

SUMMARY
MLKL, a key component downstream of RIPK3, is suggested to be a terminal executor of necroptosis. Genetic studies have revealed that Ripk3 ablation rescues embryonic lethality in Fadd- or Caspase-8-deficient mice. Given that RIPK3 has also been implicated in non-necroptotic pathways including apoptosis and inflammatory signaling, it remains unclear whether the lethality in Fadd−/− mice is indeed caused by necropotosis. Here, we show that genetic deletion of Mlkl rescues the developmental defect in Fadd-deficient mice and that Fadd−/−Mlkl−/− mice are viable and fertile. Mlkl−/−Fadd−/− mice display significantly accelerated lymphoproliferative disease characterized by lymphadenopathy and splenomegaly when compared to Ripk3−/−Fadd−/− mice. Mlkl−/−Fadd−/− bone-marrow-derived macrophages and dendritic cells have impaired NLRP3 inflammasome activation associated with defects in ASC speck formation and NF-κB-dependent NLRP3 transcription. Our findings reveal that MLKL and FADD play critical roles in preventing lymphoproliferative disease and activating the NLRP3 inflammasome.
Graphical Abstract
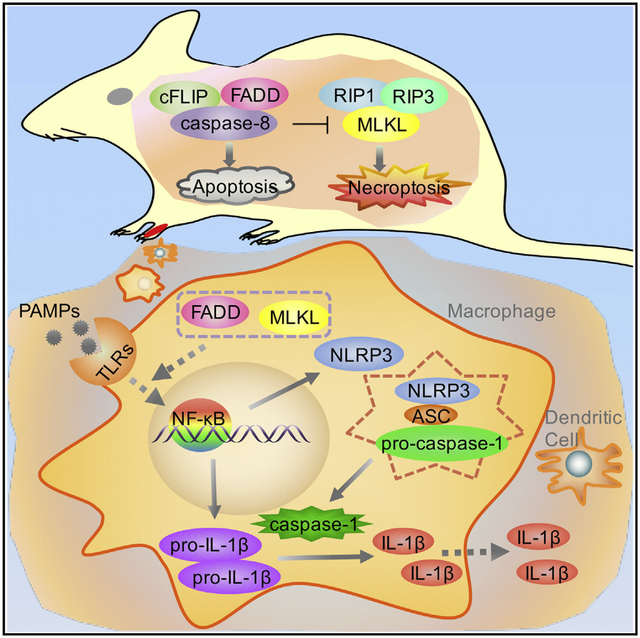
In Brief
Zhang et al. find that RIPK3-MLKL-mediated signaling results in embryonic lethality in Fadd-deficient mice. Mlkl−/−Fadd−/− mice have more severe lymphoproliferative disease than Fadd−/−Ripk3−/− mice. Further studies reveal that MLKL and FADD are critical for activating the NLRP3 inflammasome through regulation of ASC speck formation and NF-κB-dependent NLRP3 transcription.
INTRODUCTION
Regulated cell death including apoptosis and necroptosis plays essential roles in the development, homeostasis, and pathological processes of mammals. Apoptosis can be triggered through the death receptors (DRs), including tumor necrosis factor receptor-1 (TNFR1) and FAS (Nagata, 1997). Activated by their ligands, DRs recruit the adaptor protein Fas-associated death domain (FADD), which binds and activates caspase-8 to initiate apoptosis (Boldin et al., 1996; Muzio et al., 1996; Zhang and Winoto, 1996). In the absence of active caspase-8, receptor-inter acting protein kinase 1 (RIPK1) and RIPK3 interact with each other, forming a microfilament-like complex that is known as necrosome (Li et al., 2012; Orozco et al., 2014; Vercammen et al.,1998). MLKL, the mixed lineage kinase domain-like protein, has been identified as a key component downstream of RIPK3 (Sun et al., 2012; Zhao et al., 2012). Recent studies have demonstrated that MLKL is phosphorylated by RIPK3 and form oligomers that translocate to cell membrane, leading to the disruption of membrane integrity (Cai et al., 2014; Chen et al., 2014; Dondelinger et al., 2014; Orozco et al., 2014; Su et al., 2014;Wang et al., 2014).
The in vivo mechanism of RIPK1-RIPK3 necrosome-mediated necroptosis has been elucidated through intensive genetic studies. Targeted deletion of Fadd or Caspase-8 in mice causes embryonic lethality around embryonic day 11.5 (Varfolomeev et al., 1998; Yeh et al., 1998), which is probably due to vascular, cardiac, and hematopoietic defects (Kang et al., 2004, 2008; Sakamaki et al., 2002). The death of Fadd−/− or Casp8−/− embryo can be rescued by co-deletion of Ripk1; however, similar to Ripk1−/− mice, these mice die perinatally (Zhang et al., 2011; Rickard et al., 2014; Dillon et al., 2014; Kaiser et al., 2014; Kelliher et al., 1998). Deletion of Ripk3 in Fadd−/− or Casp8−/− mice also prevents the embryonic lethality (Dillon et al., 2012; Kaiser et al.,2011; Oberst et al., 2011); by contrast, Ripk3−/−Fadd−/− and Ripk3−/−Casp8−/− mice grow into fertile adults. Furthermore, co-deletion of Ripk3 and either Fadd or Caspase-8 can rescue Ripk1−/− mice into adulthood (Rickard et al., 2014; Dillon et al.,2014; Kaiser et al., 2014; Dowling et al., 2015). During necroptosis, the pseudokinase MLKL acts as a terminal executor downstream of necrosome formation. Thus, the strong evidence regarding the early embryonic death of Fadd or Caspase-8-deficient mice caused by RIPK3-dependent necroptosis could come from investigating putative MLKL functions in the Fadd or Caspase-8-deficient background.
Although the mice deficient in both Fadd and Ripk3 are viable, they display a profound lymphoproliferative disease over time with a large accumulation of B220+ CD3+ T lymphocytes. Similar phenomena are observed in Ripk3−/−Casp8−/− mice and Ripk3D161N/D161NCasp8−/− mice (Dillon et al., 2012; Kaiser et al., 2011; Newton et al., 2014; Oberst et al., 2011). Meanwhile, this disease also occurred in mice and humans containing mutations in FAS or FASL (Wilson et al., 2009). It is notable that Caspase-8−/−Ripk3−/−Ripk1−/− mice that are backcrossed to C57BL/6 mice background show milder lymphoproliferative disease compared to Caspase-8−/−Ripk3−/− mice (Rickard et al., 2014;Dillon et al., 2014; Dowling et al., 2015), whereas the TKO mice generated without backcrossing display an opposite phenotype (Kaiser et al., 2014). These results indicate that the extent of the backcross of knockout mice may have to do with the phenotypes. Interestingly, genetic loss of both FASL and TRAIL in mice displays more severe disease than in FASL mutant mice (Sedger et al., 2010), which indicates that FAS may be one of the death receptors involving in progressive lymphoproliferative disease. Together, the precise contributions of cell death pathways in the pathogenesis of lymphoproliferative disease remain unclear.
Another important process closely related to necrosis is inflammatory response. Damage-associated molecular patterns (DAMPs) released from necrotic cells stimulate immune cells through ligation of pattern recognition receptors (PRRs) to secret pro-inflammatory cytokines such as interleukin-1b (IL-1β) and IL-18, which subsequently activate host immune defenses against various pathogens. The maturation of IL-1β and IL-18 is mediated by cytosolic NOD-like receptors (NLRs) and HIN domain-containing family member AIM2, which associate with other proteins to form large multimeric complex that is known as the inflammasome (Agostini et al., 2004; Davis et al., 2011; Schroder and Tschopp, 2010). The NLRP3 inflammasome, the most extensively studied one, consists of NLRP3, the apoptosis-associated speck-like protein containing a CARD (ASC) and pro-caspase-1. Processed by the NLRP3 inflammasome, active caspase-1 subsequently promotes the maturation of the pro-inflammatory cytokines IL-1β and IL-18 (Fernandes-Alnemri et al.,2007; Latz et al., 2013; Schroder et al., 2012).
In Caspase-8-deficient bone-marrow-derived dendritic cells (BMDCs), the NLRP3 inflammasome can be activated by lipopolysaccharide (LPS) treatment alone in a RIPK3-dependent manner, which is consistent with the results from cells deficient in XIAP, cIAP1, and cIAP2. Similarly, treatment of bone-marrow-derived macrophages (BMDMs) or BMDCs with Smac mimetics leads to the NLRP3 inflammasome activation, which requires RIPK3 (Kang et al., 2013; Silke et al., 2015; Vince et al., 2012). These results indicate that cIAP or caspase-8 inhibits RIPK3 mediated the NLRP3 inflammasome activation. Nevertheless, recent studies show that Fadd−/−Ripk3−/− and Caspase-8−/−Ripk3−/− BMDMs are unable to activate the NLRP3 inflammasome upon stimulation while activation in Ripk3−/− BMDMs is normal. This result suggests that caspase-8 or FADD, instead of RIPK3, may play an important role in the NLRP3 inflammasome activation (Gurung et al., 2014; Silke et al., 2015). These seemingly contradictory results interest us in investigating the NLRP3 activation in MLKL and FADD double-knockout background.
