

Abstract
Preeclampsia is a pregnancy-related disorder characterized by hypertension, vascular dysfunction and an increase in circulating inflammatory factors including the cytokine, tumor necrosis factor-α (TNF-α). Studies have shown that placental ischemia is associated with 1) increased circulating TNF-α, 2) attenuated pressure-induced cerebral vascular tone, and 3) suppression of β-epithelial Na+ channel (βENaC) protein in cerebral vessels. In addition to its role in epithelial Na+ and water transport, βENaC is an essential signaling element in transduction of pressure-induced (aka “myogenic”) constriction, a critical mechanism of blood flow autoregulation. While cytokines inhibit expression of certain ENaC proteins in epithelial tissue, it is unknown if the increased circulating TNF-α associated with placental ischemia mediates the loss of cerebrovascular βENaC and cerebral blood flow regulation. Therefore, the purpose of this study was to test the hypothesis that increasing plasma TNF-α in normal pregnant rats reduces cerebrovascular βENaC expression and impairs cerebral blood flow (CBF) regulation. In vivo TNF-α infusion (200 ng/day, 5 days) inhibited cerebrovascular expression of βENaC and impaired CBF regulation in pregnant rats. To determine the direct effects of TNF-α and underlying pathways mediating vascular smooth muscle cell βENaC reduction, we exposed cultured VSMCs (A10 cell line) to TNF-α (1–100 ng/mL) for 16–24 h. TNF-α reduced βENaC protein expression in a concentration-dependent fashion from 0.1 to 100 ng/mL, without affecting cell death. To assess the role of canonical MAPK signaling in this response, VSMCs were treated with p38MAPK or c-Jun kinase (JNK) inhibitors in the presence of TNF-α. We found that both p38MAPK and JNK blockade prevented TNF-α-mediated βENaC protein suppression. These data provide evidence that disorders associated with increased circulating TNF-α could lead to impaired cerebrovascular regulation, possibly due to reduced βENaC-mediated vascular function.
NEW & NOTEWORTHY This manuscript identifies TNF-α as a possible placental-derived cytokine that could be involved in declining cerebrovascular health observed in preeclampsia. We found that infusion of TNF-α during pregnancy impaired cerebral blood flow control in rats at high arterial pressures. We further discovered that cerebrovascular β-epithelial sodium channel (βENaC) protein, a degenerin protein involved in mechanotransduction, was reduced by TNF-α in pregnant rats, indicating a potential link between impaired blood flow and this myogenic player. We next examined this effect in vitro using a rat vascular smooth muscle cell line. TNF-α reduced βENaC through canonical MAPK-signaling pathways and was not dependent on cell death. This study demonstrates the pejorative effects of TNF-α on cerebrovascular function during pregnancy and warrants future investigations to study the role of cytokines on vascular function during pregnancy.
Keywords: cerebral blood flow, βENaC, JNK, p38MAPK, TNF-α, vascular smooth muscle cells
INTRODUCTION
The ischemic placenta is widely considered to have a central role in the pathophysiology of preeclampsia, where proinflammatory cytokines and anti-angiogenic factors are released into the circulation. Preeclamptic patients often present with neurological symptoms and cerebrovascular events are reported to contribute to ~40% of all preeclampsia/eclampsia-related deaths; however, the biological factors involved in these events are unknown (6, 39). In both preeclamptic patients and in a rodent model of reduced uterine perfusion pressure (RUPP), placental ischemia leads to elevated levels of circulating proinflammatory cytokines, impaired cerebral myogenic tone, and impaired cerebral blood flow (CBF) autoregulation (6, 10, 31, 37, 43, 51, 63). Tumor necrosis factor-α (TNF-α) is among the most well-characterized cytokines in preeclampsia and has been shown to increase mean arterial pressure (MAP), cerebral edema, and blood-brain-barrier permeability in normal pregnant rats (62). These effects are reversed in placental ischemic rats treated with the TNF-α inhibitor, etanercept (62). While these findings suggest a link between placental ischemia-induced cerebrovascular events and TNF-α (proinflammatory cytokine), the mechanistic role of TNF-α on cerebrovascular regulation during pregnancy is not known.
Epithelial sodium channel (ENaC) proteins are members of the degenerin protein family and form non-voltage-gated Na+ channels (3, 14, 15, 23). ENaC proteins are abundantly expressed in epithelial cells where they serve a rate-limiting role in Na+ and water transport (20). ENaC proteins are also expressed in vascular smooth muscle cells (VSMCs) and participate in signaling pressure-induced changes in vascular tone (2, 14–17, 21, 22, 24, 28–30, 33–35, 45, 56, 58, 60). While three subunits (α, β, γ) form the canonical ENaC channel present in epithelial tissue, individual subunits are also capable of forming channels that have been found to generate current (4, 5, 32, 65). Among the subunits, βENaC appears to be most highly expressed in VSMCs and has been shown to mediate pressure-induced changes in vascular tone in renal afferent arterioles, renal interlobar arteries, and cerebral arteries (21, 28, 34, 58). Both rats with RUPP and women with preeclampsia express reduced levels of βENaC in the cerebral vasculature and placental, respectively, suggesting a potential association between placental factors and ENaC (51, 59). However, a direct link between placental ischemia-induced factors and inhibition of vascular βENaC expression remains unclear.
The role of individual placental-derived factors on vascular βENaC expression has not been previously studied, but there is evidence that epithelial ENaC expression is regulated by a number of hormones, growth factors, and inflammatory cytokines, including TNF-α (9, 12, 13, 19, 66). While TNF-α has been implicated in several vascular and cardiovascular disorders (27, 38, 42, 54, 55), whether TNF-α alters the expression of VSMC βENaC or impairs cerebral blood flow regulation is unknown. Therefore, the goals of this study were to test the hypotheses that TNF-α 1) impairs CBF regulation in pregnant rats and causes a loss of cerebrovascular βENaC and 2) inhibits βENaC protein expression via mitogen-activated protein kinase (MAPK) signaling in VSMCs. The results of this study demonstrate that TNF-α impairs CBF responses at high levels of arterial pressure and reduces cerebrovascular βENaC in pregnant rats. We also found that VSMC βENaC expression is reduced by TNF-α through the p38 mitogen-activated protein kinase (p38MAPK) and c-Jun kinase (JNK)-signaling pathways.
METHODS
Animals.
Timed-pregnant Sprague-Dawley (CD) rats were purchased from Charles Rivers Laboratories and arrived at the Laboratory Animal Facilities at the University of Mississippi Medical Center (UMMC) on gestational days 10–13. All animal procedures were approved by the Institutional Animal Care and Use Committee at UMMC. Rats were randomized into groups and maintained on a 12-h:12-h light-dark cycle, provided standard rodent chow and had access to water ad libitum.
TNFα infusion.
On gestational day 14, rats were anesthetized with isoflurane, and osmotic minipumps were surgically implanted intraperitoneally for the constant infusion of recombinant human TNF-α (R & D Biosystems; Cat. No. 510-RT, 210-TA) (62). TNF-α was reconstituted with 0.7% BSA as a carrier in sterile PBS according to the manufacturer’s instructions. As previously described, TNF-α was then loaded into osmotic minipumps containing sterile saline and delivered at a rate of 200 ng/day, whereas vehicle pumps contained sterile saline only (62). Plasma levels of TNF-α found in our treated pregnant rats approximate levels observed in preeclamptic women but are approximately twofold greater than what is found in RUPP rats (37, 44). Plasma TNF-α levels were measured using a standard ELISA (MyBioSource). Vehicle pregnant rats (n = 10) were surgically implanted with osmotic minipumps containing saline only. Maternal cerebral vessels and plasma samples were collected gestational day 19, frozen, and stored at −70°C for later use.
