

Summary
S-adenosylmethionine (SAM) is the methyl-donor substrate for DNA and histone methyltransferases that regulate epigenetic states and subsequent gene expression. This metabolism-epigenome link sensitizes chromatin methylation to altered SAM abundance, yet the mechanisms that allow organisms to adapt and protect epigenetic information during life-experienced fluctuations in SAM availability are unknown. We identified a robust response to SAM depletion that is highlighted by preferential cytoplasmic and nuclear mono-methylation of H3 Lys 9 (H3K9) at the expense of broad losses in histone di- and tri-methylation. Under SAM-depleted conditions, H3K9 mono-methylation preserves heterochromatin stability and supports global epigenetic persistence upon metabolic recovery. This unique chromatin response was robust across the mouse lifespan and correlated with improved metabolic health, supporting a significant role for epigenetic adaptation to SAM depletion in vivo. Together, these studies provide evidence for an adaptive response that enable epigenetic persistence to metabolic stress.
eTOC blurb
Haws et al. reveal depletion of the universal methyl-donor SAM stimulates an adaptive epigenetic response that is cell-autonomous and highly conserved across cell and whole-organism model systems. This response is highlighted by preferential methylation of H3 Lys 9 to support heterochromatin stability and long-term epigenetic persistence upon metabolic recovery.
Graphical Abstract
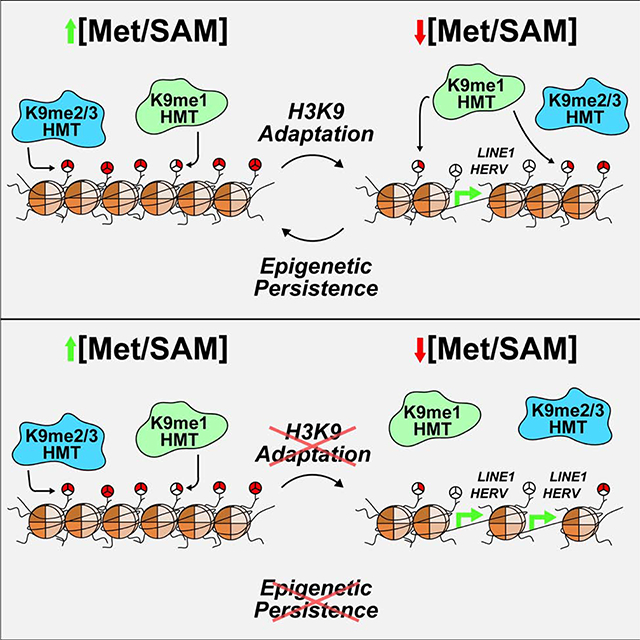
Introduction
Metabolism and the epigenome are connected by central metabolites that act as co-substrates for chromatin modifying enzymes, enabling fluctuations in metabolite availability to directly tune a cell’s ability to ‘write’ and ‘erase’ chromatin post-translational modifications (PTMs) (Fan et al., 2015; Li et al., 2018). Chromatin methylation has been shown to be particularly susceptible to metabolic perturbations as methyltransferases require a single methyl-donor cofactor, S-adenosylmethionine (SAM) (Ducker and Rabinowitz, 2017; Sanderson et al., 2019). For example, accumulation of SAM in S. cerevisiae results in global increases in histone methylation levels while reduced SAM availability has been linked with site-specific losses in histone methylation as well as global depression of DNA methylation in several organisms (Ding et al., 2015; Hayashi et al., 2018; Kera et al., 2013; Mentch et al., 2015; Shiraki et al., 2014; Shyh-Chang et al., 2013; Strekalova et al., 2019; Tang et al., 2017; Towbin et al., 2012; Ye et al., 2017).
Intracellular SAM abundance is dependent on the catalysis of its obligatory precursor metabolite, the essential amino acid methionine (Met). Consumption of diets containing low quantities of Met (e.g. vegan diets) have been shown to significantly correlate with decreased plasma Met concentrations (Farmer, 2014; Schmidt et al., 2016). This link provides an opportunity for diet to directly influence intracellular SAM availability. Intracellular Met and SAM concentrations have also been shown to fluctuate naturally in tune with circadian rhythms (Krishnaiah et al., 2017). Therefore, individuals consuming adequate amounts of dietary Met likely experience transient periods of decreased methyl-metabolite availability. It is currently unknown whether adaptive mechanisms exist that allow cells to actively respond to – and recover their functional epigenomes from – such metabolic perturbations. Here, we define the ability of a cell and/or organism to re-establish the epigenome that was present prior to onset of an environmental perturbation or metabolic stress as epigenetic persistence.
Adaptive mechanisms that respond to methyl-metabolite depletion may support critical cellular functions under these conditions. Interestingly, dietary Met-restriction is associated with beneficial metabolic reprogramming and lifespan extension in mammals (Brown-Borg et al., 2018; Green and Lamming, 2019; Lees et al., 2014; Miller et al., 2005; Orentreich et al., 1993; Yu et al., 2018). These phenotypes run counter to negative pathologies associated with dysregulated chromatin methylation (e.g., intellectual disability syndromes, cancers, and premature as well as natural aging (Greer and Shi, 2012)), suggesting cells possess responsive mechanisms capable of maintaining regulation of site specific and/or global chromatin methylation events during SAM depletion. The goal of this study is to identify and characterize such mechanisms to better understand how organisms adapt and protect their epigenomes in response to fluctuations in central metabolite availability.
Here, we identify a robust, conserved chromatin response to metabolically-depleted SAM levels. This response is characterized by broad decreases in higher-state histone methylation (di- and tri-methyl) and simultaneous preferential H3K9 mono-methylation involving newly-synthesized and chromatin-bound histone H3. Under extreme conditions of both reduced SAM levels and inhibited H3K9 mono-methylation, heterochromatin instability and de-repression of constitutively silenced DNA elements are exacerbated. Acute inhibition of H3K9 mono-methylation also leads to the dysregulation of global histone PTM states upon metabolic recovery, disrupting epigenetic persistence to decreased SAM availability. Importantly, chronic metabolic depletion of SAM in both young and old mice recapitulates these in vitro findings, revealing a conserved, age-independent chromatin response. These results implicate adaptive H3K9 mono-methylation as an indispensable mechanism to support heterochromatin stability and global epigenetic persistence in response to SAM depletion.
Results
Methionine Restriction Stimulates Global, Dynamic Histone PTM Response
To comprehensively investigate how disruption of methyl-donor metabolism affects chromatin methylation states, global histone proteomics and DNA methylation (5mC) analyses were performed on two distinct in vitro and in vivo systems. These included Met-restricted HCT116 human colorectal cancer cells and C57BL/6J mouse liver. Methionine metabolism was severely repressed in both systems, highlighted by significant reductions in Met and SAM abundance (Figure 1A and Figure S1A–S1D). Reduced methyl-metabolite availability did not affect global DNA 5mC abundance in either system (Figure 1B–1C). However, LC-MS/MS analysis of more than 40 unique histone H3 peptide proteoforms revealed dynamic PTM responses to Met-restriction in both HCT116 cells and C57BL/6J liver (Figure 1D–1E). In HCT116 cells, Met-restriction stimulated distinct biphasic changes in global PTM profiles (Figure 1D). Phase I (0 hr-45 min) was rapid and marked by a trending upregulation of H3K4me2/3, PTMs known to mark transcriptionally active promoters. Phase II (90 min-24 hrs) was characterized by broad decreases in di- and tri- histone methylation. Decreased levels of di- and tri-methylated peptides were accompanied by increases in acetylated and unmodified peptide species. Similarly, in C57BL/6J mice, histone PTM responses were marked by significant decreases in histone di- and tri-methylation that essentially matched the patterns found in HCT116 cells during the prolonged Phase II response (Figure 1E and Figure S1E–S1J). Decreased higher-state (di- and tri-) histone methylation both in vitro and in vivo highlight a decreased methylation capacity resulting from prolonged Met and/or SAM depletion. Together, these observations suggest histone methylation is more dynamically regulated by methyl-metabolite availability than global 5mC DNA methylation. Furthermore, similarities between the metabolic and epigenetic responses to Met-restriction across both systems support the use of in vitro Met-restriction as a model for mechanistic follow-up studies.
Figure 1. Methionine Restriction Stimulates Global, Dynamic Histone PTM Response.

(A) Metabolic pathway diagram illustrating changes in Met-cycle metabolite abundance after 24 hours and 5 weeks of Met-restriction in HCT116 cells and C57BL/6J liver, respectively. n=5, error bars represent SD, *p<0.05 (Welch’s t-Test). (B-C) Plots illustrating global, relative 5mC DNA methylation levels during Met-restriction. n≥3, error bars represent SD, *p<0.05 (Welch’s t-Test) (D-E) Hierarchical clustered heatmaps of LC-MS/MS generated log2 fold-change stoichiometric histone H3 peptide proteoform values relative to 0-hour or chow diet controls in HCT116 cells and C57BL/6J liver, respectively. n≥3, *p<0.05, **p<.01 (Welch’s t-Test). See also Table S1 and Figure S1.
SAM Availability Drives Robust Histone Methylation Response
Dramatic reduction of intracellular Met and SAM correlated with onset of the in vitro Phase I and II histone PTM changes, respectively (Figure 1D and Figure S1A–S1B). This implies depletion of individual methyl-metabolites may be capable of stimulating distinct histone modifying pathways. To determine if the global losses in di- and tri- histone methylation in vitro are driven specifically by SAM depletion, two alternative approaches were employed to deplete cellular SAM independent of Met (Figure 2A). The first approach utilized RNAi knockdown of the mammalian SAM synthetase MAT2A (methionine adenosyltransferase II alpha). Knockdown via MAT2A-RNAi treatment decreased MAT2A transcript abundance by greater than 97%, resulting in a near complete deprivation of SAM availability with no effect on Met levels (Figure 2B–2D). Histone proteomics analysis of MAT2A-RNAi treated cells identified site-specific histone PTM changes that mirrored those identified in HCT116 cells under Phase II Met-restriction conditions (r=0.778, p=8.59e−10) (Figure 2E). Using a complementary approach, SAM levels were depleted by overexpression of a major SAM consumer, PEMT (phosphatidylethanolamine N-methyltransferase). Ye et al., 2017 have previously shown PEMT overexpression significantly reduces SAM availability in HEK293T human embryonic kidney cells. Histone proteomics analysis of PEMT overexpressing HEK293T cells identified site-specific PTM changes that paralleled those of Met-restricted controls (r=0.803, p=3.30e−8) (Figure 2F). Together, two separate means of reducing SAM levels demonstrate SAM reduction alone is sufficient to inhibit the methylation capacity of a cell and induce subsequent changes in histone PTM abundance. Furthermore, these findings support the use of prolonged Met-restriction as a model of SAM depletion.
Figure 2. SAM Availability Drives Robust Histone Methylation Response.

(A) Pathway diagram illustrating experimental approaches to depleting intracellular SAM availability. (B) Bar graph of relative MAT2A mRNA abundance in HCT116 cells as measured by RT-qPCR. n≥2, error bars represent SD, *p<0.05 (Welch’s t-Test). (C-D) Dot plots of relative Met and SAM levels in HCT116 cells. n≥4, *p<0.05 (Welch’s t-test). (E-F) Correlation plot of LC-MS/MS generated log2 fold-change stoichiometric values for individual histone H3 peptide proteoforms. n=3. (G-J) Bar graphs illustrating LC-MS/MS generated log2 fold-changes for individual H3K9 PTMs. n≥2. *p<0.05 (Welch’s t-test) See also Table S1, Table S2, and Figure S2.
The similar changes in global histone PTM abundance across four SAM depletion systems were analyzed for the presence of methyl PTMs whose abundances either increased or were unaffected by this metabolic perturbation. Such PTMs would be candidate mediators of an adaptive response to SAM depletion. Unexpectedly, all four SAM depletion systems exhibited identical changes in H3K9 methylation (Figure 2G–2J). SAM depletion stimulated a decrease in H3K9me2/3 with a corresponding increase in H3K9ac/14ac and unmodified peptides while net H3K9me1 abundance was unchanged. Preservation of net H3K9me1 levels in response to SAM depletion was reproducible in Met-restricted MCF7, HEK-293, Panc1, and HepG2 cell lines, as well as in MAT2A-RNAi treated HepA1 cells (Figure S2A–S2F). Therefore, in diverse systems marked by reduced methylation capacity, a robust and consistent maintenance of net H3K9me1 levels suggest adaptive epigenetic mechanisms may support the abundance of this PTM.
Preferential Methylation Supports Global H3K9me1 Under SAM Depletion
Global levels of H3K9me1 could be maintained through three distinct mechanisms during SAM depletion: 1.) protection from PTM turnover, 2.) de-methylation of H3K9me2/3, and/or 3.) methylation of unmodified H3K9. The decrease in H3K9me2/3 and increase in unmodified H3K9 peptide levels across SAM depletion systems suggest H3K9 is subjected to demethylation while new methylation events appear enzymatically unfavorable. However, preferential H3K9 mono-methylation activity could constitute an adaptive response to this metabolic stress that is capable of supporting critical chromatin functions.