In this study, we demonstrate that Mlkl deletion rescued embryonic lethality caused by Fadd deficiency. Mlkl−/−Fadd−/− mice survived to adulthood but displayed severer lymphoproliferative disease compared to the Ripk3−/−Fadd−/− mice. Furthermore, we found that the NLRP3 inflammasome activation was inhibited in Mlkl−/−Fadd−/− BMDMs and BMDCs when challenged with LPS or poly(I:C). Thus, our study revealed indispensable roles of MLKL in mediating embryonic lethality of Fadd-deficient mice and suppressing progressive lymphoproliferative disease, more importantly, the potential function of MLKL and FADD in regulating inflammasome activation.
RESULTS
Embryonic Lethality Caused by Fadd Deficiency Is Rescued by Mlkl Deletion
Ablation of Fadd in mice causes embryonic death in mice between E10.5 and E11.5, which is probably due to the massive necrosis (Figure 1A). To evaluate the in vivo function of MLKL in embryonic development, we performed targeted disruption of Mlkl by CRISPR/Cas9 gene mutation system and crossed the Mlkl knockout alleles into Fadd knockout mice (Figures S1A and S1B). As expected, Mlkl deletion rescued embryonic lethality caused by Fadd deficiency. The resulting Mlkl−/−Fadd−/− mice were viable and fertile, and the newborns were weaned at expected frequencies (Figures 1B and 1C). Mlkl−/−Fadd−/− mice were almost indistinguishable from their Mlkl−/− and wild-type littermates in physical appearance and growth (Figures 1B and 1D). The immunoblot results showed undetectable expression of MLKL and FADD in multiple organs of Mlkl−/−Fadd−/− mice (Figures 1E and S1D). Previous studies have reported that the mice with specific deficiency of Fadd or Caspase-8 in epithelial cells resulted in terminal ileitis or chronic intestinal inflammation, which was prevented by the co-deletion of Ripk3 (Welz et al., 2011). Besides, keratinocyte-specific Fadd deficiency promoted RIPK3-dependent necroptosis and skin inflammation in mice (Bonnet et al., 2011). Consistent with these results, Mlkl−/−Fadd−/− mice exhibited no sign of chronic inflammation in skin or intestine when observed for over 5 months (Figures 1B and S1C), suggesting that RIPK3-MLKL-mediated necroptosis in epithelial cells is likely to participate in the pro-inflammatory signaling pathways when caspase-8 or FADD is inhibited. In conclusion, these results demonstrate that MLKL functionally mediates the consequences of Fadd deficiency in vivo.
Figure 1. Embryonic Lethality in Fadd Deficiency Mice Is Rescued by Mlkl Deletion.
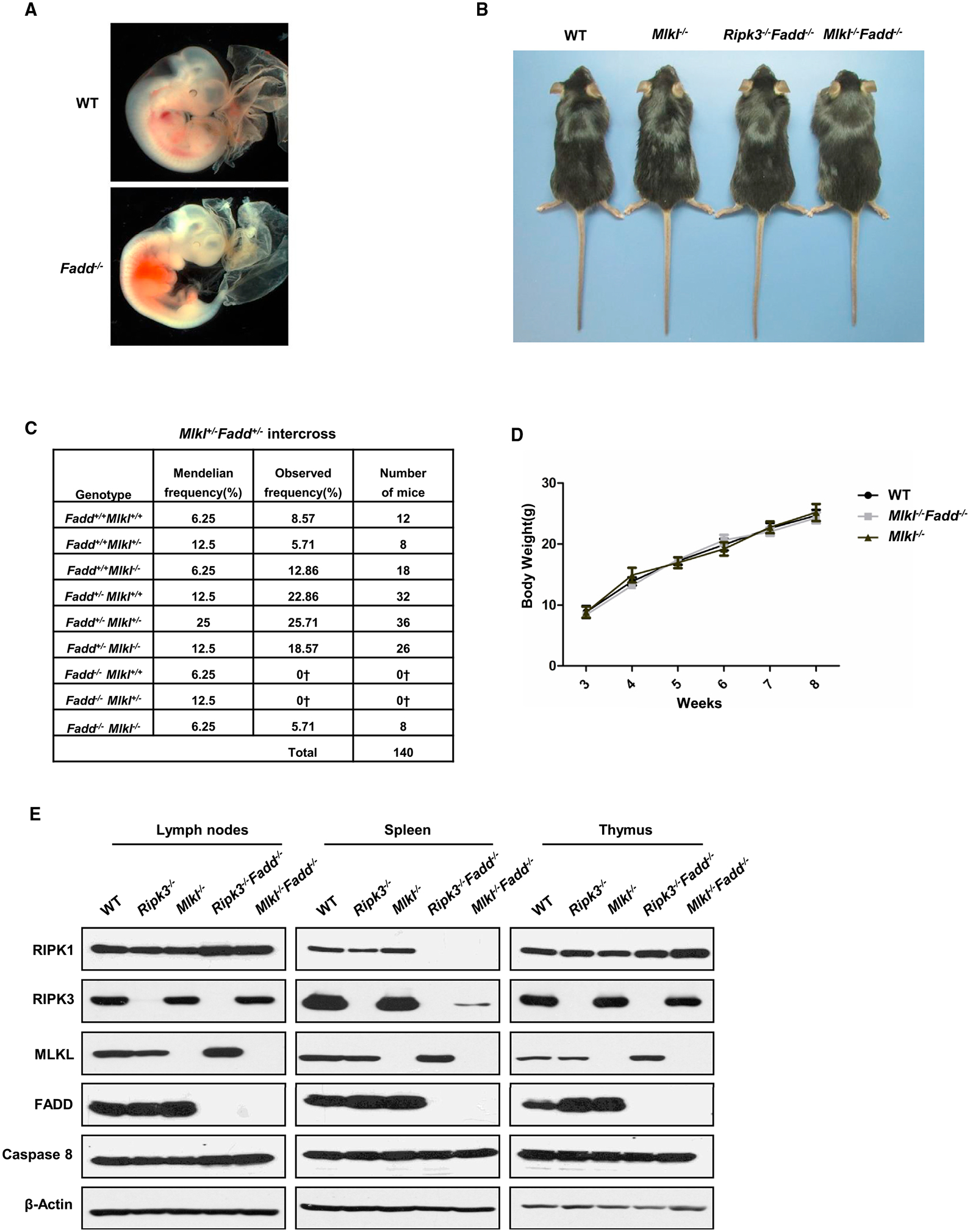
(A) Photographs of embryos of the indicated genotypes at E11.5.
(B) Photographs of 12-week-old mice of the indicated genotypes.
(C) Predicted and observed frequencies of mice born following Mlkl+/−Fadd+/− intercross. †Predicted embryonic lethal.
(D) Plot of weight of littermate wild-type, Mlkl−/−, and Mlkl−/−Fadd−/− mice.
(E) Immunoblot of RIPK1, RIPK3, MLKL, FADD, caspase-8, and β-actin of lymph nodes, spleen, and thymus from mice of the indicated genotypes.
Mlkl−/−Fadd−/− Mice Are Resistant to DR-Induced Apoptosis and Necroptosis
Cell death mostly initiates through the ligation of DRs and the signaling is relayed through a series of protein-protein interactions. To verify the disrupted functions of FADD and MLKL in Mlkl−/−Fadd−/− mice, we tested several types of cells derived from wild-type (WT) and double-knockout (DKO) mice for their sensitivities to DR-induced cell death. As expected, WT mouse dermal fibroblasts (MDFs) had remarkable loss in response to the combined treatment with TNF and the cIAP inhibitor Smac mimetic. In the presence of caspase inhibitor zVAD-fmk, almost half of the WT MDFs underwent necroptosis which was inhibited by Necrostatin-1 (Figure 2A). Thymocytes were killed by treatment with anti-Fas antibodies (Figure 2B). In addition, BMDMs from WT mice were sensitive to LPS or Poly(I:C)-induced necroptosis (Figure 2C). In contrast to WT cells, cells from Mlkl−/−Fadd−/− mice were completely resistant to the extrinsic apoptosis and necroptosis triggered by the stimuli described above (Figures 2A–2C). Furthermore, Mlkl−/−Fadd−/− mice were resistant to Fas-induced lethal hepatitis and survived for over 24 hr (Figure 2D) with normal liver architecture (Figure 2E). Together, these results show that FADD-dependent extrinsic apoptosis and MLKL-dependent necroptosis are blocked in Mlkl−/−Fadd−/− cells, indicating that the functions of FADD and MLKL are eliminated in Mlkl−/− Fadd−/− mice.
Figure 2. Sensitivities to DR-Induced Apoptosis and Necroptosis Both In Vitro and In Vivo.
