist] vibrating mesh nebulizer, and mice were exposed for a set time period (120 s).
Treatments.
Fluorescein-labeled (FITC) dextran (mol wt 10,000, FITC-dextran) was used to model delivery of rmGM-CSF (15 kDa). Phosphate-buffered saline (PBS) was used as the solvent for all solutions and as the vehicle control. Carrier-free rmGM-CSF protein was purchased from Bio-Techne (Minneapolis, MN). Each lot was divided into 5-μg aliquots at 0.5 mg/mL. We tested the bioactivity of each lot by examining colony growth and STAT5 phosphorylation of mouse bone marrow cells. GM-CSF (5 μg) was administered twice daily via the inExpose tower from 5 to 9 days after IAV infection. Finally, 1 ng of GM-CSF, the equivalent of the inhaled delivered dose, was administered intraperitoneally twice daily to a subgroup of mice to determine if site of delivery was important for biological effect.
Infections.
For intranasal infections, mice were anesthetized by intraperitoneal injection with ketamine (100 mg/kg)-xylazine (10 mg/kg). Anesthetized mice (9) were intranasally infected with 100 fluorescent focus units (FFU) of the H1N1 mouse-adapted A/Puerto Rico/8/34 (PR8) strain of IAV in 40 μL of PBS. At 7 days after IAV infection, the mice were again anesthetized and then intranasally infected with 100 colony-forming units [CFU, 0.1 median lethal dose (LD50)] of the A66.1 strain of S. pneumoniae (a kind gift from the laboratory of J. A. McCullers) in 40 μL of PBS.
Euthanasia.
For euthanasia, mice were sedated with ketamine (100 mg/kg)-xylazine (10 mg/kg) and then exsanguinated, as previously described (9). Infected mice were observed twice daily and euthanized if their body condition score (39) fell to ≤2.
Bronchoalveolar lavage fluid analysis.
Mice were sedated and exsanguinated as described above, and bronchoalveolar lavage (BAL) fluid was recovered after intratracheal instillation of 0.5 mL of PBS with 1 mM EDTA, pH 8.0; lavage was repeated five times (total volume 2.5 ml). The FITC-dextran concentration in BAL fluid was quantified by spectrophotometry using a fluorescence (excitation at 485 nm, absorbance at 530 nm) microplate (SpectraMax M5, Molecular Devices). Mouse GM-CSF concentrations were measured by Quantikine ELISA (R & D Systems, Minneapolis, MN).
Bacterial cultures.
Organs were aseptically harvested from mice and homogenized in 2.5 mL of PBS. BAL fluid and lung and liver homogenates were serially diluted on blood agar plates, bacteria were grown in the presence of 5% CO2, and backcalculation was performed to determine the number of S. pneumoniae CFUs.
Lung GM-CSF-overexpressing transgenic mice and lung epithelial permeability.
The DTGM mice use the Tet-On system to stimulate GM-CSF expression by bronchiolar club cells. DTGM mice are defined as expressing both the SCGB1A1-rTA and CMV-GM-CSF transgenes, while littermate control (LM) mice express only one of the two required transgenes. Mice were bred as previously described (9). After IAV infection, both DTGM mice and LM controls were exposed to 1 mg/mL doxycycline in drinking water starting 3 days after IAV infection; doxycycline-containing drinking water was replenished every 2–3 days. Mice were anesthetized and tracheotomized as described above (see Invasive aerosolization exposures). FITC-dextran (250 μg in 50 μL of PBS) was instilled into the tracheostomy needle followed by 400 μL of air. Mice were then ventilated for 15 min using the ventilator settings described above. After euthanasia, blood was drawn from the inferior vena cava and placed in Eppendorf tubes. After blood was clotted for 90 min at room temperature in darkness, the tubes were centrifuged at 2,000 g for 10 min at room temperature. Serum supernatants were transferred to fresh tubes and diluted 1:1 with PBS. Serum FITC-dextran concentrations were determined by direct fluorimetry.
Statistical analysis.
All statistical analysis was performed using JMP 12.0.1 software (SAS, Cary, NC). Normally distributed data were analyzed using two-sample t tests, and nonnormally distributed data were analyzed using Wilcoxon’s rank-sum tests. Univariate correlation analysis was conducted using Pearson’s correlation coefficient. Survival analysis was calculated by Kaplan-Meier curves, and the comparison was based on the log-rank test. All data points are means (SD) unless otherwise stated. Graphs were created using Prism 8 for Mac OS X (GraphPad, La Jolla, CA).
RESULTS
Noninvasive aerosol delivery of FITC-dextran.
Using the inExpose aerosolization tower (Fig. 1A), we sought to determine the efficiency of delivery of inhaled agents to the lower respiratory tract of mice lightly restrained by soft restraints (Fig. 1B). After mice were exposed to various concentrations (200–20,000 μg/mL in PBS) of aerosolized FITC-dextran (mol wt 10,000 kDa) for various durations, BAL fluid was recovered and FITC-dextran concentration was measured. The amount of FITC-dextran recovered was proportional to the amount theoretically delivered (Fig. 1C). The efficiency of delivery (%recovery) was calculated as follows: recovered FITC-dextran (μg)/theoretically delivered FITC-dextran (μg) × 100. The efficiency of delivery stabilized over longer exposure times, and the distribution was nonnormal, with a median of 0.0105% (interquartile range 0.0077–0.0177%) (Fig. 1D).
Fig. 1.