Blood pressure measurements.
Arterial blood pressure was recorded in a subset of animals. On gestational day 18, arterial catheters were surgically implanted into the right carotid artery of pregnant rats to measure arterial pressure on gestational day 19. Blood pressure measurements were recorded with LabChart software following 45 min of acclimation to restrainer cages. Arterial pressures were recorded for a minimum of 30 min, and group mean arterial pressure (MAP) values are shown in (Table 1) (62). Two animals from each group were excluded from analysis because of failure to maintain catheter patency.
Table 1.
General animal characteristics at gestational day 19
| Characteristics | Control (n) | TNF-α (n) | P Value |
|---|---|---|---|
| Maternal weight, g | 308.9 ± 5.4 (12) | 314.9 ± 8.4 (16) | 0.820 |
| Maternal MAP, mmHg | 100.9 ± 1.5 (9) | 104.6 ± 1.3 (8) | 0.089 |
| Number of live pups | 11.9 ± 0.5 (9) | 12.4 ± 0.9 (11) | 0.671 |
| %Repositories | 4.28 ± 1.4 (9) | 4.32 ± 3.4 (11) | 0.992 |
Values are means ± SE; n per group are based on available data that were collected from vehicle control and TNF-α-treated animals. Maternal weight, maternal mean arterial pressure (MAP), plasma TNF-α, number of live pups, and percentage of resorbed (nonviable) pups were measured in pregnant rats. Comparisons were made using 2-tailed independent t-tests.
Measurement of cerebral blood flow.
Normal pregnant, vehicle pregnant, and TNF-α-infused pregnant rats were anesthetized using Inactin (50 mg/kg ip) and ketamine (30 mg/kg im) on gestational day 19. The left femoral vein was cannulated for graded infusion of phenylephrine. The trachea was cannulated using PE-200 tubing and connected to a ventilator to maintain constant blood CO2 levels, as previously described (36, 63). The heads of rats were secured in a stereotaxic frame, and an incision was made at the top of the head to expose the skull. As previously described, two closed, 4-mm × 4-mm cranial windows were drilled at ~3 to 4 mm lateral and 2 mm distal to bregma (63), forming a cranial window. Probe retainers were affixed over the middle cerebral artery, and blood flow was measured using Perimed dual channel laser-Doppler flowmeter by securing the probes to the retainers. Arterial pressure was measured by a carotid artery catheter surgically implanted on gestational day 18 in all rats. After a 15-min equilibration period, baseline MAP and peak expired CO2 (as a measure of arterial CO2) were obtained and recorded throughout. Expired Pco2 was maintained at 30–50 mmHg for each experiment. MAP was increased at 20-mmHg intervals from ~100–190 mmHg by graded intravenous infusion of phenylephrine, and CBF was continuously recorded. Since there was no difference in CBF responses between normal pregnant control (n = 14)- and vehicle control (n = 10)-treated pregnant rats, both groups (n = 24) were combined (individual data groups not shown). Animals that died during CBF measurements, developed skull hemorrhages, accumulated major blood loss following surgeries, or encountered errors in phenylephrine infusion were excluded from analysis. A total of 10 animals died from anesthetic/surgical complications during CBF measurement.
Western blot analysis on cerebral vessels.
Arteries on the brain surface, including the circle of Willis, middle cerebral, posterior cerebral, and cerebellar arteries, were removed from rats following CBF experiments. Vessel tissue (15 mg) was homogenized in 150 µL of Plasma Membrane Protein Extraction Kit (Biovision) lysis solution. Cell lysates were sonicated and separated into total cellular membrane, and cytosolic fractions were centrifuged at 15,000 rpm for 45 min at 4°C and stored at −20°C until analysis. Protein content was determined by Bradford assay (Thermo Fisher). Samples (15 µg) were run on a 7.5% Tris·HCl gels (Bio-Rad) for 1.5 h at 120 V. Precision Plus Protein Standards (Bio-Rad) were used to estimate molecular mass. Separated proteins were then transferred to nitrocellulose membranes for 1.5 h at 60 V, blocked for 1 h at room temperature in Odyssey Blocking Buffer (Li-Cor Biosciences), and incubated with rabbit anti-βENaCC-term (22, 23, 29, 58), mouse anti-β-actin (1:10,000, Ab6276, Abcam), and mouse anti-vinculin (1:1,000, V9131, Sigma) overnight at 4°C. Membranes were rinsed with PBS + 0.1% Tween 20 and incubated with IR700-conjugated donkey anti-rabbit and IR800-conjugated donkey anti-mouse IgG (1:10,000, Li-Cor) for 1 h at room temperature and rinsed. Antibody labeling was visualized using an Odyssey Infared Imaging System (Li-Cor).
Cell culture.
The A10 VSMC line (rat aortic/thoracic VSMCs, American Type Cell Culture) was used for cell culture experiments. Cells were grown at 37°C with 5% CO2 in DMEM (Sigma-Aldrich) containing 0.45% glucose, supplemented with 10% FBS, and 1% penicillin-streptomycin in T-75 flasks. Cells were seeded at a density of 1 × 106/mL into 120-mm dishes, 6-well, or 24-well plates.
Cell treatment.
VSMCs were treated with recombinant rat TNF-α [Cat. No. 510-RT-010 (TNF-α); R&D Systems]. While our in vivo studies used recombinant human TNF-α, the rat isoform shares 78% homology according to the U.S. National Library of Medicine, BLAST. Rat recombinant TNF-α was used in in vitro studies for the purpose of studying the direct effect of TNF-α in the absence of other inflammatory factors in rat VSMCs. Forty-eight hours after plating, cells were treated with 0–100 ng/mL TNF-α for 16 h to quantify changes in βENaC expression. To determine phosphorylation of MAPK and JNK, cells were treated with TNF-α (50 ng/mL) for 15 m, 2 h, or 8 h. To assess the associated signaling pathways, cells were preincubated with p38MAPK (10 µM) or JNK (50 µM) inhibitors [SB 203580 (Cat. No. 1202) and SP 600125 (Cat. No. 1496), respectively; Tocris] for 1 h, followed by TNF-α (50 ng/mL) treatment with or without MAPK inhibitors for 16 h. All cell studies contained at least two or more trials, and comparisons were made as a percent control of the control sample(s) in each trial.
Protein isolation from cultured cells.
Cells were rinsed with PBS, and proteins were then isolated using the Plasma Membrane Protein Extraction Kit (Biovision) to determine the effect TNF-α on βENaC expression (Figs. 3 and 6) or RIPA buffer (with protease and kinase inhibitors) to determine the effect of TNF-α on p38/JNK phosphorylation (Fig. 5). Cell lysates were then sonicated and separated into total cellular membrane and cytosolic fractions by centrifugation at 15,000 rpm for 45 min at 4°C and stored at −70°C until analysis. Protein content of soluble fractions was determined by BCA analysis following the manufacturer’s instructions (Pierce BCA Protein Assay Kit).
Fig. 3.

TNF-α inhibits cerebrovascular expression of β-epithelial sodium channel (βENaC) in pregnant rats. A: representative Western blot of 15-μg total protein from isolated cerebral vessels (2 trials). B: group data of βENaC expression at 250 kDa normalized to β-actin or vinculin, reported as %control. Comparisons made by 2-tailed independent t-tests. Data are presented as means ± SE. *Significantly different from appropriate control. MM, molecular mass; C-term, COOH-terminus; R, rabbit; M, mouse.
Fig. 6.

