

Abstract

The low tumor permeability of nanomedicines is a major challenge for their application in tumor therapy. Reducing the size of nanomedicines or integrating penetrating peptides has been demonstrated to be very helpful to improve the tumor permeability of nanomedicines. In this paper, poly(amidoamine) (PAMAM) functionalized with the penetrating peptide CRGDK was designed as a drug carrier with a diameter of ∼5 nm. Paclitaxel (PTX) was used as a model drug and covalently linked to the carrier via a biocleavable ester bond. The CRGDK-functionalized drug-loaded nanoparticle exhibited a higher cellular uptake and a higher tumor accumulation and penetration than its nontargeted counterpart, which also endowed the functionalized nanomedicine with a higher antitumor efficiency than its nontargeted counterpart and the clinical Taxol formulation. The good performance of the peptide-bearing PAMAM-based nanomedicine indicates that our strategy is feasible to improve the tumor accumulation and penetration of nanomedicines.
Introduction
Nanoscale drug-delivery systems are very helpful in cancer therapy due to their desirable abilities to increase the water solubility of hydrophobic drugs, carry different drugs for synergistic therapy, reduce unwanted side effects, and enhance the tumor targeting of chemotherapeutic agents.1,2 However, these superior attributes are greatly counteracted by their poor tissue permeability. After reaching tumors, most nanocarriers are confined to the space around tumor vessels and cannot transport drugs uniformly into tumor tissues, greatly limiting their therapeutic effectiveness. The poor permeability of nanocarriers in tumors is mainly caused by the abnormal physiological characteristics of tumors3,4 such as high interstitial pressure, dense interstitial matrix, and dysfunctional vasculature, as well as the relatively large sizes of the nanocarriers.5
Peptides have been widely used for the construction of drug vehicles.6−8 Several types of tumor-penetrating peptides containing a C-terminal R/KXXR/K motif have been developed to improve the tumor penetration of nanocarriers.9 These peptides can specifically bind to neuropilin-1 (Nrp-1), which is overexpressed in many types of tumors.10 It has been demonstrated that either coadministering or conjugating tumor-penetrating peptides to nanocarriers can significantly increase the permeability of nanocarriers into tumor tissues.11−13 For instance, Wei et al. found that the conjugation of CRGDK (a peptide that interacts specifically with Nrp-1) to a doxorubicin-loaded micelle could increase the penetration distance from 0.44 ± 0.38 to 7.16 ± 5.08 μm.14
To achieve a high therapeutic effect, it is important to develop new drug-delivery systems with a high tumor permeability. The in vivo behavior of nanodrug delivery systems is closely related to their morphology and size.15 It is generally considered that the tumor permeability of nanocarriers increases with their decreasing size.16,17 For example, 30 nm CRGDK-bound C-SN38 nanoparticles exhibit significantly enhanced tumor permeability compared to that of their 100 nm counterpart.18 Poly(amidoamine) (PAMAM) is a commercially available dendrimer with a well-defined structure and size of few nanometers. The high tumor permeability of PAMAM has been demonstrated by several published works, and this together with its narrow size distribution and abundant reactive groups makes it very useful in drug- and gene-delivery systems.19−21 Wang et al. improved the tumor accumulation and penetration properties of DOX-loaded PAMAM by conjugating the peptide iRGD to these nanoparticles,22 indicating that for further improvement of the tumor permeability and targeting abilities of PAMAM, combination with penetrating peptides can be a suitable method.
In this work, we developed paclitaxel (PTX)-loaded PAMAM nanoparticles (CK-PAMAM-PTX) bearing the CRGDK peptide on the surface with a diameter of 5.4 ± 1.8 nm. We assumed that the tumor-penetrating peptide and small size of the nanoparticles would endow the nanoparticles with a good tumor penetration ability. As expected, the prepared CK-PAMAM-PTX exhibited a greater cellular uptake, a greater tumor accumulation and penetration, and a greater antitumor effect than the control sample Cys-PAMAM-PTX without the CRGDK peptide.