To investigate the contribution of HMT activity to global H3K9me1 levels, RNAi-mediated repression of five H3K9 HMTs was performed in Met-replete conditions to determine which enzymes catalyzed a majority of H3K9 mono-methylation reactions in HCT116 cells. Repression of HMT expression had significant yet restricted effects, given technical limitations, on H3K9 PTM abundance and suggested EHMT1/EHMT2 were the primary H3K9 mono-methyltransferases in this system (Figure S3A–S3D). Because repression of HMT expression was intended to be coupled with Met-restriction, RNAi treatment could not be extended to elicit larger changes in PTM abundance as HCT116 cells no longer proliferate beyond 24 hours of Met-restriction (Figure S3E). Therefore, small molecule (UNC0642) inhibition of H3K9 HMTs EHMT1/EHMT2 was used as the best approach to acutely block nuclear H3K9 mono-methylation (Liu et al., 2013). UNC0642 treatment of Met-replete cells reduced H3K9me1 abundance to a greater extent than RNAi repression of EHMT1/EHMT2 (Figure 3A and Figure S3D). Coupling UNC0642 treatment with Met-restriction also stimulated a significant decrease in H3K9me1 levels (Figure 3B and Figure S3F). Comparatively, these data suggest EHMT1/EHMT2 retain nearly 60% of their H3K9 mono-methylation activity during severe in situ SAM deprivation and that this activity is critical for preserving global H3K9me1 abundance (Figure 3C). Furthermore, H3K9me2 abundance decreased significantly in Met-replete cells upon UNC0642 treatment but was unaffected when UNC0642 was coupled with Met-restriction (Figure 3A–3B). This indicates only the mono-methylation activity of EHMT1 and EHMT2 is preferentially supported during SAM depletion.
Figure 3. Preferential Methylation Supports Global H3K9me1 Under SAM Depletion.

(A-B) Bar graphs illustrating LC-MS/MS generated log2 fold-changes for individual H3K9 PTMs in HCT116 cells. n≥2, error bars represent SD, *p<0.05 (Welch’s t-Test) within PTM, #p<0.05 (Welch’s t-Test) across peptides. (C) Bar graph illustrating percent loss in global H3K9me1, as measured by LC-MS/MS, in response to 5μm UNC0642 treatment. n≥2, error bars represent SD, *p<0.05 (Welch’s t-Test) (D) Plot of relative, total H3 and H3K9me1 cytoplasmic protein abundance in HCT116 cells as measured by western blot. Error bars represent SD, n=3, *p<0.05 (Welch’s t-Test). (E) Representative western blot images of those used for the quantification presented in Figure 3E. Alpha-Tubulin is used as a cytoplasm marker to confirm efficient sub-cellular fractionation. See Data S1 for REVERT Total Protein Stain images used for normalization. (F) Diagram illustrating the SILAC experimental design. (G) Bar graph illustrating contribution of newly synthesized histone H3 to the total chromatin bound pool after 24 hours of Met-restriction in HCT116 cells as measured by LC-MS/MS. n≥3, error bars represent SD, *p<0.05 (Welch’s t-test). (G) Bar graph illustrating ratio of light:heavy L-Arg isotope presence in H3K9me1 containing-peptides after 24 hours of Met-restriction in HCT116 cells as measured by LC-MS/MS. n≥3, Error bars represent SD. (H) Pie chart depicting sources of H3K9me1 during SAM depletion. (I) Scatter plot of 373 cytoplasmic proteins identified by LC-MS/MS with significant incorporation of L-Arg +0 that were also significantly upregulated after 24 hours of Met-restriction in 0.1% DMSO treated HCT116 cells. n=4, Statistical significance of p<0.05 (Welch’s t-Test). See also Table S1, Table S3, and Figure S3.
In addition to methylation of H3K9 by nuclear enzymes, mono-methylation is known to be enzymatically added on cytoplasmic histones prior to nuclear import and deposition onto chromatin (Loyola et al., 2006; Pinheiro et al., 2012). Subcellular fractionation of Met-restricted HCT116 cells coupled with western blot analyses indicated that cytoplasmic H3K9me1 levels increase 3-fold after 24 hours of Met-restriction, with a similar increase in total H3 protein levels (Figure 3D–3E). To determine the contribution of cytoplasmic H3K9me1 to the chromatin-bound nuclear pool after 24 hours of Met-restriction, a pulse-chase, SILAC experiment was performed (Figure 3F). L-Arg+10 isotope incorporation prior to Met-restriction was nearly complete with average incorporation for H3K9 peptides greater than 98% (Figure S3H). After 24 hours of Met-restriction, newly synthesized H3 was calculated to comprise 24% of the chromatin bound histone pool and contributed 18% of the global H3K9me1 signal (Figure 3G–3H). UNC0642 treatment did not significantly lessen the contribution of cytoplasmic H3K9me1 to the total pool, suggesting cytoplasmic H3K9me1 may be largely protected from enzyme catalyzed turnover once deposited onto chromatin under these conditions (Figure 3G). Modest, insignificant decreases in L-Arg+0 H3K9me1 signal after UNC0642 treatment can be attributed to a slower cell growth rate and subsequent reduction in the incorporation of newly synthesized H3 onto chromatin (Figure 3G, S3E, and S3I). Together, these results suggest ~40% of global H3K9me1 abundance is driven by preferential activity of H3K9 mono-methyltransferases during Met-restriction with nearly equal contributions from both the cytoplasm and nucleus. The remaining ~60% of H3K9me1 levels are likely protected from turnover and/or are generated via H3K9me2/3 de-methylation during SAM depletion (Figure 3I).
The significant upregulation of cytoplasmic H3K9me1 and total H3, coupled with their incorporation onto chromatin during SAM depletion, were unexpected. These findings suggest cells adapt to this metabolic stress by utilizing scarcely available Met and SAM to support increased histone abundance and H3K9 methylation. Shotgun proteomics was performed on cytoplasmic protein fractions of the previously described SILAC experiment to determine if histone protein abundance is uniquely responsive to SAM depletion. Of the more than 2,400 identified proteins, only 8 possessed significant incorporation of the light L-Arg+0 isotope and also increased in absolute abundance by a log2 fold-change value greater than 2.0 (Figure 3I). Frequency of Met residues in the primary amino acid sequence did not affect L-Arg+0 isotope incorporation (Figure S3J). Remarkably, all four core histones, MAT2A, and the methionine importer SLC3A2 (Bröer et al., 2001) were among the 8 proteins (Figure 3J). These results suggest cells respond to metabolically-induced SAM depletion by upregulating the abundance of proteins critical for chromatin stability and methyl-donor metabolism. To our knowledge, this is also the first example of a metabolic stress stimulating increased histone protein abundance, highlighting the importance of chromatin regulation under these conditions.
H3K9me1 is Redistributed Over Repetitive and Transposable Elements During SAM Depletion
Multiple, non-redundant mechanisms for H3K9me1 maintenance suggest strong biological pressure exists to retain this PTM under SAM depleted conditions. One critical function for H3K9me1 is to serve as a primer for H3K9me2/3 methylation as methyltransferases that yield these higher methylation states require a mono-methyl substrate (Loyola et al., 2009, 2006; Peters et al., 2001). Accordingly, installment of H3K9me2/3 for constitutive heterochromatin-mediated repression of telomeric, pericentromeric, and centromeric repetitive/transposable elements requires readily available H3K9me1 (Bannister et al., 2001; Lachner et al., 2001; Rea et al., 2000).
To determine if H3K9me1 is being maintained globally or redistributed to regions of susceptible heterochromatin during SAM depletion, ChIP-sequencing was performed. Met-restriction stimulated a 35% loss in total H3K9me3 peak number (Figure 4A). A higher percentage of H3K9me3 peaks were lost at LTR, intergenic, and intron annotated loci relative to the total, while H3K9me3 peaks at satellite, simple repeat, and SINE annotated loci were slightly less sensitive to SAM depletion (Figure 4A). Interestingly, H3K9me1 enrichment at repetitive and transposable loci was either largely unaffected by SAM depletion or increased (Figure 4B). Only intergenic and intron annotated H3K9me1 peaks were lost to a greater extent than the total peak number, suggesting H3K9me1 is more susceptible to de-methylation at these genomic loci.
Figure 4. H3K9me1 is Redistributed Over Repetitive and Transposable Elements During SAM Depletion.

(A-B) Bar graphs illustrating percent changes in H3K9 PTM peak number at various annotated loci after 24 hours of Met-restriction in HCT116 cells. (C-D) Plots depicting changes in normalized H3K9 PTM ChIP-sequencing reads to loci of lost H3K9me3 after 24 hours of Met-restriction in HCT116 cells. (E) Bar graphs illustrating percent changes in loci annotation for regions of lost H3K9me3 experiencing increased relative to decreased H3K9me1 ChIP-sequencing read enrichment after 24 hours of Met-restriction in HCT116 cells. (F) Representative ChIP-sequencing track image.
Maintained or increased H3K9me1 peaks at constitutively repressed regions during SAM depletion could result from the replacement of bulk losses in higher-state H3K9 methylation with H3K9me1 at these loci. To investigate this possibility, H3K9me1 ChIP-sequencing reads were mapped to nearly 16e4 loci determined to lose H3K9me3 peak enrichment upon SAM depletion. This analysis determined H3K9me1 enrichment increased at 66.4% of these sites, with a preference for LINE, LTR, simple repeat, and satellite element annotated loci (Figure 4C–4E). Figure 4F provides a representative image of H3K9me3 replacement by H3K9me1 at a pericentric, repetitive element. Together, these ChIP-sequencing analyses suggest SAM depletion stimulates the redistribution of H3K9me1 methylation patterns to repetitive and transposable genomic loci which are susceptible to higher-state H3K9 demethylation.
Preferential H3K9 Mono-methylation Preserves Heterochromatin Stability
Replacement of H3K9me3 by H3K9me1 may function to preserve heterochromatin stability and pericentric chromatin repression during SAM depletion by providing the primer for site-specific H3K9me2/3 methylation and/or by preventing H3K9 acetylation. An MNase accessibility assay was used to evaluate global heterochromatin stability under SAM depleted conditions as MNase can more readily digest euchromatic DNA compared to heterochromatic DNA. Coupling Met-restriction with UNC0642 treatment resulted in a significantly greater loss and accumulation of di- and mono-nucleosome species, respectively, compared to Met-restriction alone (Figure 5A–5B). This indicates inhibition of H3K9 mono-methylation during SAM depletion has a more detrimental effect on global heterochromatin stability than SAM depletion in isolation. To determine if global decreases in heterochromatin stability resulted in de-repression of transposable DNA elements, RT-qPCR was employed. Transcript abundance of LINE1, HERV-K, and HERV-R were all significantly elevated after SAM depletion. Coupling SAM depletion with UNC0642 treatment resulted in the further elevation of LINE1 and HERV-K transcript abundance while HERV-R expression was unaffected (Figure 5C–5E). Because SAM depletion-induced losses in heterochromatin stability and retrotransposon repression are exacerbated with inhibition of nuclear EHMT1/EHMT2 activity, these data suggest adaptive H3K9 methylation functions, in part, to preserve heterochromatin stability.
Figure 5. Preferential H3K9 Mono-methylation Preserves Heterochromatin Stability.

(A) Bar graph illustrating the percent abundances of 5 distinct nucleosome species following MNase digestion of HCT116 cells. Met-restriction coupled with mock (DMSO) or UNC0642 treatment was performed for 24 hours. n≥3, error bars represent SD, *p<0.05 (Welch’s t-Test). (B) Representative DNA agarose gel image of those used for the quantification presented in Figure 5A. (C-E) Bar graphs illustrating relative mRNA abundances of LINE1, HERV-K, and HERV-R in HCT116 cells as measured by RT-qPCR. Met-restriction coupled with mock (DMSO) or UNC0642 treatment was performed for 24 hours. Recovery in Met-replete media from either treatment occurred for 24 hours before harvest. n 4, error bars represent SD, *p<0.05 (Welch’s t-Test). See also Table S2.
Acute Adaptation of H3K9 Methylation Supports Epigenetic Persistence Upon Metabolic Recovery
Dysregulation of cellular response mechanisms under SAM depletion may produce aberrant changes in histone PTMs that compromise long-term epigenetic persistence upon metabolic recovery. To determine if adaptive regulation of H3K9 methylation is required for epigenetic persistence to SAM depletion, HCT116 cells were allowed to recover in Met-replete media after being subjected to Met-restriction coupled with acute mock (DMSO) or UNC0642 treatment (Figure 6A). Histone proteomics analysis confirmed that inhibition of EHMT1/EHMT2 activity during SAM depletion results in a significant loss of H3K9me1 abundance (Figure 6B–6C). Upon 5 and 24 hours of Met-repletion, mock-treated cells largely regained the original histone PTM state (Figure 6B). However, histone PTM states of cells acutely treated with UNC0642 only during Met-restriction remained dysregulated after both 5 and 24 hours of Met-repletion (Figure 6B). Although significant global dysregulation was apparent, the H3K9 peptide proteoforms displayed the most dynamic differences in abundance (Figure 6D–6E). Interestingly, the signature H3K9 PTM response to SAM depletion described in Figure 2 was largely retained upon Met-repletion in cells acutely treated with UNC0642 (Figure 6D–6E). H3K9me3 abundance was an exception, recovering to levels consistent with regained LINE1, HERV-K, and HERV-R repression (Figure 5C–5E). This suggests a subset of H3K9me1 that is unaffected by acute UNC0642 treatment may mark critical genomic loci (e.g. transposable DNA elements) for higher-state methylation once SAM availability is restored.