A: Scireq inExpose aerosolization tower system wherein aerosolized drugs can be administered simultaneously to up to 12 mice. B: mice are held in place, with their noses exposed to the aerosol, using a soft restraint system that prevents repeated sedation. C: FITC-dextran (mol wt 10,000) was diluted in PBS at 200, 2,000, and 20,000 μg/mL, aerosolized, and administered to mice (n = 12–18 mice per group). At 0–400 s during aerosolization, mice were removed from the tower, the lower respiratory tract was immediately sampled via tracheostomy and bronchoalveolar lavage, and the amount of FITC-dextran was quantified by spectrophotometry. D: lower respiratory tract recovery (delivery) in the mouse was low, with %recovery [recovery (%) = measured (μg)/theoretical delivery (μg) × 100] stabilizing at ~0.01%. E: optimal exposure time was determined by administration of a fixed amount of FITC-labeled dextran (5 μg) to mice (n = 4–6 per group) over various exposure times and aerosol solution concentrations shown in Table 1. Mice treated with FITC-dextran over 22.5 s had the highest mean recovery (3.71 ± 0.21 ng) that was significantly (*P < 0.05) increased from all durations of exposure >45 s; yet the 22.5-s duration group had the lowest coefficient of variance (5.5%).
Optimization of duration of aerosol exposure.
The benefit of the inExpose aerosolization system is the ability to administer therapeutics to the lower respiratory tract of mice, repeatedly, without use of anesthesia. To minimize stress, yet maximize delivery, we sought to determine the minimum exposure time required to deliver a fixed amount of drug to mice effectively and reproducibly. When a given amount of drug is administered via aerosolization, there is one constant, the nebulization rate (2.22 μL/s), and two interdependent variables, the concentration of the drug in solvent (μg/mL) and the exposure time (s) (Table 1). Previously, we demonstrated that ~500–1,000 pg of inducibly airway-expressed GM-CSF conferred protection against mortality from primary IAV infection (9). To reach this level of GM-CSF in the airways, we calculated, given an efficiency of delivery of 0.01%, that administration of 5 μg of GM-CSF would equate to a delivered lower respiratory tract dose of ~500 pg of GM-CSF. To test this, we administered a fixed dose of 5 μg of FITC-dextran to the mice over various durations of exposure (Fig. 1E, Table 1) and then measured FITC-dextran in BAL fluid immediately after the end of exposure. The amount of FITC-dextran recovered was more variable when mice were exposed for only 11.25 s (3.06 ± 2.33 ng), but the recovery was more reproducible and maximized when mice were exposed for 22.5 s (3.71 ± 0.21 ng) compared with longer-duration exposures (Fig. 1E). Averaged across all durations/concentrations, the efficiency of recovery of 5 μg of FITC-dextran was 0.0574 ± 0.0259%.
Table 1.
Variables for administration of aerosols to mice via the inExpose system
| Dose, μg | Aerosol Solution, μg/mL | Volume, μL | Nebulization Rate, μL/s | Exposure Time, s |
|---|---|---|---|---|
| 5 | 200 | 25 | 2.22 | 11.25 |
| 5 | 100 | 50 | 2.22 | 22.5 |
| 5 | 50 | 100 | 2.22 | 45 |
| 5 | 25 | 200 | 2.22 | 90 |
| 5 | 20 | 250 | 2.22 | 112.5 |
| 5 | 10 | 500 | 2.22 | 225 |
| 5 | 5 | 1,000 | 2.22 | 450 |
Dosing of a fixed amount of aerosolized drug (μg) is a function of the fixed solution nebulization rate (2.22 μL/s) and 2 variables: duration of exposure (s) and volume of the solution (μL).
Invasive aerosol delivery of FITC-dextran with flexiVent.
We also investigated the efficiency of delivery when mice were tracheotomized and the aerosol was delivered directly to the lower respiratory tract via the flexiVent rodent ventilator. For these invasive experiments, we kept the exposure time constant at 120 s. Aerosolized FITC-dextran (20–2,000 μg/mL) was administered over 120 s and then recovered from BAL fluid and measured. Again, FITC-dextran delivery was proportional to the concentration of the aerosol solution (r = 0.969, P < 0.001; Fig. 2A). Recovery across the three concentrations in tracheotomized mice, however, was 30-fold higher (0.355 ± 0.102%; Fig. 2B) with the flexiVent than with the inExpose system (0.0105%), as described in Fig. 1D. We also questioned whether more aerosol delivery would occur in larger, older mice due to their theoretically larger airways. To investigate this possibility, we administered various amounts of FITC-dextran to adult (10- to 14-wk-old) and young (4- to 6-wk-old) mice and compared delivery/recovery. FITC-dextran delivery/recovery was significantly reduced in younger mice across the aerosol concentrations (Fig. 2C). Lastly, we examined whether, within the adult mouse group, body weight would correlate with delivery/recovery. Indeed, FITC-dextran delivery/recovery significantly correlated with body weight in adult mice (P < 0.01; Fig. 2D).
Fig. 2.

A: lower respiratory tract recovery of FITC-dextran after tracheostomy and aerosol administration using the Scireq flexiVent rodent ventilator. During mechanical ventilation, various concentrations (20, 200, and 2,000 μg/mL) of aerosols were generated and administered over a fixed time period (120 s) to mice (n = 5–14 mice per group). B: recovered FITC-dextran correlated with the concentration administered (r = 0.969, P < 0.001) via the flexiVent, and recovery (%) was similar across all aerosol concentrations. C: FITC-dextran delivery/recovery was significantly reduced in younger mice, regardless of aerosol concentration. *P < 0.05, **P < 0.01, ***P < 0.001. D: size effect in C was also observed when delivery/recovery was correlated with body weight (BW) in adult mice.
Aerosol delivery and airway pharmacokinetics of GM-CSF.