Results and Discussion
Preparation and Characterization
The synthetic route of CK-PAMAM-PTX is schematically shown in Scheme 1. PTX was covalently linked to PAMAM via the amidation reaction of a succinate-based PTX ester derivative (PTX-COOH) with part of the amide groups in PAMAM. Thereafter, heterobifunctional poly(ethylene glycol) (PEG) Mal-PEG-NHS was introduced by reacting with the remaining amine groups to give PEG-PAMAM-PTX. Finally, the CRGDK peptide was linked to PEG-PAMAM-PTX via reaction of the thiol group in CRGDK with the maleimide groups in PEG-PAMAM-PTX, and the NRP-1-targeted CK-PAMAM-PTX was then obtained after purification by ultrafiltration. The control PAMAM-based drug-delivery system without CRGDK, named Cys-PAMAM-PTX, was synthesized by replacing CRGDK with cysteine, and the PTX-free carrier CK-PAMAM was prepared by omitting the PTX-linking procedure.
Scheme 1. Schematic Preparation Route of PAMAM-PTX, PEG-PAMAM-PTX, CK-PAMAM-PTX, and Fluorescently Labeled CK-PAMAM-PTX.

CK-PAMAM-PTX was structurally characterized by 1H NMR spectroscopy (Figure 1). The peaks at 2.40, 3.51, 2.1–2.2, and 7–8.2 ppm indicate the presence of the PAMAM, PEG, CRGDK, and PTX moieties in the nanoparticles, respectively. It is notable that the proton signals in the spectrum can hardly be resolved due to substantial overlap. We estimated the amounts of PTX, PEG, and CRGDK in CK-PAMAM-PTX by measuring the decrease in the corresponding reactants after each coupling step using high-performance liquid chromatography (HPLC). It was determined that there were approximately 7.5 PTX, 32 PEG, and 10 CRGDK in one PAMAM molecule, and the PTX loading of CK-PAMAM-PTX was calculated to be 7.0 wt %. The presence of PTX in CK-PAMAM-PTX was further confirmed by UV–vis absorption spectroscopy (Figure 2A). The absorption band at approximately 233 nm in the spectrum of CK-PAMAM-PTX is attributable to the absorption of PTX since the spectrum obtained by subtracting the spectrum of CK-PAMAM from that of CK-PAMAM-PTX is consistent with the spectrum of PTX, as shown in Figure 2A. The spectrum after subtraction was used to evaluate the PTX loading of CK-PAMAM-PTX (∼7.6 wt %) based on a preestablished calibration curve, which was close to the loading measured by HPLC.
Figure 1.
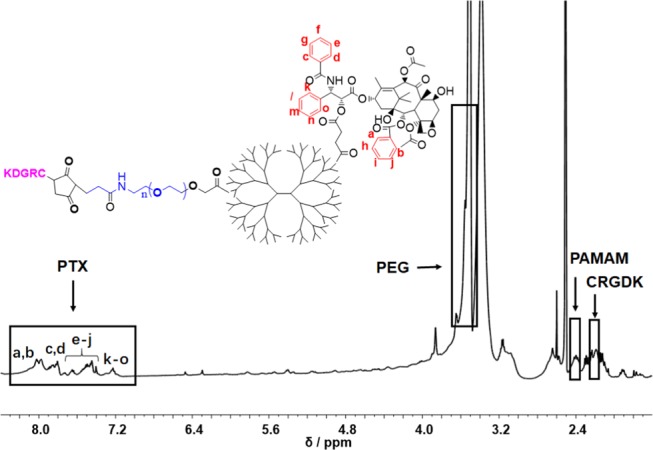
1H NMR spectrum of CK-PAMAM-PTX.
Figure 2.

(A) UV–vis absorbance spectra of CK-PAMAM-PTX, CK-PAMAM, and Taxol in water and the curve obtained by subtracting the spectrum of CK-PAMAM from that of CK-PAMAM-PTX. (B) Hydrodynamic diameter distributions of CK-PAMAM-PTX and PAMAM in distilled water. (C) Typical TEM image of CK-PAMAM-PTX.
The hydrodynamic diameters of the PAMAM-based system in an aqueous medium were measured by DLS. The diameter of CK-PAMAM-PTX was 5.4 ± 1.8 nm, which is approximately 3 nm larger than that of unmodified PAMAM (2.4 ± 1.7 nm) (Figure 2B). The increase in the diameter of CK-PAMAM-PTX versus PAMAM should be due to the attachment of PTX, PEG, and CRGDK on the surface of PAMAM. The morphological structure of CK-PAMAM-PTX was characterized by transmission electron microscopy (TEM, Figure 2C). The TEM image shows that CK-PAMAM-PTX has a spherical shape and shows a mean diameter of ∼3 nm. There are some aggregates as observed by TEM, which may be due to the aggregation caused by the evaporation of water during the preparation of the TEM sample. The ζ-potential of CK-PAMAM-PTX in deionized water was 3.32 ± 0.01. The slightly positive charge may be due to the lysine unit in the CRGDK peptide.