Figure 6. Acute Adaptation of H3K9 Methylation Supports Epigenetic Persistence Upon Metabolic Recovery.

(A) Diagram illustrating the experimental design for replenishing Met and SAM availability after 24 hours of Met-restriction coupled with acute 5 μM UNC0642 or 0.1% DMSO treatment in HCT116 cells. (B) Heatmap of log2 fold-change stoichiometric values for histone H3 and H4 peptides as measured by LC-MS/MS. n=4, *p<0.05, **p<0.01 (Welch’s t-Test). (C-E) Bar graphs illustrating log2 fold-changes for individual H3K9 PTMs in HCT116 cells as measured by LC-MS/MS. n=4, error bars represent SD, *p<0.05 (Welch’s t-Test) within PTM, #p<0.05 (Welch’s t-Test) across PTMs. (F) PCA diagram of TPM values. Cells allowed to recover for 24 hours from Met-restriction coupled with acute DMSO or UNC0642 treatment were used for this analysis. n=4. (G-H) Plots illustrating NES and FDR q-values for GSEA identified GO terms when comparing UNC0642 to mock recovery transcripts and mock to UNC0642 recovery transcripts, respectively. See also Table S1, Table S4, TableS5, Figure S4, Figure S5, and Figure S6.
Met-replete cells allowed to recover from acute UNC0642 treatment also experienced prolonged changes in H3K9 PTM abundance after inhibitor removal (Figure S4A). Because inhibition of recombinant EHMT1/EHMT2 catalytic activity by UNC0642 is transient (Figure S4B), disrupted epigenetic persistence – in either isolation from or conjunction with SAM depletion – is likely a direct consequence of acute losses in EHMT1/EHMT2 H3K9 mono-methylation activity.
To assess whether there were broad transcriptional changes indicative of lost epigenetic persistence, RNA-sequencing was performed on pre-Met-restriction cells as well as those allowed to recover in Met-replete media after acute mock or UNC0642 treatment coupled with Met-Restriction. Principle Component Analysis (PCA) of normalized Transcripts Per Million (TPM) values produced 3 distinct clusters (Figure 6F), suggesting the global perturbations in epigenomes (Figure 6B) correlate with altered transcript profiles in these cells. A multiple condition comparison analysis was used to determine which transcripts distinguished the clusters from one another. Gene Set Enrichment Analysis (GSEA) of transcripts specific to UNC0642- relative to mock-treated recovery cells revealed 71 significantly enriched gene ontology (GO) terms possessing an FDR q-value less than 0.25 (Figure 6H and Table S5). GO terms covered a wide range of biological processes including “Nucleic Acid Binding Transcription Factor Activity” and “Negative Regulation of Protein Metabolic Processes” among others (Figure S5A–S5B and S6A–S6B). An identical analysis of mock- relative to UNC0642-treated recovery genes did not identify any significantly enriched GO terms (Figure 6I and Table S5).
Residually dysregulated gene-expression patterns in UNC0642-treated recovery cells further suggest adaptation of H3K9 methylation to SAM depletion is essential for genome-wide epigenetic persistence upon metabolic recovery. Thus, blocking H3K9 mono-methylation – a critical component of the cellular responses under this condition – has two significant consequences for the cell: 1.) exacerbated heterochromatin instability and 2.) global loss of epigenetic persistence.
Responses to Ensure Epigenetic Persistence Under SAM Depletion are Robust in vivo, Independent of Age
To determine if robust responses to SAM depletion exist in vivo and are age-independent, 6- and 22-month C57BL/6J mice were subjected to 3 weeks of Met-restriction followed by 5 weeks of Met-repletion (Figure 7A). Several previous reports have correlated decreased H3K9me2/3 abundance with aging in numerous organisms (Larson et al., 2012; Ni et al., 2012; Scaffidi and Misteli, 2006; Tvardovskiy et al., 2017; Wood et al., 2010). Age-dependent dysregulation of heterochromatin has also been associated with de-repression of repetitive DNA and transposable elements, as well as DNA damage, which may contribute to disease progression (Booth and Brunet, 2016; De Cecco et al., 2019, 2013; Oberdoerffer and Sinclair, 2007; Wood et al., 2016). Therefore, conservation of the SAM depletion responses that enable epigenetic persistence across the lifespan would further support the functional importance of these mechanisms as an “older” chromatin environment is thought to be inherently dysregulated.
Figure 7. Responses to Ensure Epigenetic Persistence Under SAM Depletion are Robust in vivo, Independent of Age.

(A) Diagram depicting the experimental design for in vivo Met-restriction of 6- and 22-month C57BL/6J mice followed by dietary Met-reintroduction. (B) Heatmap of log2 fold-change stoichiometric values for histone H3 and H4 peptides in C57BL/6J liver relative to age-matched controls as measured by LC-MS/MS. n≥5, *p<0.05, **p<0.01 (Welch’s t-Test). (C) Bar graph illustrating log2 fold-changes for individual H3K9 PTMs in liver from Met-restricted C57BL/6J mice relative to age-matched controls as measured by LC-MS/MS. n≥5, error bars represent SD, *p<0.05 (Welch’s t-Test) within PTM, #p<0.05 (Welch’s t-Test) across PTMs. (D-E) Bar graphs illustrating relative mRNA abundances of LINE1, SINE B1, and SINE B2 in C57BL/6J liver as measured by RT-qPCR. n≥5, error bars represent SD, *p<0.05 (Student’s t-Test). (F) Bar graph illustrating log2 fold-changes for individual H3K9 PTMs in liver from C57BL/6J relative to age-matched controls as measured by LC-MS/MS. n≥5, error bars represent SD, *p<0.05, (Welch’s t-Test). (G-H) Bar graphs illustrating relative mRNA abundances of LINE1, SINE B1, and SINE B2 in C57BL/6J liver as measured by RT-qPCR. n≥5, error bars represent SD, *p<0.05 (Student’s t-Test). See also Table S1, Table S2, and Figure S7.
Remarkably, histone proteomics analysis of liver tissue revealed H3K9 was the only residue on which PTMs responded similarly to SAM depletion in both 6- and 22-month mice relative to age-matched controls (Figure 7B). This PTM response at H3K9 phenocopied those in the previously described in vitro and whole-organism SAM depletion studies, further highlighting the robust regulation of this residue (Figure 7C). Significant age-dependent responses to SAM depletion were identified on all remaining histone residues (Figure 7B and S7A–S7E). Furthermore, significantly elevated LINE1, SINE B1, and SINE B2 transcript abundance in both 6- and 22-month mice suggest the chromatin instability phenotype characterized in Figure 5 also occurs during metabolic depletion of SAM in vivo (Figure 7D–7E). After 5 weeks of dietary Met-reintroduction, both young and old mice displayed the ability to re-establish an epigenome similar to that possessed by age-matched controls (Figure 7B). Interestingly, 22-month mice appeared to more rapidly adopt the control histone PTM state, while the 6-month mice still exhibited residual losses in H3K9me3 and increases in H3K9me1 after 5 weeks of recovery (Figure 7F). Consistent with the general recovery of chromatin states, both age groups largely re-established repression of LINE1, SINE B1, and SINE B2 expression (Figure 7G–7H).
Metabolic changes in young and old mice tracked with H3K9 methylation patterns. Both 6- and 22-month mice in the Met-restriction group experienced significant losses in total body weight relative to age matched controls (Figure S7F). Decreases in overall body weight were comprised of significant losses in both lean and fat mass (Figure S7F). Altered body-weight and compositions were accompanied by significantly improved glucose tolerance in both young and old mice (Figure S7G). Upon Met-repletion, mice re-acquired their initial metabolic state in an age-independent manner. All animals experienced significant increases in total body weight, comprised of elevated lean and fat mass, as well normalized glucose tolerance relative to age-matched controls (Figure S7H–S7I).
Together, strong association of the H3K9 chromatin response to SAM depletion with metabolic reprogramming in young and old mice suggest H3K9 adaptation mechanisms are not only robust across lifespan but may facilitate these metabolic phenotypes. Further studies will be needed to determine whether the molecular events described here in response to SAM depletion are directly responsible for metabolic remodeling. Most importantly, these data show the ability of epigenetic states to persist upon recovery from a metabolic challenge is an inherent property of both isolated cells and complex mammals.
Discussion
Here, we provide a comprehensive investigation into the regulation of chromatin during a metabolic stress. A highly conserved, robust histone methylation response to SAM depletion was discovered, characterized by broad losses in histone H3 di- and tri-methylation while H3K9me1 is maintained to safeguard against exacerbated heterochromatin instability and support epigenetic persistence upon metabolic recovery. Preferential maintenance of H3K9me1 abundance is facilitated by nuclear and cytoplasmic H3K9 mono-methylation events. Moreover, the highly selective upregulation of all four core histone proteins, the SAM synthetase MAT2A, and the methionine importer SLC3A2 provide compelling evidence that cells mount a coordinated, adaptive response to preserve chromatin states. To our knowledge, this is the first identified mechanism in which scarce, diet-derived nutrients are funneled toward the preservation of specific chromatin PTM states.
It has previously been determined H3K9me1 has the shortest half-life of 17 measured PTMs (Zee et al., 2010), highlighting the importance of adaptive, preferential methylation for supporting global H3K9me1 abundance in a SAM depleted environment. Here, epigenetic adaptation was found to be critical for more than simply sustaining global H3K9me1 levels under these conditions. ChIP-sequencing analyses determined H3K9me1 patterns experience a significant redistribution across heterochromatin that results in H3K9me1 enrichment at repetitive and transposable DNA elements, particularly in regions where H3K9me3 is lost. Targeting of H3K9me1 to these regions is likely critical for preserving heterochromatin as inhibition of nuclear H3K9 mono-methylation resulted in global losses in heterochromatin stability and site-specific retrotransposon de-repression. We hypothesize H3K9me1 facilitates repression of retrotransposons during SAM depletion through two simultaneous mechanisms: 1.) acting as the primed substrate for HMTs which deposit di- and tri-methyl PTMs onto H3K9 and 2.) preventing the acetylation of newly demethylated H3K9 residues. Increased H3K9 acetylation through impaired H3K9 deacetylation pathways is associated with global genomic instability as well as de-repression of LINE1, providing support for the second mechanism (Mostoslavsky et al., 2006; Simon et al., 2019; Van Meter et al., 2014).
Ensuring sufficient amounts of H3K9me1 are available to facilitate heterochromatin stability is not limited to nuclear derived H3K9me1. Pinheiro et al., 2012 have previously shown inhibition of cytoplasmic H3K9 mono-methylation through shRNA knockdown of Prdm3 and Prdm16 stimulates pericentric heterochromatin loss and disruption of the nuclear lamina. Our study demonstrates cytoplasmic derived H3K9me1 comprises a significant portion (18%) of the total chromatin bound pool after 24 hours of Met-restriction in vitro. Adaptive regulation of H3K9me1 during SAM depletion is also critical for long-term epigenetic persistence to this metabolic stress. Inhibition of nuclear H3K9 mono-methylation under this condition resulted in sustained dysregulation of global histone PTMs upon metabolic recovery, significantly influencing global gene expression patterns. This requirement for an adaptive histone PTM response to support epigenetic persistence upon metabolic recovery is the first identified mechanism of its kind.
Replication of in vitro results in vivo, independent of age, strongly supports the discovery of a universal response to SAM depletion. Here, we demonstrate that the acute mechanisms required to support H3K9me1-mediated heterochromatin stability are inducible in both 6- and 22-month mice after dietary Met-restriction. Furthermore, H3K9me1 maintenance under SAM depleted conditions uniquely correlated with beneficial metabolic reprogramming (i.e. reduced fat mass and improved glucose tolerance) in both young and old mice. This suggests the robust regulation of H3K9 PTMs in response to SAM depletion may be required to support the metabolic reprogramming stimulated by dietary Met-restriction. Future studies will be needed to determine if the chromatin-directed responses upon acute SAM depletion are directly responsible for this metabolic reprogramming. Global epigenetic persistence to metabolic SAM depletion was observed in both 6- and 22-month mice. Validation of this phenomenon in vivo, independent of age, suggests similar mechanisms critical for maintaining proper regulation of the epigenome during life-experienced fluctuations in the availability of other metabolic cofactors may exist as well. Such fluctuations could be stimulated by periods of prolonged fasts, chronic intake of foods lacking essential cofactors, and circadian regulated changes in metabolite availability (Farmer, 2014; Krishnaiah et al., 2017; Schmidt et al., 2016).
Notably, we did not observe an age-associated decrease in H3K9 methylation between 6- and 22-month control diet mice (Table S1). Similar regulation of H3K9 PTMs and mirrored metabolic responses to SAM depletion across age groups suggest our 22-month mice, the equivalent to a 60–70 year old human, have not yet experienced a decline in health-span (Flurkey et al., 2007). It will be interesting to determine if the acute responses to ensure epigenetic persistence revealed here are functional in very old animals (i.e. >30 months) where H3K9 methylation is thought to be inherently dysregulated. Disruption of these mechanisms later in an organism’s lifespan could support age-associated de-repression of transposable elements (Booth and Brunet, 2016; De Cecco et al., 2013; Oberdoerffer and Sinclair, 2007; Wood et al., 2016), contributing to negative phenotypes that accelerate the aging process (De Cecco et al., 2019; Simon et al., 2019).