Using the inExpose aerosolization system, we examined the delivery of the actual therapeutic, rmGM-CSF, to mice and the pharmacokinetics of airway clearance of GM-CSF after administration. Fortunately, the baseline levels of mouse GM-CSF in the lower respiratory tract (2.41 ± 3.14 pg/mL in 2.5 mL of BAL fluid) (9) are near the limit of detection of the GM-CSF Quantikine ELISA kit (minimum standard 7.8125 pg/mL). Therefore, we could safely assume that any GM-CSF measured by ELISA was exogenously administered. We repeated the experiment described in Fig. 1E with GM-CSF to determine the effect of the duration of exposure and solute concentration on drug delivery. rmGM-CSF (5 μg) was dissolved in PBS and administered as shown in Table 1 with a fixed nebulization rate of 2.22 μL/s. Our results agreed with the FITC-dextran experiment: high-concentration aerosols with short exposure times (analyzed as a categorical variable) led to increased GM-CSF deposition in the lower respiratory tract compared with longer exposure times (Fig. 3A; P < 0.05). Exposure to 100 μg/mL GM-CSF for 22.5 s yielded the highest level of recovered GM-CSF (1,040 ± 337 pg) (Fig. 3A); the efficiency of delivery for the 5-μg dose of GM-CSF was slightly lower (0.0208 ± 0.00674%) than for FITC-dextran. When exposure time was analyzed as a continuous variable, there was a significant negative correlation (r = −0.649, P < 0.01), with the amount recovered decreasing with longer exposure times (Fig. 3B).
Fig. 3.

A: lower respiratory tract delivery/recovery of mouse granulocyte/macrophage colony-stimulating factor (GM-CSF) following aerosolization and administration of recombinant mouse GM-CSF (rmGM-CSF, 5 μg) to mice (n = 4–6 per group) over various exposure times and aerosol concentrations via the inExpose system. Immediately after aerosol exposure, bronchoalveolar lavage (BAL) fluid was recovered, and mouse GM-CSF levels were measured by ELISA. High-concentration aerosols with short exposure times (analyzed as a categorical variable) led to increased GM-CSF deposition in the lower respiratory tract compared with longer exposure times (*P < 0.05). B: analysis of exposure time as a continuous variable showed a significant negative correlation between exposure time and GM-CSF recovery. C: to assay the airway pharmacokinetics, aerosolized rmGM-CSF (5 μg, 100 μg/mL in PBS) was administered to mice (n = 5–6 per group) over 22.5 s, BAL fluid was recovered 0–4 h after exposure, and mouse GM-CSF levels were measured by ELISA. Airway pharmacokinetics of inhaled GM-CSF demonstrated a 2-compartment model with an initial (distribution) phase [half-life (t1/2) = 0.88 h] followed by a less-steep (terminal) phase (t1/2 = 1.3 h).
We next sought to determine the airway pharmacokinetics of GM-CSF following administration. Again 5 μg of GM-CSF at 100 μg/mL in PBS was administered over 22.5 s to adult male and female mice, and BAL fluid was recovered at 0–5 h after exposure (Fig. 1C). After aerosol administration, airway GM-CSF levels decreased in a two-phase process. The first (distribution) phase, from 0 to 0.5 h, showed an accelerated clearance with airway half-life (t1/2) of 0.88 h, whereas the second (terminal) phase, from 0.5 to 4 h, showed a slower clearance with an airway t1/2 of 1.3 h. Interestingly, subset analysis revealed lower GM-CSF recovery in female than male mice (19.1 ± 0.853 vs. 27.3 ± 0.401 g, P < 0.05; Supplemental Fig. S1, available at https://doi.org/10.6084/m9.figshare.11351630.v1).
Effect of inhaled GM-CSF on susceptibility to pneumococcus after influenza infection.
We recently reported that airway GM-CSF overexpression after IAV infection markedly decreased mortality (9) but that GM-CSF did not affect the initial weight loss associated with virus infection but, instead, conferred protection during the recovery phase through unclear mechanisms (9). We hypothesized that elevated airway levels of GM-CSF conferred protection by preventing SBP. However, our published transgenic model did not lend itself to answering this question, since we used an antibiotic, doxycycline, to induce airway GM-CSF secretion by club cells using a Tet-On system (9). Therefore, we investigated whether administration of GM-CSF via noninvasive aerosolization, twice daily, to mice during IAV infection could confer a survival benefit to mice that were secondarily infected with S. pneumoniae (A66.1 strain) (31, 33) to model ARDS (8). Mice were anesthetized and infected intranasally with 100 FFU (<0.1 LD50) of the PR8 strain of IAV. From 5 to 9 days postinfection, one group of mice received inhaled GM-CSF (5 μg) over 22.5 s via the inExpose tower twice daily (Fig. 4A). Control mice received inhaled vehicle only. A third group received GM-CSF (1 ng) via intraperitoneal injection twice daily from 5 to 9 days postinfection to determine if the effect of GM-CSF was systemic or locally mediated. At 7 days postinfection, mice were anesthetized again and infected intranasally with 100 CFU of the A66.1 strain of S. pneumoniae. Inhaled GM-CSF conferred a significant survival benefit to mice compared with control (Fig. 4B; P < 0.05), whereas intraperitoneal injection of GM-CSF did not confer a survival benefit. We tested whether inhaled GM-CSF reduced levels of S. pneumoniae in various organ compartments 18 h after intranasal infection (8 days postinfection) (Fig. 4C). Inhaled GM-CSF did not affect pneumococcal growth in either the airways (BAL) or the lung parenchyma (lung homogenates) but significantly reduced systemic bacteremia as determined by growth from homogenized liver (Fig. 4C; P < 0.01).
Fig. 4.

A and B: effect of administration of recombinant mouse granulocyte/macrophage colony-stimulating factor [rmGM-CSF (Rx)] during influenza A virus (IAV) infection and secondary bacterial pneumonia with Streptococcus pneumoniae was examined in terms of body weight loss/recovery and survival. rmGM-CSF (5 μg) was administered twice daily via inhalation using the inExpose system (iGM-CSF), or 1 ng was administered via intraperitoneal injection at the same time points (ip GM-CSF), whereas control mice only received inhaled PBS (iPBS). Numbers of mice per group surviving or still at risk are listed below days postinjection (DPI) in B. Inhaled rmGM-CSF conferred a significant survival (*P < 0.05) compared with the iPBS control group. C: to determine whether iGM-CSF enhanced bacterial clearance in various organ compartments, mice were harvested 18 h after secondary infection with S. pneumoniae, and bronchoalveolar lavage (BAL) fluid, lung homogenates, and liver homogenates were grown on blood agar plates. Inhaled GM-CSF did not affect bacterial counts in airways or lung parenchyma but lowered bacterial growth from liver homogenates (**P < 0.01), suggesting less systemic bacteremia.
