

Abstract
Spinal motor neurons (MNs) are susceptible to glutamatergic excitotoxicity, an effect associated with lumbar MN degeneration in amyotrophic lateral sclerosis (ALS). MN susceptibility to environmental toxicant exposure, one prospective contributor to sporadic ALS, has not been systematically studied. The goal of this study was to test the ability of a well-known environmental neurotoxicant to induce hyperexcitability in mouse lumbar MNs. Methylmercury (MeHg) causes neurotoxicity through mechanisms involving elevated intracellular Ca2+ concentration ([Ca2+]i), a hallmark of excitotoxicity. We tested whether acute exposure to MeHg induces hyperexcitability in MNs by altering synaptic transmission, using whole cell patch-clamp recordings of lumbar spinal MNs in vitro. Acute MeHg exposure (20 μM) led to an increase in the frequency of both spontaneous excitatory postsynaptic currents (EPSCs) and miniature EPSCs. The frequency of inhibitory postsynaptic currents (IPSCs) was also increased by MeHg. Action potential firing rates, both spontaneous and evoked, were increased by MeHg, despite increases in both EPSCs and IPSCs, indicating a shift toward hyperexcitability. Also consistent with hyperexcitability, fluo 4-AM microfluorimetry indicated that MeHg exposure induced an increase in [Ca2+]i. Spinal cord hyperexcitability is partially mediated by Ca2+-permeable AMPA receptors, as MeHg-dependent increases in EPSCs were blocked by 1-napthyl spermine. Therefore, spinal MNs appear highly susceptible to MeHg exposure, leading to significant increases in spontaneous network excitability and disruption of normal function. Prolonged hyperexcitability could lead to eventual neurodegeneration and loss of motor function as observed in spinal cord after MeHg exposure in vivo and may contribute to MeHg-induced acceleration of ALS symptoms.
NEW & NOTEWORTHY Spinal motor neurons (MN) are susceptible to glutamatergic excitotoxicity, an effect associated with lumbar MN degeneration in amyotrophic lateral sclerosis (ALS). This study investigated MN susceptibility to environmental toxicant exposure, one prospective contributor to sporadic ALS. Spinal MNs appear highly susceptible to methylmercury exposure, leading to significant increases in spontaneous network excitability and disruption of normal function. Prolonged hyperexcitability could lead to neurodegeneration and loss of motor function as observed in ALS spinal cord symptoms.
Keywords: excitotoxicity, glutamate, methylmercury, motor neuron, spinal cord
INTRODUCTION
Motor neurons (MNs) are required for communication between the brain and skeletal muscles. Degeneration of MNs leads to progressive, debilitating neurological disorders, collectively known as MN diseases (MNDs). Amyotrophic lateral sclerosis (ALS) is the most common type of MND. Progressive bulbar palsy, progressive muscular atrophy, and primary lateral sclerosis are less common forms of MND. In the majority of MND cases, the causes of motor neuron degenerations are not yet known (Bäumer et al. 2014; Kanning et al. 2010).
Spinal MN cell bodies reside in the ventral horn of the spinal cord, and their axons project outside of the spinal cord to control effector organs, including skeletal muscles. These neurons have unique physical and functional properties, including large somas and long large caliber axons that extend as much as 1 m or more in adult humans (Nijssen et al. 2017; Ragagnin et al. 2019). Because of their large size and complexity, spinal MNs have high energy requirements. These morphological features and high metabolic activity contribute to the unique susceptibility of spinal MNs to degeneration in MNDs (Nijssen et al. 2017; Ragagnin et al. 2019). Glutamate-mediated excitotoxicity is a common feature of ALS, leading to elevated intracellular Ca2+ concentration ([Ca2+]i) and mitochondrial damage in central nervous system (CNS) neurons (Nijssen et al. 2017). Recent reports indicate that there is hypoexcitability in spinal motor neurons in ALS (Martínez-Silva et al. 2018) that appears to correlate with symptom severity (Marchand-Pauvert et al. 2019). However, the mechanism of spinal MN decline in function in ALS remains unknown and may vary between sporadic and familial forms. In the present study, we investigated the susceptibility of spinal MNs to hyperexcitability from neurotoxicants as a precursor to excitotoxicity.
Spinal MNs receive excitatory inputs from upper MNs as well as sensory neurons in the brain (Nijssen et al. 2017; Ragagnin et al. 2019). In postsynaptic spinal MNs, glutamatergic N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) ionotropic receptors are both found in abundance. In addition, spinal MNs lack the calcium binding proteins calbindin and calretinin (Berg et al. 2018; Shaw and Eggett 2000), contributing to a poor capacity for regulating [Ca2+]i in MNs. These properties put spinal MNs at risk of excessive glutamatergic stimulation, which in turn can lead to increased [Ca2+]i and subsequent high demands on mitochondrial function, as they attempt to regulate excess [Ca2+]i. Normally, glutamate-mediated Ca2+ enters mainly through NMDA receptors, whereas MN AMPA receptors (AMPARs) are typically Ca2+ impermeant, a property conferred by a posttranscriptional modification of the GluA2 subunit referred to as RNA editing (Bailey et al. 2017). When RNA editing is impaired, or GluA2 subunits are missing, AMPARs become highly Ca2+ permeable. Such excessive increases in [Ca2+]i have been observed in ALS as well as exposure to environmental neurotoxicants such as methylmercury (MeHg) (Bailey et al. 2017; Nijssen et al. 2017; Ragagnin et al. 2019).
Worldwide, mercury exposure in the form of methylmercury (MeHg), a persistent and contemporaneous environmental neurotoxicant, remains a major health concern (Clarkson and Magos 2006; Horowitz et al. 2014). The amount of mercury in the environment continues to grow as a result of metal processing and the use of fossil fuels, including coal (Clarkson 1995, 2002; Clarkson and Magos 2006; Colón-Rodríguez et al. 2017). Exposure to MeHg through contaminated food and water has been linked to severe neurological disorders including motor, cognitive, and developmental deficits (Clarkson 1995, 1997; Colón-Rodríguez et al. 2017). It has been shown that MeHg exposure may lead to or exacerbate motor impairment (Cao et al. 2013; Cavanagh and Chen 1971a; Colón-Rodríguez et al. 2017; Takaoka et al. 2008), but little is known about the cellular and molecular mechanisms of MeHg action on the spinal cord.
In sporadic MNDs, environmental or toxic factors may play a role in disease pathogenesis. A majority (~90%) of ALS cases have no known genetic basis, but the disease has been linked to environmental exposure since regional outbreaks have been observed across nonrelatives (Mitchell 2000; Wang et al. 2017). Environmental risks factors associated with MNDs include exposure to pesticides, solvents, and heavy metals such as lead and mercury (Wang et al. 2017). Consistent with this, MeHg exposure leads to MN degeneration, and the symptoms of MeHg toxicity are similar to those of ALS (Adams et al. 1983; Callaghan et al. 2011; Colón-Rodríguez et al. 2017; Hansen et al. 1989; Johnson and Atchison 2009; Praline et al. 2007). As a result, mercury exposure has been used as a model to understand the contribution of an environmental agent to excitotoxicity in MN degeneration, as seen in ALS and other spinal degenerative diseases (Bailey et al. 2017; Johnson et al. 2011; Johnson and Atchison 2009).
As a model for MN degeneration, it is important to understand how mercury affects MN physiology. MeHg exposure has been linked to severe motor impairment, as observed in acute and chronic poisoning episodes (Bakir et al. 1973; Cariccio et al. 2019; Cavanagh and Chen 1971a; McAlpine and Araki 1958), and damage to MNs may contribute to MeHg-induced motor impairment since MeHg exposure leads to loss of MNs in vivo (Cao et al. 2013; Cavanagh and Chen 1971b; Colón-Rodríguez et al. 2017; Takaoka et al. 2008). However, little is known about the cellular and molecular mechanisms of MeHg action on the spinal cord. In addition, understanding the targets of action of MeHg on spinal MNs will greatly enhance our ability to design interventions to slow the neurotoxic effects of MeHg exposure.