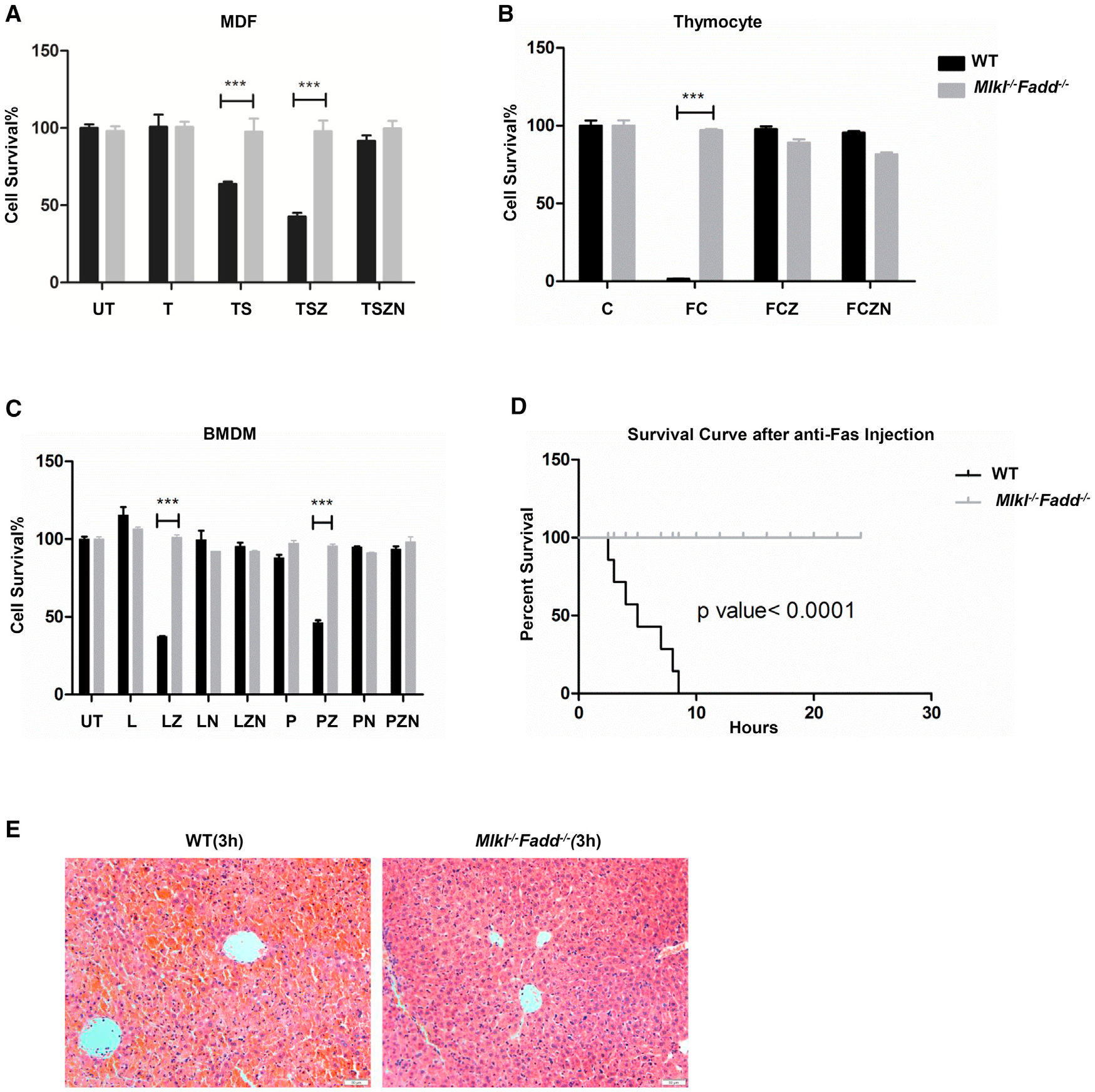
(A) Wild-type and littermate Mlkl−/−Fadd−/− MDFs were treated with mouse TNF-α (30 ng/ml), TNF-α +Smac mimetic (100 nM), TNF-α+Smac mimetic+zVAD (20 μM), and TNF-α+ Smac mimetic+zVAD+Necrostatin1(30 μM), respectively.
(B) Thymuses from wild-type and littermate Mlkl−/−Fadd−/− mice were milled and filtered into single cells. Thymocytes were treated with CHX (30 μg/ml), Fas ligand (100 ng/ml)+CHX, Fas ligand+CHX+zVAD (20 μM), and Fas ligand+CHX+zVAD+ Necrostatin1 (30 μM), respectively.
(C) Bone marrow from wild-type and littermate Mlkl−/−Fadd−/− mice were induced by M-CSF (50 ng/ml) for 1 week. BMDMs from indicated genotypes were challenged with LPS (100 ng/ml), LPS+zVAD (20 μM), LPS+zVAD+Necrostatin1 (30 μM), Poly(I:C) (100 μg/ml), and Poly(I:C)+z VAD, Poly(I:C)+z VAD+Necrostatin-1, respectively.
(A–C) Cell viability was determined using the CellTiter-Glo kit. The data are represented as the mean ± SD of triplicate wells. ***p < 0.001(Student’s t test). Abbreviations are as follows: UT, untreated; T, TNF-α; S, Smac mimetic; Z, zVAD; N, necrostatin. F, Fas ligand; C, CHX; L, LPS; P, Poly(I:C).
(D) Kaplan-Meier survival plot of 8-week-old wild-type (n = 7) and Mlkl−/−Fadd−/− (n = 7) mice injected intraperitoneally with 12.5 μg of anti-Fas Jo-2 antibody.
(E) Histology of liver sections from wild-type (left panel) and Mlkl−/−Fadd−/− (right panel) mice 3 hr after Jo-2 antibody injection. Scale bars, 50 μm.
Mlkl−/−Fadd−/− Mice Display More Severe Progressive Lymphoproliferative Disease Compared to Ripk3−/−Fadd−/− Mice
In all settings where Caspase-8 or Fadd deficiency has been rescued by elimination of Ripk3 or the kinase activity of RIPK3, resulting viable mice exhibit lymphoid hyperplasia as they age (Dillon et al., 2012; Kaiser et al., 2011; Newton et al., 2014; Oberst et al., 2011). Similar to Ripk3−/−Fadd−/− or Ripk3−/−Caspase-8−/− mice, Mlkl−/−Fadd−/− mice displayed swollen spleen and lymph nodes leading to a systematic lymphoproliferative disease over time but normal T cell population in thymus (Figures 3A, S2A, and S2C). Histology staining indicated excessive cell accumulation and abnormal structure in spleen and lymph nodes from Mlkl−/−Fadd−/− mice (Figures 3B and S2B). Cells accumulated in spleen and lymph nodes were shown to be a population of B220+CD3+ T lymphocytes (Figure 3C). Surprisingly, we observed that Mlkl−/−Fadd−/− mice showed more severe lymphadenopathy and splenomegaly than Ripk3−/−Fadd−/− mice did (Figures 3A, 3B, S2A, and S2B). Consistent with these phenotypes, a larger population of B220+CD3+ T lymphocytes was accumulated in peripheral lymphoid organs (especially in lymph nodes) of Mlkl−/−Fadd−/− mice in comparison with contemporary Ripk3−/−Fadd−/−mice (Figure 3C). It seems that the remarkable distinction, comparing to Ripk3−/−Fadd−/− mice, should owe to the praecox lymphoaccumulation (started around 9 weeks old, data not shown) and more rapid development of the disease in Mlkl−/−Fadd−/− mice. Differences in organ sizes and B220+CD3+ T lymphocytes percentage between two types of DKO mice became less significant over time (Figures 3A, 3C, and S2A). Aging mice to about 16 weeks old, we found enlarged parathymic lymph nodes emerged beside the thymus (it appeared earlier in Mlkl−/−Fadd−/− mice than in Ripk3−/−Fadd−/− mice), which were confirmed to be accumulations of B220+CD3+ T lymphocytes similar to those shown in spleen and other lymph nodes (Figures 3A, S2A, and S2C). The faster accumulation of B220+CD3+ T lymphocytes we observed in Mlkl−/−Fadd−/− mice indicated a divergence between these two genotypes in cell proliferation. To test our hypothesis, we checked the cell proliferation in peripheral lymph organs using Ki67 staining. Compared with Ripk3 Fadd−/− mice Fadd−/− mice and WT controls, more Ki67 positive cells were detected in peripheral lymph organs of Mlkl−/−Fadd−/− mice, indicating more active cell proliferation (Figure S2D). Thus, our data implicate that hyperproliferation of B220+CD3+ T lymphocytes contributes to more severe lymphoproliferative disease in Mlkl−/−Fadd−/−mice than in Ripk3−/−Fadd−/− mice.
Figure 3. Mlkl−/−Fadd−/− Mice Display More Severe Systemic Lymphoproliferative Disease Than Ripk3−/−Fadd−/− Mice.