Effect of GM-CSF overexpression on lung epithelial permeability during IAV infection.
We hypothesized that GM-CSF enhanced lung epithelial barrier function in vivo during IAV infection. To test this hypothesis, we used our inducible conditional transgenic DTGM model, wherein we can induce GM-CSF in the airways (BAL levels ~1,250 pg) with doxycycline administration during active influenza infection (9). DTGM and LM mice were infected with IAV (1 LD50), and doxycycline was added to drinking water at 3 days postinfection. At 10 days postinfection, mice were tracheotomized, FITC-dextran was administered to their airways, and the mice were ventilated for 15 min. Lung epithelial permeability from the airway to the bloodstream was measured by quantifying serum levels of FITC-dextran. In our preliminary experiment, our negative control (naïve WT mouse) demonstrated a FITC-dextran level of 0.1 μg/mL, whereas our positive control [a WT mouse 24 h after intranasal administration of LPS (100 μg) to induce acute lung injury] demonstrated a serum FITC-dextran level of 23.6 μg/mL. Uninfected mice demonstrated some baseline epithelial permeability, with serum FITC-dextran levels of 0.342 ± 0.268 μg/mL (Fig. 5). In contrast, in IAV-infected mice, lung permeability at 10 days postinfection did not differ, with GM-CSF overexpression of 8.86 ± 6.33 and 8.69 ± 7.08 μg/mL for LM and DTGM, respectively. Interestingly, GM-CSF overexpression in the absence of IAV infection led to increased permeability (4.27 ± 2.05 μg/mL).
Fig. 5.
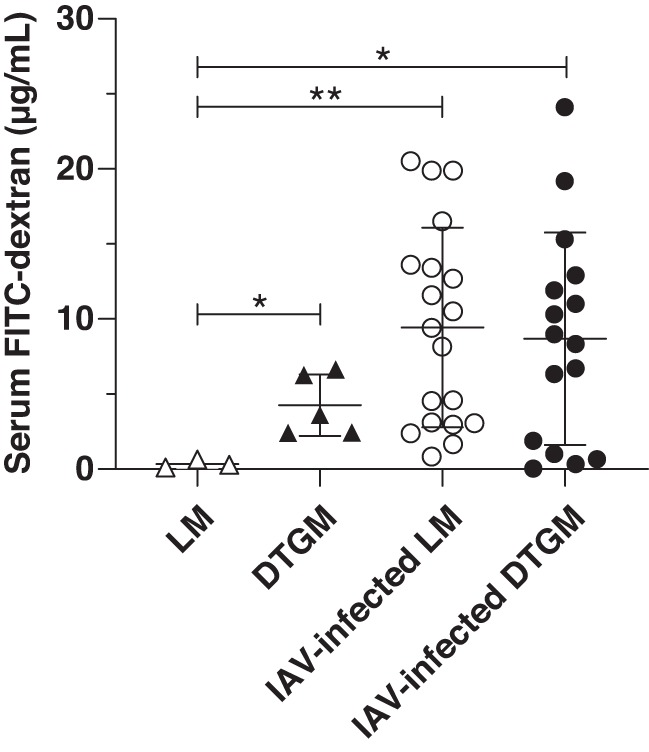
Effect of granulocyte/macrophage colony-stimulating factor (GM-CSF) overexpression during influenza A virus (IAV) infection on lung epithelial permeability was examined using FITC-dextran. Airway GM-CSF-overexpressing transgenic (DTGM) or littermate control (LM) mice were infected with IAV, and doxycycline was administered in drinking water starting at 3 days postinfection. At 10 days postinfection, mice were tracheotomized, received FITC-dextran via the tracheostomy needle, and were ventilated for 15 min. Serum FITC-dextran levels were measured by spectrophotometry in uninfected LM (△) and DTGM (▲) mice and at 10 days postinfection in LM (○) and DTGM (●) mice. GM-CSF overexpression in the absence of IAV infection significantly increased lung permeability (*P < 0.05). While IAV infection increased lung permeability compared with uninfected LM mice (**P < 0.01), in IAV-infected animals (○ and ●), GM-CSF overexpression did not affect serum levels of FITC-dextran.
DISCUSSION
While GM-CSF may be a viable therapeutic option for viral/bacterial pneumonia, the precise mechanisms of protection are unclear. Previous studies have used either intranasal administration of GM-CSF (4, 32) or constitutive overexpression models (12, 40) to investigate mechanisms of GM-CSF-mediated protection. In this study we generated a mouse model wherein we could repeatedly administer aerosolized GM-CSF to mice to investigate its therapeutic potential during viral and bacterial pneumonias. We wanted to characterize this aerosolization method to determine the delivery and airway pharmacokinetics of inhaled GM-CSF. While lower respiratory tract delivery of inhaled GM-CSF was low, aerosol treatment with GM-CSF did confer protection against SBP with S. pneumoniae during IAV infection. We plan future studies to examine the benefit of inhaled GM-CSF on various transgenic strains to better determine the mechanism of protection.