TNF-α induces the phosphorylation of p38MAPK, JNK, and NF-κB in vascular smooth muscle cells. A, top: representative immunoblots depicting p38MAPK and phospho-p38MAPK at 15 min, 2 h, and 8 h following TNF-α treatment. A, bottom: quantification of the ratio of phospho-p38MAPK to native p38MAPK following 15 m (n = 9), 2 h (n = 10), or 8 h (n = 10) of TNF-α treatment compared with control cells (n = 17). B, top: representative immunoblots depicting JNK and phospho-JNK at 15 min, 2 h, and 8 h following TNF-α treatment. B, bottom: quantification of the ratio of phospho-JNK to native JNK protein expression following 15 m (n = 9), 2 h (n = 6), or 8 h (n = 10) of TNF-α treatment compared with control cells (n = 9). C, left: representative immunoblot depicting NF-κB and phospho-NF-κB following 16 h of TNF-α treatment (50 ng/mL). C, right: quantification of the ratio of phospho-NF-κB to native NF-κB protein expression following 16-h TNF-α treatment (n = 9) compared with control cells (n = 9). Comparisons made by 1-way ANOVA, followed by the Holms-Sidak post hoc test. All data are presented as means ± SE. *P < 0.05, significantly different vs. control; #P < 0.01, significantly different vs. control (CON).
Fig. 5.

TNF-α does not induce cell death in vascular smooth muscle cells (VSMCs). To determine if the decrease in β-epithelial sodium channel (βENaC) by TNF-α was associated with cell death or reduced proliferation, cultured VSMCs were treated with TNF-α for 16 h to determine the amount of live and dead cells. Control cells (n = 16) and cells treated with TNF-α at 1 (n = 16), 10 (n = 16), and 50 (n = 16) ng/mL were examined. MeOH (n = 16) was used as negative and positive controls for live and dead signals, respectively. A: ratio of live-to-dead cells expressed as a percentage of control. B: quantification of live cells following TNF-α treatment. C: quantification of dead cells following TNF-α treatment. Comparisons were made by 1-way ANOVA, followed by the Holms-Sidak post hoc test. All data are presented as means ± SE. *P < 0.01, significantly different from control. RFU, relative fluorescence units.
Western blot on A10 cell lysates.
Protein samples (15 µg) were separated using 10% (βENaC), 12.5% (p38MAPK/JNK), or graded 4–15% (NF-κB) Tris·HCl criterion gels (Bio-Rad) for 1.5 h at 120 V. Precision Plus Protein Standards (Bio-Rad) were used to estimate molecular mass. Separated proteins were transferred to nitrocellulose membranes for 1.5 h at 60 V. Membranes were blocked for 1 h at room temperature in Odyssey Blocking Buffer (Li-Cor Biosciences), and incubated with rabbit anti-βENaCC-term (1:2,500) for 48–72 h at 4°C (22, 23, 29, 58). Separate membranes were probed with rabbit anti-p38MAPK (Cat. No. 9212), mouse anti-phospho-p38MAPK (Cat. No. 9216), rabbit anti-JNK (Cat. No. 9252), mouse anti-phospho-JNK (Cat. No. 9255), mouse anti-NFkB (Cat. No. 6956), or rabbit anti-phospho-NF-κB (Cat. No. 3033) (1:1,000; Cell Signaling Technology) overnight at 4°C. Mouse anti-β-actin (1:10,000, Ab6276, Abcam) was used as a loading control. Following primary antibody incubation, membranes were rinsed with PBS + 0.1% Tween 20 and incubated with IR700-conjugated goat anti-rabbit and IR800-conjugated goat anti-mouse IgG (1:10,000; Li-Cor) for 1 h at room temperature. Membranes were rinsed with PBS + 0.1% Tween 20 and a final rinse of PBS, and antibody labeling was visualized with an Odyssey Infrared Imaging System (Li-Cor). Samples that exhibited inefficient transferring or artifact or identified as outliers by ROUT (Prism, San Diego, CA) were discarded.
Live/dead viability/cytotoxicity assay.
To determine the effect of TNF-α on VSMC viability, the LIVE/DEAD Viability/Cytotoxicity kit (Invitrogen Molecular Probes, Carlsbad, CA) was used according to the manufacturer’s instructions. Simultaneous detection of live and dead cells is assayed using two fluorescence probes. Calcein-AM, a probe for intracellular esterase activity, is used to label live cells, whereas ethidium homodimer-1, a probe for loss of plasma membrane integrity, labels dead cells. VSMC monolayers were cultured in 96-well plates and treated with TNF-α (1–50 ng/mL) for 16 h. Untreated control cells and cells treated with 70% methanol served as controls for live and dead cells, respectively. Following treatment, cells were labeled with calcein-AM and ethidium homodimer-1, and fluorescence was measured using a fluorescence microplate reader. The live-to-dead ratio was calculated by dividing the fluorescence of calcein-AM by ethidium homodimer-1 and then normalized into percentage of control cells.
Statistical analysis.
CBF values were plotted as percent change from baseline versus MAP. Differences in values measured at each MAP interval were determined. For CBF studies, groups were compared using two-way ANOVA followed by Holm-Sidak post hoc analysis, corrected for multiple comparisons. Repeated-measures analysis was not performed due to incomplete data sets for a few animals in each group. For all other data, groups were compared using a two-tailed independent t-test or one-way or two-way ANOVA followed by Holm-Sidak post hoc analysis where appropriate. Figure legends identify specific analyses conducted. All statistical analyses were performed using Prism software. All data are presented as means ± SE. A value of P ≤ 0.05 was considered statistically significant.
RESULTS
Impact of TNF-α infusion on maternal and fetal characteristics.
Maternal body weight, number of live pups, and percent resorption were measured in a subset of animals and did not differ between control and TNF-α-infused pregnant rats (Table 1). Infusion of recombinant TNF-α from gestational days 14–19 resulted in an increase in plasma TNF-α (83.5 ± 21.3 vs. 26 ± 4.9 pg/mL; P = 0.0358; Fig. 1) compared with control pregnant rats on gestational day 19. There was a subtle increase in MAP by TNF-α infusion (~5 mmHg) that trended toward statistical significance (104.6 ± 1.3 vs. 100.9 ± 1.5; P = 0.089; Table 1) in pregnant rats on gestational day 19, which is comparable with our previous report showing infusion of TNF-α at this concentration increased MAP by ~9 mmHg (62). However, TNF-α can have varying effects on blood pressure depending on disease/model (41, 48), and in this study, TNF-α infusion at 200 ng/day in pregnant rats had no significant effect on blood pressure in this study.
Fig. 1.

Infusion of TNF-α increases plasma levels of TNF-α. Animals were infused with TNF-α (n = 14) via a mini-osmotic pump (intravenous) at a rate of 200 ng/day or with saline only (n = 10) on gestational days 14–19. Two-tailed independent t-test was used for comparison. Data are presented as means ± SE. *P = 0.0358, significantly different from vehicle.
TNF-α infusion impairs cerebral blood flow control at high MAPs.
To determine the effect of TNF-α on CBF control during pregnancy, the relationship between the percent change in CBF and stepwise increases in MAP was recorded. Two-way ANOVA revealed a significant main effect of perfusion pressure (P < 0.0001), treatment (TNF-α+/−, P < 0.0001), and interaction of pressure and treatment (P < 0.0001). Although there was no significant difference in CBF between control and TNF-α-treated animals at 100–160 mmHg pressure steps, CBF significantly increased at 180 mmHg (220 ± 17 vs. 166 ± 7; P < 0.0001) and 190 mmHg (261 ± 26 vs. 181 ± 9; P < 0.0001) in TNF-α-infused pregnant rats relative to control pregnant rats (Fig. 2A). These findings suggest control over CBF is disrupted in TNF-α-treated dams at higher pressures, but not at pressures within the autoregulatory range (~50–150 mmHg). Because arterial CO2 can dramatically influence vasodilation, we examined constant expired CO2 levels throughout and found no significant difference in the groups at any perfusion pressure, indicating the changes in CBF are not due to carbon dioxide (Fig. 2B). The P values for effect of treatment and the interaction of perfusion pressure × treatment on expired CO2 levels were 0.3824 and 0.7167, respectively. Perfusion pressure itself did significantly affect CO2 (P = 0.0333), indicating that CO2 levels increased with increases in MAP by phenylephrine in pregnant rats.