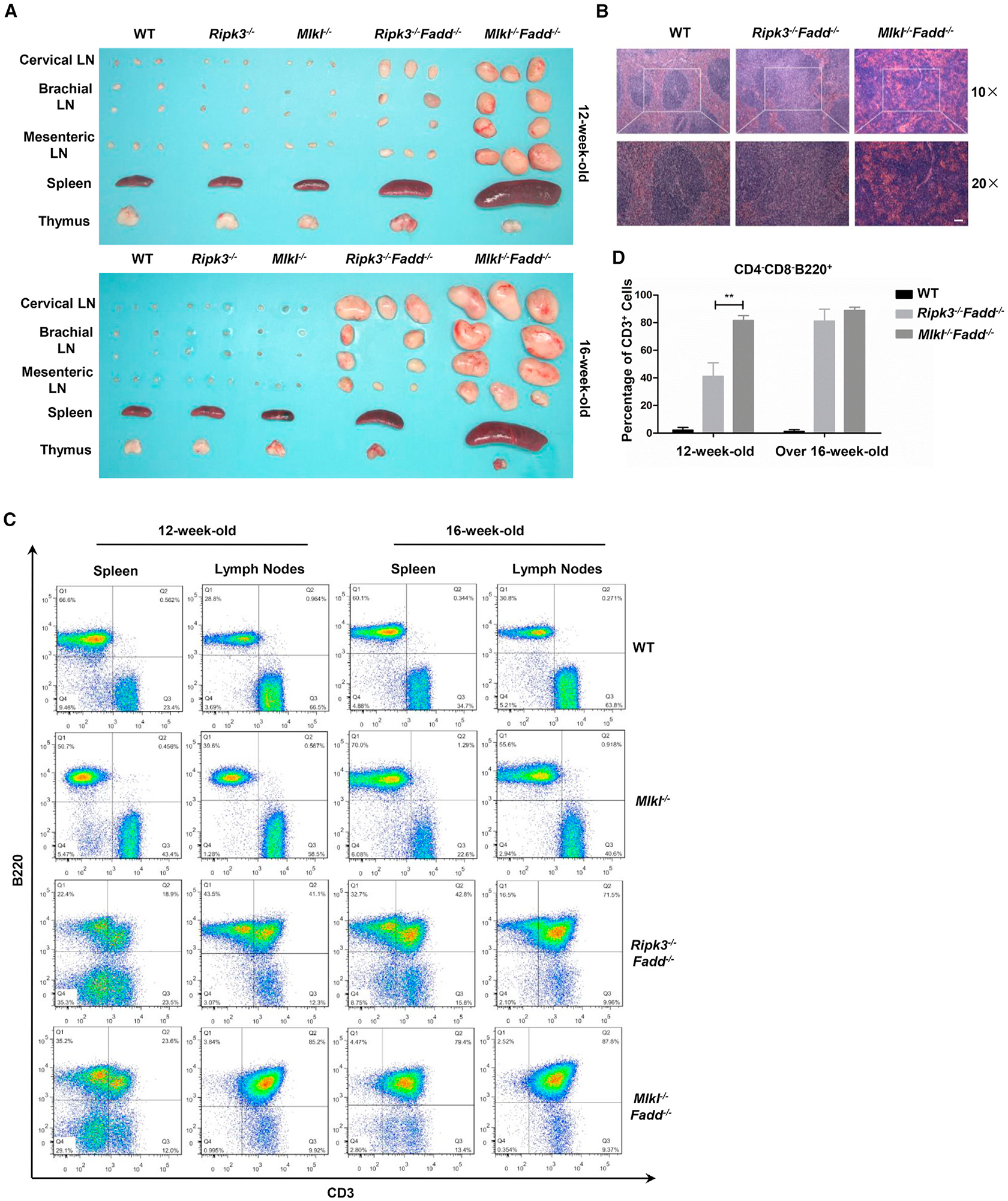
(A) Lymph nodes, spleens, and thymus removed from 12- and 16-week-old mice of indicated genotypes.
(B) H&E-stained sections of fixed spleen from mice of indicated genotypes. Scale bars, 50 μm
(C) Percentage of B220+CD3+ cells from spleen and lymph nodes in 12- mice or 16-week-old mice of the indicated genotypes.
(D) Percentage of CD3+ cells in CD4−CD8−B220+ populations from lymph nodes of mice with indicated genotypes and ages. Error bars, SD for three wild-type, three Ripk3−/−Fadd−/− and three Mlkl−/−Fadd−/− mice.
Mlkl−/−Fadd−/− BMDMs Are Impaired in the NLRP3 Inflammasome Activation When Challenged with LPS or Poly(I:C)
Inflammasomes are large intracellular multiprotein complexes that play a central role in innate immunity. Recent studies have provided some evidence that FADD, RIPK3, and MLKL may be involved in regulating the NLRP3 inflammasome activation (Gurung et al., 2014; Kang et al., 2013, 2015; Lawlor et al., 2015; Silke et al., 2015; Vince et al., 2012). To investigate the function of these proteins in the NLRP3 inflammasome activation, we challenged Ripk3−/−Fadd−/−, Mlkl−/−Fadd−/−, Ripk3−/−, and Mlkl−/− BMDMs with LPS for priming and subsequently stimulated them with ATP to activate the canonical NLRP3 inflammasome. In comparison with Ripk3−/− and Mlkl−/− BMDMs, caspase-1 processing was significantly reduced in Ripk3−/−Fadd−/− and Mlkl−/−Fadd−/− BMDMs (Figure 4A). Consistently, IL-1β secretion was significantly decreased in Ripk3−/− Fadd−/− and Mlkl−/−Fadd−/− BMDMs (Figure 4B). These BMDMs were also primed with poly(I:C) and treated with ATP for the inflammasome activation. Similar to the aforementioned results, Mlkl−/−Fadd−/−BMDMs had little response to the treatment while the WT BMDMs had normal inflammasome activation (Figures 4C and 4D). Furthermore, the similar set of experiments was also performed in BMDCs. Mlkl−/−Fadd−/− BMDCs were defective in NLRP3 inflammasome activation as well (Figures S4B and S4C). Previous studies have revealed that FADD and caspase-8 were apical mediators of canonical and noncanonical NLRP3 inflammasome activation (Kang et al., 2013; Silke et al., 2015; Vince et al., 2012; Gurung et al., 2014). To investigate the cause of the defect in DKO BMDMs, we examined the NLRP3 inflammasome activation in BMDMs derived from LysM-cre-mediated cell-specific Fadd deletion mice, and the deletion efficiency of FADD was tested by immunoblot (Figure S4A). The results showed that LPS or poly(I:C) primed BMDMs with low FADD expression had normal caspase-1 processing and IL-1b secretion upon stimulation as WT control (Figures 4E and 4F).
Figure 4. Activation of Canonical NLRP3 Inflammasome in BMDMs.