In Vitro Drug Release
The in vitro PTX release profiles of CK-PAMAM-PTX in different media are shown in Figure 3. It can be seen that in phosphate-buffered saline (PBS) at pH 7.4, PTX is released very slowly, and less than 5% of PTX is released over the 72 h monitoring duration, indicating the relatively high stability of the ester linkage in PBS. In contrast, a much faster release of PTX was found in fetal bovine serum (FBS), and approximately 68% of PTX is released within 72 h, which may be attributable to the presence of esterase in FBS.23
Figure 3.

In vitro PTX release profiles of CK-PAMAM-PTX in PBS (0.1 M, pH 7.4) and fetal bovine serum at 37 °C.
In Vitro Cellular Uptake
To study the in vitro cellular uptake of the PAMAM-based drug-delivery system, CK-PAMAM-PTX and Cys-PAMAM-PTX were covalently labeled with fluorescein isothiocyanate (FITC) (Scheme 1), followed by tracing their cellular uptakes in the human breast cancer cell line MDA-MB-231 by confocal laser scanning microscopy (CLSM) after 1 h of incubation. The CLSM images show that the fluorescence intensity of CK-PAMAM-PTX in MDA-MB-231 cells is remarkably higher than that of Cys-PAMAM-PTX and the competition group (Figure 4A). The cellular association of CK-PAMAM-PTX was further quantitatively analyzed by flow cytometry (Figure 4B). MDA-MB-231 cells treated with CK-PAMAM-PTX showed a rightward shift compared to the cells treated with Cys-PAMAM-PTX, further demonstrating the higher cellular association of CK-PAMAM-PTX. The mean fluorescence intensity of the cells treated with CK-PAMAM-PTX was ∼2.2-fold higher than that of Cys-PAMAM-PTX-treated cells. After treatment with free CRGDK peptide, the cellular association of CK-PAMAM-PTX decreased remarkably compared to that in the noncompetition group, which indicates that the CRGDK peptide contributed to the targeting effect of CK-PAMAM-PTX (Figure 4C). These results demonstrate that the CRGDK peptide can significantly improve the cellular uptake of the PAMAM vehicle.
Figure 4.
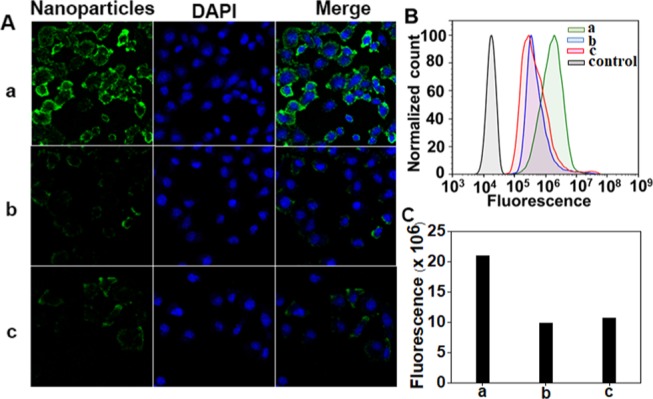
(A) CLSM images and (B) flow cytometry data of the in vitro cellular association of CK-PAMAM-PTX, Cys-PAMAM-PTX, and CRGDK + CK-PAMAM-PTX in MDA-MB-231 cells. (C) Fluorescence intensity of CK-PAMAM-PTX, Cys-PAMAM-PTX, and CK-PAMAM-PTX in cells quantified by flow cytometry: (a) CK-PAMAM-PTX, (b) Cys-PAMAM-PTX, and (c) CRGDK + CK-PAMAM-PTX.