Fig. 2.

TNF-α impairs cerebral blood flow (CBF) control at high arterial pressures. CBF responses to stepwise increases of mean arterial pressure (MAP) were recorded. A: TNF-α (n = 15) increased CBF at pressures at 180 and 190 mmHg compared with control (n = 24) pregnant rats. B: exhaled CO2 was consistently maintained throughout the experiments. Comparisons were made by 2-way ANOVA with Holm-Sidak post hoc test, showing a significant effect on perfusion pressure (P < 0.0001), treatment (P < 0.0001), and interaction of pressure and treatment (P < 0.0001). Data are presented as means ± SE. *P < 0.000, significantly different from vehicle.
Increased circulating TNF-α reduces cerebrovascular βENaC protein expression.
Cytosolic βENaC protein expression isolated from cerebral vessels, normalized to either β-actin (P = 0.022) or vinculin (P = 0.024), was reduced by ~35% in TNF-α-infused pregnant rats, suggesting a possible mechanism by which TNF-α impairs cerebral blood flow control in vivo. A representative blot and group data are shown in Fig. 3, A and B.
TNF-α induces a concentration-dependent decrease in βENaC expression in cultured VSMCs.
To confirm a direct effect of TNF-α on βENaC expression, cultured VSMCs were exposed to TNF-α independent of other circulating factors. TNF-α induced a concentration-dependent reduction in membrane-bound βENaC expression from 0.1 to 100 ng/mL (slope −9.85; P = 0.003). Exposure to TNF-α reduced βENaC to 90 and 53% of controls at 0.1 and 100 ng/mL, respectively. Representative blots for βENaC and β-actin are shown in Fig. 4A and group data in Fig. 4B.
Fig. 4.
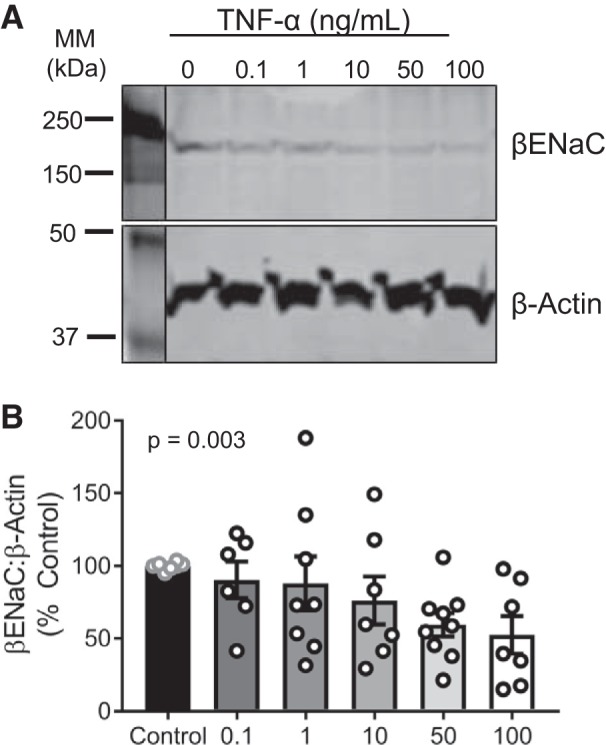
TNF-α induces a concentration-dependent reduction in β-epithelial sodium channel (βENaC) in cultured vascular smooth muscle cells. A: representative immunoblot depicting βENaC and β-actin. B: quantification of βENaC following 16-h treatment of TNF-α at the following concentrations: 0 (control; n = 8), 0.1 (n = 6), 1 (n = 8), 10 (n = 7), 50 (n = 9), and 100 (n = 7) ng/mL. Comparisons made by 1-way ANOVA with posttest for linear trend (P = 0.003). Data are presented as means ± SE. MM, molecular mass.
Reduction in βENaC by TNF-α is not likely due to cell death.
To determine whether in vitro cell death/toxicity accounts for the decrease in βENaC protein with TNF-α exposure, we examined cell viability in cultured VSMCs. Although TNF-α treatment resulted in a modest decrease in the live-to-dead fluorescence ratio (Fig. 5A), this was due to a reduction in the calcein-AM fluorescence (“live” signal) (Fig. 5B). There was no increase in ethidium homodimer-1 fluorescence (“dead” signal) in TNF-α-treated VSMCs (Fig. 5C). These data suggest that while TNF-α treatment may have modestly reduced VSMC viability, it did not induce cell death/toxicity. Thus, it is unlikely that the TNF-α-induced reduction in VSMC βENaC is due to cell death.
TNF-α induces the phosphorylation of p38MAPK, JNK and NF-κB.
Exposure to TNF-α (50 ng/mL) rapidly and robustly induced p38MAPK and JNK phosphorylation by 15 min, and NF-κB phosphorylation was elevated by 16 h. Phospo-p38MAPK-to-p38MAPK increased 228 ± 76% by 15 min and remained significantly increased at 2 (275 ± 32%) and 8 (314 ± 53%) h after exposure (Fig. 6A). TNF-α also induced a time-dependent increase on phosphorylation of p38MAPK (P = 0.0005). Phospho-JNK-to-JNK increased to 290 ± 16% by 15 min and remained significantly elevated through 8 h (246 ± 26%), but peaked at 2 h (521 ± 44) (Fig. 6B). Phospho-NF-κB-to-NF-κB was also increased 262.1 ± 70.4% at 16 h following TNF-α treatment (Fig. 6C). These results suggest that the MAPK-NF-κB-signaling pathway is activated by TNF-α in VSMCs and persists through 8–16 h, with the percent change in JNK phosphorylation almost 2 times greater than that of p38MAPK and NF-κB.
Role of p38MAPK and JNK in the reduction of βENaC by TNF-α.
To determine whether p38MAPK and/or JNK pathways mediate βENaC inhibition from TNF-α, we treated VSMCs with TNF-α (50 ng/mL, 16 h) in the presence of specific blockers SB203580 (p38MAPK) and SP600125 (JNK). Representative blots and group data are shown in Fig. 7, A and B. A two-way ANOVA indicated a significant main effect of TNF-α (P = 0.036) and kinase inhibitor (P = 0.007). Under control conditions (no added TNF-α), inhibition of either p38MAPK or JNK had no significant effect on βENaC expression (to 212 ± 50.3%, P = 0.0001) compared with untreated control cells. TNF-α exposure reduced βENaC to 54 ± 6% of control cells (100 ± 6.6%; P = 0.037). In contrast, both inhibition of p38MAPK (89.5 ± 14.5 vs. 54 ± 6%, P = 0.085) or JNK prevented the reduction of βENaC protein (99 ± 17.4 vs. 54 ± 6%, P = 0.06) in TNF-α-treated cells (Fig. 7B). This suggests that both p38 and JNK pathways could be involved in the regulation of βENaC protein under a proinflammatory stimulus such as TNF-α.
Fig. 7.