While some groups have estimated the delivery of aerosols using the inExpose system (2), this study is the first, to our knowledge, to directly measure the delivery of aerosolized therapeutics to mice. While the efficiency of delivery to the lower respiratory tract was low (0.01% with the inExpose system and 0.3% with the flexiVent), the delivery was reproducible. A recent study detailed the deposition of nanoparticles in murine lungs after aerosolization using the flexiVent and, with light sheet fluorescence microscopy, determined a deposition efficiency of 5%, which is much higher than the value we measured (43). We cannot ascertain the reason for the discrepancy; however, we are confident in our value for the percent recovery, as it was similar for both FITC-dextran (mol wt 10,000) and rmGM-CSF and it fit our pharmacokinetic modeling of GM-CSF clearance from the airways. Ventilation and aerosol protocol variations can lead to differences in aerosol delivery efficiency and, consequently, in the dose delivered to the subjects (27). Our low efficiency of delivery likely is due to multiple factors, including the high bias flow with the systems compared with the small minute ventilation of the mice: 150 breaths/min × 10 μL/g tidal volume × 20 g body wt = 30,000 μL/min = 30 mL/min = 0.03 L/min minute ventilation (compared with 1.5 L/min bias flow with the inExpose system). However, the strength of our study is that we examine the “real-world” delivery of aerosolized GM-CSF using the flexiVent and inExpose systems.
As determined for humans, aerosol particle size also has tremendous importance in whether a drug is inhaled and subsequently exhaled without delivery (diffusion, 0.05–0.4 μm), is delivered to the lower respiratory tract (sedimentation, 0.5–5 μm), or is too large to even be delivered to the respiratory system (inertial impaction, >5 μm) (18). Of course, the airways of mice are much smaller than those of humans. The airways of C57BL/6 mice are 1.5–1.9 mm diameter (trachea) down to 0.6–0.9 mm diameter after six branching generations (36). Therefore, the mouse trachea is approximately the same diameter as a 10th-generation human terminal bronchiole (19). This likely leads to preferential deposition of aerosolized drug in the proximal airways, a phenomenon likely even more prevalent in mice (17) than humans (16). Indeed, delivery/recovery of aerosolized drug was lower in younger (Fig. 2C), smaller (Fig. 2D), and female (Supplemental Fig. S1) mice, all likely due to smaller airway size. The smaller/younger mice likely have a longer airway time constant (10), and it is possible that, with longer exposure times, this difference would be mitigated. Finally, even in the larger adult mice, the amount of FITC-dextran recovered correlated with body weight (r = 0.689, P < 0.01), whereas, theoretically, one would assume that it would remain constant across body weight (Fig. 2D). This has interesting implications for how aerosolized drugs are dosed for pediatric patients, as the typical weight- or body surface area-based approach may not be sufficient for aerosolized therapeutics (1). Indeed, in a pediatric cystic fibrosis population pharmacokinetic study of inhaled tobramycin, serum peaks were lower with decreasing age, even though the same dose (300 mg/5 mL, twice daily) was administered to all patients (41).
Interestingly, the optimal duration of exposure of a fixed amount of drug was relatively short, only 22.5 s. We do not have a satisfactory explanation as to why shorter-duration exposures with higher concentrations of drug would lead to better lower respiratory tract delivery. It is most likely that higher concentrations of drug in the Aeroneb chamber are less susceptible to adsorption on the various surfaces of the equipment before delivery to the mice. It is also possible that, with longer exposure times, evaporative cooling of the nasopharynx created more liquid films in the airways and impeded delivery. Finally, it is possible that the solute concentration in the aerosol droplets and the epithelial lining fluid (ELF) quickly reaches a dynamic equilibrium and that the main determinant of ELF concentration is the concentration of the solute in the aerosol droplet. Regardless of the mechanism, this short exposure time was fortuitous, as it lessened the stress of the twice-daily inExpose treatments on the mice.
The 5-μg dose of aerosolized GM-CSF led to a peak airway level of ~1,250 pg/2.5 mL of BAL fluid (500 pg/mL), and the airway half-life of GM-CSF was relatively short (~1 h) in uninfected mice. We previously reported that inducible transgenic overexpression of airway GM-CSF at a level of 300 pg/mL after IAV infection conferred protection against mortality (9). We were concerned that twice-daily treatment with aerosolized GM-CSF may not achieve sufficient airway levels to yield a biologic effect. However, inhaled GM-CSF did confer protection against SBP with S. pneumoniae, indicating that we had provided sufficient exogenous amounts. However, the mechanism of protection is still unclear. Intraperitoneal administration of rmGM-CSF did not affect mortality in our SBP model, while inhaled rmGM-CSF did confer protection. This result suggests that the protective effect conferred by GM-CSF is mediated locally in the lung. Interestingly, a 2012 clinical trial of intravenous recombinant human GM-CSF for patients with acute lung injury did not demonstrate any clinical benefit as measured by ventilator-free days (23). Along this line of thought, the multicenter clinical trial of inhaled GM-CSF (GI-HOPE) is underway in Europe; however, no clinical trial of inhaled GM-CSF for pneumonia has been approved by the US Food and Drug Administration.
We were surprised that, instead of enhancing airway bacterial clearance, as has been previously described (32), inhaled GM-CSF led to decreased systemic bacteremia, while it did not decrease bacterial recovery from lung tissue (Fig. 4C). To translocate from the air space to the bloodstream, bacteria must traverse several barriers. Bacteria must first pass the epithelial barrier generated by type I and type II alveolar epithelial cells (AECs) to enter the interstitium. Then bacteria can enter the bloodstream by lymphatic drainage of the interstitium, or they need to pass the endothelial barrier to enter the bloodstream directly. GM-CSF has been shown to enhance lung barrier function by its actions on NF-κB (42). Given that virtually all (>92%) resistance to albumin flux across the alveolocapillary membrane lies in the epithelial barrier (7), our original hypothesis was that GM-CSF enhanced nonspecific epithelial barrier function, presumably due to direct actions on type I and type II AECs and blocked paracellular translocation of pneumococcus from the airway to the lymphatics and/or bloodstream. Type II AECs express the GM-CSF receptor α-subunit (13), and while type I and type II AECs may have separate embryological origins, type II AECs can serve as type I progenitors (6). However, given that GM-CSF overexpression during IAV did not affect epithelial barrier function against FITC-dextran permeation in the bloodstream (Fig. 5), it seems that GM-CSF does not enhance nonspecific epithelial barrier function but, instead, specifically affects active lung barrier function against bacterial invasion.