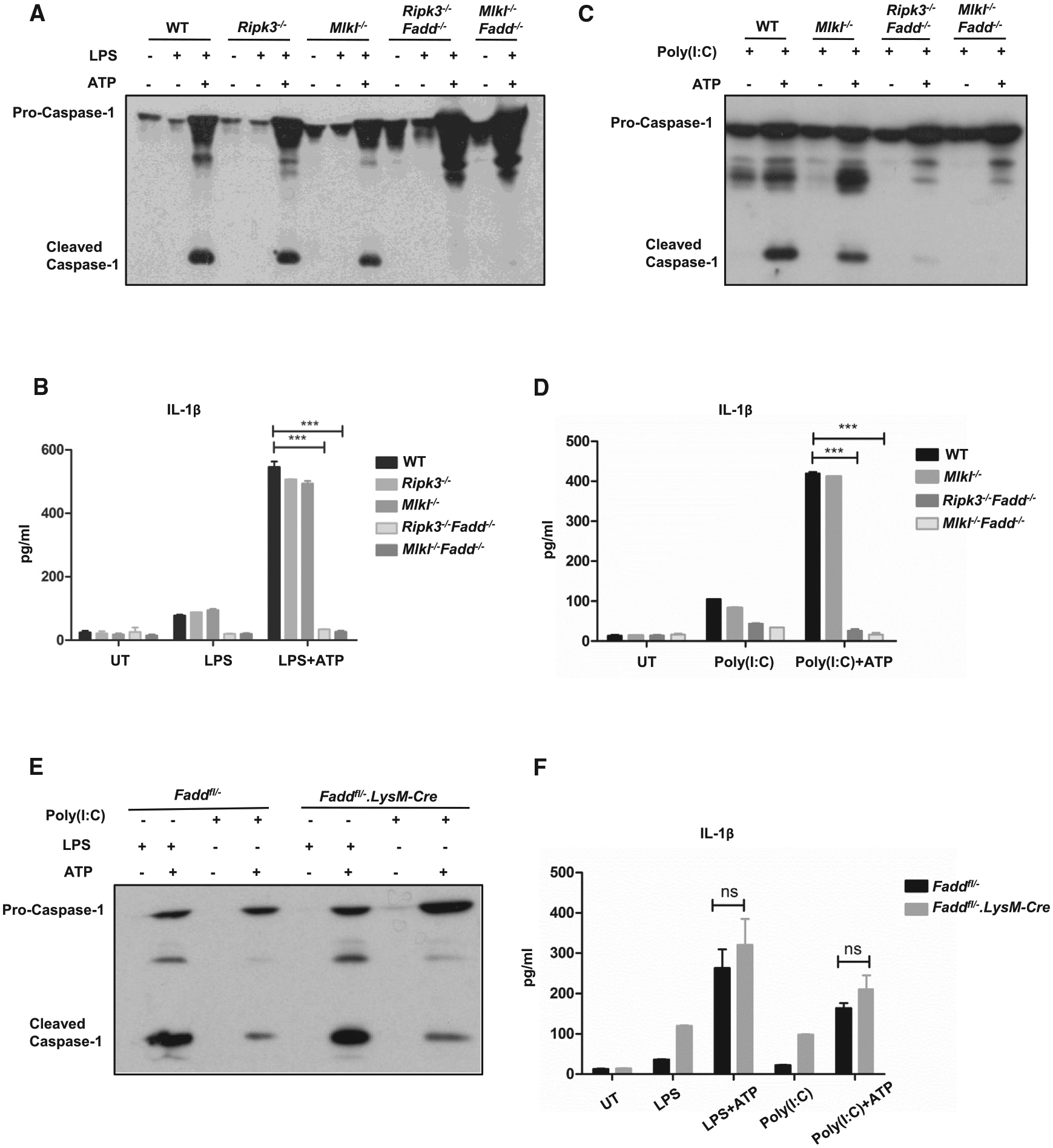
(A and C) Wild-type, Ripk3−/−, Mlkl−/−, Ripk3−/−Fadd−/−, and Mlkl−/−Fadd−/− BMDMs were primed with (A) 20 ng/ml LPS or (C) 100 mg/ml Poly(I:C) for 5 hr prior to 30-min treatment of 5 mM ATP. Supernatants were analyzed by immunoblot for caspase-1 activation.
(B and D) Wild-type, Ripk3−/−, Mlkl−/−, Ripk3−/−Fadd−/−, and Mlkl−/−Fadd−/− BMDMs were stimulated with (B) 20 ng/ml LPS or (D) 100 μg/ml Poly(I:C) for 12 hr prior to 15-min treatment of 5 mM ATP. Supernatants were analyzed by ELISA for IL-1b secretion.
(E) Wild-type and FADD insufficient BMDMs were primed with 20 ng/ml LPS or (C) 100 μg/ml Poly(I:C) for 5 hr prior to 30-min treatment of 5 mM ATP. Supernatants were analyzed by immunoblot for caspase-1 activation and (F) were analyzed by ELISA for IL-1β secretion. ***p < 0.001 (Student’s t test). n.s., not significant.
In conclusion, in contrast to the Ripk3−/−, Mlkl−/− and FADD insufficient BMDMs, Ripk3−/−Fadd−/− and Mlkl−/−Fadd−/− BMDMs or BMDCs have impaired canonical NLRP3 inflammasome activation when challenged with LPS or Poly(I:C). Our data suggest that MLKL is likely to function together with FADD to co-regulate the canonical NLRP3 inflammasome activation engaged through TLR3 or TLR4.
NLRP3 Inflammasome Activation Failure in Mlkl−/−Fadd−/− BMDMs Is Due to Defects of ASC Speck Formation and NF-κB-Dependent NLRP3 Transcription
Formation of NLRP3 inflammasome requires the pyrin domain (PYD) of NLRP3 to recruit the adaptor protein ASC through PYD-PYD interaction. This interaction further facilitates the recruitment of pro-caspase-1 to assemble the inflammasome complex which is necessary for caspase-1 activation (Fernandes-Alnemri et al., 2007; Sutterwala et al., 2014).
To investigate whether the impaired inflammasome activation in Mlkl−/−Fadd−/− or Ripk3−/−Fadd−/− BMDMs was due to a defect in the complex formation, we analyzed the ASC polymerization after LPS stimulation in Mlkl−/−Fadd−/−, Ripk3−/−Fadd−/− BMDMs, and controls. As expected, in contrast to controls, Mlkl−/−Fadd−/− and Ripk3−/−Fadd−/− BMDMs had significantly reduced ASC polymerization in both the pellet and the supernatant of cell lysates (Figure 5A). The same defects were shown in Mlkl−/−Fadd−/− and Ripk3−/−Fadd−/− BMDCs (Figure S4D). In line with the results above, significantly fewer ASC specks formed in Mlkl−/−Fadd−/− BMDMs than in WT BMDMs (Figures 5B and 5C), suggesting that MLKL and FADD may mediate the NLRP3 inflammasome activation through regulating ASC speck formation.
Figure 5. ASC Polymerization in BMDMs upon LPS Stimulation.
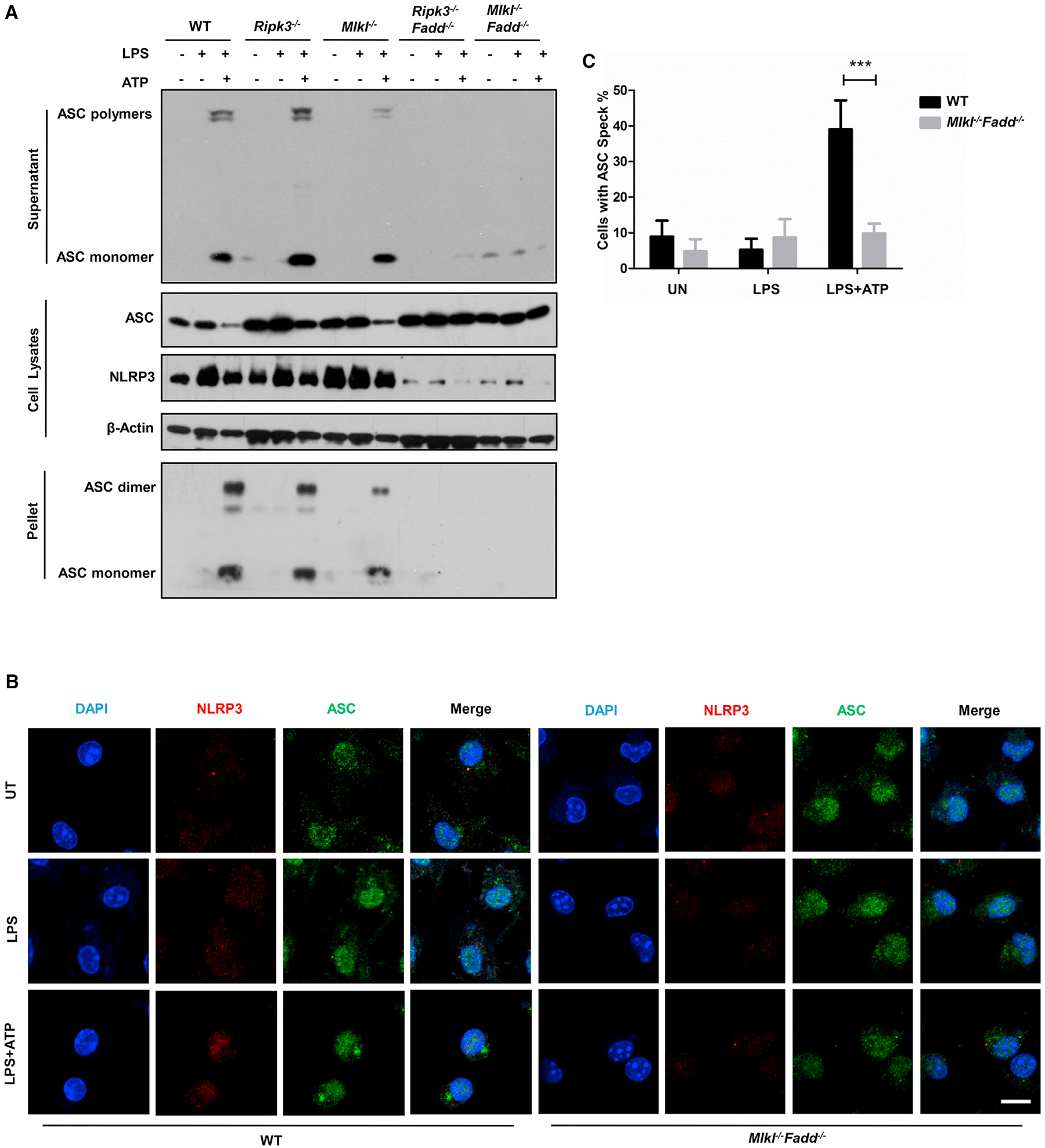
(A) Wild-type, Ripk3−/−, Mlkl−/−, Ripk3−/−Fadd−/−, and Mlkl−/−Fadd−/− BMDMs or for 5 hr prior to 30-min treatment of 5 mM ATP. Supernatants and cell pellets were analyzed by immunoblot for ASC polymerization. Cell lysates were analyzed by immunoblot for the expression of ASC and NLRP3.