It is not known whether spinal MNs are direct targets of MeHg toxicity or whether spinal MN degeneration occurs secondarily as a result of distal MeHg effects on peripheral or central targets within the CNS. MeHg exposure in vivo leads to motor dysfunction and bioaccumulation of mercury in motor brain regions (Adams et al. 1983; Schwarz et al. 1996; Su et al. 1998). In the cerebellum and hippocampus, MeHg acts primarily through Ca2+-dependent mechanisms in the cerebellum and hippocampus (Yuan and Atchison 1995, 2007, 2016); however, it is not known if MeHg acts similarly on spinal MNs. Cerebellar neurons display distinct transient increases in excitability that appear to be Ca2+ dependent (Yuan and Atchison 2016). In addition, spinal MNs in culture are highly sensitive targets of MeHg (Ramanathan and Atchison 2011), displaying a distinct increase in [Ca2+]i similar to those occurring at identical concentrations in cerebellar granule cells (Limke et al. 2004; Marty and Atchison 1997, 1998). However, it is not yet known whether MeHg produces similar increases in intracellular Ca2+ for MNs in the context of spinal circuitry or whether this increase in Ca2+ correlates with changes in action potential activity in MNs. Furthermore, while Ca2+ influx in cultured spinal neurons depends at least partially on excitatory amino acid receptor activation (Ramanathan and Atchison 2011), it is unknown if synaptic activity is changed when spinal MNs receive appropriate premotor synaptic inputs.
In this study, we focused on the cellular and synaptic properties of spinal MNs to MeHg exposure in lumbar spinal cord slices using Ca2+ imaging and whole cell patch-clamp recording. MeHg exposure directly increased spinal MN excitability as a result of changes in both intrinsic membrane properties and synaptic inputs. Prolonged hyperexcitability in response to MeHg exposure has been linked to neurodegeneration (Colón-Rodríguez et al. 2017). In addition, this hyperexcitability appears to be mediated at least partly by Ca2+-dependent mechanisms. Neurons across the brain appear to experience similar MeHg-induced hyperexcitability, indicating that a common locus of action might underlie MeHg induced cell death. Therefore, therapeutic interventions that disrupt this hyperexcitability have the potential for reducing symptoms across the nervous system.
METHODS
All procedures and protocols used in this study adhere to published guidelines of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee at Michigan State University.
Preparation of spinal cord slices.
To sample a homogeneous population, spinal cord slices were cut from the lumbar region of young adult (25–28 days postnatal) male mice. Mice of this age produced reliably health spinal cord slices necessary for performing long (>30 min) recordings. Spinal cord slices were prepared using a series of artificial cerebral spinal fluid (ACSF) buffers (pH 7.3–7.4, 305 ± 5 mosmol/kgH2O) that were either N-methyl-d-glucamine (NMDG) based or 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) based. NMDG-based ACSF (Ting et al. 2018) was composed of the following (in mM): 92 NMDG, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 d-glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 0.5 CaCl2, and 10 MgSO4. HEPES-based ACSF contained the following (in mM): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 D-glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 2 CaCl2, and 2 MgSO4.
Initially, mice were deeply anesthetized with isoflurane (>5%) and received a transcardioperfusion with ACSF containing NMDG-based buffer before dissection of the spinal cord. The spinal cord was temporarily (<30 s) placed in ice-cold (1–4°C) oxygenated (95% O2-5% CO2) NMDG-based ACSF. Next, the spinal cord segment containing the lumbar region was embedded in low-gelling-temperature agar (4% (w/v)). Spinal cord slices were then cut in oxygenated ice-cold NMDG-based ACSF into 250-µm-thick slices and placed in a holding chamber with 30–35°C HEPES-based ACSF that gradually (<30 min) equilibrated to room temperature (25°C). Spinal cord slices were transferred from the holding chamber after 1–2 h and placed in a submersion recording chamber. Slices were perfused with oxygenated 30–35°C gravity-fed normal ACSF (2–4 mL/min) composed of the following (in mM): 126 NaCl, 1 NaH2PO4, 25 NaHCO3, 25 d-glucose, 3 KCl, 2 MgSO4 and 2 CaCl2. Neurons were visualized using a Nikon Eclipse E600FN microscope (Nikon Instruments Inc., Melville, NY) equipped with a water-immersion objective (×40) with infrared wavelength illumination, differential interference contrast optics (DIC), and a SONY IR-1000 charge-coupled device camera (DAGE MTI, Michigan City, IN) with contrast enhancement.
Motor neuron identification.
Spinal motor neurons were visualized in the lumbar region of the spinal cord ventral horn, using IR-DIC. Neuron selection was based on visual inspection of large cell body size. MN identification was also confirmed electrophysiologically with average input resistance (95 ± 8.2 MΩ) and time constant (13 ± 2.3 ms) consistent with large cell bodies of motor neurons. In addition, current-clamp measures displayed large afterdepolarizations across all neurons included, consistent with prior reports of spinal MNs (Mitra and Brownstone 2012).
Whole cell recording in spinal cord slices.
Membrane potentials and currents were collected using a Multiclamp 700A patch-clamp amplifier (Axon Instruments, Foster City, CA) and digitized using a Digidata 1440A analog-to-digital converter, which was controlled by the pClamp 10.7 software package (Axon Instruments). Whole cell voltage and current recordings were digitized at 20 kHz, and synaptic current recordings were filtered at 3 kHz.
Membrane potential recordings of action potentials were recorded in current-clamp mode. To measure intrinsic membrane properties and spiking activity, recording electrodes were filled with a K-gluconate-based internal solution (Sceniak and Maciver 2006; Sceniak and Sabo 2010) composed of the following (in mM): 100 K-gluconate, 20 KCl, 10 phosphocreatine, 5 MgCl2, 10 HEPES, 4 Na-ATP, 0.3 Na-GTP (pH 7.3 and 290–300 mosmol/kgH2O).
Synaptic currents were recorded in voltage-clamp mode. Spontaneous excitatory postsynaptic currents (EPSCs) were recorded (Sceniak et al. 2016) using the following Cs-methyl sulfonate internal electrode solution (in mM): 120 Cs-methyl sulfonate, 12 CsCl, 0.1 EGTA, 2 MgCl2, 10 HEPES, 2 Na-ATP, 0.25 Na-GTP, 10 phosphocreatine, and 5 QX-314 (pH 7.3 with CsOH and 290–300 mosmol/kgH2O). Spontaneous inhibitory postsynaptic currents (IPSCs) were recorded (Sceniak et al. 2016) using the following CsCl-based internal electrode solution (in mM): 145 CsCl, 2 MgCl2, 40 HEPES, 0.1 EGTA, 2 Na2ATP, and 1.5 Na2GTP (pH 7.3 with CsOH and 290–300 mosmol/kgH2O). This CsCl-based internal solution was used to reverse the Cl− concentration gradient and amplify the signal-to-noise ratio, providing optimal collection of IPSCs (Sceniak and Maciver 2008).
Electrode resistance ranged from 4 to 8 MΩ when electrodes were filled with internal pipette solution. Recordings were considered successful if the initial resting membrane potential was −57 mV or more negative to ensure stable recordings of sufficient duration (>30 min). Recordings with seals <1 GΩ, resting potentials more positive than −57 mV, or series resistance higher than 30 MΩ were not included in the analysis. Only one cell was recorded per slice. All recordings were continuous before and during MeHg application. All cells were recorded for at least 40 min total, allowing us to make within-cell comparisons for before and after MeHg exposure.
EPSCs and IPSCs.
Recordings of AMPA receptor-mediated currents were made using whole cell voltage-clamp techniques (Vhold = −55 mV) in normal ACSF in the presence of the GABAergic antagonist picrotoxin (100 μM). Miniature excitatory postsynaptic currents (mEPSCs) were collected in the presence of tetrodotoxin (TTX; 1 μM) in addition to picrotoxin. IPSCs were collected in the presence of glutamate receptor antagonists 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μM) and (2R)-amino-5-phosphonovaleric acid (APV; 50 μM).
MeHg was added to normal ACSF to achieve a final concentration of 20 μM. After the baseline recordings were collected, the perfusion was switched to MeHg-containing ACSF. After 15 min of MeHg exposure, recordings were made for comparison to control (pre-MeHg exposure). To determine the contribution of Ca2+-permeable AMPA currents to the effects of MeHg, 100 μM 1-napthyl acetyl spermine (NAS), a selective Ca2+-permeable AMPA receptor antagonist (Blaschke et al. 1993), was bath applied. After initial characterization, neurons were exposed to NAS (100 μM) for 15 min and then mEPSCs were collected. Next, MeHg (20 μM) was added with NAS and mEPSCs were recorded after an additional 15 min.
Fluo 4-AM calcium imaging.