Inhibition of JNK-signaling pathway prevents TNF-α-induced decreases in β-epithelial sodium channel (βENaC) protein expression in vascular smooth muscle cells (VSMCs). VSMCs were treated for 16 h with TNF-α (50 ng/mL) with or without SB203580 (MAPK inhibitor) or SP600125 (JNK inhibitor). A: representative immunoblot depicting βENaC and β-actin from control groups (left) and TNF-α-treated groups (right) with or without SB203580 (p38MAPK inhibitor) or SP600125 (JNK inhibitor). Images were acquired from the same blot under the same experimental conditions, demarcated, and arranged according to the order data are presented. βENaC was normalized to β-actin, which varied and contributed to quantitative findings. B: quantification of βENaC in control (n = 26) and control + SB203580 (n = 9)-, control + SP600125 (n = 8)-, TNF-α (n = 21)-, TNF-α + SB203580 (n = 16)-, and TNF-α + SP600125 (n = 16)-treated VSMCs. Comparisons made by 2-way ANOVA (main effect of TNF-α, P = 0.036; main effect of kinase inhibitor, P < 0.007) followed by the Holms-Sidak post hoc test. All data are presented as means ± SE. MM, molecular mass.
DISCUSSION
Altered CBF regulation is thought to contribute to cerebrovascular dysfunction and neurophysiological impairments in preeclampsia/eclampsia (26). We previously found that placental ischemia impairs cerebrovascular myogenic tone and CBF autoregulation in rats (51, 63). However, the mechanism(s) for altered CBF autoregulation during preeclampsia is unclear. The cytokine TNF-α, an ischemic placental-derived proinflammatory cytokine, is likely a contributing factor. We previously reported that TNF-α infusion in pregnant rats increases BBB permeability and cerebral edema, whereas TNF-α blockade in placental ischemic rats prevented these effects. However, the effect of chronic increases in circulating TNF-α, independent of other placental-derived factors, during pregnancy on CBF autoregulation has not been addressed. Thus, the purpose of this study was to determine if TNF-α infusion during gestational days 14–19 contributes to altered CBF regulation during pregnancy.
Our main finding suggests that TNF-α infusion during pregnancy impairs CBF regulation at high pressures that are considered outside the normal autoregulatory range (~50–150 mmHg), suggesting modest elevations in plasma TNF-α alone may not fully account for placental ischemia-induced CBF impairment previously observed (53). There are several potential explanations for this finding. First, increases in TNF-α alone, isolated from other placental-derived factors, may not be sufficient to impair CBF regulation. Placental-ischemia alters the expression of multiple inflammatory cytokines that may act synergistically with TNF-α to disrupt CBF control. Further studies addressing the importance of other placental-derived factors, alone and in combination with TNF-α, are needed to address this possibility. Second, our assay may not be sensitive enough to detect subtle changes in CBF. A limitation of our approach is that we can only measure relative, not absolute, changes in cerebral blood flow in response to increases in pressure. Therefore, we are unable to determine if there are differences in baseline blood flow in control versus TNF-α-treated rats. However, the impairment of CBF regulation at high perfusion pressures is clinically meaningful because it is consistent with an increased susceptibility to neurological complications (e.g., seizures) in patients with hypertension and preeclampsia or in patients with preeclampsia and with transient spikes in blood pressure.
ENaC proteins are most widely known for their role in regulating epithelial Na+ and water transport (20). Studies from our laboratory and others demonstrate that βENaC also mediates the myogenic response in small renal and cerebral arteries and renal arterioles (14–17, 21, 22, 24, 28, 30, 33, 34, 45, 56, 60). Several cytokines and hormones reduce ENaC expression in epithelial cells (66). For example, certain interleukins (IL-1β, IL-4, and IL-13) decrease ENaC expression via MAPK-signaling pathways (9, 13, 19, 50). Transforming growth factor (TGF)-β reduces αENaC expression via ERK1/2 and causes internalization of the channel by interaction with βENaC (18, 47). Moreover, TNF-α reduces expression of α-, β-, and γ-ENaC subunits and transepithelial current in AT2 cells (12, 67). While TNF-α reduces ENaC in epithelial cells, this is the first study to measure the effect of TNF-α on βENaC in VSMCs and cerebral vessels. βENaC mRNA and protein expression is decreased in the placenta of women with preeclampsia (59) and in the middle cerebral artery in a rodent model of placental ischemia (51), suggesting βENaC expression may be reduced by placental ischemia in several vascular/organ systems. Thus, it is possible that placental-derived TNF-α could mediate changes in vascular βENaC.
In this study, we examined the effect of TNF-α on βENaC expression in cerebral vessels to determine if reduced βENaC expression is associated with impaired CBF control. We found that βENaC protein expression was significantly reduced in cerebral vessels from isolated TNF-α-infused pregnant rats. While the in vivo experiments do not prove that TNF-α-mediated alteration in CBF is caused by reduced βENaC expression, this study links impaired CBF control in TNF-α-treated pregnant rats and reduced cerebrovascular βENaC expression. Alternatively, it is possible that TNF-α alters other pathways involved in signaling pathways which contribute to myogenic control.
The direct effect of TNF-α on βENaC expression was assessed using an in vitro approach, and we found that 50–100 ng/mL TNF-α inhibits βENaC expression in cultured rat VSMCs (A10 cell line). While these concentrations are higher than the plasma concentrations with TNF-α infusion, they are consistent with other studies examining the effect of cytokine activated-signaling pathways in vitro (7, 12). To determine whether cell death contributes to the decrease in βENaC protein by TNF-α, we also measured cell viability following cytokine treatment. TNF-α did not induce VSMC death, indicating that the decrease in βENaC is not a result of cell death processes. We also observed no concentration-response of TNF-α on cell death at the concentrations studied (1–50 ng/mL). However, TNF-α did induce a modest decrease in VSMC viability in a concentration-dependent fashion. This is consistent with other reports showing that certain cytokines reduce viability/proliferation in VSMCs (11, 40, 57). Thus, it is possible that βENaC is associated with cell viability-signaling pathways.
To examine the underlying signaling pathways involved in regulating βENaC protein, we treated cells with TNF-α and found a rapid induction of phosphorylation of p38MAPK and JNK starting at 15 m. The phosphorylation of p38MAPK and JNK in response to TNF-α was sustained through 8 h of exposure in VSMCs. While both p38MAPK- and JNK-signaling pathways were activated by TNF-α in VSMCs, JNK phosphorylation response was more robust with more than a twofold increase in phosphorylation at 2 h relative to p38. We also found that NF-κB phosphorylation was elevated following 16 h of TNF-α treatment, indicating that p38MAPK and JNK pathways may elicit the downstream activation of NF-κB in VSMCs following TNF-α treatment. The reduction in βENaC by TNF-α was fully prevented by inhibiting p38MAPK or JNK, suggesting that TNF-α may mediate this response via multiple MAPK pathways, possibly to regulate NF-κB that could impact βENaC protein expression under a stimulus. However, activated JNK and p38 MAPK have been shown to phosphorylate numerous substrates, including c-Jun that is specifically phosphorylated by JNK and ATF-2 that is phosphorylated by both JNK and p38 MAPK (64). Thus, it could be possible that βENaC is largely regulated by other downstream factors under a stimulus such as TNF-α, which requires dramatic activation and convergence of MAPK pathways. Furthermore, while both p38MAPK and JNK activation can regulate NF-κB activity in several cell lines (1, 8, 52, 53), it is unclear how p38MAPK and JNK regulates NF-κB under TNF-α exposure in A10 cells and, furthermore, whether NF-κB mediates changes to βENaC protein in VSMCs. TNF-α has been shown to increase p38MAPK/JNK and NF-κB phosphorylation by other independent mechanisms, such as via ROS. Future studies to isolate the role individual components of the MAPK-signaling pathway are necessary to fully understand how TNF-α reduces βENaC in VSMCs due to the fact that MAPKs regulate ENaC by signaling pathways specific to cell type. There is also evidence that MAPKs can influence mRNA stability (25, 46); whether this occurs with TNF-α-mediated changes in βENaC has not been examined. A full assessment of this signaling pathway, including NF-kB phosphorylation, transcription factors, coactivators/repressors, and mRNA stability, is warranted.