(B) Immunofluorescence microscopy of LPS-primed wild-type Mlkl−/−Fadd−/− BMDMs without or with 5 mM ATP treatment, top (untreated), middle (LPS primed), and bottom (ATP stimulation after LPS priming). Cells were fixed and immunostained for ASC (green) and NLRP3 (red). DAPI (blue) was used for nuclear staining. Scale bars, 5 mm.
(C) Quantification of results from Figure 4B. Total cells and cells with an ASC polymer (speck) were counted, and the percentages of cells with ASC polymer were calculated. The data are represented as the mean ± SD of five microscopic fields per group.
As the key component of the inflammasome complex, NLRP3 can be upregulated by the priming signal. Thus, we examined both NLRP3 protein level and mRNA level by immunoblot and qRT-PCR, respectively. In Mlkl−/−Fadd−/− and Ripk3−/−Fadd−/− BMDMs treated with LPS, NLRP3 can hardly respond to the priming signal compared with WT control while ASC had normal transcription and expression levels in all BMDMs (Figures 5A, 6A, and 6C). Besides, pro-IL1β, the substrate of the activated inflammasome, was unable to be upregu lated in Mlkl−/−Fadd−/− and Ripk3−/−Fadd−/− BMDMs either (Figure 6B).
Figure 6. NF-kB Activation in BMDMs upon LPS Stimulation.
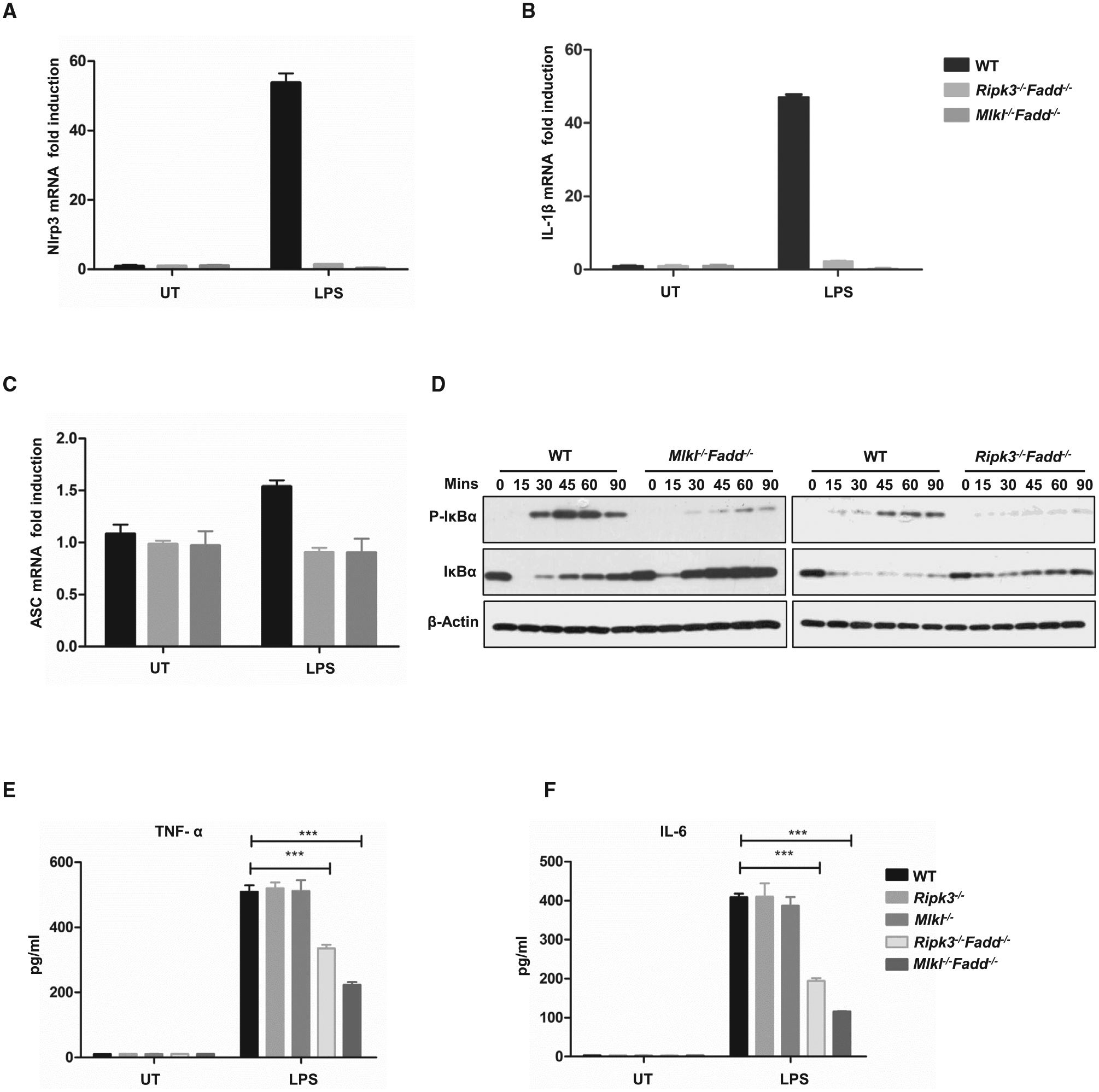
(A–C) Wild-type, Ripk3−/−Fadd−/−, and Mlkl−/−Fadd−/− BMDMs were treated with 20 ng/ml LPS for 6 hr, and mRNA levels of NLRP3 (A), IL-1β (B), and ASC (C) were determined by qPCR.
(D)Wild-type and Mlkl−/−Fadd−/− BMDMs were treated with 100 ng/ml LPS for 15, 30, 60, and 90 min, respectively. The expression of indicated the proteins were detected by western blot.
(E and F) Wild-type, Ripk3−/−, Mlkl−/−, Ripk3−/−Fadd−/−, and Mlkl−/−Fadd−/− BMDMs were treated with 20 ng/ml LPS for 12 hr. Supernatants were collected for the detections of TNF-α and IL-6 by ELISA.
Given that NLRP3 and pro-IL-1βare transcriptionally regulated by NF-κB, we therefore examined NF-κB signals following LPS stimulation in Mlkl−/−Fadd−/− and wild-type BMDMs by immunoblots (Bauernfeind et al., 2009; Schroder et al., 2012; Sutterwala et al., 2014). We found that NF-κB activation was significantly decreased associated with less phosphorylation and degradation of IκB in both Mlkl−/−Fadd−/− and Ripk3−/−Fadd−/− BMDMs than in WT BMDMs (Figure 6D). Furthermore, we examined the secretion levels of TNF-α and IL-6, which are the target genes of NF-κB. It was shown that Mlkl−/−Fadd−/− BMDMs had notable reduction of TNF-α and IL-6 levels in comparison to WT controls (Figures 6E and 6F).
In summary, our findings suggest that the NLRP3 inflammasome formation is insensitive to LPS/ATP stimulation in Mlkl−/−Fadd−/− and Ripk3−/−Fadd−/− BMDMs. This is likely due to the defects in NF-κB-dependent Nlrp3 and pro-IL-1β transcriptions.Consequently, the lack of NLRP3 and pro-IL-1β expression renders the Mlkl−/−Fadd−/− and Ripk3−/−Fadd−/− BMDMs incapable of forming ASC speck and recruiting other components to assemble the NLRP3 inflammasome complex, which results in the failure of caspase-1 processing and mature IL-1β secretion.
DISCUSSION
The findings of our study provide genetic evidence that MLKL mediates the embryonic lethality of Fadd deficiency in vivo. Combined with previous studies (Dillon et al., 2012; Kaiser et al., 2011; Oberst et al., 2011; Varfolomeev et al., 1998; Yeh et al., 1998), we speculate that, during the embryonic development, RIPK3-MLKL-mediated necroptotic signaling is tightly regulated by the caspase-8-FADD-cFLIP complex although the function of MLKL in the condition of Caspase-8 genetic ablation remains to be identified. Several studies have intimated that Fadd or Caspase-8 deficiency induced RIPK3-dependent pathway may target the yolk sac vasculature and hematopoietic cells during embryogenesis. However, direct evidence showing that embryonic lethality of Fadd-deficient mice is caused by RIPK3-MLKL-mediating necroptosis must still be obtained.
Considering that the CRISPR/Cas9 system could introduce off-target effects in the Mlkl knockout mice, we confirmed the phenotypes using another Mlkl knockout mouse line that was generated by an independent single guide (sg)RNA (Figures S3A and S3B). The two independent lines of Mlkl knockout mice were individually crossed into the same line of Fadd-deficient mice, and the two line-ages displayed equivalent properties, excluding the possibility that off-target effects contributed to the phenotypes of Mlkl−/−Fadd−/− mice (Figure S3C).