GM-CSF may affect the transcellular translocation of pneumococcus by changing the phenotype of type II AECs. Indeed, type II AECs do express lysozyme and are associated with both lysosome and lamellar body organelles (5, 22), although whether lysosomes serve an antibacterial function in type II AECs is unknown. However, it is entirely plausible that the actions of GM-CSF are due only to its effects on leukocytes and do not involve epithelial cells. GM-CSF may increase the number and/or antibacterial activity of local AMs (37), or it may affect the differentiation of newly recruited monocytes during IAV infection to a more functional antibacterial phenotype. With this new aerosolization model, we will be able to investigate these possibilities.
This study does have several limitations. Our delivery efficacy results are based on the assumption that recovery is interchangeable with delivery of the therapeutic of interest. Of course, this may not be the case, as the drug may adsorb to the lung mucosal surface and may not be recoverable by simple lung lavage. However, the fact that the efficiency of “delivery” of inhaled GM-CSF agreed well with that of inhaled FITC-dextran supports the idea that this may be a reasonable assumption. While we were surprised at the difference between noninvasive inhalation using the inExpose system and invasive delivery via tracheostomy and the flexiVent (30-fold higher), we cannot surmise too much from these results, as the Aeroneb chambers were slightly different between the two systems: delivery via the flexiVent was via a fine (2- to 4-μm droplet size) mist Aeroneb, whereas delivery via the inExpose was performed using a standard (4- to 6-μm droplet size) mist Aeroneb. This result should be investigated further, however, as it does have implications in the clinic, where similar dosages of inhaled therapeutics are used, regardless of whether a patient is intubated or not. Finally, while the airway half-life of GM-CSF in naïve, uninflamed murine lungs was ~1 h, it is likely that, during inflammation, the influx of leukocytes accelerates the airway clearance of GM-CSF and, thereby, shortens the half-life. Future studies are needed to determine 1) the level of airway GM-CSF needed to elicit the desired effect and 2) whether, during inflammation, GM-CSF must be administered more often to remain above critical levels, as these GM-CSF levels may influence cellular differentiation (34).
GRANTS
This work was funded by the Children’s Miracle Network and the Department of Pediatrics, Penn State University College of Medicine. E. S. Halstead is currently supported by the National Institute of Allergy and Infectious Diseases through the National Institutes of Health Loan Repayment Program.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.M.U., Z.C.C., and E.S.H. conceived and designed research; T.M.U., E.K.H., M.M., and S.B. performed experiments; T.M.U., E.K.H., M.M., S.B., M.W., and E.S.H. analyzed data; T.M.U., Z.C.C., and E.S.H. interpreted results of experiments; E.S.H. prepared figures; E.S.H. drafted manuscript; T.M.U., E.K.H., M.M., Z.C.C., M.W., and E.S.H. edited and revised manuscript; T.M.U., E.K.H., M.M., S.B., Z.C.C., M.W., and E.S.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Kevan Hartshorn and Mitchell White for assistance in preparing the influenza virus used in all experiments.
REFERENCES
- 1.Amirav I, Balanov I, Gorenberg M, Luder AS, Newhouse MT, Groshar D. β-Agonist aerosol distribution in respiratory syncytial virus bronchiolitis in infants. J Nucl Med 43: 487–491, 2002. [PubMed] [Google Scholar]
- 2.Fulzele SV, Chatterjee A, Shaik MS, Jackson T, Singh M. Inhalation delivery and anti-tumor activity of celecoxib in human orthotopic non-small cell lung cancer xenograft model. Pharm Res 23: 2094–2106, 2006. doi: 10.1007/s11095-006-9074-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghoneim HE, McCullers JA. Adjunctive corticosteroid therapy improves lung immunopathology and survival during severe secondary pneumococcal pneumonia in mice. J Infect Dis 209: 1459–1468, 2014. doi: 10.1093/infdis/jit653. [DOI] [PubMed] [Google Scholar]
- 4.Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J Immunol 191: 1250–1259, 2013. doi: 10.4049/jimmunol.1300014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gibson KF, Phadke S. Intracellular distribution of lysozyme in rat alveolar type II epithelial cells. Exp Lung Res 20: 595–611, 1994. doi: 10.3109/01902149409031739. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez R, Leaffer D, Chapin C, Gillespie AM, Eckalbar W, Dobbs L. Cell fate analysis in fetal mouse lung reveals distinct pathways for TI and TII cell development. Am J Physiol Lung Cell Mol Physiol 317: L653–L666, 2019. doi: 10.1152/ajplung.00503.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorin AB, Stewart PA. Differential permeability of endothelial and epithelial barriers to albumin flux. J Appl Physiol 47: 1315–1324, 1979. doi: 10.1152/jappl.1979.47.6.1315. [DOI] [PubMed] [Google Scholar]