Future studies will be needed to determine how H3K9 mono-methyltransferases are able to maintain their catalytic activity under SAM depleted environments. One interesting possibility is that H3K9 mono-methyltransferases have evolved to possess inherent kinetic properties, such as an extremely high affinity for SAM, that allow these enzymes to function at very low SAM levels. The existing literature on H3K9 HMT enzymology is incomplete, making such a conclusion only possible after a comprehensive in vitro analysis is performed. Direct shuttling of SAM to H3K9me1 HMTs by MAT2A could also support H3K9me1 HMT catalytic activity. As SAM is energetically expensive to produce, due to the 1:1 requirement of Met and ATP molecules for its synthesis, in addition to being highly unstable (Wu et al., 1983), it would be advantageous for the cell to possess mechanisms which permit preferential shuttling of SAM during methyl-metabolite depletion. Directed SAM production and utilization have been proposed in other metabolic contexts (Katoh et al., 2011; Kera et al., 2013; Li et al., 2015).
Star Methods Text
Lead Contact and Materials Availability
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, John M. Denu (john.denu@wisc.edu). This study did not generate new unique reagents, all resources utilized for this study were acquired from outside sources.
Experimental Model and Subject Details
Animals and Diets
All experiments involving animals were approved by the Institutional Animal Care and Use Committee of the William S. Middleton Memorial Veterans Hospital, Madison WI. To test the effect of Met deprivation in the context of young mice, 9-week-old male C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and placed on either an amino acid defined Control diet (TD.01084, Envigo, Madison, WI, USA) or a Met-deficient diet (TD.140119, Envigo, Madison, WI, USA) their respective diets at 14 weeks of age; full diet compositions for these diets have been previously described (PMID: 29401631). After 5 weeks on the diet, mice were sacrificed following an overnight fast and liver was flash frozen for analysis as described.
To compare the effect of Met restriction on young and old mice, C57BL/6J. Nia male mice were obtained from the National Institute on Aging Aged Rodent Colony, and housed in our animal facility for 3–4 months until reaching 5 and 21 months of age. All animals were then placed on the Control diet (TD.01084) for 4 weeks, until the mice were 6 and 22 months of age. Mice were then randomized to either remain on the Control diet, or were placed on the Met-deficient diet (TD.140119) for three weeks. Liver was collected and flash-frozen in liquid nitrogen harvested from mice euthanized after an approximately 16-hour fast.
Cell Lines
HCT116, HEK-293, HEK-293T, HMCF7, Panc1, HepG2, and HepA1 cell lines were cultured at 37°C with 5.0% CO2.
Method Details
Metabolite Extraction
For HCT116 cells, ~1.5×10^6 cells were quickly rinsed 3 times with 1.5 ml ice-cold PBS pH 7.4 before the addition of 1.5 ml of −80°C 80:20 MeOH:H2O extraction solvent. Cells were incubated with extraction solvent at −80°C for 15 minutes. Next, cells were scraped off their dish and transferred into a 2 ml microcentrifuge Eppendorf tube which was followed by a 5-minute, maximum speed centrifugation at 4°C. The supernatant was transferred to a new 2 ml microcentrifuge Eppendorf tube. Next, 0.5 ml extraction solvent was added to the remaining pellet, vortexed, and again centrifugated at maximum speed for 5 minutes at 4°C. The 2 supernatants from each extraction were pooled and dried completely using a Thermo Fisher Savant ISS110 SpeedVac. Dried metabolite extracts were resuspended in 80 μl of 85% acetonitrile following microcentrifugation for 5 minutes at maximum speed at 4°C to pellet any remaining insoluble debris. The supernatant was then transferred to a glass vial for LC-MS analysis.
For M. musculus liver tissue, an average tissue weight of 75 mg was powdered in liquid nitrogen using a mortar and pestle. Powdered tissue was transferred to an individual 1.5 ml microcentrifuge Eppendorf tube and incubated with 1 ml −80°C 80:20 MeOH:H2O extraction solvent on dry ice for 5 minutes post-vortexing. Tissue homogenate was centrifugated at maximum speed for 5 minutes at 4°C. Supernatant was transferred to a 15 ml tube after which the remaining pellet was resuspended in 0.8 ml −20°C 40:40:20 ACN:MeOH:H2O extraction solvent and incubated on ice for 5 minutes. Tissue homogenate was again centrifugated at maximum speed for 5 minutes at 4°C after which the supernatant was pooled with the previously isolated metabolite fraction. The 40:40:20 ACN:MeOH:H2O extraction was then repeated as previously described. Next, 550 μl of pooled metabolite extract for each sample was transferred to a 1.5 ml microcentrifuge Eppendorf tube and completely dried using a Thermo Fisher Savant ISS110 SpeedVac. Dried metabolite extracts were resuspended in 450 μl of 85% ACN following microcentrifugation for 5 minutes at maximum speed at 4°C to pellet any remaining insoluble debris. Supernatant was then transferred to a glass vial for LC-MS analysis.
LC-MS Metabolite Analysis
Each prepared metabolite sample was injected onto a Thermo Fisher Scientific Vanquish UHPLC with a Waters XBridge BEH Amide column (100 mm x 2.1 mm, 3.5 μm) coupled to a Thermo Fisher Q-Exactive mass spectrometer. Mobile phase (A) consisted of 97% H2O, 3% ACN, 20 mM ammonium acetate, and 15 mM ammonium hydroxide pH 9.6. Organic phase (B) consisted of 100% ACN. Metabolites were resolved using the following linear gradient: 0 min, 85% B, 0.15 ml/min; 1.5 min, 85% B, 0.15 ml/min; 5.5 min, 40% B, 0.15 ml/min; 10 min, 40% B, 0.15 ml/min; 10.5 min, 40% B, 0.3 ml/min; 14.5 min, 40% B, 0.3 ml/min; 15 min, 85% B, 0.15 ml/min; 20 min, 85% B, 0.15 ml/min. The mass spectrometer was operated in positive ionization mode with a MS1 scan at resolution = 70,000, automatic gain control target = 3 × 106, and scan range = 60–186 m/z and 187–900 m/z. This protocol was adapted from Mentch et al., 2015. Individual metabolite data were called using MAVEN (Clasquin et al., 2012; Melamud et al., 2010) with retention times empirically determined in-house. Peak Area Top values were analyzed to determine metabolite expression.
LC-MS DNA Methylation Assay
500 ng of cytosine, 5mC, and 5hmC double-stranded DNA standards from Zymo Research were incubated with Zymo Research DNA Degradase Plus for 3 hours at 37°C following enzyme inactivation at 70°C for 20 minutes. 75 μl of ACN was added to each standard reaction to bring the final volume to 100 μl. DNA standards were centrifugated at 21,100xg for 5 minutes at 4°C after which 75 μl of supernatant was transferred to a glass vial for LC-MS analysis. A range of 0.1 to 100 ng of digested DNA was analyzed via LC-MS to determine each nucleoside’s linear range of detection.
Genomic DNA extracted using a Promega Wizard Genomic DNA Purification Kit after which 1 μg was incubated with Zymo Research DNA Degradase Plus at 37°C following enzyme inactivation at 70°C for 20 minutes. 175 μl of ACN was added to each reaction to bring the final volume to 200 μl. DNA samples were centrifugated at 21,100xg for 5 minutes at 4°C after which 150 μl of supernatant was transferred to a glass vial for LC-MS analysis.
Each prepared nucleoside sample was injected onto a Thermo Fisher Scientific Vanquish UHPLC with a Waters XBridge BEH Amide column (100 m x 2.1 mm, 3.5 μm) coupled to a Thermo Fisher Q-Exactive mass spectrometer run in positive ionization mode. Mobile phase (B) consisted of H2O, 3% ACN, 20 mM ammonium acetate, and 15 mM ammonium hydroxide pH 9.6. Organic phase (B) consisted of 100% ACN. Nucleosides were resolved using the following linear gradient: 0 min, 85% B, 0.15 ml/min; 1.5 min, 85% B, 0.15 ml/min; 5.5 min, 40% B, 0.15 ml/min; 10 min, 40% B, 0.15 ml/min; 10.5 min, 40% B, 0.3 ml/min; 14.5 min, 40% B, 0.3 ml/min; 15 min, 85% B, 0.15 ml/min; 20 min, 85% B, 0.15 ml/min. The mass spectrometer was operated in positive ionization mode with a MS1 scan at resolution = 70,000, automatic gain control target = 3 × 106, and scan range = 200–300 m/z, followed by a DDA scan with a loop count of 5. DDA settings were as follows: window size = 2.0 m/z, resolution = 17,500, automatic gain control target = 1 × 105, DDA maximum fill time = 50 ms, and normalized collision energy = 30. This protocol was adapted from Mentch et al., 2015. Individual metabolite data were called using MAVEN (Clasquin et al., 2012; Melamud et al., 2010) with retention times empirically determined in-house. Peak Area Top values were analyzed to determine metabolite expression with dAdenosine being utilized for internal normalization (m/z dCytosine: 228.0978, m/z d5mC: 242.1135, m/z d5hmC: 258.108, m/z dAdenosine: 252.109).
Histone Isolation and Chemical Derivatization
Tissue culture cell pellets were resuspended in 800 μl of ice-cold Buffer A (10 mM Tris-HCl pH 7.4, 10 mM NaCl, and 3 mM MgCl2) supplied with protease and histone deacetylase inhibitors (10 μg/ml leupeptin, 10 μg/ml aprotinin, 100 μM phenylmethylsulfonyl fluoride, 10 mM nicotinamide, 1 mM sodium-butyrate, and 4 μM trichostatin A) followed by 80 strokes of light pestle homogenization in a 1 ml Wheaton dounce homogenizer. Cell homogenate was then transferred to a 1.5 ml microcentrifuge Eppendorf tube and centrifugated at 800xg for 10 minutes at 4°C to pellet nuclei. The supernatant was either transferred to a fresh 1.5 ml Eppendorf tube or discarded. The nuclei pellet was resuspended in 500 μl ice-cold PBS pH 7.4 followed by centrifugation at 800xg for 10 minutes at 4°C. The supernatant was discarded and nuclei were again washed with 500 μl ice-cold PBS pH 7.4. Pelleted nuclei were then resuspended in 500 μl of 0.4N H2SO4 and rotated at 4°C for 4 hours. Samples were centrifugated at 3,400xg for 5 minutes at 4°C to pellet nuclear debris and precipitated non-histone proteins. The supernatant was transferred to a new 1.5 ml Eppendorf tube after which 125 μl of 100% trichloroacetic acid was added and incubated overnight on ice at 4°C. Samples were centrifugated at 3,400xg for 5 minutes at 4°C to pellet precipitated histone proteins. The supernatant was discarded, after which the precipitant was washed with 1 ml ice-cold acetone +0.1% HCl. Samples were centrifugated at 3,400xg for 2 minutes at 4°C and the supernatant was discarded. This process was repeated except precipitant was washed with 100% ice-cold acetone. Residual acetone was allowed to evaporate at room temperature for 10 minutes after which the dried precipitant was dissolved in 125 μl H2O. Samples were then centrifugated at 21,100xg for 5 minutes at 4°C to pellet any remaining insoluble debris. Supernatant containing purified histone was then transferred to a new 1.5 ml Eppendorf tube and stored at −20°C until needed for future analyses.
For M. musculus liver tissue, an average tissue weight of 50 mg was homogenized in 800μl ice-cold Buffer supplemented with protease and HDAC inhibitors (listed above) using a 1 ml Wheaton dounce homogenizer (20 strokes of the loose pestle followed by 20 strokes of the tight pestle) and strained through a 100 μM filter before being transferred to a new 1.5 ml Eppendorf tube. All remaining steps for completing the histone isolation were performed as described previously in the tissue culture isolation protocol.
To prepare purified histone proteins for LC-MS/MS analysis, 5 μg of histone was diluted with H2O to a final volume of 9 μl. 1 μl of 1 M triethylammonium bicarbonate was added to each sample to buffer the solution to a final pH of 7–9. Next, 1 μl of 1:100 propionic anhydride:H2O was added to each sample and allowed to incubate for 2 minutes at room temperature. The propionylation reaction was then quenched via the addition of 1 μl 80 mM hydroxylamine which was allowed to incubate for 20 minutes at room temperature. Next, propionylated histones were digested with 0.1 μg trypsin for 4 hours at 37°C. Upon completion of trypsin digestion, 5 μl of .02 M NaOH was added to bring the final pH between 9–10. Propionylated, trypsin digested histone peptides were then N-terminally modified with 1 μl 1:50 phenyl isocyanate:ACN for 1-hour at 37°C. Modified peptides were desalted and eluted-off of Empore™C18 extraction membrane. Eluted samples were dried completely using a Thermo Fisher Scientific Savant ISS110 SpeedVac, resuspended in 40 μl sample diluent (94.9% H2O, 5% ACN, .1% TFA), and transferred into glass vials for LC-MS/MS analysis.