There are several limitations to this study which should be considered. First, this study does not provide direct evidence that TNF-α-induced changes in CBF is mediated by inhibition of βENaC expression. Use of ENaC inhibitors, such as amiloride and benzamil, would not be useful since ENaC expression is already inhibited. A direct role for βENaC can only be determined by rescuing βENaC expression during TNF-α treatment. Unfortunately, ENaC agonists or animal models with overexpression of βENaC in VSMCs are not available. Thus, it is currently impossible to prove that βENaC mediates the TNF-α impairment of CBF autoregulation. It is important to also note that TNF-α could potentially regulate other proteins important in the myogenic signaling cascade in our pregnancy model. Future studies to examine the effect of ENaC inhibition on CBF during pregnancy could help provide insight into the role of TNF-α-mediated changes on CBF in this study. Second, while the A10 cell line is a commonly used model of VSMCs that displays many characteristics of VSMCs, including the expression of α-actin and myosin heavy chains (49), they are derived from the aorta of neonatal rats, not the cerebrovasculature of adult pregnant rats that are myogenically active. Although our laboratory has previously shown that A10 cells express βENaC (23, 61), future studies to examine expression of βENaC in isolated VSMC from cerebral vessels of pregnant rats would be ideal. Lastly, because βENaC can undergo internalization and trafficking from the membrane to cytosol, it is ideal to measure total βENaC from both soluble and insoluble fractions when evaluating changes to βENaC protein. However, overall, our data suggest that both cytosolic and membrane-bound βENaC is reduced by TNF-α.
This study highlights the role of inflammatory cytokines on CBF control and the putative mechanosensory protein, βENaC. TNF-α-mediated suppression of βENaC expression could be a contributing factor to impaired cerebrovascular function due to inhibition of the myogenic constrictor response. These findings suggest a need for further investigation of the role of other placental ischemia-derived cytokines on inhibition of degenerin-mediated myogenic constriction and blood flow autoregulation.
GRANTS
This work was supported by National Institutes of Health Grants R01-HL-12186106, R01-HL-136684-02, R01-HL-136684, P01-HL-051971, P20-GM-104357, P20-GM-121334, and 5U54-GM-115428, National Research Service Award Institutional Training Grant T32-HL-105324, and the American Heart Association Grant 19POST34450074.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or American Heart Association.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.D., M.J.R., J.P.G., and H.A.D. conceived and designed research; J.D., S.T.Y., and E.H. performed experiments; J.D., E.H., and H.A.D. analyzed data; J.D., M.J.R., J.P.G., and H.A.D. interpreted results of experiments; J.D. and E.H. prepared figures; J.D. and H.A.D. drafted manuscript; J.D., M.J.R., J.P.G., and H.A.D. edited and revised manuscript; J.D., S.T.Y., E.H., M.J.R., J.P.G., and H.A.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Marietta Arany for excellent technical support.
REFERENCES
- 1.Aggarwal BB. Tumour necrosis factors receptor associated signalling molecules and their role in activation of apoptosis, JNK and NF-kappaB. Ann Rheum Dis 59, Suppl 1: i6–i16, 2000. doi: 10.1136/ard.59.suppl_1.i6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ben-Shahar Y. Sensory functions for degenerin/epithelial sodium channels (DEG/ENaC). Adv Genet 76: 1–26, 2011. doi: 10.1016/B978-0-12-386481-9.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benos DJ, Stanton BA. Functional domains within the degenerin/epithelial sodium channel (Deg/ENaC) superfamily of ion channels. J Physiol 520: 631–644, 1999. doi: 10.1111/j.1469-7793.1999.00631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonny O, Chraibi A, Loffing J, Jaeger NF, Gründer S, Horisberger JD, Rossier BC. Functional expression of a pseudohypoaldosteronism type I mutated epithelial Na+ channel lacking the pore-forming region of its alpha subunit. J Clin Invest 104: 967–974, 1999. doi: 10.1172/JCI6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367: 463–467, 1994. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 6.Chakravarty A, Chakrabarti SD. The neurology of eclampsia : some observations. Neurol India 50: 128–135, 2002. [PubMed] [Google Scholar]
- 7.Cheng G, Wei L, Xiurong W, Xiangzhen L, Shiguang Z, Songbin F. IL-17 stimulates migration of carotid artery vascular smooth muscle cells in an MMP-9 dependent manner via p38 MAPK and ERK1/2-dependent NF-kappaB and AP-1 activation. Cell Mol Neurobiol 29: 1161–1168, 2009. doi: 10.1007/s10571-009-9409-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi H, Dikalova A, Stark RJ, Lamb FS. c-Jun N-terminal kinase attenuates TNFα signaling by reducing Nox1-dependent endosomal ROS production in vascular smooth muscle cells. Free Radic Biol Med 86: 219–227, 2015. doi: 10.1016/j.freeradbiomed.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 9.Choi JY, Choi YS, Kim SJ, Son EJ, Choi HS, Yoon JH. Interleukin-1beta suppresses epithelial sodium channel beta-subunit expression and ENaC-dependent fluid absorption in human middle ear epithelial cells. Eur J Pharmacol 567: 19–25, 2007. doi: 10.1016/j.ejphar.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 10.Cornelius DC, Amaral LM, Harmon A, Wallace K, Thomas AJ, Campbell N, Scott J, Herse F, Haase N, Moseley J, Wallukat G, Dechend R, LaMarca B. An increased population of regulatory T cells improves the pathophysiology of placental ischemia in a rat model of preeclampsia. Am J Physiol Regul Integr Comp Physiol 309: R884–R891, 2015. doi: 10.1152/ajpregu.00154.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuneo AA, Herrick D, Autieri MV. Il-19 reduces VSMC activation by regulation of mRNA regulatory factor HuR and reduction of mRNA stability. J Mol Cell Cardiol 49: 647–654, 2010. doi: 10.1016/j.yjmcc.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dagenais A, Fréchette R, Yamagata Y, Yamagata T, Carmel JF, Clermont ME, Brochiero E, Massé C, Berthiaume Y. Downregulation of ENaC activity and expression by TNF-alpha in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 286: L301–L311, 2004. doi: 10.1152/ajplung.00326.2002. [DOI] [PubMed] [Google Scholar]
- 13.Dames P, Bergann T, Fromm A, Bücker R, Barmeyer C, Krug SM, Fromm M, Schulzke JD. Interleukin-13 affects the epithelial sodium channel in the intestine by coordinated modulation of STAT6 and p38 MAPK activity. J Physiol 593: 5269–5282, 2015. doi: 10.1113/JP271156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drummond HA. βENaC is a molecular component of a VSMC mechanotransducer that contributes to renal blood flow regulation, protection from renal injury, and hypertension. Front Physiol 3: 341, 2012. doi: 10.3389/fphys.2012.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drummond HA, Gebremedhin D, Harder DR. Degenerin/epithelial Na+ channel proteins: components of a vascular mechanosensor. Hypertension 44: 643–648, 2004. doi: 10.1161/01.HYP.0000144465.56360.ad. [DOI] [PubMed] [Google Scholar]