Spinal cord slices, prepared as described above, were incubated in fluo 4-AM (4 μM) (catalog no. F14201; Molecular Probes) in the presence of 0.02% (v/v) Pluronic F-127 solution in ACSF at 34°C for 40–60 min in the dark and then placed in normal room temperature ACSF for 1 h to recover and washed with ACSF to reduce background fluorescence (Cossart et al. 2005; MacLean and Yuste 2009). Spinal cord slices were imaged using a Leica TCS SL laser scanning confocal microscope (Leica Microsystem, Heidelberg, Germany), equipped with Nomarski optics (×40 water-immersion objective, numerical aperture 0.75). Fluo 4-AM fluorescence was excited by 488-nm light from an argon laser. Images were collected before and during exposure to 20 μM MeHg (baseline and then every 5 min for 25 min). Changes in fluo 4-AM fluorescence intensity in a region of interest of the ventral MN area of the lumbar spinal cord were analyzed using Leica software. Individual neurons were identified using custom scripts written in ImageJ (National Institutes of Health, Bethesda, MD). The change in fluorescence for each identified spinal MN was calculated as deviations in fluorescence (ΔF) normalized to baseline (F0), or ΔF/F0, using MATLAB (The MathWorks, Natick, MA).
Data analysis.
Time-locked current responses were analyzed offline. Intrinsic membrane properties were estimated through pClamp and postprocessing in MATLAB. Clampex 10.7 (pClamp analysis program) was used to identify synaptic events, using the event detection algorithm with the template search feature. All events were manually verified once the template search was performed. Data were collected in 30-s chunks. Four 30-s chunks were analyzed for a total of 2 min. All recordings were verified to contain stable baselines and to be free of artifacts before synaptic analysis was performed. Every identifiable event within a 30-s chunk was included in analysis and for all four chunks.
Synaptic events were analyzed to determine the intersynaptic interval (ISI), frequency (1/ISI), and amplitude. ISI was calculated as the difference from the peak of temporally consecutive synaptic events across the time series of events. During analysis (see Fig. 4, 5, 6, and 8), measures were pooled for all events across the population of cells within each group (control or treated), for each synaptic parameter (ISI, frequency, or amplitude). Action potential arrival times were determined using custom algorithms in MATLAB. All synaptic current data were analyzed and quantified using custom-written analysis routines in MATLAB. All data are means ± SE unless otherwise stated. Statistical significance was determined using the paired t test, the Mann–Whitney Wilcoxon rank sum test for nonparametric data, or the Kolmogorov–Smirnov test for cumulative distributions. Estimates were considered significant at P ≤ 0.05. All statistical analyses were performed using MATLAB.
Fig. 4.

Methylmercury (MeHg) significantly increased excitatory postsynaptic currents (EPSCs) in lumbar spinal motor neurons (MNs). A: representative voltage-clamp recordings of a single lumbar spinal MN before and after 20 μM MeHg exposure. After 15 min of MeHg exposure, there was a noticeable increase in spontaneous EPSC (sEPSC) frequency. B and C: comparison of control and MeHg-exposed conditions (solid and dashed lines, respectively) across the population of sampled neurons (n = 10 neurons). The intersynaptic interval (ISI) cumulative distribution is significantly shifted to the left (P = 2e-209, Kolmogorov–Smirnov test). D and E: there was also a significant rightward shift in the cumulative amplitude distribution (P = 1.4e-104, Kolmogorov–Smirnov test). F–H: across the population there was also a significant (**P = 0.0091, Wilcoxon rank sum test) decrease in the mean ISI (132 ± 12 and 82 ± 8 ms for control and MeHg-treated neurons, respectively), corresponding to an increase in sEPSC mean frequency from 7.5 ± 0.7 Hz (control) to 12.1 ± 1.2 Hz (MeHg). The mean population amplitudes (16.6 ± 1.3 and 21.6 ± 3 pA, control and MeHg, respectively) were not significantly different, despite the trend. Open and shaded bars represent control and MeHg conditions, respectively. con, Control. Black arrows indicate the geometric mean of the distribution.
Fig. 5.

Methylmercury (MeHg) exposure significantly increased TTX-insensitive miniature excitatory postsynaptic currents (mEPSCs) in lumbar spinal motor neurons. A: representative voltage-clamp recordings of mEPSCs collected in the presence of TTX (1 μM), illustrating an increase in events in response to MeHg exposure (20 μM). B and C: comparison of control and MeHg-exposed conditions (solid and dashed lines, respectively) across the population of sampled lumbar spinal MNs (n = 8). There was a significant leftward shift in the cumulative mEPSC ISI distribution after MeHg exposure compared with control (P = 2.38e-120, Kolmogorov–Smirnov test). D and E: the cumulative mEPSC amplitude distribution was significantly different for control vs. MeHg-treated groups (P = 1.3e-31, Kolmogorov–Smirnov test, with an increase primarily in intermediate amplitude events after MeHg exposure). F–H: across the population (n = 8), the mean mEPSC frequency was significantly increased after MeHg exposure (5.5 ± 0.39 and 8.3 ± 1.3 Hz, and 180 ± 12 and 121 ± 19 ms, respectively; *P = 0.03, Wilcoxon rank sum test). Although there was a trend toward increased mEPSC amplitude after MeHg exposure (18.8 ± 2.0 and 24.3 ± 2.7 pA respectively), the population means were not significantly different. con, Control. Black arrows indicate the geometric mean of the distribution.
Fig. 6.

Methylmercury (MeHg) induced an increase in inhibitory postsynaptic current (IPSC) frequency in lumbar spinal motor neurons (MNs). A: representative current recordings in lumbar spinal MNs before and after MeHg exposure (20 μM, 15 min) illustrate increased IPSC frequency. B and C: comparison of control and MeHg-exposed conditions (solid and dashed lines, respectively) across the population of sampled lumbar spinal MNs (n = 11). MeHg induced a significant (P = 2.41e-41, Kolmogorov–Smirnov test) leftward shift in the cumulative intersynaptic interval (ISI) distribution compared with control, indicating an increase in IPSC frequency. D and E: despite the increased number of events, there was no significant change in IPSC amplitude. F–H: across the population, the IPSC mean ISI (430.5 ± 52 and 273.2 ± 43 ms, control and MeHg-treated, respectively) decreased significantly (P = 0.03, Wilcoxon rank sum test) compared with control. The corresponding frequencies were 2.3 ± 0.4 Hz for control and 3.7 ± 0.5 Hz after MeHg exposure. The IPSC amplitudes for control and MeHg-treated neurons (47 ± 7 and 51 ± 8 pA, respectively) were not significantly different. con, Control. Black arrows indicate the geometric mean of the distribution.
Fig. 8.