Histone Proteomics Analysis
Derivatized histone peptides were injected onto a Dionex Ultimate3000 nanoflow HPLC with a Waters nanoEase UPLC C18 column (100 m x 150 mm, 3μm) coupled to a Thermo Fisher Q-Exactive mass spectrometer at 700 nL/ min. Aqueous phase (A) consisted of H2O + 0.1% formic acid while the mobile phase (B) consisted of acetonitrile + 0.1% formic acid (B). Histone peptides were resolved using the following linear gradient: 0 min, 2.0% B; 5 min, 2.0% B; 65 min, 35% B; 67 min, 95% B; 77 min, 95% B; 79 min, 2.0% B; 89 min, 2.0% B. Data was acquired using data-independent acquisition (DIA) mode. The mass spectrometer was operated with a MS1 scan at resolution = 35,000, automatic gain control target = 1 × 106, and scan range = 390–910 m/z, followed by a DIA scan with a loop count of 10. DIA settings were as follows: window size = 10 m/z, resolution = 17,500, automatic gain control target = 1 × 106, DIA maximum fill time = AUTO, and normalized collision energy = 30.
DIA Thermo .raw files were imported into Skyline open source proteomics software (MacLean et al., 2010) and matched against a pre-constructed peptide database containing unique peptide proteoforms from H3 and H4. The identity of each individually called peak was verified by hand after which total MS1 peak area values were exported for downstream analyses. To control for differences in ionization efficiencies across histone peptides, all peptides possessing an identical primary sequence were analyzed as a peptide family which enabled the calculation of stoichiometric values for each unique peptide proteoform within the family. Stoichiometric values were compared across conditions to identify altered histone peptide abundances. Furthermore, combinatorial peptide stoichiometry values were deconvoluted by totaling the stoichiometry values for all peptides possessing a given PTM independent of any other PTMs that may be present on other residues of the same primary peptide. A Welch’s T-Test with a p-value cutoff of 0.05 was used to determine statistical significance.
Hierarchical clustering was performed in MATLAB R2016a using the ‘clustergram’ command. Pearson’s Correlation Coefficients were calculated in MATLAB R2016a using the ‘corr’ command.
RNAi Mediated Inhibition of MAT2A and H3K9 HMT Expression
For 35 mm tissue culture dishes, 1.5×105 HCT116 cells were reverse transfected with 30 pmol of targeted or control RNAi using 5 μl of Lipofectamine RNAiMAX in addition to 500 μl Opti-MEM and 2.5 ml RPMI-1640 +10% FBS. MAT2A-RNAi treated HCT116 cells used for LC-MS metabolite analysis were cultured using dialyzed FBS. MAT2A-RNAi and H3K9 HMT-RNAi treatments were performed for 48 and 24 hours, respectively.
For 100 mm tissue culture dishes, 2.0×106 HCT116 cells were reverse transfected with 180 pmol of targeted or control RNAi using 30 μl of Lipofectamine RNAiMAX in addition to 2 ml Opti-MEM and 8 ml RPMI-1640 +10% FBS. MAT2A-RNAi and H3K9 HMT-RNAi treatments were performed for 48 and 24 hours, respectively.
RNAi mediated knockdown of MAT2A in HepA1 cells was performed as described in Katoh et al., 2011.
Methionine Restriction of Tissue Culture Cells
HCT116, HEK-293, MCF7, Panc1, and HepG2 culture cells were initially cultured in RPMI-1640 +10% FBS at 37°C, 5% CO2. One day before Met-restriction, 3.5×106 cells were seeded into 100 mm tissue culture dishes. One-hour before the restriction began, RPMI-1640 was replaced with fresh, Met-replete media. At the time of Met-restriction, cells were rinsed 2x with 3 ml of sterile PBS pH 7.4 followed by addition of RPMI-1640 void of Met. Any small molecule inhibitors or vehicle controls would be added at this time.
For experiments in which Met-restriction was followed by Met repletion, HCT116 cells were washed with 3 ml of sterile PBS pH 7.4 2x to maximize small molecule inhibitor and DMSO removal before reintroduction of Met replete RPMI-1640.
HCT116 cells cultured for LC-MS metabolite analysis were cultured in RPMI-1640 containing 10% dialyzed FBS beginning 1-hour prior to beginning Met-restriction.
Methionine restriction of HEK-293T cells (data presented in Figure 2) was performed as previously described by Ye et al., 2017.
SILAC in Tissue Culture
197 ml of SILAC RPMI-1640 +10% DFBS void of L-Arg and L-Lys was supplemented with 50mg isotopically heavy 3C6 15N4 L-Arg HCl and 9.43mg isotopically light L-Lys to obtain final concentrations of 1.149 mM and 0.219 mM, respectively. HCT116 cells were cultured in the SILAC media for 7 days (or 9 doubling cycles at a reported doubling time of 21 hours) after which cells were washed 3x with 5 ml PBS pH 7.4 before replacing the SILAC media with isotopically light L-Arg and L-Lys RPMI-1640 +10% DFBS void of Met. Half of the cells were treated with 5 μM UNC0642 or 0.1% DMSO at this time. Furthermore, 0-hour control HCT116 cells which were never exposed to isotopically light L-Arg, Met-restricted RPMI-1640 were harvested. HCT116 cells cultured in the Met-restricted media were harvested after 24 hours.
SILAC Shotgun Proteomics Analysis
50 μg of cytoplasmic protein extract from the previously described SILAC Met-restriction experiment were diluted with H2O into a final volume of 75 μl. Next, dithiothreitol was added to each sample at a final concentration of 10 mM, followed by a 15-minute incubation at 95°C. A sonication water bath was used to resolubilize precipitated proteins post-heat denaturation. Iodoacetamide was then added to a final concentration of 20 mM, followed by a 20-minute incubation at room temperature protected from light. Alkylated proteins were precipitated with 337 μl ice-cold acetone for 20 minutes at −20°C. Samples were centrifugated at 21,000xg for 15 minutes at 4°C, the supernatant was discarded, and precipitated protein was resuspended in 400 μl of urea buffer (2 M urea and 100 mM ammonium bicarbonate pH 8). Precipitated proteins were again solubilized using a sonication water bath. Solubilized proteins were digested into peptides using 0.8 μg trypsin at 37°C overnight. Upon digest completion, samples were desalted and eluted-off Empore™C18 extraction membrane. Eluted samples were dried completely using a Thermo Fisher Scientific Savant ISS110 SpeedVac, after which being resuspended in 50 μl sample diluent (94.9% H2O, 5% ACN, .1% TFA) and transferred to glass vials for LC-MS/MS analysis.
Each prepared cytoplasmic peptide sample was injected onto a Dionex Ultimate3000 nanoflow HPLC with a Waters nanoEase UPLC C18 column (100 m x 150 mm, 3μm) coupled to a Thermo Fisher Q-Exactive mass spectrometer at 800 nL/ min. Aqueous phase (A) consisted of H2O + 0.1% formic acid while the mobile phase (B) consisted of acetonitrile + 0.1% formic acid (B). Histone peptides were resolved using the following linear gradient: 0 min, 5.0% B; 5 min, 5.0% B; 120 min, 30% B; 122 min, 95% B; 127 min, 95% B; 129 min, 5.0% B; 145 min, 5.0% B. Data was acquired using data-dependent acquisition (DDA) mode. The mass spectrometer was operated with a MS1 scan at resolution = 70,000, automatic gain control target = 1 × 106, and scan range = 400–1000 m/z, followed by a DDA scan with a loop count of 20. DDA settings were as follows: window size = 2.0 m/z, resolution = 17,500, automatic gain control target = 2 × 105, DDA maximum fill time = 150 ms, and normalized collision energy = 28.
MaxQuant Proteomics Analysis
Thermo .raw DDA files were analyzed using MaxQuant v1.6.2.6 (Cox et al., 2014) using the default settings with the following exceptions: Labels: 1, Arg10; Instrument: Orbitrap; LFQ: LFQ min. ratio count: 2, LFQ min. number of neighbors: 3, LFQ average number of neighbors: 6; FASTQ File: Proteome: H. Sapiens Proteome ID UP000005640.
Identified proteins with a Q-value of 0.01 or above were removed. For the remaining proteins, LFQ light (L-Arg+0) and heavy (L-Arg+10) values were summed to generate LFQ total protein abundance values. LFQ light values were divided by the corresponding LFQ total value to generate a percent abundance of LFQ light species for each identified protein. Welch’s t-Tests with a p-value cut off of 0.05 were used to determine significant accumulation of LFQ light species in Met-restricted HCT116 samples relative to 0-hour controls. For proteins determined to have significant LFQ light species abundance after Met-restriction, LFQ total values were used to determine the corresponding change in overall protein abundance. Up to 2 null-values were removed from replicates of an experimental group to account for missed identifications. If 3 of the 4 replicates for a single experimental group contained null-values, that protein was removed from all experiment groups and therefore excluded from any downstream analyses. A Welch’s t-Test with a p-value cut off of 0.05 was used to determine if an identified protein was significantly more abundant after Met-restriction relative to 0-hour controls.
SILAC Histone Proteomics Analysis
Histones were isolated, chemically derivatized, and analyzed by LC-MS/MS as described in “Histone Isolation and Chemical Derivatization” and “Histone Proteomics Analysis.” DDA Thermo .raw files of histone proteins with incorporated isotopically heavy 3C6 15N4 L-Arg were used to construct an H3K9/K14 peptide library using Skyline opensource proteomics software (MacLean et al., 2010). DIA files were then matched against both isotopically heavy and light L-Arg peptide libraries after which peptide identities were confirmed by hand. MS1 intensity values for both isotopically heavy and light L-Arg peptides were summed to generate total MS1 peptide proteoform intensity values which were then used to calculate the percent of total MS1 peptide proteoform signal which was contributed by isotopically heavy peptide proteoforms. Welch’s T-Tests with a p-value cutoff of 0.05 were used to determine significant differences in total MS1 peptide proteoform stoichiometric values as well as the percent contribution of isotopically heavy peptide proteoforms to total peptide proteoform intensity values.
Chromatin Immunoprecipitation and Sequencing
30×106 HCT116 cells were resuspended in 1 ml MNase digestion buffer (50 mM Tris-HCl pH 7.6, 1 mM CaCl2, and 0.2% Triton X-100) after which 330 μl of the cell suspension was aliquoted into individual 1.5 ml tubes. Each tube was treated with either 45, 52.5, or 60 units of Worthington MNase for 5 minutes at 37°C shaking at 400 rpm to generate mono-nucleosomes. MNase digestions were stopped via the addition of EDTA to a final concentration of 5mM. Digested cells were then pooled and lysed at 4°C using a Covaris S220 Focused-ultrasonicator with the following settings: Peak Power = 75, Duty Factor = 5, Cycles/Burst = 100, Duration = 30” on/30” off x2. Insoluble debris was centrifugated at 10,000 rpm for 10 minutes at 4°C after which the supernatant containing prepared nucleosomes was transferred to a new tube. Glycerol was added to a final concentration of 5% prior to −80°C storage. For input controls, 43 μl of prepared nucleosomes were aliquoted and saved for further processing at −80°C.
Next, 860 μl of prepared nucleosomes was incubated with 5 μg of the desired antibody, rotating overnight at 4°C. The nucleosome/antibody solution was added to 50 μl of prepared M-280 Sheep Anti-Rabbit IgG superparamagnetic Dynabeads™ and allowed to rotate at 4°C for 4 hours. Conjugated beads were then washed for 10 minutes, rotating at 4°C under the following conditions: 3× 1 ml RIPA (10 mM Tris pH 7.6, 1 mM EDTA, 0.1% SDS, 0.1% Na-deoxycholate, 1% Triton X-100) buffer, 2× 1 ml RIPA buffer +0.3 M NaCl, 2× 1 ml LiCl buffer (0.25 M LiCl, 0.5% NP-40, 0.5% Na-deoxycholate), 1x with 1 ml 1xTE +50 mM NaCl. Following the final wash, beads were centrifugated at 4°C for 3 minutes at 960xg after which the supernatant was carefully removed and discarded.
To elute immunoprecipitated nucleosome species, beads were resuspended in 210 μl of elution buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS) and incubated at 65°C for 30 minutes shaking at 400 rpm. Beads were resuspended every 2 minutes via brief vortexing. Next samples were centrifugated at 16,000xg for 1 minute at room temperature after which 200 μl of supernatant was transferred to a new 1.5 ml Eppendorf tube. Eluted nucleosome species were then treated with 2 μl Thermo Fisher Scientific Proteinase K for 2 hours at 55°C. DNA was recovered from the Proteinase K digestion using the QIAquick PCR Purification Kit.
DNA libraries were prepared by the University of Wisconsin-Madison Biotechnology Center using the Illumina TruSeq DNA Nano kit and then sequenced across 4 lanes of an Illumina 1×100 HiSeq2500 High Throughput flowcell.
FASTQ files were aligned unpaired using bowtie2–2.1.0 (Langmead and Salzberg, 2012) against human genome 19. Bowtie2 output SAM files were converted to BAM files, sorted and indexed using samtools 1.5 (Li et al., 2009). Broad peaks were called from sorted BAM files using MACS2–2.1.1.20160309 (Zhang et al., 2008) with a Q-value cutoff of 0.1. Input DNA was used as the control file. All identified peaks possessing a log2 fold-change below 1.75 were discarded prior to downstream analyses. Bedtools v2.25.0 (Quinlan and Hall, 2010) was used to identify unique and overlapping across samples and also used to assign raw mapped reads to pre-specified genomic loci. Genomic loci were annotated using HOMERv4.9 (Heinz et al., 2010).