In Vitro Cytotoxicity
To estimate the pharmacological activity of CK-PAMAM-PTX, the in vitro cytotoxicity of CK-PAMAM-PTX against MDA-MB-231 cells was measured with the positive controls of Cys-PAMAM-PTX and commercially available Taxol (Figure 5A). As shown in Figure 5A, both CK-PAMAM-PTX and Cys-PAMAM-PTX exhibited a lower cytotoxicity than Taxol at equal PTX concentrations, which may be attributed to the slow release of PTX from the PAMAM-based drug-delivery systems. Furthermore, the cytotoxicity of Cys-PAMAM-PTX is slightly lower than that of CK-PAMAM-PTX, which may be due to the enhanced cellular uptake of CK-PAMAM-PTX. In addition, the cytotoxicity of the empty carrier CK-PAMAM was measured against MDA-MB-231 tumor cells and noncancerous L929 fibroblasts. As shown in Figure 5B, CK-PAMAM did not show significant cytotoxicity to either cell line at concentrations up to 400 μg/mL, indicating the desirable cytocompatibility of the carrier.
Figure 5.

(A) In vitro cytotoxicity of CK-PAMAM-PTX, Cys-PAMAM-PTX, and Taxol against MDA-MB-231 cells. (B) In vitro cytotoxicity of CK-PAMAM against MDA-MB-231 and L929 cells.
In Vivo Tumor Accumulation
To further evaluate the tumor-targeting ability of CK-PAMAM-PTX in vivo, CK-PAMAM-PTX and Cys-PAMAM-PTX were labeled with Cy5.5 and injected into MDA-MB-231 tumor-bearing mice via the tail vein, respectively. Noninvasive near-infrared (NIR) fluorescence imaging was conducted 1, 2, 4, 8, and 24 h after injection of the nanoparticles. As shown in Figure 6A, fluorescence signals could be observed in tumors at 1 h post injection and sustained for up to 24 h for both CK-PAMAM-PTX and Cys-PAMAM-PTX. For both samples, the fluorescence signals in tumors increased to a maximum at approximately 4 h post injection, which is similar to the cases of PAMAM modified with iRGD and BSA.24 The ratios of the signal intensities of tumor to normal tissue (T/N ratios) at different time points after injection of CK-PAMAM or Cys-PAMAM-PTX were measured. As shown in Figure 6B, the T/N ratios from CK-PAMAM-PTX in the tumor were significantly higher than those from Cys-PAMAM-PTX from 2 to 24 h post injection, indicating that the CRGDK peptide can truly improve the tumor-targeting ability of the PAMAM-based drug-delivery system in vivo.
Figure 6.

(A) In vivo NIR fluorescence imaging of nude mice bearing MDA-MB-231 tumors at 1, 2, 4, 8, and 24 h post injection of CK-PAMAM-PTX and Cys-PAMAM-PTX with a dose of 7.5 mg/kg PTX equiv. Fluorescence intensity was normalized to the same scale. (B) T/N ratios at different time points after injection of CK-PAMAM-PTX and Cys-PAMAM-PTX with a dose of 7.5 mg/kg PTX equiv. (*P < 0.05, n = 3).
To further study the tumor accumulation and biodistribution of the nanoparticles, the mice were sacrificed immediately after in vivo imaging at 24 h post injection, and the tumor tissues, livers, kidneys, spleens, hearts, and lungs were excised and imaged (Figure 7A). For the mice treated with CK-PAMAM-PTX, the tumor showed the strongest fluorescence intensity among all of the tested samples, and the liver and kidney showed stronger fluorescence intensity than other organs, indicating that CK-PAMAM-PTX was excreted mainly by the renal and hepatobiliary pathways. The total fluorescence intensity of each organ and tissue was measured to evaluate the biodistribution of the nanoparticles (Figure 7B). The fluorescence intensity of the tumors treated with CK-PAMAM-PTX was ∼2-fold higher than that of the tumors treated with Cys-PAMAM-PTX, confirming the higher tumor accumulation in CK-PAMAM-PTX compared to Cys-PAMAM-PTX.
Figure 7.

(A) Ex vivo NIR fluorescence imaging of dissected tumors and main organs of the nude mice bearing MDA-MB-231 tumors after 24 h of in vivo imaging. (B) Fluorescence signals of the excised organs (*P < 0.05, n = 3).