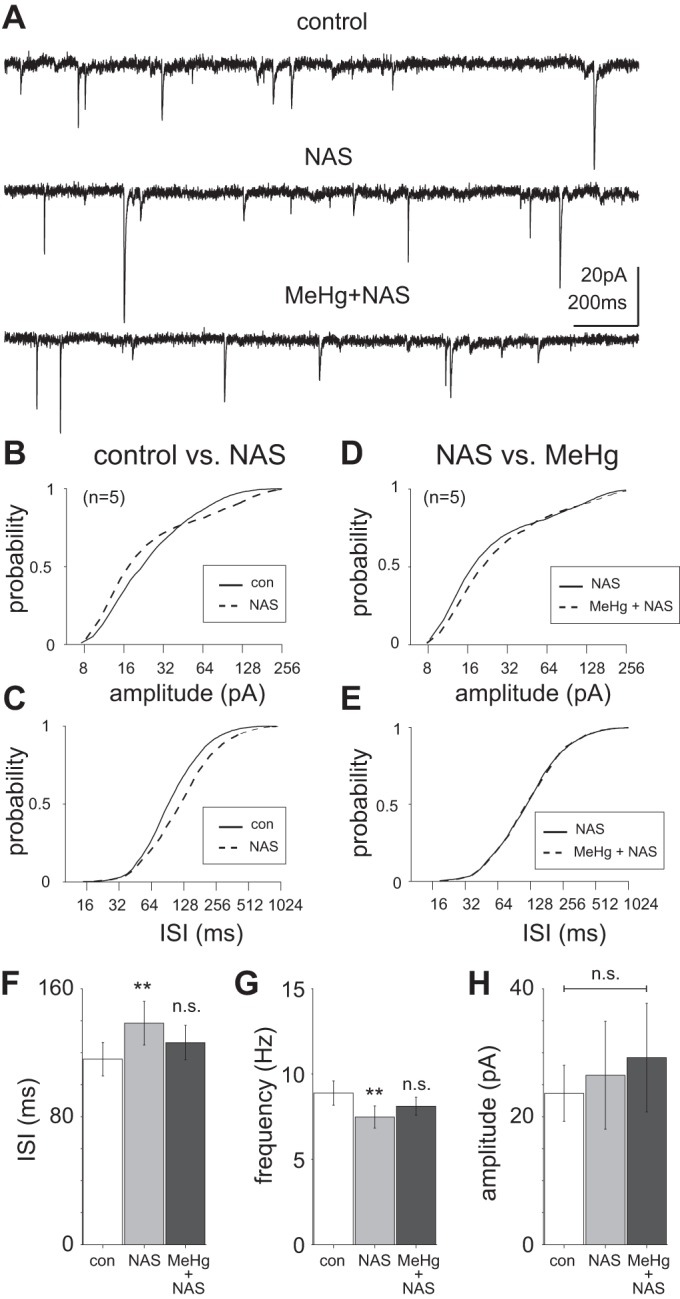
Role of Ca2+-permeable AMPA receptor-mediated currents in methylmercury (MeHg)-induced hyperexcitability. A: representative current recordings of excitatory postsynaptic currents (EPSCs) in lumbar spinal motor neurons in a control slice compared with exposure to 1-napthyl acetyl spermine (NAS; 100 μM) and to NAS combined with MeHg (20 μM). B: the amplitude of EPSCs recorded in the presence of NAS were significantly different from those in control (P = 1.51e-11, Kolmogorov–Smirnov test). C: there was also a significant rightward shift in the cumulative distribution of EPSC intersynaptic intervals (ISIs; P = 1.42e-30, Kolmogorov–Smirnov test). D: the EPSC amplitude cumulative distribution was significantly different between NAS alone and NAS with MeHg (MeHg + NAS; P = 2.2e-10, Kolmogorov–Smirnov test). E: there was not a significant difference between the ISI cumulative distributions of EPSCs recorded in NAS alone vs. MeHg + NAS. F and G: across the population (n = 5), there was a significant increase (**P = 0.008, Wilcoxon rank sum test) in EPSC ISI under control (115.9 ± 10 ms) compared with NAS-exposed conditions (138.42 ± 13 ms), corresponding to a decrease in frequency (8.6 ± 0.7 and 7.2 ± 0.6 Hz, respectively). However, there was no significant difference in event occurrence between NAS alone and MeHg + NAS (126.3 ± 11 ms, 7.9 ± 0.6 Hz). H: across the population, there was no significant difference between pre-NAS EPSC amplitude (23.64 ± 4 pA) compared with NAS exposure (26.5 ± 8 pA) or between NAS alone or MeHg + NAS (29.22 ± 8 pA). con, Control; n.s., no significant difference.
RESULTS
To begin to understand how MeHg directly affects spinal MNs, we assessed population activity in acute slices of lumbar spinal cord. To determine the population response of spinal MNs to MeHg exposure, we collected images of ventral horn of lumbar spinal cord slices labeled with fluo 4-AM (see methods; Fig. 1A). Confocal images were collected both before and during MeHg (20 μM) exposure every 5 min for a total of 25 min (Fig. 1B). MeHg exposure consistently increased fluo 4-AM fluorescence intensity relative to the control over the times sampled (5–25 min; Fig. 1B). Across the population of sampled neurons (n = 15 neurons, 5 slices), the relative intensity (ΔF/F0) within individual cells displayed a statistically significant increase (P = 4.1e-6, Wilcoxon rank sum test) after 15 min of MeHg exposure compared with control (Fig. 1C). Therefore, MeHg increased [Ca2+]i in lumbar spinal cord gray matter as well as in individual spinal MNs.
Fig. 1.
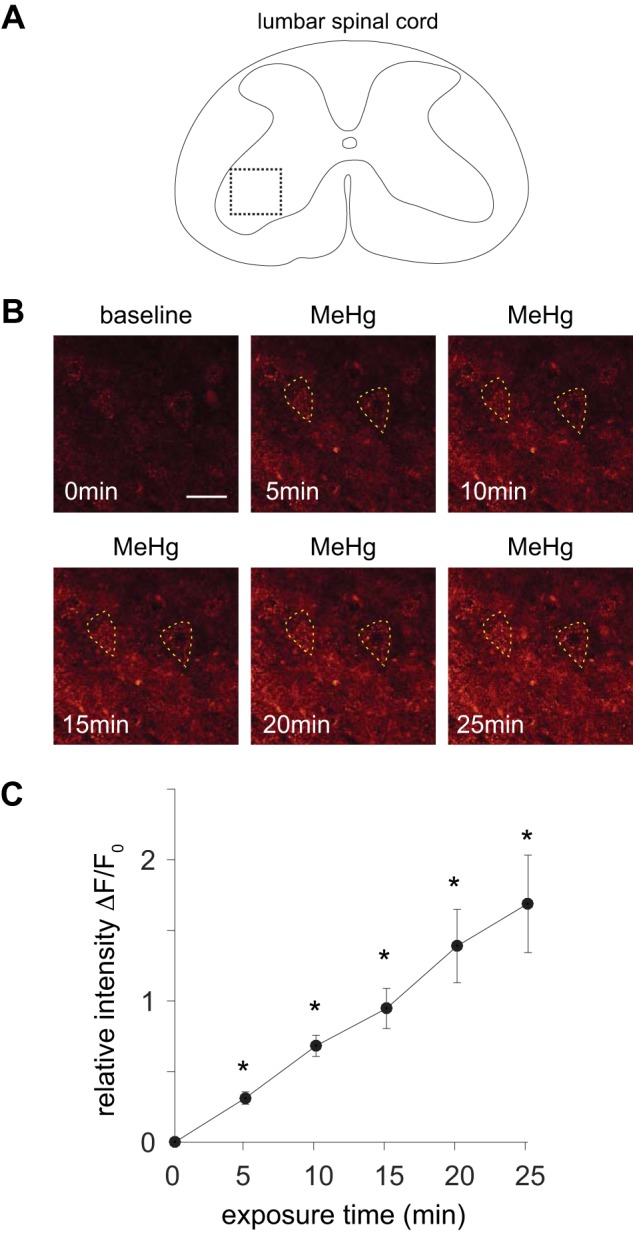
Methylmercury (MeHg) induced an increase in fluo 4 fluorescence in lumbar spinal motor neurons (MNs). A: illustration of a lumbar spinal cord slice imaged with fluo 4-AM in the presence of 2 mM Ca2+. Dashed inset reflects the region of the spinal cord ventral horn shown in B. B: microfluorometric images (scale bar, 50 μm) of lumbar spinal cord motor region labeled with fluo 4-AM before (0 min) and after exposure to 20 μM MeHg (5–25 min). Individual spinal MNs are outlined with dashed line. C: relative fluorescence intensity (ΔF/F0) is shown vs. exposure time to 20 μM MeHg across the imaged spinal cord slices (n = 5) for the population of identified spinal MNs (n = 15). There was a significant increase (*P = 4.1e-6) in ΔF/F0 across the sampled MNs of the ventral spinal cord after MeHg exposure (t > 10 min).
Intrinsic action potential firing was also determined in the presence of MeHg, using whole cell current-clamp recording of spinal MNs. Responses from representative neurons are shown in Fig. 2. Firing rate vs. current responses (f-I) were collected across a range of amplitudes of injected current steps (3-s duration). All cells displayed an increase in firing rate as the current amplitude increased (Fig. 2, A–C). However, the initial spontaneous firing rate varied across the sampled cells (n = 7). All cells were recorded at their resting potentials (Vm = −63 ± 4 mV) with no holding current, using a K-gluconate-based internal solution (see methods). After 15 min of MeHg exposure, there was a consistent increase in the firing rate in response to current steps as well as in the spontaneous firing. Even in neurons with a low spontaneous firing rate (Fig. 2A), spontaneous firing increased. While some cells displayed an increase in f-I slope (Fig. 2, D and E) after exposure (15 min) to MeHg, others exhibited an upward shift with no change in slope (Fig. 2F). Therefore, MeHg exposure increased neuronal excitability. There was no significant change in either action potential amplitude (81.2 ± 6.6 and 81.5 ± 7.2 mV for control and MeHg-treated, respectively) or resting membrane potential (−62.2 ± 5.2 and −60.1 ± 6.3 mV for control and MeHg-treated, respectively) in response to MeHg exposure. Fluctuations of the resting membrane potential or background baseline synaptic noise were noticeably increased after MeHg treatment (see Fig. 2A, insets).