Previous studies have demonstrated that FASL-FAS signaling kills unwanted cells through FADD/caspase-8-mediated apoptosis during the development of lymphocytes. Conditional deletion of Fadd or Caspase-8 in T cell results in proliferation defects in response to activation signals (Zhang et al., 1998; Kabra et al., 2001; Salmena et al., 2003; Ch’en et al., 2008), while combined deletion of either Ripk3 or Mlkl and Fadd in an intact germline leads to accumulation of abnormal CD3+B220+ T cells. Given that MLKL works downstream of RIPK3, we hypothesize that the lack of MLKL may block other possible undefined cell signaling pathways independent of RIPK3, resulting in the praecox accumulation of CD3+B220+ T cells in spleen and lymph nodes in Mlkl−/−Fadd−/− mice. Another possibility is that more cell proliferation occurring in Mlkl−/−Fadd−/− mice leads to severer lymphoproliferative disease. However, the precise mechanisms behind the interesting phenotypes remain to be explored.
A high dose of LPS results in endotoxemia in mice and subsequently leads to the massive systemic release of pro-inflammatory cytokines. When we challenged Mlkl−/−Fadd−/− mice with LPS to assess the capacity of initiating innate immunity, we observed a dramatic reduction of cytokine release in Mlkl−/−Fadd−/− mice (Figures 7A–7C). We speculate that the reduced cytokine secretion was due to impaired activation of the NLRP3 inflammasome and NF-kB signaling as shown in the previous investigations in Ripk3−/−Fadd−/− mice and Ripk3−/−Casp8−/− mice (Gurung et al., 2014; Kang et al., 2013). The defects in both DKO mice raise an important issue about how the NF-κB signaling in DKO BMDMs is regulated. Although conditional deletion of Fadd in BMDMs have negligible effects on the activation of inflammasome upon the stimulation of LPS or Poly(I:C) as Mlkl−/− or Rip3−/− BMDMs (Figures 4E and 4F), it remains likely that FADD alone determines the NLRP3 inflammasome activation due to its incomplete deletion (Figure S4A). Thus, we hypothesized that RIPK3/MLKL and FADD/caspase-8 may co-regulate a target or targets that mediate NF-κB signaling upon LPS or Poly(I:C) stimulation. Nevertheless, the functions of these proteins in regulating the targets need to be clarified by sophisticated biochemical studies, and their functions in activating noncanonical NLRP3 inflammasome and other types of inflammasomes should be further investigated.
Figure 7. Decreasing Cytokine Secretion in Mlkl−/−Fadd−/− Mice.
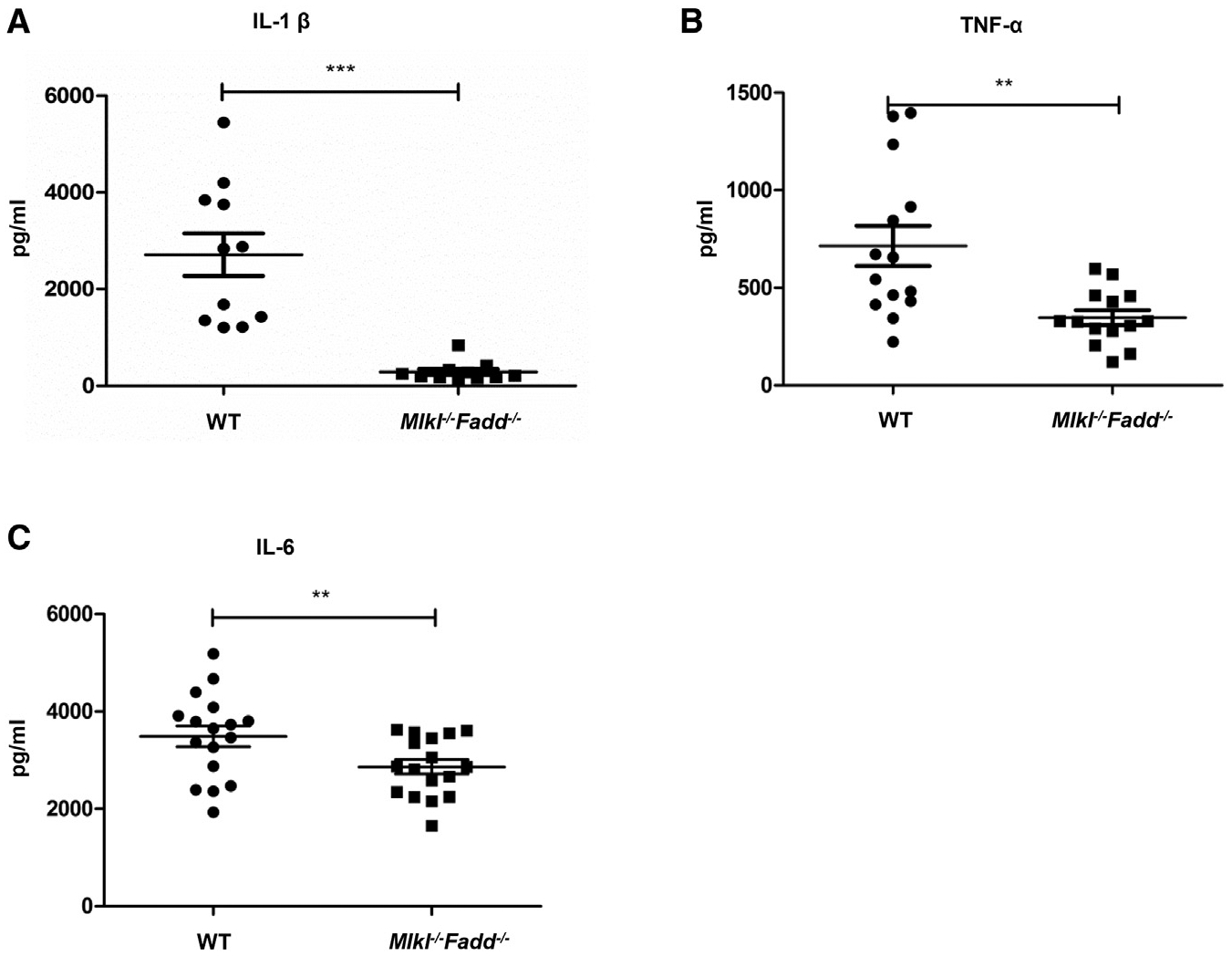
(A–C) Wild-type and Mlkl−/−Fadd−/− mice were challenged with LPS (i.p., 54 mg/kg), and the productions of serum IL-1β (A), TNF-α (B), and IL-6 (C) were detected by ELISA 12 hr later. Each dot represents one mouse.
The findings that FADD restricted MLKL-dependent pathway during embryonic development and in maintaining the homeostasis of T cell suggest the importance of the interplay between FADD-dependent apoptosis and MLKL-dependent necroptosis. More importantly, the key components of the two cell death pathways may function as regulators in other situations as we have observed in stimulating BMDMs or BMDCs. In this light, it is noteworthy that our findings here point out MLKL and FADD, which are multifunctional in regulating cell death and innate immune response, have broad implications for the related investigations as well as approaches to therapies of cancer and immune disorders.
EXPERIMENTAL PROCEDURES
Reagents
LPS, ATP, and PolyI:C were obtained from Sigma. TNF-a and z-VAD were from R&D Systems and Calbiochem, respectively. Necrostatin-1 was purchased from Enzo Life Sciences. The following antibodies were used for western blotting and immunofluorescence experiments: RIPK1 (BD Biosciences), RIPK3 (Prosci), IκBα and p- IkBa (Cell Signaling Technology), ASC (Santa Cruz Biotechnology), IL-1β (Santa Cruz), mouse caspase-1 (AdipoGen), NLRP3 (R&D). MLKL antibody was a gift from Dr. Jiahuai Han (Xiamen University). FADD antibody was from Dr. Jianke Zhang (Thomas Jefferson University). Mouse TNF-α, IL-6, and IL-1β ELISA Kits were from MultiSciences. Cell viability was determined by measuring ATP levels using the Cell Titer-Glo kit (Promega).
Mice
Mice were housed in a specific pathogen-free (SPF) facility. Ripk3 knockout mice were provided by Dr. Xiaodong Wang (NIBS, Beijing, China), and Fadd−/−Tg mice were provided by Dr. Jianke Zhang (Thomas Jefferson University). Mlkl knockout mice were generated by CRISPR-Cas9 mutation system (Bioray Laboratories). A 301-bp deletion in the Mlkl gene was introduced in the Mlkl locus, and genotyping results of mice are presented in Figures S1A and S1B. Additional information is provided upon request. Animal experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (CAS).
Cell Survival Assay
Cell survival was determined using the CellTiter-Glo Luminescent Cell Viability Assay kit, and the luminescence was recorded with a microplate luminometer (Thermo Scientific).