- 8.Gotts JE, Bernard O, Chun L, Croze RH, Ross JT, Nesseler N, Wu X, Abbott J, Fang X, Calfee CS, Matthay MA. Clinically relevant model of pneumococcal pneumonia, ARDS, and nonpulmonary organ dysfunction in mice. Am J Physiol Lung Cell Mol Physiol 317: L717–L736, 2019. doi: 10.1152/ajplung.00132.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halstead ES, Umstead TM, Davies ML, Kawasawa YI, Silveyra P, Howyrlak J, Yang L, Guo W, Hu S, Hewage EK, Chroneos ZC. GM-CSF overexpression after influenza a virus infection prevents mortality and moderates M1-like airway monocyte/macrophage polarization. Respir Res 19: 3, 2018. doi: 10.1186/s12931-017-0708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hammer J, Newth CJ. Infant lung function testing in the intensive care unit. Intensive Care Med 21: 744–752, 1995. doi: 10.1007/BF01704742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herold S, Hoegner K, Vadász I, Gessler T, Wilhelm J, Mayer K, Morty RE, Walmrath HD, Seeger W, Lohmeyer J. Inhaled granulocyte/macrophage colony-stimulating factor as treatment of pneumonia-associated acute respiratory distress syndrome. Am J Respir Crit Care Med 189: 609–611, 2014. doi: 10.1164/rccm.201311-2041LE. [DOI] [PubMed] [Google Scholar]
- 12.Huang FF, Barnes PF, Feng Y, Donis R, Chroneos ZC, Idell S, Allen T, Perez DR, Whitsett JA, Dunussi-Joannopoulos K, Shams H. GM-CSF in the lung protects against lethal influenza infection. Am J Respir Crit Care Med 184: 259–268, 2011. doi: 10.1164/rccm.201012-2036OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huffman Reed JA, Rice WR, Zsengellér ZK, Wert SE, Dranoff G, Whitsett JA. GM-CSF enhances lung growth and causes alveolar type II epithelial cell hyperplasia in transgenic mice. Am J Physiol Lung Cell Mol Physiol 273: L715–L725, 1997. doi: 10.1152/ajplung.1997.273.4.L715. [DOI] [PubMed] [Google Scholar]
- 14.Jain S, Kamimoto L, Bramley AM, Schmitz AM, Benoit SR, Louie J, Sugerman DE, Druckenmiller JK, Ritger KA, Chugh R, Jasuja S, Deutscher M, Chen S, Walker JD, Duchin JS, Lett S, Soliva S, Wells EV, Swerdlow D, Uyeki TM, Fiore AE, Olsen SJ, Fry AM, Bridges CB, Finelli L; 2009 Pandemic Influenza A (H1N1) Virus Hospitalizations Investigation Team . Hospitalized patients with 2009 H1N1 influenza in the United States, April–June 2009. N Engl J Med 361: 1935–1944, 2009. doi: 10.1056/NEJMoa0906695. [DOI] [PubMed] [Google Scholar]
- 15.Karlström A, Heston SM, Boyd KL, Tuomanen EI, McCullers JA. Toll-like receptor 2 mediates fatal immunopathology in mice during treatment of secondary pneumococcal pneumonia following influenza. J Infect Dis 204: 1358–1366, 2011. doi: 10.1093/infdis/jir522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim CS, Hu SC, DeWitt P, Gerrity TR. Assessment of regional deposition of inhaled particles in human lungs by serial bolus delivery method. J Appl Physiol (1985) 81: 2203–2213, 1996. doi: 10.1152/jappl.1996.81.5.2203. [DOI] [PubMed] [Google Scholar]
- 17.Kuehl PJ, Anderson TL, Candelaria G, Gershman B, Harlin K, Hesterman JY, Holmes T, Hoppin J, Lackas C, Norenberg JP, Yu H, McDonald JD. Regional particle size dependent deposition of inhaled aerosols in rats and mice. Inhal Toxicol 24: 27–35, 2012. doi: 10.3109/08958378.2011.632787. [DOI] [PubMed] [Google Scholar]
- 18.Labiris NR, Dolovich MB. Pulmonary drug delivery. I. Physiological factors affecting therapeutic effectiveness of aerosolized medications. Br J Clin Pharmacol 56: 588–599, 2003. doi: 10.1046/j.1365-2125.2003.01892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levitzky MG. Function and structure of the respiratory system In: Pulmonary Physiology (8th ed.). New York: McGraw-Hill, 2013, p. 1–10. [Google Scholar]
- 20.Marcelin G, Aldridge JR, Duan S, Ghoneim HE, Rehg J, Marjuki H, Boon AC, McCullers JA, Webby RJ. Fatal outcome of pandemic H1N1 2009 influenza virus infection is associated with immunopathology and impaired lung repair, not enhanced viral burden, in pregnant mice. J Virol 85: 11208–11219, 2011. doi: 10.1128/JVI.00654-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol 12: 252–262, 2014. doi: 10.1038/nrmicro3231. [DOI] [PubMed] [Google Scholar]
- 22.Miyake Y, Kaise H, Isono K, Koseki H, Kohno K, Tanaka M. Protective role of macrophages in noninflammatory lung injury caused by selective ablation of alveolar epithelial type II cells. J Immunol 178: 5001–5009, 2007. doi: 10.4049/jimmunol.178.8.5001. [DOI] [PubMed] [Google Scholar]
- 23.Paine R III, Standiford TJ, Dechert RE, Moss M, Martin GS, Rosenberg AL, Thannickal VJ, Burnham EL, Brown MB, Hyzy RC. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med 40: 90–97, 2012. doi: 10.1097/CCM.0b013e31822d7bf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Randolph AG, Vaughn F, Sullivan R, Rubinson L, Thompson BT, Yoon G, Smoot E, Rice TW, Loftis LL, Helfaer M, Doctor A, Paden M, Flori H, Babbitt C, Graciano AL, Gedeit R, Sanders RC, Giuliano JS, Zimmerman J, Uyeki TM; Pediatric Acute Lung Injury and Sepsis Investigator’s Network and the National Heart, Lung, and Blood Institute ARDS Clinical Trials Network . Critically ill children during the 2009–2010 influenza pandemic in the United States. Pediatrics 128: e1450–e1458, 2011. doi: 10.1542/peds.2011-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reed JA, Ikegami M, Cianciolo ER, Lu W, Cho PS, Hull W, Jobe AH, Whitsett JA. Aerosolized GM-CSF ameliorates pulmonary alveolar proteinosis in GM-CSF-deficient mice. Am J Physiol Lung Cell Mol Physiol 276: L556–L563, 1999. doi: 10.1152/ajplung.1999.276.4.L556. [DOI] [PubMed] [Google Scholar]