Fig. 2.

Methylmercury (MeHg) increased spontaneous and evoked action potential generation in lumbar spinal motor neurons (MNs). Current injections were used in current-clamp mode to estimate firing rate vs. current (f-I) relationships. A–C: representative voltage recordings (10 s) of spinal MNs in response to increasing current-step (3 s) amplitudes before and after MeHg exposure. MeHg induced both an increase in the firing rate to the current step as well as the spontaneous spiking (occurring outside of the time of the current pulse). Insets (bottom) indicate close-up views of membrane potential during spontaneous (nonstimulated) period. The amplitude of fluctuations in voltage are significantly enhanced in the presence of MeHg (asterisk). D and E: MeHg induced a significant increase in the responsiveness to current injections. While some cells (cell 1 and cell 2; D and E, respectively) showed an increase in f-I slope, others (cell 3; F) showed an upward shift in f-I without a change in slope. Estimates of action potential rate are shown as black circles for control and gray circles for MeHg-exposed conditions. Solid and dashed lines represent linear regression fits for control and MeHg responses, respectively.
Next, action potential firing properties of spinal MNs were quantified across the population of all cells recorded (Fig. 3). The spontaneous firing rate (measured with no current injection) in MeHg exposed (1.90 ± 0.69 spikes/s) compared with control conditions (0.48 ± 0.20 spikes/s) was significantly increased (n = 7; P = 0.0006, Wilcoxon rank sum test; Fig. 3, A and B). There was also a significant increase (P = 0.037, Wilcoxon rank sum test) in the response gain or slope of the f-I curve for control compared with MeHg exposed condition (0.78 ± 0.05 and 1.10 ± 0.16 spikes·s−1·pA, respectively; Fig. 3, C and D). MeHg appears not only to increase excitability, as seen through the spontaneous action potential firing rate, but also to alter the intrinsic membrane f-I slope; this change is similar to that observed when there is an increase in the standard deviations of background synaptic noise fluctuations of cortical neurons (Sceniak and Sabo 2010). Overall, the effects of MeHg on action potential firing are consistent with a population network increase in excitability similar to what was observed through Ca2+ imaging of lumbar spinal gray matter (see Fig. 1).
Fig. 3.
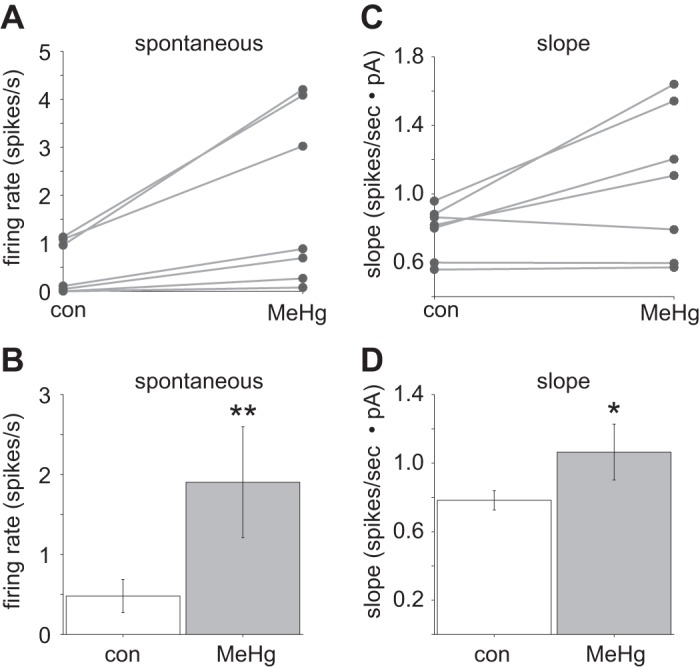
Methylmercury (MeHg) induced a significant change in intrinsic spiking generation across the population (n = 7). A and B: MeHg exposure induced a significant (**P = 0.0006, Wilcoxon rank sum test) increase in the spontaneous firing rate in spinal motor neurons (MNs) compared with control (1.90 ± 0.69 and 0.48 ± 0.20 spikes/s, respectively). C and D: there was also a significant (*P = 0.037, Wilcoxon rank sum test) trend toward MeHg increasing the slope of the firing rate vs. current relationship compared with control (1.1 ± 0.16 and 0.80 ± 0.05 spikes·s−1·pA, respectively). con, Control.
We next examined changes in spontaneous EPSCs (sEPSCs), to investigate the effects of MeHg on the synaptic properties of spinal MNs. The sEPSCs were recorded in the presence of picrotoxin (100 μM) before and after 15-min MeHg exposure. MeHg (15 min) caused an apparent increase in the frequency and amplitude of events (Fig. 4A). Spontaneous EPSCs were pooled across the population of recorded neurons (n = 10) for each condition (control and MeHg exposed). Quantification of sEPSCs revealed a highly significant leftward shift in the sEPSC intersynaptic interval (ISI) cumulative distribution (P = 1.9e-200, Kolmogorov–Smirnov test; Fig. 4, B and C), indicating a decrease in population ISI or an increase in event frequency. There was also a significant (P = 1.4e-104, Kolmogorov–Smirnov test; Fig. 4, D and E) rightward shift in the sEPSC amplitude cumulative distribution, indicating an overall increase in sEPSC amplitude, especially for intermediate amplitude events. Across the population (n = 10 neurons; Fig. 4, F–H), the mean ISI in the presence of MeHg (82 ± 8.5 ms) was significantly lower (P = 0.009, paired t test) than control (132 ± 12 ms), indicating an increase in sEPSC frequency in MeHg-treated vs. control neurons (12 ± 1.2 and 7.6 ± 0.7 Hz, respectively). However, the mean EPSC amplitude was not significantly different across the population (Fig. 4H).
MeHg had a similar effect on tetrodotoxin (TTX)-insensitive mEPSCs. Across the recorded neurons (n = 8), mEPSCs were pooled to compare control to MeHg-exposed conditions (Fig. 5). The dominant effect of MeHg was to increase mEPSC frequency (Fig. 5A). The cumulative distribution of mEPSC ISIs was significantly shifted to the left (P = 2.4e-120, Kolmogorov–Smirnov test; Fig. 5, B and C), indicating an increase in mEPSC frequency. The mEPSC amplitude was significantly different between control and MeHg-exposed neurons (P = 1.3e-31, Kolmogorov–Smirnov test; Fig. 5, D and E), with an overall increase in intermediate amplitude events. Across the population of cells sampled (n = 8), there was a significant (P = 0.03, paired t test) increase in mEPSC frequency between control and MeHg-exposed slices (5.5 ± 0.4 and 8.2 ± 1.3 Hz, or 180 ± 12 and 121 ± 19 ms, respectively; Fig. 5, F–H). Although there was a significant difference in mEPSC amplitude cumulative distributions between control and MeHg-exposed slices, the mean amplitudes were not significantly different (Fig. 5H).
To determine the effects of MeHg on spinal MN inhibitory synaptic transmission, sIPSCs were recorded before and after 15-min exposure to MeHg (Fig. 6). All identified sIPSCs were combined for the recorded neurons (n = 11) for control and treated groups (Fig. 6). MeHg increased in the rate of IPSCs with no apparent effect on IPSC amplitude. The IPSC cumulative ISI distribution was significantly shifted to the left (P = 2.6e-41, Kolmogorov–Smirnov test), indicating an overall increase in IPSC frequency (Fig. 6, B and C). There was no significant difference in the IPSC cumulative amplitude distributions for control compared with MeHg-treated slices (Fig. 6, D and E). Across the population (n = 11) there was a significant difference (P = 0.03, paired t test) in the mean IPSC frequency of control and MeHg-treated neurons (2.3 ± 0.4 and 3.7 ± 0.5 Hz, respectively; Fig. 6, F–H). The IPSC amplitudes were essentially identical for control and MeHg-treated groups (47 ± 7 and 50 ± 8 pA, respectively, Fig. 6H).
MeHg treatment of spinal motor neurons not only increased the frequency of inhibitory synaptic transmission but also induced giant inhibitory currents (Fig. 7). Figure 7 illustrates giant inhibitory currents recorded using CsCl-based internal solution in the presence of CNQX and APV. Giant IPSCs were observed in neurons in three separate spinal cord slices after MeHg treatment for 15 min or greater. These giant currents were not present in the control condition and only occurred in 4 of 11 cells sampled. In addition, giant currents did not occur when recording using K-gluconate-based internal solution.
Fig. 7.