Isolation of Mouse BMDMs and Inflammasome Activation
Bone marrow cells were collected from mouse femurs and tibias and were cultured for 7 days in RPMI medium containing 10% fetal bovine serum (FBS), penicillin, streptomycin, and 50 ng/ml M-CSF. Medium was changed every 2 days. BMDMs were induced to differentiate in vitro from isolated bone marrow cells. For inflammasome assays, BMDMs were pretreated with LPS or Poly(I:C) for 5 hr before stimulated with ATP (5 mM) for 30 min. The media supernatant and cell lysates were collected for precipitation and immunoblot analysis.
ASC Oligomerization Detection
ASC pyroptosomes were detected as previously reported (Juliana et al., 2010). Macrophages were seeded in 6-well plates (2 × 106 cells per well) and treated with different stimuli. The cells were pelleted by centrifugation and resuspended in 0.5 ml of ice-cold buffer containing 20 mM HEPES-KOH (pH 7.5), 150 mM KCl, 1% Nonidet P-40, 0.1 mM PMSF, and a protease inhibitor mixture. Cell lysates were centrifuged at 5,000 × g for 10 min at 4°C, and pellets were washed twice with PBS before being resuspended in 500 μl of PBS. The pellets were crosslinked with fresh DSS (4 mM) for 30 min and pelleted by centrifugation at 5,000 × g for 10 min. Crosslinked pellets were resuspended in 30 μl of SDS sample buffer and subjected to western blot with the anti-mouse ASC antibodies.
Statistical Analysis
Data presented in this article are representative results of at least three independent experiments. The in vitro results were presented as the mean ± SD of triplicate wells. The statistical significance of data was evaluated by Student’s t test in which p < 0.01 was considered significant and p < 0.001 was highly significant. The statistical calculations were performed with GraphPad Prism software.
Supplementary Material
Highlights.
Ablation of Mlkl rescues embryonic lethality in Fadd-deficient mice
Mlkl−/−Fadd−/− mice show severe lymphoproliferative disease
Mlkl−/−Fadd−/− BMDMs and BMDCs have impaired NLRP3 inflammasome activation
MLKL and FADD regulate ASC speck formation and NF-κB-dependent NLRP3 transcription
ACKNOWLEDGMENTS
We thank Dr. Xiaodong Wang (National Institute of Biological Sciences) for providing RIPK3−/− mice and Dr. Jiahuai Han (Xiamen University) for providing mouse MLKL antibody. We thank Dr. Yu Sun (UCLA) for editing the manuscript and helpful discussion. We also thank Animal Facility of Institute for Nutritional Sciences for mouse care. This work was supported by grants from the National Natural Science Foundation of China (31571426 and 81502548). Haibing Zhang was supported by Thousand Young Talents Program of the Chinese government.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.06.103.
REFERENCES
- Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, and Tschopp J (2004). NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325. [DOI] [PubMed] [Google Scholar]
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, et al. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol 183, 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, and Wallach D (1996). Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1-and TNF receptor-induced cell death. Cell 85, 803–815. [DOI] [PubMed] [Google Scholar]
- Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, Haase I, and Pasparakis M (2011). The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 35, 572–582. [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, and Liu ZG (2014). Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol 16, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch’en IL, Beisner DR, Degterev A, Lynch C, Yuan J, Hoffmann A, and Hedrick SM (2008). Antigen-mediated T cell expansion regulated by parallel pathways of death. Proc. Natl. Acad. Sci. USA 105, 17463–17468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, and Han J (2014). Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 24, 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BK, Wen H, and Ting JPY (2011). The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol 29, 707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, and Green DR (2012). Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 1, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, et al. (2014). RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, et al. (2014). MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7, 971–981. [DOI] [PubMed] [Google Scholar]
- Dowling JP, Nair A, and Zhang J (2015). A novel function of RIP1 in postnatal development and immune homeostasis by protecting against RIP3-dependent necroptosis and FADD-mediated apoptosis. Front. Cell Dev. Biol 3, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, and Alnemri ES (2007). The pyroptosome: a supra-molecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung P, Anand PK, Malireddi RKS, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, and Kanneganti TD (2014). FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol 192, 1835–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, Meng R, Quong AA, Latz E, Scott CP, and Alnemri ES (2010). Anti-inflammatory compounds parthenolide and Bay 11–7082 are direct inhibitors of the inflammasome. J. Biol. Chem 285, 9792–9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabra NH, Kang C, Hsing LC, Zhang J, and Winoto A (2001). T cell-specific FADD-deficient mice: FADD is required for early T cell development. Proc. Natl. Acad. Sci. USA 98, 6307–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, and Mocarski ES (2011). RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH, et al. (2014). RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. USA 111, 7753–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TB, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A, Waisman A, Brenner O, Haffner R, Gustafsson E, et al. (2004). Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol 173, 2976–2984. [DOI] [PubMed] [Google Scholar]
- Kang TB, Oh GS, Scandella E, Bolinger B, Ludewig B, Kovalenko A, and Wallach D (2008). Mutation of a self-processing site in caspase-8 compromises its apoptotic but not its nonapoptotic functions in bacterial artificial chromosome-transgenic mice. J. Immunol 181, 2522–2532. [DOI] [PubMed] [Google Scholar]
- Kang TB, Yang SH, Toth B, Kovalenko A, and Wallach D (2013). Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38, 27–40. [DOI] [PubMed] [Google Scholar]
- Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, Roback L, Kaiser W, Oberst A, Sagara J, et al. (2015). Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun 6, 7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, and Leder P (1998). The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8, 297–303. [DOI] [PubMed] [Google Scholar]
- Latz E, Xiao TS, and Stutz A (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, et al. (2015). RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun 6, 6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et al. (2012). The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. (1996). FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death–inducing signaling complex. Cell 85, 817–827. [DOI] [PubMed] [Google Scholar]
- Nagata S (1997). Apoptosis by death factor. Cell 88, 355–365. [DOI] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, et al. (2014). Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360. [DOI] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, and Green DR (2011). Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, Albert ML, Green DR, and Oberst A (2014). RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 21, 1511–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, et al. (2014). RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188. [DOI] [PubMed] [Google Scholar]
- Sakamaki K, Inoue T, Asano M, Sudo K, Kazama H, Sakagami J, Sakata S, Ozaki M, Nakamura S, Toyokuni S, et al. (2002). Ex vivo whole-embryo culture of caspase-8-deficient embryos normalize their aberrant phenotypes in the developing neural tube and heart. Cell Death Differ. 9, 1196–1206. [DOI] [PubMed] [Google Scholar]
- Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au PYB, Berry DM, Tamblyn L, Shehabeldin A, Migon E, et al. (2003). Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 17, 883–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, and Tschopp J (2010). The inflammasomes. Cell 140, 821–832. [DOI] [PubMed] [Google Scholar]
- Schroder K, Sagulenko V, Zamoshnikova A, Richards AA, Cridland JA, Irvine KM, Stacey KJ, and Sweet MJ (2012). Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 217, 1325–1329. [DOI] [PubMed] [Google Scholar]
- Sedger LM, Katewa A, Pettersen AK, Osvath SR, Farrell GC, Stewart GJ, Bendall LJ, and Alexander SI (2010). Extreme lymphoproliferative disease and fatal autoimmune thrombocytopenia in FasL and TRAIL double-deficient mice. Blood 115, 3258–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silke J, Rickard JA, and Gerlic M (2015). The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol 16, 689–697. [DOI] [PubMed] [Google Scholar]
- Su L, Quade B, Wang H, Sun L, Wang X, and Rizo J (2014). A plug release mechanism for membrane permeation by MLKL. Structure 22, 1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, and Wang X (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Haasken S, and Cassel SL (2014). Mechanism of NLRP3 inflammasome activation. Ann. N Y Acad. Sci 1319, 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, et al. (1998). Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9, 267–276. [DOI] [PubMed] [Google Scholar]
- Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, and Vandenabeele P (1998). Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med 187, 1477–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vince JE, Wong WWL, Gentle I, Lawlor KE, Allam R, O’Reilly L, Mason K, Gross O, Ma S, Guarda G, et al. (2012). Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36, 215–227. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, and Wang X (2014). Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146. [DOI] [PubMed] [Google Scholar]
- Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernández-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, and Pasparakis M (2011). FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477, 330–334. [DOI] [PubMed] [Google Scholar]
- Wilson NS, Dixit V, and Ashkenazi A (2009). Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat. Immunol 10, 348–355. [DOI] [PubMed] [Google Scholar]
- Yeh WC, de la Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, et al. (1998). FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279, 1954–1958. [DOI] [PubMed] [Google Scholar]
- Zhang J, and Winoto A (1996). A mouse Fas-associated protein with homology to the human Mort1/FADD protein is essential for Fas-induced apoptosis. Mol. Cell. Biol 16, 2756–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Cado D, Chen A, Kabra NH, and Winoto A (1998). Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392, 296–300. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhou X, McQuade T, Li J, Chan FKM, and Zhang J (2011). Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471, 373–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, and Liu ZG (2012). Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 109, 5322–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.