- 26.Rice TW, Rubinson L, Uyeki TM, Vaughn FL, John BB, Miller RR III, Higgs E, Randolph AG, Smoot BE, Thompson BT; NHLBI ARDS Network . Critical illness from 2009 pandemic influenza A virus and bacterial coinfection in the United States. Crit Care Med 40: 1487–1498, 2012. doi: 10.1097/CCM.0b013e3182416f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robichaud A, Fereydoonzad L, Schuessler TF. Delivered dose estimate to standardize airway hyperresponsiveness assessment in mice. Am J Physiol Lung Cell Mol Physiol 308: L837–L846, 2015. doi: 10.1152/ajplung.00343.2014. [DOI] [PubMed] [Google Scholar]
- 28.Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, Vogel J, Kopf M. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog 10: e1004053, 2014. doi: 10.1371/journal.ppat.1004053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sever-Chroneos Z, Murthy A, Davis J, Florence JM, Kurdowska A, Krupa A, Tichelaar JW, White MR, Hartshorn KL, Kobzik L, Whitsett JA, Chroneos ZC. GM-CSF modulates pulmonary resistance to influenza A infection. Antiviral Res 92: 319–328, 2011. doi: 10.1016/j.antiviral.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skarbinski J, Jain S, Bramley A, Lee EJ, Huang J, Kirschke D, Stone A, Wedlake T, Richards SM, Page S, Ragan P, Bullion L, Neises D, Williams RM, Petruccelli BP, Vandermeer M, Lofy KH, Gindler J, Finelli L; 2009 Pandemic Influenza A H1N1 Virus Fall Hospitalizations Investigation Team . Hospitalized patients with 2009 pandemic influenza A (H1N1) virus infection in the United States—September–October 2009. Clin Infect Dis 52, Suppl 1: S50–S59, 2011. doi: 10.1093/cid/ciq021. [DOI] [PubMed] [Google Scholar]
- 31.Smith AM, Adler FR, Ribeiro RM, Gutenkunst RN, McAuley JL, McCullers JA, Perelson AS. Kinetics of coinfection with influenza A virus and Streptococcus pneumoniae. PLoS Pathog 9: e1003238, 2013. doi: 10.1371/journal.ppat.1003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Subramaniam R, Barnes PF, Fletcher K, Boggaram V, Hillberry Z, Neuenschwander P, Shams H. Protecting against post-influenza bacterial pneumonia by increasing phagocyte recruitment and ROS production. J Infect Dis 209: 1827–1836, 2014. doi: 10.1093/infdis/jit830. [DOI] [PubMed] [Google Scholar]
- 33.Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-γ during recovery from influenza infection. Nat Med 14: 558–564, 2008. doi: 10.1038/nm1765. [DOI] [PubMed] [Google Scholar]
- 34.Sun L, Rautela J, Delconte RB, Souza-fonseca-guimaraes F, Lew AM, Xu Y, Zhan Y. GM-CSF quantity has a selective effect on granulocytic vs. monocytic myeloid development and function. Front Immunol 9: 1922, 2018. doi: 10.3389/fimmu.2018.01922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tazawa R, Trapnell BC, Inoue Y, Arai T, Takada T, Nasuhara Y, Hizawa N, Kasahara Y, Tatsumi K, Hojo M, Ishii H, Yokoba M, Tanaka N, Yamaguchi E, Eda R, Tsuchihashi Y, Morimoto K, Akira M, Terada M, Otsuka J, Ebina M, Kaneko C, Nukiwa T, Krischer JP, Akazawa K, Nakata K. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med 181: 1345–1354, 2010. doi: 10.1164/rccm.200906-0978OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thiesse J, Namati E, Sieren JC, Smith AR, Reinhardt JM, Hoffman EA, McLennan G. Lung structure phenotype variation in inbred mouse strains revealed through in vivo micro-CT imaging. J Appl Physiol (1985) 109: 1960–1968, 2010. doi: 10.1152/japplphysiol.01322.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Todd EM, Ramani R, Szasz TP, Morley SC. Inhaled GM-CSF in neonatal mice provides durable protection against bacterial pneumonia. Sci Adv 5: eaax3387, 2019. doi: 10.1126/sciadv.aax3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trapnell BC, Carey BC, Uchida K, Suzuki T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol 21: 514–521, 2009. doi: 10.1016/j.coi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ullman-Culleré MH, Foltz CJ. Body condition scoring: a rapid and accurate method for assessing health status in mice. Lab Anim Sci 49: 319–323, 1999. [PubMed] [Google Scholar]
- 40.Unkel B, Hoegner K, Clausen BE, Lewe-Schlosser P, Bodner J, Gattenloehner S, Janßen H, Seeger W, Lohmeyer J, Herold S. Alveolar epithelial cells orchestrate DC function in murine viral pneumonia. J Clin Invest 122: 3652–3664, 2012. doi: 10.1172/JCI62139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X, Koehne-Voss S, Anumolu SS, Yu J. Population pharmacokinetics of tobramycin inhalation solution in pediatric patients with cystic fibrosis. J Pharm Sci 106: 3402–3409, 2017. doi: 10.1016/j.xphs.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 42.Ward C, Schlingmann B, Stecenko AA, Guidot DM, Koval M. NF-κB inhibitors impair lung epithelial tight junctions in the absence of inflammation. Tissue Barriers 3: e982424, 2015. doi: 10.4161/21688370.2014.982424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang L, Feuchtinger A, Möller W, Ding Y, Kutschke D, Möller G, Schittny JC, Burgstaller G, Hofmann W, Stoeger T, Daniel Razansky, Walch A, Schmid O. Three-dimensional quantitative co-mapping of pulmonary morphology and nanoparticle distribution with cellular resolution in nondissected murine lungs. ACS Nano 13: 1029–1041, 2019. doi: 10.1021/acsnano.8b07524. [DOI] [PubMed] [Google Scholar]