Methylmercury (MeHg) exposure elicited giant inhibitory postsynaptic currents (IPSCs) in lumbar spinal motor neurons. Representative current recordings of 3 distinct neurons show giant IPSCs compared with spontaneous IPSCs. The amplitude, kinetics, and total charge (integrated area under the curve) were significantly larger for the giant IPSCs compared with the normal IPSC currents, only occurred in the presence of MeHg, and were not seen in every slice.
To test the hypothesis that the increase in glutamatergic EPSC frequency was due to a change in Ca2+-permeable AMPAR function, we attempted to block the increase in excitability induced by MeHg using NAS (Fig. 8), which selectively inhibits AMPARs containing Ca2+-permeable GluA2 (Blaschke et al. 1993). EPSCs were first recorded in the absence and presence of NAS (100 μM) for 15 min (Fig. 8A). The EPSC frequency cumulative distributions (combined across all neurons recorded, n = 5) of control and NAS-treated neurons were statistically different (P = 1.4e-30, Kolmogorov–Smirnov test; Fig. 8B), with a slight (115 ± 10 and 138 ± 13 ms, respectively) but significant (P = 0.008, paired t test) decrease in EPSC frequency after NAS treatment (8.7 ± 0.7 and 7.3 ± 0.6 Hz, respectively). The EPSC amplitudes were not significantly different before and after NAS treatment (24 ± 4 and 26 ± 8 pA, respectively; Fig. 8, C and H). Addition of MeHg in the presence of NAS (after an additional 15-min period) did not significantly increase either the frequency (127 ± 11 ms or 7.9 ± 0.5 Hz; Fig. 8, D, F, and G) or amplitude (29 ± 8 pA; Fig. 8, E and H). Therefore, the elevation in EPSC frequency induced by MeHg exposure, as shown in Fig. 4, was blocked in the presence of NAS pretreatment. Together, these results suggest that MeHg and NAS act through a common mechanism. The selectivity of NAS suggests that Ca2+-permeable AMPA receptors are involved in hyperexcitability associated with MeHg exposure in spinal motor neurons.
DISCUSSION
Mercury bioaccumulates in spinal MNs (Su et al. 1998; Wakabayashi et al. 1995); however, this does not necessarily lead to cytotoxicity. In the cerebellum, MeHg levels are higher in Purkinje cells than in cerebellar granule cells and yet Purkinje cells are less susceptible to MeHg-induced cell death (Edwards et al. 2005; Leyshon-Sørland et al. 1994). Spinal MNs are a primary target of cell death in ALS and MeHg exposure (Su et al. 1998). However, studies have not examined the direct action of MeHg on spinal MNs. This is important because the majority of ALS cases examined have no known genetic link (Bailey et al. 2017). Therefore, it is important to understand the direct effects of environmental insults on spinal MNs.
Our results indicate that spinal MNs exhibit direct effects of MeHg exposure. Overall, there was an increase in excitability in the motor regions of lumbar spinal cord as observed through Ca2+ imaging and action potential measurements. The ventral horn of the lumbar region of the spinal cord displayed a significant increase in Ca2+ influx. In addition, MNs in this region displayed significant increases in spontaneous action potential firing. There was also increased sensitivity to current injection, indicated by increased slope in the f-I curve (firing rate as a function of injected current). Together, these results point to MeHg producing an overall increase in network excitability through synaptic barrages that increase the membrane fluctuations of individual cells. The effects of background synaptic noise on the f-I relationship are similar to those observed when noise is simulated with the dynamic clamp system (Sceniak and Sabo 2010)
Spinal MNs displayed increases in the frequency of excitatory glutamatergic synaptic currents in the presence of MeHg, indicating a presynaptic action on the probability of release. An increase in [Ca2+]i is consistent with an increase in probability of release. In addition, the frequency of inhibitory synaptic inputs was also increased. Although there was an increase in both excitatory and inhibitory synaptic drive, the spontaneous action potential firing was elevated in response to MeHg, indicating that increases in excitation and inhibition did not cancel each other, but rather increased membrane potential fluctuations similar to those observed in background synaptic noise (Sceniak and Sabo 2010). The frequency of synaptic barrages was significantly elevated in response to MeHg exposure, indicating a significant drive toward increased excitability. In addition, we observed giant synaptic potentials, which have been linked in cerebellum to inward Cl− currents induced by MeHg exposure (Yuan and Atchison 1999, 2005). Although the mechanism of the giant (hundreds of picoamperes to a few nanoamperes) slow inward current is unknown, as in the cerebellum (Yuan et al. 2005; Yuan and Atchison 1999), it appears to be linked to MeHg exposure times (>15 min) and likely represents a major disruption of synaptic function and network connectivity (E/I balance shift) that is induced by MeHg. In the future, it would be interesting to know whether the MeHg-induced increases in sEPSC and sIPSC frequency lead to production of the giant inward currents or whether a nonsynaptic mechanism is responsible for the giant hyperpolarizations.
The effects of MeHg on spinal MNs appeared to be at least partially mediated by Ca2+-dependent AMPARs. AMPAR permeability to cations is controlled by the GluA2 subunit. The majority of AMPARs in the CNS are GluA2 containing. It has been shown that RNA editing, switching a Q to an R in the GluA2 amino acid sequence, is necessary to render GluA2-containing AMPARs impermeable to Ca2+ (Liu and Savtchouk 2012). When AMPARs contain either an unedited form of GluA2 (GluA2Q) or a “Flip” alternative splice, they can exhibit prolonged Ca2+ permeability leading to potential excitotoxicity (Colón-Rodríguez et al. 2017; Liu and Savtchouk 2012). Alterations of mRNA associated with GluA2 have been proposed as potential sites of action of MeHg (Colón-Rodríguez A, Colon-Carrion NM, Atchison WD unpublished observations). Our present results are consistent with an increase in Ca2+ permeability of AMPARs in response to MeHg exposure, because there was a significant increase in [Ca2+]i in motor regions of the spinal cord after MeHg exposure. Previously, similar Ca2+ increases were observed in the hippocampus and cerebellum (Yuan and Atchison 1993, 2007, 2016). Further supporting a role for Ca2+ influx via AMPARs, MeHg-induced increases in excitatory synaptic responses were blocked by NAS, which antagonizes Ca2+-permeable AMPARs (Koike et al. 1997). This demonstrates that MeHg targets Ca2+-permeable AMPARs in MNs, suggesting that the action of MeHg on Ca2+-dependent AMPARs is a contributor to the hyperexcitability. These results indicate a ubiquitous action of MeHg through Ca2+-mediated mechanisms to induce excitotoxicity.
Although our results are consistent with MeHg acting on AMPARs, we cannot rule out other sources of Ca2+ influx. Both organic and inorganic mercury act on nonspecific cation channels (Arakawa et al. 1991; Huang and Narahashi 1997a, 1997b; Narahashi et al. 1994; Xu et al. 1998; Xu and Atchison 1996; Yuan et al. 2005). In spinal cord, we found that MeHg exposure increased the rate of spontaneous action potentials and response gain (f-I slope) to current step injections, independent of a significant membrane potential depolarization.
We studied the acute effects of MeHg, which reflect the initial insult, and found acute hyperexcitability. Hyperexcitability leads to eventual neurodegeneration. A recent study reported that there is an absence of hyperexcitability in the spinal MNs of ALS patients (Marchand-Pauvert et al. 2019). Hypoexcitability of spinal MNs appears to correlate with the progression of ALS symptoms (Marchand-Pauvert et al. 2019; Martínez-Silva et al. 2018). This is consistent with neurotoxicant exposure: neurons within the network would initially experience hyperexcitability followed by some cell death from excitotoxicity. Loss of neurons would reduce synaptic connectivity within the network and ultimately lead to hypoexcitability. Our results reported here suggest that spinal MNs exhibit similar effects as those observed in cerebellum and brain stem (Yuan and Atchison 1993, 2003).
In conclusion, when applied acutely, MeHg affects spinal MNs in a manner similar to that of neurons in hippocampus and cerebellum (Yuan and Atchison 1995, 1999). The hyperexcitability of spinal MNs presented in this study would significantly and negatively affect spinal MN function and network balance if similar increases in excitability were present in vivo. Progression of MND in response to MeHg or other neurotoxicants likely results, in part, from initial hyperexcitability that leads to excitotoxicity.