Tumor Penetration
To study the tumor permeability of CK-PAMAM-PTX, the microdistribution of CK-PAMAM-PTX and Cys-PAMAM-PTX in ex vivo tumor sections was measured (Figure 8A). A higher fluorescence intensity was observed on the interface of the tumor and muscle tissues for both nanoparticles, indicating that the nanoparticles preferred to accumulate on the surface of the tumor. Furthermore, the fluorescence intensity in the tumors treated with CK-PAMAM-PTX was obviously higher than the intensity in the tumors treated with Cys-PAMAM-PTX, and the signal distribution was more homogeneous. To further assess the tissue permeability of CK-PAMAM-PTX, immunofluorescence analysis of the treated tumor tissue was performed. As shown in Figure 8B, CK-PAMAM-PTX can penetrate deeply into the tumor after extravasation from the tumor blood vessels, whereas Cys-PAMAM-PTX mostly stays around the tumor vessels, indicating that conjugation with the penetrating peptide CRGDK can significantly improve the tissue permeability of the nanomedicine.
Figure 8.
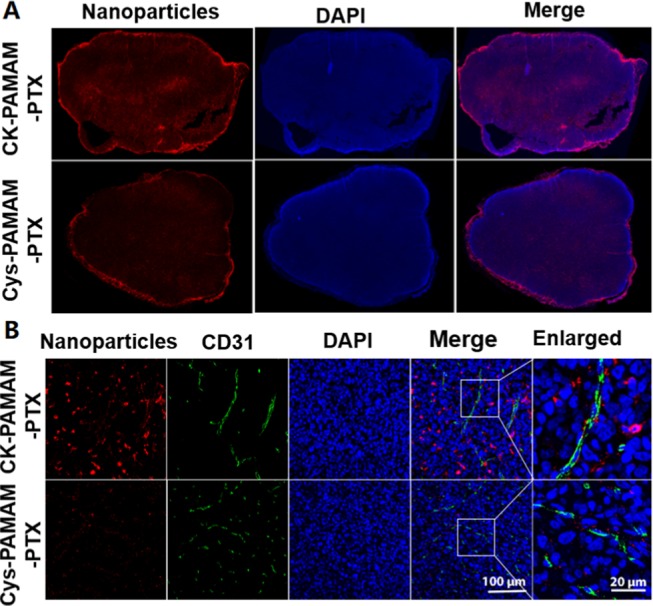
(A) Fluorescence images of MDA-MB-231 tumor sections from nude mice 24 h post injection of CK-PAMAM-PTX and Cys-PAMAM-PTX. (B) Immunofluorescence staining of CD31 in the tumor tissues.
In Vivo Antitumor Effects
The in vivo antitumor effect of CK-PAMAM-PTX was studied with subcutaneous MDA-MB-31 tumor-bearing BALB/c nude mice as model animals. The antitumor performance of CK-PAMAM-PTX was compared to the performance of Cys-PAMAM-PTX, and the clinical Taxol formulation with the PTX dose normalized to be 7.5 mg/kg of body weight in each group. The CK-PAMAM- and PBS-treated groups were used as negative controls. The antitumor effects of the nanoparticles were quantified based on the relative tumor volume. As shown in Figure 9A, all three PTX formulations exhibited a significant tumor inhibition compared to the control groups (CK-PAMAM and saline injection). Among all of the animal groups, the group treated with CK-PAMAM-PTX showed the slowest tumor growth rate. The relative tumor growth inhibition (TGI) of CK-PAMAM-PTX was calculated to be 56% on the 13th day, which was significantly higher than that of Cys-PAMAM-PTX (39%) or clinical Taxol (33%). The improved TGI of CK-PAMAM-PTX should be attributed to its higher tumor accumulation and penetration. Based on the body weights of the mouse groups over time (Figure 9B), it can be concluded that the doses of the PTX formulations were well tolerated and that the PAMAM-based carriers do not show significant toxicity to the test mice.
Figure 9.

(A) In vivo antitumor effectiveness of each treated group, expressed as the average value of the relative tumor volume v/v0 (*P < 0.05 versus Cys-PAMAM-PTX injection group, n = 6; the day of injection was set as day 0). (B) Evolution over time of the body weights of each group during the experiments.
Conclusions
In summary, a strategy for improving the tumor accumulation and penetration of chemotherapeutic drugs was developed using CRGDK-functionalized PAMAM as a nanovehicle. In this work, PTX was employed as a model drug and covalently conjugated to the surface of PAMAM via a biocleavable ester bond. The penetrating peptide CRGDK was introduced through PEG. The introduction of CRGDK significantly improved the cellular uptake and tumor accumulation and penetration of the nanomedicine, which were fully demonstrated by CLSM, flow cytometry, and in vivo and tumor section fluorescence imaging, and endowed CK-PAMAM-PTX with a significantly higher antitumor effect than Cys-PAMAM-PTX and clinical Taxol.