- 16.Drummond HA, Grifoni SC, Jernigan NL. A new trick for an old dogma: ENaC proteins as mechanotransducers in vascular smooth muscle. Physiology (Bethesda) 23: 23–31, 2008. doi: 10.1152/physiol.00034.2007. [DOI] [PubMed] [Google Scholar]
- 17.Drummond HA, Stec DE. βENaC acts as a mechanosensor in renal vascular smooth muscle cells that contributes to renal myogenic blood flow regulation, protection from renal injury and hypertension. J Nephrol Res 1: 1–9, 2015. doi: 10.17554/j.issn.2410-0579.2015.01.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank J, Roux J, Kawakatsu H, Su G, Dagenais A, Berthiaume Y, Howard M, Canessa CM, Fang X, Sheppard D, Matthay MA, Pittet JF. Transforming growth factor-beta1 decreases expression of the epithelial sodium channel alphaENaC and alveolar epithelial vectorial sodium and fluid transport via an ERK1/2-dependent mechanism. J Biol Chem 278: 43939–43950, 2003. doi: 10.1074/jbc.M304882200. [DOI] [PubMed] [Google Scholar]
- 19.Galietta LJ, Pagesy P, Folli C, Caci E, Romio L, Costes B, Nicolis E, Cabrini G, Goossens M, Ravazzolo R, Zegarra-Moran O. IL-4 is a potent modulator of ion transport in the human bronchial epithelium in vitro. J Immunol 168: 839–845, 2002. doi: 10.4049/jimmunol.168.2.839. [DOI] [PubMed] [Google Scholar]
- 20.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396, 1997. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 21.Ge Y, Gannon K, Gousset M, Liu R, Murphey B, Drummond HA. Impaired myogenic constriction of the renal afferent arteriole in a mouse model of reduced βENaC expression. Am J Physiol Renal Physiol 302: F1486–F1493, 2012. doi: 10.1152/ajprenal.00638.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grifoni SC, Chiposi R, McKey SE, Ryan MJ, Drummond HA. Altered whole kidney blood flow autoregulation in a mouse model of reduced beta-ENaC. Am J Physiol Renal Physiol 298: F285–F292, 2010. doi: 10.1152/ajprenal.00496.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grifoni SC, Gannon KP, Stec DE, Drummond HA. ENaC proteins contribute to VSMC migration. Am J Physiol Heart Circ Physiol 291: H3076–H3086, 2006. doi: 10.1152/ajpheart.00333.2006. [DOI] [PubMed] [Google Scholar]
- 24.Guan Z, Pollock JS, Cook AK, Hobbs JL, Inscho EW. Effect of epithelial sodium channel blockade on the myogenic response of rat juxtamedullary afferent arterioles. Hypertension 54: 1062–1069, 2009. doi: 10.1161/HYPERTENSIONAHA.109.137992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guhaniyogi J, Brewer G. Regulation of mRNA stability in mammalian cells. Gene 265: 11–23, 2001. doi: 10.1016/S0378-1119(01)00350-X. [DOI] [PubMed] [Google Scholar]
- 26.Hammer ES, Cipolla MJ. Cerebrovascular Dysfunction in Preeclamptic Pregnancies. Curr Hypertens Rep 17: 64, 2015. doi: 10.1007/s11906-015-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashmi S, Zeng QT. Role of interleukin-17 and interleukin-17-induced cytokines interleukin-6 and interleukin-8 in unstable coronary artery disease. Coron Artery Dis 17: 699–706, 2006. doi: 10.1097/01.mca.0000236288.94553.b4. [DOI] [PubMed] [Google Scholar]
- 28.Jernigan NL, Drummond HA. Myogenic vasoconstriction in mouse renal interlobar arteries: role of endogenous beta and gammaENaC. Am J Physiol Renal Physiol 291: F1184–F1191, 2006. doi: 10.1152/ajprenal.00177.2006. [DOI] [PubMed] [Google Scholar]
- 29.Jernigan NL, Drummond HA. Vascular ENaC proteins are required for renal myogenic constriction. Am J Physiol Renal Physiol 289: F891–F901, 2005. doi: 10.1152/ajprenal.00019.2005. [DOI] [PubMed] [Google Scholar]
- 30.Jernigan NL, LaMarca B, Speed J, Galmiche L, Granger JP, Drummond HA. Dietary salt enhances benzamil-sensitive component of myogenic constriction in mesenteric arteries. Am J Physiol Heart Circ Physiol 294: H409–H420, 2008. doi: 10.1152/ajpheart.00571.2007. [DOI] [PubMed] [Google Scholar]
- 31.Kalantar F, Rajaei S, Heidari AB, Mansouri R, Rashidi N, Izad MH, Mirahmadian M. Serum levels of tumor necrosis factor-α, interleukin-15 and interleukin-10 in patients with pre-eclampsia in comparison with normotensive pregnant women. Iran J Nurs Midwifery Res 18: 463–466, 2013. [PMC free article] [PubMed] [Google Scholar]
- 32.Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82: 735–767, 2002. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 33.Kim EC, Ahn DS, Yeon SI, Lim M, Lee YH. Epithelial Na+ channel proteins are mechanotransducers of myogenic constriction in rat posterior cerebral arteries. Exp Physiol 97: 544–555, 2012. doi: 10.1113/expphysiol.2011.062232. [DOI] [PubMed] [Google Scholar]
- 34.Kim EC, Choi SK, Lim M, Yeon SI, Lee YH. Role of endogenous ENaC and TRP channels in the myogenic response of rat posterior cerebral arteries. PLoS One 8: e84194, 2013. doi: 10.1371/journal.pone.0084194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimes BW, Brandt BL. Characterization of two putative smooth muscle cell lines from rat thoracic aorta. Exp Cell Res 98: 349–366, 1976. doi: 10.1016/0014-4827(76)90446-8. [DOI] [PubMed] [Google Scholar]
- 36.Koehler RC, Roman RJ, Harder DR. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci 32: 160–169, 2009. doi: 10.1016/j.tins.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 37.LaMarca B, Speed J, Fournier L, Babcock SA, Berry H, Cockrell K, Granger JP. Hypertension in response to chronic reductions in uterine perfusion in pregnant rats: effect of tumor necrosis factor-alpha blockade. Hypertension 52: 1161–1167, 2008. doi: 10.1161/HYPERTENSIONAHA.108.120881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liuzzo G, Trotta F, Pedicino D. Interleukin-17 in atherosclerosis and cardiovascular disease: the good, the bad, and the unknown. Eur Heart J 34: 556–559, 2013. doi: 10.1093/eurheartj/ehs399. [DOI] [PubMed] [Google Scholar]
- 39.MacKay AP, Berg CJ, Atrash HK. Pregnancy-related mortality from preeclampsia and eclampsia. Obstet Gynecol 97: 533–538, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Mazighi M, Pellé A, Gonzalez W, Mtairag M, Philippe M, Hénin D, Michel JB, Feldman LJ. IL-10 inhibits vascular smooth muscle cell activation in vitro and in vivo. Am J Physiol Heart Circ Physiol 287: H866–H871, 2004. doi: 10.1152/ajpheart.00918.2003. [DOI] [PubMed] [Google Scholar]
- 41.Mehaffey E, Majid DSA. Tumor necrosis factor-α, kidney function, and hypertension. Am J Physiol Renal Physiol 313: F1005–F1008, 2017. doi: 10.1152/ajprenal.00535.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mendall MA, Patel P, Asante M, Ballam L, Morris J, Strachan DP, Camm AJ, Northfield TC. Relation of serum cytokine concentrations to cardiovascular risk factors and coronary heart disease. Heart 78: 273–277, 1997. doi: 10.1136/hrt.78.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molvarec A, Czegle I, Szijártó J, Rigó J Jr. Increased circulating interleukin-17 levels in preeclampsia. J Reprod Immunol 112: 53–57, 2015. doi: 10.1016/j.jri.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 44.Muzammil S, Singhal U, Gulati R, Bano I. Serum tumor necrosis factor-alpha in pre eclampsia. Indian J Physiol Pharmacol 49: 236–240, 2005. [PubMed] [Google Scholar]