GRANTS
This work was supported by National Institute of Environmental Health Sciences Grant R01ES024064 (to W. D. Atchison).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.P.S., Y.Y., and W.D.A. conceived and designed research; M.P.S. and J.B.S. performed experiments; M.P.S. analyzed data; M.P.S., J.B.S., S.L.S., and Y.Y. interpreted results of experiments; M.P.S. prepared figures; M.P.S. drafted manuscript; M.P.S., S.L.S., Y.Y., and W.D.A. edited and revised manuscript; M.P.S., S.L.S., Y.Y., and W.D.A. approved final version of manuscript.
REFERENCES
- Adams CR, Ziegler DK, Lin JT. Mercury intoxication simulating amyotrophic lateral sclerosis. JAMA 250: 642–643, 1983. doi: 10.1001/jama.1983.03340050054029. [DOI] [PubMed] [Google Scholar]
- Arakawa O, Nakahiro M, Narahashi T. Mercury modulation of GABA-activated chloride channels and non-specific cation channels in rat dorsal root ganglion neurons. Brain Res 551: 58–63, 1991. doi: 10.1016/0006-8993(91)90913-G. [DOI] [PubMed] [Google Scholar]
- Bailey JM, Colón-Rodríguez A, Atchison WD. Evaluating a gene-environment interaction in amyotrophic lateral sclerosis: methylmercury exposure and mutated SOD1. Curr Environ Health Rep 4: 200–207, 2017. doi: 10.1007/s40572-017-0144-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakir F, Damluji SF, Amin-Zaki L, Murtadha M, Khalidi A, al-Rawi NY, Tikriti S, Dahir HI, Clarkson TW, Smith JC, Doherty RA. Methylmercury poisoning in Iraq. Science 181: 230–241, 1973. doi: 10.1126/science.181.4096.230. [DOI] [PubMed] [Google Scholar]
- Bäumer D, Talbot K, Turner MR. Advances in motor neurone disease. J R Soc Med 107: 14–21, 2014. doi: 10.1177/0141076813511451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg EM, Bertuzzi M, Ampatzis K. Complementary expression of calcium binding proteins delineates the functional organization of the locomotor network. Brain Struct Funct 223: 2181–2196, 2018. doi: 10.1007/s00429-018-1622-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke M, Keller BU, Rivosecchi R, Hollmann M, Heinemann S, Konnerth A. A single amino acid determines the subunit-specific spider toxin block of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate receptor channels. Proc Natl Acad Sci USA 90: 6528–6532, 1993. doi: 10.1073/pnas.90.14.6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan B, Feldman D, Gruis K, Feldman E. The association of exposure to lead, mercury, and selenium and the development of amyotrophic lateral sclerosis and the epigenetic implications. Neurodegener Dis 8: 1–8, 2011. doi: 10.1159/000315405. [DOI] [PubMed] [Google Scholar]
- Cao B, Lv W, Jin S, Tang J, Wang S, Zhao H, Guo H, Su J, Cao X. Degeneration of peripheral nervous system in rats experimentally induced by methylmercury intoxication. Neurol Sci 34: 663–669, 2013. doi: 10.1007/s10072-012-1100-3. [DOI] [PubMed] [Google Scholar]
- Cariccio VL, Samà A, Bramanti P, Mazzon E. Mercury involvement in neuronal damage and in neurodegenerative diseases. Biol Trace Elem Res 187: 341–356, 2019. doi: 10.1007/s12011-018-1380-4. [DOI] [PubMed] [Google Scholar]
- Cavanagh JB, Chen FC. The effects of methyl-mercury-dicyandiamide on the peripheral nerves and spinal cord of rats. Acta Neuropathol 19: 208–215, 1971a. doi: 10.1007/BF00684597. [DOI] [PubMed] [Google Scholar]
- Cavanagh JB, Chen FC. Amino acid incorporation in protein during the “silent phase” before organo-mercury and p-bromophenylacetylurea neuropathy in the rat. Acta Neuropathol 19: 216–224, 1971b. doi: 10.1007/BF00684598. [DOI] [PubMed] [Google Scholar]
- Clarkson TW. Environmental contaminants in the food chain. Am J Clin Nutr 61, Suppl: 682S–686S, 1995. doi: 10.1093/ajcn/61.3.682S. [DOI] [PubMed] [Google Scholar]
- Clarkson TW. The toxicology of mercury. Crit Rev Clin Lab Sci 34: 369–403, 1997. doi: 10.3109/10408369708998098. [DOI] [PubMed] [Google Scholar]
- Clarkson TW. The three modern faces of mercury. Environ Health Perspect 110, Suppl 1: 11–23, 2002. doi: 10.1289/ehp.02110s111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Crit Rev Toxicol 36: 609–662, 2006. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- Colón-Rodríguez A, Hannon HE, Atchison WD. Effects of methylmercury on spinal cord afferents and efferents–a review. Neurotoxicology 60: 308–320, 2017. doi: 10.1016/j.neuro.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Ikegaya Y, Yuste R. Calcium imaging of cortical networks dynamics. Cell Calcium 37: 451–457, 2005. doi: 10.1016/j.ceca.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Edwards JR, Marty MS, Atchison WD. Comparative sensitivity of rat cerebellar neurons to dysregulation of divalent cation homeostasis and cytotoxicity caused by methylmercury. Toxicol Appl Pharmacol 208: 222–232, 2005. doi: 10.1016/j.taap.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Hansen JC, Reske-Nielsen E, Thorlacius-Ussing O, Rungby J, Danscher G. Distribution of dietary mercury in a dog. Quantitation and localization of total mercury in organs and central nervous system. Sci Total Environ 78: 23–43, 1989. doi: 10.1016/0048-9697(89)90020-X. [DOI] [PubMed] [Google Scholar]
- Horowitz HM, Jacob DJ, Amos HM, Streets DG, Sunderland EM. Historical mercury releases from commercial products: global environmental implications. Environ Sci Technol 48: 10242–10250, 2014. doi: 10.1021/es501337j. [DOI] [PubMed] [Google Scholar]
- Huang CS, Narahashi T. The role of G proteins in the activity and mercury modulation of GABA-induced currents in rat neurons. Neuropharmacology 36: 1623–1630, 1997a. doi: 10.1016/S0028-3908(97)00173-1. [DOI] [PubMed] [Google Scholar]
- Huang CS, Narahashi T. The role of phosphorylation in the activity and mercury modulation of GABA-induced currents in rat neurons. Neuropharmacology 36: 1631–1640, 1997b. doi: 10.1016/S0028-3908(97)00172-X. [DOI] [PubMed] [Google Scholar]
- Johnson FO, Atchison WD. The role of environmental mercury, lead and pesticide exposure in development of amyotrophic lateral sclerosis. Neurotoxicology 30: 761–765, 2009. doi: 10.1016/j.neuro.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson FO, Yuan Y, Hajela RK, Chitrakar A, Parsell DM, Atchison WD. Exposure to an environmental neurotoxicant hastens the onset of amyotrophic lateral sclerosis-like phenotype in human Cu2+/Zn2+ superoxide dismutase 1 G93A mice: glutamate-mediated excitotoxicity. J Pharmacol Exp Ther 338: 518–527, 2011. doi: 10.1124/jpet.110.174466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Annu Rev Neurosci 33: 409–440, 2010. doi: 10.1146/annurev.neuro.051508.135722. [DOI] [PubMed] [Google Scholar]
- Koike M, Iino M, Ozawa S. Blocking effect of 1-naphthyl acetyl spermine on Ca2+-permeable AMPA receptors in cultured rat hippocampal neurons. Neurosci Res 29: 27–36, 1997. doi: 10.1016/S0168-0102(97)00067-9. [DOI] [PubMed] [Google Scholar]
- Leyshon-Sørland K, Jasani B, Morgan AJ. The localization of mercury and metallothionein in the cerebellum of rats experimentally exposed to methylmercury. Histochem J 26: 161–169, 1994. doi: 10.1007/BF00157965. [DOI] [PubMed] [Google Scholar]
- Limke TL, Bearss JJ, Atchison WD. Acute exposure to methylmercury causes Ca2+ dysregulation and neuronal death in rat cerebellar granule cells through an M3 muscarinic receptor-linked pathway. Toxicol Sci 80: 60–68, 2004. doi: 10.1093/toxsci/kfh131. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Savtchouk I. Ca2+ permeable AMPA receptors switch allegiances: mechanisms and consequences. J Physiol 590: 13–20, 2012. doi: 10.1113/jphysiol.2011.213926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean JN, Yuste R. Imaging action potentials with calcium indicators. Cold Spring Harb Protoc 2009: pdb.prot5316, 2009. [DOI] [PubMed] [Google Scholar]