Quantitative PCR
RNA was extracted from HCT116 cells using Thermo Fisher Scientific TRIzol and from liver tissue using Sigma TRI Reagent, both by following the manufacturer’s instructions. The concentration and purity of RNA were determined by absorbance at 260/280 nm.
cDNA was prepared from 1 μg of RNA using RevertAid Reverse Transcriptase for HCT116 RNA and Superscipt III for liver RNA. Oligo dT primers were used in both cDNA synthesizing reactions.
HCT116 cDNA was analyzed on a Roche 480 Light Cycler System with PerfeCTa SYBR Green SuperMix while cDNA generated from liver was analyzed on an Applied Biosystems StepOne Plus instrument with Sybr Green PCR Master Mix. HCT116 cDNA qPCR reactions were normalized internally using GAPDH while liver cDNA qPCR reactions were normalized internally using actin. Primer sequences are available in Table S3.
RNA sequencing
RNA libraries were prepared by Novogene using NEBNext adaptors followed by paired end 2×150 sequencing on their Illumina Platform. FASTQ files were trimmed using Trimmomatic v0.39 (Bolger et al., 2014) and aligned using RSEM v1.2.4 (Li and Dewey, 2011) against the human genome 18 reference transcriptome. RSEM .genes output files were used for Ebseq EBMultiTest multiple comparison analysis (Leng et al., 2013). Gene lists associated with individual EBMultiTest patterns were further analyzed with GSEA 2 (Mootha et al., 2003; Subramanian et al., 2005).
MNase Accessibility Assay
6×106 HCT116 cells were resuspended in 1 ml of digestion buffer (50 mM Tris-HCl pH 7.6, 1 mM CaCl2, and 0.2% Triton X-100) in addition to 15 unites of Worthington MNase. MNase treated cell suspensions were incubated for 5 minutes at 37°C shaking at 400 rpm. MNase digestions were stopped via the addition of EDTA to a final concentration of 5 mM. Digested cells were then pooled and lysed at 4°C using a Covaris® S220 Focused-ultrasonicator with the following settings: Peak Power = 75, Duty Factor = 5, Cycles/Burst = 100, Duration = 30” on/30” off x2. Insoluble debris was pelleted via centrifugation at 10,000 rpm for 10 minutes at 4°C after which the supernatant was transferred to a new tube. 150 μl of digested chromatin was treated with 2 μl of Thermo Fisher Scientific Proteinase K for 90 minutes at 55°C. DNA was then purified using the QIAquick PCR Purification Kit. Next, 1 μg of purified DNA was separated on a 2% agarose gel and quantified using GelAnalyzer 2010a.
Western-Blotting and Image Quantification
Protein separated on SDS-PAGE gels were transferred onto 0.2 μm nitrocellulose membrane and stained with 5 ml of LI-COR REVERT Total Protein Stain for 5 minutes at room temperature before being blocked in 0.1% fat-free milk PBS pH 7.4 for 1 hour. Primary antibodies were diluted 1:1000 in 5% fat-free milk PBST pH 7.4 and incubated with the membrane overnight at 4°C. LI-COR secondary antibodies were used at a 1:15,000 dilution in 5% fat-free milk PBST pH 7.4 and incubated with the membrane for 1 hour at room temperature. Membranes were imaged using an Odyssey Infrared Imager (model no. 9120). Densitometry was performed using Image Studio™ Lite software. All data was normalized to REVERT™ Total Protein Stain.
In vivo Tests
For glucose tolerance tests, mice were fasted overnight for 16 hours and then injected with glucose (1 g/kg) intraperitoneally as previously described (Arriola Apelo et al., 2016). Glucose measurements were taken using a Bayer Contour blood glucose meter and test strips. Mouse body composition was determined using an EchoMRI Body Composition Analyzer (EchoMRI™, Houston, TX, USA) according to the manufacturer’s procedures.
Protein Purification
The H. sapiens EHMT1 catalytic subunit E. coli expression plasmid used in this study (Addgene plasmid # 51314) was acquired from addgene, made possible through a gift by Cheryl Arrowsmith. The H. sapiens EHMT2 catalytic subunit E. coli expression plasmid used in this study was provided by the laboratory of Peter W. Lewis at the University of Wisconsin-Madison.
Transformed Rosetta™ E. coli competent cells were cultured in 1L of 2XYT media at 37°C to an O.D. 600 of 0.8. IPTG was then added to each culture at a final concentration of 1mM. Cultures were allowed to grow for 16 hours at 18°C before being harvested and stored at −80°C. Pellets were resuspended in 30ml Buffer A (50 mM NaPi, 250 mM NaCl, 10mM imidazole, pH 7.2) and lysed using sonicated in the presence of lysozyme. Lysate was centrifugated and the supernatant was collected. The supernatant was then loaded onto a HisTrap FF nickel column in line with a GE AKTA FPLC. Protein was eluted off the column using a linear gradient ending in 100% Buffer B (50 mM NaPi, 250 mM NaCl, 250 mM imidazole, pH 7.2) and collected into time-dependent fractions. Fractions were analyzed by coomassie staining of SDS-PAGE gels to assess purity.
Histone Methyltransferase Activity Assay
The methyltransferase activity of recombinantly purified EHMT1 and EHMT2 catalytic subunits was assessed using Promega Corporation’s MTase-Glo™ Methyltransferase Assay. Assays were performed using 12.5 nM enzyme in the presence of saturating concentrations of SAM (30 μM) and unmodified H3K9 peptide (30 μM, WARTKQTARKSTGGKAPR – 3’) as well as either saturating levels of UNC0642 (5 μM) or 0.1% DMSO. Reactions proceeded for 1 hour at 30°C before addition of MTase-Glo™ reagent following the Promega protocol. Luminescence was detected using a Biotek Synergy H4 Hybrid plate reader. All values detected were determined to be within the instrument’s linear range of detection via the use of a SAH standard curve. To test for reversibility of enzyme inhibition by small molecule, 1 μM EHMT1 and EHMT2 were incubated with saturating concentrations of UNC0642 (5 μM) or 0.1% DMSO followed overnight 1,000x or 10,000x dialysis with a molecular weight cutoff of 10,000 Da against 1x Reaction Buffer without BSA (20mM Tris, pH 8.0, 50mM NaCl, 1mM EDTA, 3mM MgCl2, 1mM DTT). Following dialysis, enzyme and reaction components were diluted using the appropriate dialysis buffers to the working concentrations described above. The MTase-Glo™ assay was then 1043 repeated using identical conditions as described above.
Quantification and Statistical Analysis
Explanations of statistical analyses and parameters are present in the main and supplemental figure legends as well as in ‘Method Details” where appropriate. Dixon’s Q test for statistical outliers was used as exclusion criteria.
Data and Software Availability
RNA-sequencing as well as H3K9me1 and H3K9me3 ChIP-sequencing data have been uploaded to Gene Expression Omnibus (GEO). GEO accession numbers are available in the Key Resource Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-H3K9me1 | Abcam | Cat#ab176880; RRID#AB_2751009 |
| Rabbit polyclonal anti-H3K9me3 | Abcam | Cat#ab8898; RRID#AB_306848 |
| Rabbit polyclonal anti-Histone H3 | Abcam | Cat#ab46765; RRID#AB_880439 |
| Rabbit monoclonal anti-alpha Tubulin | Cell Signaling Technologies | Cat#cst2125; RRID#AB_2619646 |
| IRDye® 800CW Goat anti-Rabbit | LI-COR Biosciences | Cat#925-32211; RRID#AB_2651127 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| UNC0642 G9a/GLP Inhibitor | R&D Systems | Cat#5132/10 |
| DNA Degradase Plus | Zymo Research | Cat#E2020 |
| Lipofectamine RNAiMax | Thermo Fisher | Cat#13778030 |
| L-Lysine-2HCl | Thermo Fisher Scientific | Cat#PI89987 |
| L-Arginine-HCl 13C6 15N4 | Thermo Fisher | Cat#PI89990 |
| Dimethyl Sulfoxide (DMSO) | Sant Cruz Biotechnology | Cat#sc-358801 |
| Sequencing Grade Modified Trypsin | Promega | Cat#V5113 |
| Propionic Anhydride | Sigma-Aldrich | Cat#240311 |
| Phenyl Isocyanate | Sigma-Aldrich | Cat#78750-25ML |
| Leupeptin Hemisulfate | Thermo Fisher | Cat#AAJ61188MC |
| Aprotinin | Gold Biotechnology | Cat#A-655-100 |
| Phenylmethylsulfonyl Fluoride (PMSF) | Gold Biotechnology | Cat#P-470-50 |
| Trichostatin A | Thermo Fisher | Cat#501012404 |
| Sodium Butyrate | Sigma-Aldrich | Cat#303410-100G |
| Nicotinamide | Sigma-Aldrich | Cat#N3376-500G |
| Urea | Acros Organics | Cat#197460050 |
| Dithiothreitol (DTT) | Gold Biotechnology | Cat#DTT100 |
| Iodoacetamide | Sigma-Aldrich | Cat#I1149-5G |
| Ammonium Bicarbonate | Sigma-Aldrich | Cat#09830-500G |
| Micrococcal Nuclease (MNase) | Worthington Biochemical | Cat#LS004798 |
| Proteinase K | Thermo Fisher | Cat#EO0491 |
| TRIzol™ | Thermo Fisher | Cat#15596018 |
| TRI Reagent | Sigma-Aldrich | Cat#T9424-25ML |
| 3M Empore C18 47mm extraction disks | Empore | Cat#2215 |
| Atlantis dC18 NanoEase Column (3 μM, 100 μM, 150 mm) | Waters | Cat#186002209 |
| XBridge BEH Amide Column (3.5 μM, 2.1 mm, 150mm) | Waters | Cat#186004860 |
| Superscript III Reverse Transcriptase | Thermo Fisher | Cat#18080044 |
| RPMI-1640 | Thermo Fisher | Cat#11875119 |
| RPMI-1640 for SILAC | Thermo Fisher | Cat#88365 |
| RPMI-1640, no methionine | Thermo Fisher | Cat#A1451701 |
| Fetal Bovine Serum (FBS) | Thermo Fisher | Cat#16000044 |
| Dialyzed Fetal Bovine Serum (DFBS) | Thermo Fisher | Cat#88212 |
| H3K9 unmodified peptide | This paper | N/A |
| pET28a-LIC-4H4H | Addgene | Plasmid # 51314; RRID#Addgene_51314 |
| pET28a-LIC-EHMT2-913-1193 | Peter W. Lewis Laboratory | N/A |
| Critical Commercial Assays | ||
| PerfeCTa SYBR® Green SuperMix | Quanta Bio | Cat#95054-02K |
| SYBR Green PCR Master Mix | Thermo Fisher | Cat#4309155 |
| REVERT Total Protein Stain | LI-COR Biosciences | Cat#926-11011 |
| RevertAid First Strand cDNA Synthesis Kit | Thermo Fisher | Cat#K1621 |
| Wizard® Genomic DNA Purification Kit | Promega | Cat#A1120 |
| Dynabeads™ M-280 Sheep Anti Rabbit IgG | Thermo Fisher | Cat#11203D |
| QIAquick PCR Purification Kit | QIAGEN | Cat#28104 |
| TruSeq DNA Nano | Illumina | Cat#20015964 |
| Axygen™ Light Cycler 480 Compatible PCR Plates | Thermo Fisher | Cat#14-223-212 |
| Promega MTase-Glo™ | Promega | Cat#V7602 |
| Deposited Data | ||
| H3K9me1 and H3K9me3 ChIP-sequencing Data | This paper | GEO Accession Number: GSE122565 |
| RNA-sequencing Data | This paper | GEO Accession Number: GSE138574 |
| Experimental Models: Cell Lines | ||
| H. sapiens: HCT116 | ATCC | CCL-247 |
| H. sapiens: HEK-293T | ATCC | CRL-3216 |
| H. sapiens: HEK-293 | ATCC | CRL-1573 |
| H. sapiens: HepG2 | ATCC | HB-8065 |
| H. sapiens: MCF7 | ATCC | HTB-22 |
| H. sapiens: Panc1 | ATCC | CRL-1469 |
| M. musculus: Hepa-1c1c7 | ATCC | CRL-2026 |
| Experimental Models: Organisms/Strains | ||
| M. musculus: C57BL/6J (9-weeks of age) | The Jackson Laboratory | JAX: 000664 |
| M. musculus: C57BL/6J (2- and 18-months of age) | The National Institute on Aging Rodent Colony | N/A |
| Oligonucleotides | ||
| 5-Methylcytosine and 5-Hydroxymethylcytosine DNA | Zymo Research | Cat#D505 |
| Stealth RNAi Sense Sequence: H. sapiens MAT2A: 5′AUCAAGGACAGCAUCACUGAUUUGG-3′ | Thermo Fisher | Kera et al., 2013 |
| Stealth RNAi Sense Sequence: Control: 5′-AGGAGGAUAUUACCAUGGACAAGAC-3′ | Thermo Fisher | Kera et al., 2013 |
| SUV39H1 RNAi: H. sapiens | Novus Biologicals | H00006839-R01-20nmol |
| SUV39H2 RNAi: H. sapiens | Novus Biologicals | H00079723-R01-20nmol |
| SETDB1 RNAi: H. sapiens | Novus Biologicals | H00009869-R01-20nmol |
| G9a/EHMT2 RNAi: H. sapiens | Novus Biologicals | H00010919-R02-20nmol |
| GLP/EHMT1 RNAi: H sapiens | Novus Biologicals | H00079813-R01-20nmol |
| RT-qPCR Primers | See Table S2 | See Table S2 |
| Software and Algorithms | ||
| Bowtie2-2.1.0 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools 1.5 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| MACS2-2.1.1.20160309 | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/ |
| Bedtools v2.25.0 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| HOMER v4.9 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| GelAnalyzer 2010a | Lazar Software | http://gelanalyzer.com/index.html |
| MAVEN | Melamud et al., 2010; Clasquin et al., 2012 | http://genomics-pubs.princeton.edu/mzroll/index.php |
| MAXQUANT | Cox et al., 2014 | http://www.biochem.mpg.de/5111795/maxquant |
| Trimmomatic v0.39 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| RSEM v1.2.4 | Li et al., 2011 | https://deweylab.github.io/RSEM/ |
| EBSeq v1.24.0 | Leng et al., 2013 | http://bioconductor.org/packages/release/bioc/html/EBSeq.html |
| GSEA 2 | Tamayo et al., 2005 Mootha et al., 2003 |
http://software.broadinstitute.org/gsea/index.jsp |
| Skyline | MacLean et al., 2010 | https://skyline.ms/project/home/software/Skyline/begin.view |
| Other | ||
| Control M. musculus Diet | Envigo | TD.01084 |
| Methionine Deficient M. musculus Diet | Envigo | TD.140119 |
Supplementary Material
Table S3, Related to Figure 3. SILAC Cytoplasmic Shotgun Proteomics Summary.