- 45.Nagasawa T, Imig JD. Afferent Arteriolar Responses to β,γ-methylene ATP and 20-HETE are not Blocked by ENaC Inhibition. Physiol Rep 1: e00082, 2013. doi: 10.1002/phy2.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nyati KK, Masuda K, Zaman MM, Dubey PK, Millrine D, Chalise JP, Higa M, Li S, Standley DM, Saito K, Hanieh H, Kishimoto T. TLR4-induced NF-κB and MAPK signaling regulate the IL-6 mRNA stabilizing protein Arid5a. Nucleic Acids Res 45: 2687–2703, 2017. doi: 10.1093/nar/gkx064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peters DM, Vadász I, Wujak L, Wygrecka M, Olschewski A, Becker C, Herold S, Papp R, Mayer K, Rummel S, Brandes RP, Günther A, Waldegger S, Eickelberg O, Seeger W, Morty RE. TGF-β directs trafficking of the epithelial sodium channel ENaC which has implications for ion and fluid transport in acute lung injury. Proc Natl Acad Sci USA 111: E374–E383, 2014. doi: 10.1073/pnas.1306798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramseyer VD, Garvin JL. Tumor necrosis factor-α: regulation of renal function and blood pressure. Am J Physiol Renal Physiol 304: F1231–F1242, 2013. doi: 10.1152/ajprenal.00557.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rao RS, Miano JM, Olson EN, Seidel CL. The A10 cell line: a model for neonatal, neointimal, or differentiated vascular smooth muscle cells? Cardiovasc Res 36: 118–126, 1997. doi: 10.1016/S0008-6363(97)00156-9. [DOI] [PubMed] [Google Scholar]
- 50.Roux J, Kawakatsu H, Gartland B, Pespeni M, Sheppard D, Matthay MA, Canessa CM, Pittet JF. Interleukin-1beta decreases expression of the epithelial sodium channel alpha-subunit in alveolar epithelial cells via a p38 MAPK-dependent signaling pathway. J Biol Chem 280: 18579–18589, 2005. doi: 10.1074/jbc.M410561200. [DOI] [PubMed] [Google Scholar]
- 51.Ryan MJ, Gilbert EL, Glover PH, George EM, Masterson CW, McLemore GR Jr, LaMarca B, Granger JP, Drummond HA. Placental ischemia impairs middle cerebral artery myogenic responses in the pregnant rat. Hypertension 58: 1126–1131, 2011. doi: 10.1161/HYPERTENSIONAHA.111.181453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saha RN, Jana M, Pahan K. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J Immunol 179: 7101–7109, 2007. doi: 10.4049/jimmunol.179.10.7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schulze-Osthoff K, Ferrari D, Riehemann K, Wesselborg S. Regulation of NF-kappa B activation by MAP kinase cascades. Immunobiology 198: 35–49, 1997. doi: 10.1016/S0171-2985(97)80025-3. [DOI] [PubMed] [Google Scholar]
- 54.Simon T, Taleb S, Danchin N, Laurans L, Rousseau B, Cattan S, Montely JM, Dubourg O, Tedgui A, Kotti S, Mallat Z. Circulating levels of interleukin-17 and cardiovascular outcomes in patients with acute myocardial infarction. Eur Heart J 34: 570–577, 2013. doi: 10.1093/eurheartj/ehs263. [DOI] [PubMed] [Google Scholar]
- 55.Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol 78: 539–552, 2009. doi: 10.1016/j.bcp.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Syntichaki P, Tavernarakis N. Genetic models of mechanotransduction: the nematode Caenorhabditis elegans. Physiol Rev 84: 1097–1153, 2004. doi: 10.1152/physrev.00043.2003. [DOI] [PubMed] [Google Scholar]
- 57.Tian Y, Sommerville LJ, Cuneo A, Kelemen SE, Autieri MV. Expression and suppressive effects of interleukin-19 on vascular smooth muscle cell pathophysiology and development of intimal hyperplasia. Am J Pathol 173: 901–909, 2008. doi: 10.2353/ajpath.2008.080163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.VanLandingham LG, Gannon KP, Drummond HA. Pressure-induced constriction is inhibited in a mouse model of reduced betaENaC. Am J Physiol Regul Integr Comp Physiol 297: R723–R728, 2009. doi: 10.1152/ajpregu.00212.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang S, He G, Yang Y, Liu Y, Diao R, Sheng K, Liu X, Xu W. Reduced expression of Enac in Placenta tissues of patients with severe preeclampsia is related to compromised trophoblastic cell migration and invasion during pregnancy. PLoS One 8: e72153, 2013. doi: 10.1371/journal.pone.0072153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Takeya K, Aaronson PI, Loutzenhiser K, Loutzenhiser R. Effects of amiloride, benzamil, and alterations in extracellular Na+ on the rat afferent arteriole and its myogenic response. Am J Physiol Renal Physiol 295: F272–F282, 2008. doi: 10.1152/ajprenal.00200.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Warrington JP, Coleman K, Skaggs C, Hosick PA, George EM, Stec DE, Ryan MJ, Granger JP, Drummond HA. Heme oxygenase-1 promotes migration and β-epithelial Na+ channel expression in cytotrophoblasts and ischemic placentas. Am J Physiol Regul Integr Comp Physiol 306: R641–R646, 2014. doi: 10.1152/ajpregu.00566.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warrington JP, Drummond HA, Granger JP, Ryan MJ. Placental ischemia-induced increases in brain water content and cerebrovascular permeability: role of TNF-α. Am J Physiol Regul Integr Comp Physiol 309: R1425–R1431, 2015. doi: 10.1152/ajpregu.00372.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Warrington JP, Fan F, Murphy SR, Roman RJ, Drummond HA, Granger JP, Ryan MJ. Placental ischemia in pregnant rats impairs cerebral blood flow autoregulation and increases blood-brain barrier permeability. Physiol Rep 2: 2, 2014. doi: 10.14814/phy2.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wei L, Zhu Z, Wang J, Liu J. JNK and p38 mitogen-activated protein kinase pathways contribute to porcine circovirus type 2 infection. J Virol 83: 6039–6047, 2009. doi: 10.1128/JVI.00135-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wesch D, Althaus M, Miranda P, Cruz-Muros I, Fronius M, González-Hernández T, Clauss WG, Alvarez de la Rosa D, Giraldez T. Differential N termini in epithelial Na+ channel δ-subunit isoforms modulate channel trafficking to the membrane. Am J Physiol Cell Physiol 302: C868–C879, 2012. doi: 10.1152/ajpcell.00255.2011. [DOI] [PubMed] [Google Scholar]
- 66.Wynne BM, Zou L, Linck V, Hoover RS, Ma HP, Eaton DC. Regulation of Lung Epithelial Sodium Channels by Cytokines and Chemokines. Front Immunol 8: 766, 2017. doi: 10.3389/fimmu.2017.00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamagata T, Yamagata Y, Nishimoto T, Hirano T, Nakanishi M, Minakata Y, Ichinose M, Dagenais A, Berthiaume Y. The regulation of amiloride-sensitive epithelial sodium channels by tumor necrosis factor-alpha in injured lungs and alveolar type II cells. Respir Physiol Neurobiol 166: 16–23, 2009. doi: 10.1016/j.resp.2008.12.008. [DOI] [PubMed] [Google Scholar]