- Marchand-Pauvert V, Peyre I, Lackmy-Vallee A, Querin G, Bede P, Lacomblez L, Debs R, Pradat PF. Absence of hyperexcitability of spinal motoneurons in patients with amyotrophic lateral sclerosis. J Physiol 597: 5445–5467, 2019. doi: 10.1113/JP278117. [DOI] [PubMed] [Google Scholar]
- Martínez-Silva ML, Imhoff-Manuel RD, Sharma A, Heckman CJ, Shneider NA, Roselli F, Zytnicki D, Manuel M. Hypoexcitability precedes denervation in the large fast-contracting motor units in two unrelated mouse models of ALS. eLife 7: e30955, 2018. doi: 10.7554/eLife.30955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty MS, Atchison WD. Pathways mediating Ca2+ entry in rat cerebellar granule cells following in vitro exposure to methyl mercury. Toxicol Appl Pharmacol 147: 319–330, 1997. doi: 10.1006/taap.1997.8262. [DOI] [PubMed] [Google Scholar]
- Marty MS, Atchison WD. Elevations of intracellular Ca2+ as a probable contributor to decreased viability in cerebellar granule cells following acute exposure to methylmercury. Toxicol Appl Pharmacol 150: 98–105, 1998. doi: 10.1006/taap.1998.8383. [DOI] [PubMed] [Google Scholar]
- McAlpine D, Araki S. Minamata disease: an unusual neurological disorder caused by contaminated fish. Lancet 272: 629–631, 1958. doi: 10.1016/S0140-6736(58)90348-9. [DOI] [PubMed] [Google Scholar]
- Mitchell JD. Amyotrophic lateral sclerosis: toxins and environment. Amyotroph Lateral Scler Other Motor Neuron Disord 1: 235–250, 2000. doi: 10.1080/14660820050515061. [DOI] [PubMed] [Google Scholar]
- Mitra P, Brownstone RM. An in vitro spinal cord slice preparation for recording from lumbar motoneurons of the adult mouse. J Neurophysiol 107: 728–741, 2012. doi: 10.1152/jn.00558.2011. [DOI] [PubMed] [Google Scholar]
- Narahashi T, Ma JY, Arakawa O, Reuveny E, Nakahiro M. GABA receptor-channel complex as a target site of mercury, copper, zinc, and lanthanides. Cell Mol Neurobiol 14: 599–621, 1994. doi: 10.1007/BF02088671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijssen J, Comley LH, Hedlund E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol 133: 863–885, 2017. doi: 10.1007/s00401-017-1708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praline J, Guennoc A-M, Limousin N, Hallak H, de Toffol B, Corcia P. ALS and mercury intoxication: a relationship? Clin Neurol Neurosurg 109: 880–883, 2007. doi: 10.1016/j.clineuro.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Ragagnin AMG, Shadfar S, Vidal M, Jamali MS, Atkin JD. Motor neuron susceptibility in ALS/FTD. Front Neurosci 13: 532, 2019. doi: 10.3389/fnins.2019.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan G, Atchison WD. Ca2+ entry pathways in mouse spinal motor neurons in culture following in vitro exposure to methylmercury. Neurotoxicology 32: 742–750, 2011. doi: 10.1016/j.neuro.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sceniak MP, Lang M, Enomoto AC, James Howell C, Hermes DJ, Katz DM. Mechanisms of functional hypoconnectivity in the medial prefrontal cortex of Mecp2 null mice. Cereb Cortex 26: 1938–1956, 2016. doi: 10.1093/cercor/bhv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sceniak MP, Maciver MB. Cellular actions of urethane on rat visual cortical neurons in vitro. J Neurophysiol 95: 3865–3874, 2006. doi: 10.1152/jn.01196.2005. [DOI] [PubMed] [Google Scholar]
- Sceniak MP, Maciver MB. Slow GABAA mediated synaptic transmission in rat visual cortex. BMC Neurosci 9: 8, 2008. doi: 10.1186/1471-2202-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sceniak MP, Sabo SL. Modulation of firing rate by background synaptic noise statistics in rat visual cortical neurons. J Neurophysiol 104: 2792–2805, 2010. doi: 10.1152/jn.00023.2010. [DOI] [PubMed] [Google Scholar]
- Schwarz S, Husstedt I, Bertram HP, Kuchelmeister K. Amyotrophic lateral sclerosis after accidental injection of mercury. J Neurol Neurosurg Psychiatry 60: 698, 1996. doi: 10.1136/jnnp.60.6.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Eggett CJ. Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J Neurol 247, Suppl 1: I17–I27, 2000. doi: 10.1007/BF03161151. [DOI] [PubMed] [Google Scholar]
- Su M, Wakabayashi K, Kakita A, Ikuta F, Takahashi H. Selective involvement of large motor neurons in the spinal cord of rats treated with methylmercury. J Neurol Sci 156: 12–17, 1998. doi: 10.1016/S0022-510X(98)00030-6. [DOI] [PubMed] [Google Scholar]
- Takaoka S, Kawakami Y, Fujino T, Oh-ishi F, Motokura F, Kumagai Y, Miyaoka T. Somatosensory disturbance by methylmercury exposure. Environ Res 107: 6–19, 2008. doi: 10.1016/j.envres.2007.05.012. [DOI] [PubMed] [Google Scholar]
- Ting JT, Lee BR, Chong P, Soler-Llavina G, Cobbs C, Koch C, Zeng H, Lein E. Preparation of acute brain slices using an optimized N-methyl-d-glucamine protective recovery method. J Vis Exp e53825, 2018. doi: 10.3791/53825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Kakita A, Sakamoto M, Su M, Iwanaga K, Ikuta F. Variability of brain lesions in rats administered methylmercury at various postnatal development phases. Brain Res 705: 267–272, 1995. doi: 10.1016/0006-8993(95)01208-7. [DOI] [PubMed] [Google Scholar]
- Wang M-D, Little J, Gomes J, Cashman NR, Krewski D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology 61: 101–130, 2017. doi: 10.1016/j.neuro.2016.06.015. [DOI] [PubMed] [Google Scholar]
- Xu YF, Atchison WD. Effects of omega-agatoxin-IVA and omega-conotoxin-MVIIC on perineurial Ca++ and Ca++-activated K+ currents of mouse motor nerve terminals. J Pharmacol Exp Ther 279: 1229–1236, 1996. [PubMed] [Google Scholar]
- Xu YF, Hewett SJ, Atchison WD. Passive transfer of Lambert-Eaton myasthenic syndrome induces dihydropyridine sensitivity of ICa in mouse motor nerve terminals. J Neurophysiol 80: 1056–1069, 1998. doi: 10.1152/jn.1998.80.3.1056. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Disruption by methylmercury of membrane excitability and synaptic transmission of CA1 neurons in hippocampal slices of the rat. Toxicol Appl Pharmacol 120: 203–215, 1993. doi: 10.1006/taap.1993.1104. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Methylmercury acts at multiple sites to block hippocampal synaptic transmission. J Pharmacol Exp Ther 275: 1308–1316, 1995. [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Comparative effects of methylmercury on parallel-fiber and climbing-fiber responses of rat cerebellar slices. J Pharmacol Exp Ther 288: 1015–1025, 1999. [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Electrophysiological studies of neurotoxicants on central synaptic transmission in acutely isolated brain slices. Curr Protoc Toxicol 17: 11.11.1–11.11.38, 2003. doi: 10.1002/0471140856.tx1111s17. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Methylmercury induces a spontaneous, transient slow inward chloride current in Purkinje cells of rat cerebellar slices. J Pharmacol Exp Ther 313: 751–764, 2005. doi: 10.1124/jpet.104.080721. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Methylmercury-induced increase of intracellular Ca2+ increases spontaneous synaptic current frequency in rat cerebellar slices. Mol Pharmacol 71: 1109–1121, 2007. doi: 10.1124/mol.106.031286. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Multiple sources of Ca2+ contribute to methylmercury-induced increased frequency of spontaneous inhibitory synaptic responses in cerebellar slices of rat. Toxicol Sci 150: 117–130, 2016. doi: 10.1093/toxsci/kfv314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Otero-Montañez JKL, Yao A, Herden CJ, Sirois JE, Atchison WD. Inwardly rectifying and voltage-gated outward potassium channels exhibit low sensitivity to methylmercury. Neurotoxicology 26: 439–454, 2005. doi: 10.1016/j.neuro.2005.03.005. [DOI] [PubMed] [Google Scholar]