Highlights.
SAM producing and consuming pathways dictate histone methylation profiles.
Mono-methylation of H3K9 is an adaptive epigenetic response to SAM depletion.
Epigenetic persistence to SAM depletion requires adaptive H3K9 methylation.
Epigenetic adaptation to SAM depletion is conserved in vivo, independent of age.
Acknowledgements
This research was supported in part by grants from the NIH (S.A.H. – T32 DK007665; K.A.K. – T32 DK007665; W.H.L. – T32 DK007665, F32 GM128399; R.S. – GM113033; B.P.T. – R01 GM094314; C.Y. – K99 GM129415; J.M.D. – R37 GM059785; D.W.L. – AG041765, AG050135, AG051974, AG056771, AG062328), grants-in-aid from the Japan Society for the Promotion of Science (K.I. – 18H05374), a UW-Madison SCRMC postdoctoral fellowship (C.K.W.), a New Investigator Program Award (D.W.L.) and a Collaborative Health Sciences Program Award (V.L.C.) from the Wisconsin Partnership Program, the V Foundation for Cancer Research (V.L.C.), and a Glenn Foundation Award for Research in the Biological Mechanisms of Aging (D.W.L.), as well as startup funds from the UW-Madison School of Medicine and Public Health and the UW-Madison Department of Medicine (V.L.C. and D.W.L.). This research was conducted while D.W.L. was an AFAR Research Grant recipient from the American Federation for Aging Research. D.Y. was supported in part by a fellowship from the American Heart Association (17PRE33410983). The Lamming laboratory is supported in part by the U.S. Department of Veterans Affairs (I01-BX004031), and this work was supported using facilities and resources from the William S. Middleton Memorial Veterans Hospital. This work does not represent the views of the Department of Veterans Affairs or the United States Government. We would like to thank Alexis J. Lawton for the bioinformatics analysis presented in Figure S3I. We would also like to thank the University of Wisconsin-Madison Biotechnology Center and the University of Wisconsin-Madison Center for High Throughput Computing for their help in generating and analyzing the ChIP-sequencing data presented in Figure 5, respectively.
Footnotes
Declaration of Interests
J.M.D. is a consultant for FORGE Life Sciences and co-founder of Galilei BioSciences. D.W.L has received funding from, and is a scientific advisory board member of, Aeovian Pharmaceuticals, which seeks to develop novel, selective mTOR inhibitors for the treatment of various diseases. Remaining authors declare no competing interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, Pumper CP, Kimple ME, Lamming DW, 2016. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell 15, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T, 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120–124. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B, 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma. Oxf. Engl. 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth LN, Brunet A, 2016. The Aging Epigenome. Mol. Cell 62, 728–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bröer A, Friedrich B, Wagner CA, Fillon S, Ganapathy V, Lang F, Bröer S, 2001. Association of 4F2hc with light chains LAT1, LAT2 or y+LAT2 requires different domains. Biochem. J. 355, 725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Borg HM, Rakoczy S, Wonderlich JA, Borg KE, Rojanathammanee L, 2018. Metabolic adaptation of short-living growth hormone transgenic mice to methionine restriction and supplementation. Ann. N. Y. Acad. Sci. 1418, 118–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clasquin MF, Melamud E, Rabinowitz JD, 2012. LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr. Protoc. Bioinforma. Chapter 14, Unit14.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M, 2014. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics MCP 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, Peterson AL, Kreiling JA, Neretti N, Sedivy JM, 2013. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 12, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, Le O, Beauséjour C, Ambati J, Ambati K, Simon M, Seluanov A, Gorbunova V, Slagboom PE, Helfand SL, Neretti N, Sedivy JM, 2019. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W, Smulan LJ, Hou NS, Taubert S, Watts JL, Walker AK, 2015. s-Adenosylmethionine Levels Govern Innate Immunity through Distinct Methylation-Dependent Pathways. Cell Metab. 22, 633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker GS, Rabinowitz JD, 2017. One-Carbon Metabolism in Health and Disease. Cell Metab. 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Krautkramer KA, Feldman JL, Denu JM, 2015. Metabolic regulation of histone post-translational modifications. ACS Chem. Biol. 10, 95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer B, 2014. Nutritional adequacy of plant-based diets for weight management: observations from the NHANES. Am. J. Clin. Nutr. 100, 365S–368S. [DOI] [PubMed] [Google Scholar]
- Fei Q, Shang K, Zhang J, Chuai S, Kong D, Zhou T, Fu S, Liang Y, Li C, Chen Zhi, Zhao Y, Yu Z, Huang Z, Hu M, Ying H, Chen Zhui, Zhang Y, Xing F, Zhu J, Xu H, Zhao K, Lu C, Atadja P, Xiao Z-X, Li E, Shou J, 2015. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat. Commun. 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, Currer JM, Harrison DE, 2007. The Mouse in Aging Research. In the Mouse in Biomedical Research 2nd Edition. Fox JG, et al. , editors. American College Laboratory Animal Medicine (Elsevier; ), pp. 637–671. [Google Scholar]
- Green CL, Lamming DW, 2019. Regulation of metabolic health by essential dietary amino acids. Mech. Ageing Dev., Molecular aspects of aging and longevity 177, 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Shi Y, 2012. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardies SC, Wang L, Zhou L, Zhao Y, Casavant NC, Huang S, 2000. LINE-1 (L1) lineages in the mouse. Mol. Biol. Evol. 17, 616–628. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Teruya T, Chaleckis R, Morigasaki S, Yanagida M, 2018. S-Adenosylmethionine Synthetase Is Required for Cell Growth, Maintenance of G0 Phase, and Termination of Quiescence in Fission Yeast. iScience 5, 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK, 2010. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YK, Park JS, Lee CS, Yeom YI, Chung AS, Lee KK, 1999. Efficient integration of short interspersed element-flanked foreign DNA via homologous recombination. J. Biol. Chem. 274, 36585–36591. [DOI] [PubMed] [Google Scholar]
- Katoh Y, Ikura T, Hoshikawa Y, Tashiro S, Ito T, Ohta M, Kera Y, Noda T, Igarashi K, 2011. Methionine Adenosyltransferase II Serves as a Transcriptional Corepressor of Maf Oncoprotein. Mol. Cell 41, 554–566. [DOI] [PubMed] [Google Scholar]
- Kera Y, Katoh Y, Ohta M, Matsumoto M, Takano-Yamamoto T, Igarashi K, 2013. Methionine Adenosyltransferase II-dependent Histone H3K9 Methylation at the COX-2 Gene Locus. J. Biol. Chem. 288, 13592–13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klawitter S, Fuchs NV, Upton KR, Muñoz-Lopez M, Shukla R, Wang J, Garcia-Cañadas M, Lopez-Ruiz C, Gerhardt DJ, Sebe A, Grabundzija I, Merkert S, Gerdes P, Pulgarin JA, Bock A, Held U, Witthuhn A, Haase A, Sarkadi B, Löwer J, Wolvetang EJ, Martin U, Ivics Z, Izsvák Z, Garcia-Perez JL, Faulkner GJ, Schumann GG, 2016. Reprogramming triggers endogenous L1 and Alu retrotransposition in human induced pluripotent stem cells. Nat. Commun. 7, 10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaiah SY, Wu G, Altman BJ, Growe J, Rhoades SD, Coldren F, Venkataraman A, Olarerin-George AO, Francey LJ, Mukherjee S, Girish S, Selby CP, Cal S, Er U, Sianati B, Sengupta A, Anafi RC, Kavakli IH, Sancar A, Baur JA, Dang CV, Hogenesch JB, Weljie AM, 2017. Clock Regulation of Metabolites Reveals Coupling between Transcription and Metabolism. Cell Metab. 25, 961–974.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T, 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL, 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson K, Yan S-J, Tsurumi A, Liu J, Zhou J, Gaur K, Guo D, Eickbush TH, Li WX, 2012. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet. 8, e1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees EK, Król E, Grant L, Shearer K, Wyse C, Moncur E, Bykowska AS, Mody N, Gettys TW, Delibegovic M, 2014. Methionine restriction restores a younger metabolic phenotype in adult mice with alterations in fibroblast growth factor 21. Aging Cell 13, 817–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BMG, Haag JD, Gould MN, Stewart RM, Kendziorski C, 2013. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinforma. Oxf. Engl. 29, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN, 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup, 2009. The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Swanson SK, Gogol M, Florens L, Washburn MP, Workman JL, Suganuma T, 2015. Serine and SAM Responsive Complex SESAME Regulates Histone Modification Crosstalk by Sensing Cellular Metabolism. Mol. Cell 60, 408–421. [DOI] [PubMed] [Google Scholar]
- Li X, Egervari G, Wang Y, Berger SL, Lu Z, 2018. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 19, 563–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Barsyte-Lovejoy D, Li F, Xiong Y, Korboukh V, Huang X-P, Allali-Hassani A, Janzen WP, Roth BL, Frye SV, Arrowsmith CH, Brown PJ, Vedadi M, Jin J, 2013. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Med. Chem. 56, 8931–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyola A, Bonaldi T, Roche D, Imhof A, Almouzni G, 2006. PTMs on H3 Variants before Chromatin Assembly Potentiate Their Final Epigenetic State. Mol. Cell 24, 309–316. [DOI] [PubMed] [Google Scholar]
- Loyola A, Tagami H, Bonaldi T, Roche D, Quivy JP, Imhof A, Nakatani Y, Dent SYR, Almouzni G, 2009. The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep. 10, 769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ, 2010. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinforma. Oxf. Engl. 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauger O, Klinck R, Chabot B, Muchardt C, Allemand E, Batsché E, 2015. Alternative splicing regulates the expression of G9A and SUV39H2 methyltransferases, and dramatically changes SUV39H2 functions. Nucleic Acids Res. 43, 1869–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamud E, Vastag L, Rabinowitz JD, 2010. Metabolomic analysis and visualization engine for LC-MS data. Anal. Chem. 82, 9818–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gómez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE, Nichenametla SN, Locasale JW, 2015. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 22, 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Buehner G, Chang Y, Harper JM, Sigler R, Smith-Wheelock M, 2005. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 4, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC, 2003. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng H-L, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD, Alt FW, 2006. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329. [DOI] [PubMed] [Google Scholar]
- Ni Z, Ebata A, Alipanahiramandi E, Lee SS, 2012. Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 11, 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, Ra Y-S, Zilberberg K, McLeod J, Scherer SW, Sunil Rao J, Eberhart CG, Grajkowska W, Gillespie Y, Lach B, Grundy R, Pollack IF, Hamilton RL, Van Meter T, Carlotti CG, Boop F, Bigner D, Gilbertson RJ, Rutka JT, Taylor MD, 2009. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 41, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdoerffer P, Sinclair DA, 2007. The role of nuclear architecture in genomic instability and ageing. Nat. Rev. Mol. Cell Biol. 8, 692–702. [DOI] [PubMed] [Google Scholar]
- Orentreich N, Matias JR, DeFelice A, Zimmerman JA, 1993. Low Methionine Ingestion by Rats Extends Life Span. J. Nutr. 123, 269–274. [DOI] [PubMed] [Google Scholar]
- Peters AH, O’Carroll D, Scherthan H, Mechtler K, Sauer S, Schöfer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T, 2001. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107, 323–337. [DOI] [PubMed] [Google Scholar]
- Pinheiro I, Margueron R, Shukeir N, Eisold M, Fritzsch C, Richter FM, Mittler G, Genoud C, Goyama S, Kurokawa M, Son J, Reinberg D, Lachner M, Jenuwein T, 2012. Prdm3 and Prdm16 are H3K9me1 Methyltransferases Required for Mammalian Heterochromatin Integrity. Cell 150, 948–960. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM, 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinforma. Oxf. Engl. 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T, 2000. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599. [DOI] [PubMed] [Google Scholar]
- Rhyu D-W, Kang Y-J, Ock M-S, Eo J-W, Choi Y-H, Kim W-J, Leem S-H, Yi J-M, Kim H-S, Cha H-J, 2014. Expression of human endogenous retrovirus env genes in the blood of breast cancer patients. Int. J. Mol. Sci. 15, 9173–9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson SM, Gao X, Dai Z, Locasale JW, 2019. Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat. Rev. Cancer 19, 625–637. [DOI] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, 2006. Lamin A-dependent nuclear defects in human aging. Science 312, 1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JA, Rinaldi S, Scalbert A, Ferrari P, Achaintre D, Gunter MJ, Appleby PN, Key TJ, Travis RC, 2016. Plasma concentrations and intakes of amino acids in male meat-eaters, fish-eaters, vegetarians and vegans: a cross-sectional analysis in the EPIC-Oxford cohort. Eur. J. Clin. Nutr. 70, 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, Kume S, 2014. Methionine Metabolism Regulates Maintenance and Differentiation of Human Pluripotent Stem Cells. Cell Metab. 19, 780–794. [DOI] [PubMed] [Google Scholar]
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, Asara JM, Daley GQ, Cantley LC, 2013. Influence of Threonine Metabolism on S-Adenosylmethionine and Histone Methylation. Science 339, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M, Van Meter M, Ablaeva J, Ke Z, Gonzalez RS, Taguchi T, De Cecco M, Leonova KI, Kogan V, Helfand SL, Neretti N, Roichman A, Cohen HY, Meer MV, Gladyshev VN, Antoch MP, Gudkov AV, Sedivy JM, Seluanov A, Gorbunova V, 2019. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab. 29, 871–885.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strekalova E, Malin D, Weisenhorn EMM, Russell JD, Hoelper D, Jain A, Coon JJ, Lewis PW, Cryns VL, 2019. S-adenosylmethionine biosynthesis is a targetable metabolic vulnerability of cancer stem cells. Breast Cancer Res. Treat. 175, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Fang Y, Huang G, Xu X, Padilla-Banks E, Fan W, Xu Q, Sanderson SM, Foley JF, Dowdy S, McBurney MW, Fargo DC, Williams CJ, Locasale JW, Guan Z, Li X, 2017. Methionine metabolism is essential for SIRT1-regulated mouse embryonic stem cell maintenance and embryonic development. EMBO J. 36, 3175–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin BD, González-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM, 2012. Step-Wise Methylation of Histone H3K9 Positions Heterochromatin at the Nuclear Periphery. Cell 150, 934–947. [DOI] [PubMed] [Google Scholar]
- Tvardovskiy A, Schwämmle V, Kempf SJ, Rogowska-Wrzesinska A, Jensen ON, 2017. Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 45, 9272–9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meter M, Kashyap M, Rezazadeh S, Geneva AJ, Morello TD, Seluanov A, Gorbunova V, 2014. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 5, 5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson GW, Wickramasekara S, Palomera-Sanchez Z, Black C, Maier CS, Williams DE, Dashwood RH, Ho E, 2014. SUV39H1/H3K9me3 attenuates sulforaphane-induced apoptotic signaling in PC3 prostate cancer cells. Oncogenesis 3, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Hillenmeyer S, Lawrence C, Chang C, Hosier S, Lightfoot W, Mukherjee E, Jiang N, Schorl C, Brodsky AS, Neretti N, Helfand SL, 2010. Chromatin remodeling in the aging genome of Drosophila. Aging Cell 9, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Jones BC, Jiang N, Chang C, Hosier S, Wickremesinghe P, Garcia M, Hartnett DA, Burhenn L, Neretti N, Helfand SL, 2016. Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc. Natl. Acad. Sci. U. S. A. 113, 11277–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SE, Huskey WP, Borchardt RT, Schowen RL, 1983. Chiral instability at sulfur of S-adenosylmethionine. Biochemistry 22, 2828–2832. [DOI] [PubMed] [Google Scholar]
- Ye C, Sutter BM, Wang Y, Kuang Z, Tu BP, 2017. A Metabolic Function for Phospholipid and Histone Methylation. Mol. Cell 66, 180–193.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Yang SE, Miller BR, Wisinski JA, Sherman DS, Brinkman JA, Tomasiewicz JL, Cummings NE, Kimple ME, Cryns VL, Lamming DW, 2018. Short-term methionine deprivation improves metabolic health via sexually dimorphic, mTORC1-independent mechanisms. FASEB J. 32, 3471–3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee BM, Levin RS, Xu B, LeRoy G, Wingreen NS, Garcia BA, 2010. In vivo residue-specific histone methylation dynamics. J. Biol. Chem. 285, 3341–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3, Related to Figure 3. SILAC Cytoplasmic Shotgun Proteomics Summary.
Data Availability Statement
RNA-sequencing as well as H3K9me1 and H3K9me3 ChIP-sequencing data have been uploaded to Gene Expression Omnibus (GEO). GEO accession numbers are available in the Key Resource Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-H3K9me1 | Abcam | Cat#ab176880; RRID#AB_2751009 |
| Rabbit polyclonal anti-H3K9me3 | Abcam | Cat#ab8898; RRID#AB_306848 |
| Rabbit polyclonal anti-Histone H3 | Abcam | Cat#ab46765; RRID#AB_880439 |
| Rabbit monoclonal anti-alpha Tubulin | Cell Signaling Technologies | Cat#cst2125; RRID#AB_2619646 |
| IRDye® 800CW Goat anti-Rabbit | LI-COR Biosciences | Cat#925-32211; RRID#AB_2651127 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| UNC0642 G9a/GLP Inhibitor | R&D Systems | Cat#5132/10 |
| DNA Degradase Plus | Zymo Research | Cat#E2020 |
| Lipofectamine RNAiMax | Thermo Fisher | Cat#13778030 |
| L-Lysine-2HCl | Thermo Fisher Scientific | Cat#PI89987 |
| L-Arginine-HCl 13C6 15N4 | Thermo Fisher | Cat#PI89990 |
| Dimethyl Sulfoxide (DMSO) | Sant Cruz Biotechnology | Cat#sc-358801 |
| Sequencing Grade Modified Trypsin | Promega | Cat#V5113 |
| Propionic Anhydride | Sigma-Aldrich | Cat#240311 |
| Phenyl Isocyanate | Sigma-Aldrich | Cat#78750-25ML |
| Leupeptin Hemisulfate | Thermo Fisher | Cat#AAJ61188MC |
| Aprotinin | Gold Biotechnology | Cat#A-655-100 |
| Phenylmethylsulfonyl Fluoride (PMSF) | Gold Biotechnology | Cat#P-470-50 |
| Trichostatin A | Thermo Fisher | Cat#501012404 |
| Sodium Butyrate | Sigma-Aldrich | Cat#303410-100G |
| Nicotinamide | Sigma-Aldrich | Cat#N3376-500G |
| Urea | Acros Organics | Cat#197460050 |
| Dithiothreitol (DTT) | Gold Biotechnology | Cat#DTT100 |
| Iodoacetamide | Sigma-Aldrich | Cat#I1149-5G |
| Ammonium Bicarbonate | Sigma-Aldrich | Cat#09830-500G |
| Micrococcal Nuclease (MNase) | Worthington Biochemical | Cat#LS004798 |
| Proteinase K | Thermo Fisher | Cat#EO0491 |
| TRIzol™ | Thermo Fisher | Cat#15596018 |
| TRI Reagent | Sigma-Aldrich | Cat#T9424-25ML |
| 3M Empore C18 47mm extraction disks | Empore | Cat#2215 |
| Atlantis dC18 NanoEase Column (3 μM, 100 μM, 150 mm) | Waters | Cat#186002209 |
| XBridge BEH Amide Column (3.5 μM, 2.1 mm, 150mm) | Waters | Cat#186004860 |
| Superscript III Reverse Transcriptase | Thermo Fisher | Cat#18080044 |
| RPMI-1640 | Thermo Fisher | Cat#11875119 |
| RPMI-1640 for SILAC | Thermo Fisher | Cat#88365 |
| RPMI-1640, no methionine | Thermo Fisher | Cat#A1451701 |
| Fetal Bovine Serum (FBS) | Thermo Fisher | Cat#16000044 |
| Dialyzed Fetal Bovine Serum (DFBS) | Thermo Fisher | Cat#88212 |
| H3K9 unmodified peptide | This paper | N/A |
| pET28a-LIC-4H4H | Addgene | Plasmid # 51314; RRID#Addgene_51314 |
| pET28a-LIC-EHMT2-913-1193 | Peter W. Lewis Laboratory | N/A |
| Critical Commercial Assays | ||
| PerfeCTa SYBR® Green SuperMix | Quanta Bio | Cat#95054-02K |
| SYBR Green PCR Master Mix | Thermo Fisher | Cat#4309155 |
| REVERT Total Protein Stain | LI-COR Biosciences | Cat#926-11011 |
| RevertAid First Strand cDNA Synthesis Kit | Thermo Fisher | Cat#K1621 |
| Wizard® Genomic DNA Purification Kit | Promega | Cat#A1120 |
| Dynabeads™ M-280 Sheep Anti Rabbit IgG | Thermo Fisher | Cat#11203D |
| QIAquick PCR Purification Kit | QIAGEN | Cat#28104 |
| TruSeq DNA Nano | Illumina | Cat#20015964 |
| Axygen™ Light Cycler 480 Compatible PCR Plates | Thermo Fisher | Cat#14-223-212 |
| Promega MTase-Glo™ | Promega | Cat#V7602 |
| Deposited Data | ||
| H3K9me1 and H3K9me3 ChIP-sequencing Data | This paper | GEO Accession Number: GSE122565 |
| RNA-sequencing Data | This paper | GEO Accession Number: GSE138574 |
| Experimental Models: Cell Lines | ||
| H. sapiens: HCT116 | ATCC | CCL-247 |
| H. sapiens: HEK-293T | ATCC | CRL-3216 |
| H. sapiens: HEK-293 | ATCC | CRL-1573 |
| H. sapiens: HepG2 | ATCC | HB-8065 |
| H. sapiens: MCF7 | ATCC | HTB-22 |
| H. sapiens: Panc1 | ATCC | CRL-1469 |
| M. musculus: Hepa-1c1c7 | ATCC | CRL-2026 |
| Experimental Models: Organisms/Strains | ||
| M. musculus: C57BL/6J (9-weeks of age) | The Jackson Laboratory | JAX: 000664 |
| M. musculus: C57BL/6J (2- and 18-months of age) | The National Institute on Aging Rodent Colony | N/A |
| Oligonucleotides | ||
| 5-Methylcytosine and 5-Hydroxymethylcytosine DNA | Zymo Research | Cat#D505 |
| Stealth RNAi Sense Sequence: H. sapiens MAT2A: 5′AUCAAGGACAGCAUCACUGAUUUGG-3′ | Thermo Fisher | Kera et al., 2013 |
| Stealth RNAi Sense Sequence: Control: 5′-AGGAGGAUAUUACCAUGGACAAGAC-3′ | Thermo Fisher | Kera et al., 2013 |
| SUV39H1 RNAi: H. sapiens | Novus Biologicals | H00006839-R01-20nmol |
| SUV39H2 RNAi: H. sapiens | Novus Biologicals | H00079723-R01-20nmol |
| SETDB1 RNAi: H. sapiens | Novus Biologicals | H00009869-R01-20nmol |
| G9a/EHMT2 RNAi: H. sapiens | Novus Biologicals | H00010919-R02-20nmol |
| GLP/EHMT1 RNAi: H sapiens | Novus Biologicals | H00079813-R01-20nmol |
| RT-qPCR Primers | See Table S2 | See Table S2 |
| Software and Algorithms | ||
| Bowtie2-2.1.0 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools 1.5 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| MACS2-2.1.1.20160309 | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/ |
| Bedtools v2.25.0 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| HOMER v4.9 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| GelAnalyzer 2010a | Lazar Software | http://gelanalyzer.com/index.html |
| MAVEN | Melamud et al., 2010; Clasquin et al., 2012 | http://genomics-pubs.princeton.edu/mzroll/index.php |
| MAXQUANT | Cox et al., 2014 | http://www.biochem.mpg.de/5111795/maxquant |
| Trimmomatic v0.39 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| RSEM v1.2.4 | Li et al., 2011 | https://deweylab.github.io/RSEM/ |
| EBSeq v1.24.0 | Leng et al., 2013 | http://bioconductor.org/packages/release/bioc/html/EBSeq.html |
| GSEA 2 | Tamayo et al., 2005 Mootha et al., 2003 |
http://software.broadinstitute.org/gsea/index.jsp |
| Skyline | MacLean et al., 2010 | https://skyline.ms/project/home/software/Skyline/begin.view |
| Other | ||
| Control M. musculus Diet | Envigo | TD.01084 |
| Methionine Deficient M. musculus Diet | Envigo | TD.140119 |