Experimental Section
Materials
PAMAM dendrimers (G4), FITC, 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide (EDC·HCl), and N-hydroxysuccinimide (NHS) were obtained from Sigma-Aldrich. N-hydroxysulfo-succinimide-polyoxyethylene-maleimide (NHS-PEG-MAL, MW 2000) was purchased from JenKem Technology Co., Ltd. The CRGDK peptide was obtained from GL Biochem Ltd. (Shanghai, China). Sulfo-Cyanine 5.5 NHS ester (Cy5.5-NHS) was purchased from GE Healthcare. PTX was purchased from J&K Scientific Ltd. and was modified with succinic anhydride to give a carboxyl acid group at the C–2′-OH position.25 Clinical Taxol (paclitaxel) injection was obtained from Haikou Pharmaceutical Factory Co., Ltd. Dulbecco’s modified Eagle’s medium (DMEM), FBS, trypsin-EDTA, and the antibiotic solution were purchased from Invitrogen Life Technologies.
Characterization
Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker DRX-300 spectrometer. UV–vis absorption spectra were measured with a Shimadzu UV-3600 spectrophotometer. TEM was performed using a JEM-2100 microscope operating at an accelerating voltage of 120 kV. The hydrodynamic size and ζ-potential were measured on a ζ-potential analyzer (Brookhaven ZetaPlus).
Preparation of CK-PAMAM-PTX
PTX-COOH (20 mg, 0.02 mmol) and PAMAM (20 mg, 0.0014 mmol) were dissolved in 0.5 mL of anhydrous dimethyl sulfoxide (DMSO) in the presence of EDC·HCl (4 mg, 0.02 mmol) and NHS (2.4 mg, 0.02 mmol). After stirring for 2 h, the crude product was purified by precipitation from DMSO into diethyl ether/acetone (4:1, v/v) three times. The obtained product and 180 mg of Mal-PEG-NHS (MW 2000, 0.09 mmol) were dissolved in anhydrous DMSO and stirred overnight to give PEG-PAMAM-PTX, which was purified by dialysis against PBS (0.01 M, pH 6.8) for 2 h and then by ultrafiltration (Millipore Amicon Ultra-15 centrifugal filter tube, 3000 NMWL) to remove the impurities. After lyophilization of PEG-PAMAM-PTX, 50 mg of PEG-PAMAM-PTX and 5 mg of CRGDK (0.009 mmol) were dissolved in 0.5 mL of PBS (0.01 M, pH 7.4) and stirred at room temperature overnight. The crude product was purified by ultrafiltration to give the NRP-1-targeted drug-delivery nanoparticle CK-PAMAM-PTX.
Preparation of Cys-PAMAM-PTX
PEG-PAMAM-PTX (50 mg) and l-cysteine (1 mg, 0.009 mmol) were dissolved in 0.5 mL of PBS (0.01 M, pH 7.4) and stirred at room temperature overnight. The crude product was purified by ultrafiltration to give nontargeted Cys-PAMAM-PTX.
Preparation of FITC-Labeled CK-PAMAM-PTX and Cys-PAMAM-PTX
PTX-COOH (10 mg, 0.01 mmol), PAMAM (10 mg, 0.0007 mmol), EDC·HCl (2 mg, 0.01 mmol), and NHS (1.2 mg, 0.01 mmol) were dissolved in 0.5 mL of anhydrous DMSO and stirred at room temperature for 2 h. The crude product was purified by precipitation from DMSO into diethyl ether/acetone (4:1, v/v) three times. The obtained product and 1 mg of FITC (0.0028 mmol) were dissolved in 0.5 mL of anhydrous DMSO and stirred for 1 h. Then, 90 mg of Mal-PEG-NHS (MW 2000, 0.045 mmol) was added to the resulting solution, and the mixture was stirred overnight to give FITC-labeled PEG-PAMAM-PTX, which was purified by dialysis against PBS (pH 6.8) for 2 h and then by ultrafiltration (Millipore Amicon Ultra-15 centrifugal filter tube, 3000 NMWL) to remove the impurities. FITC-labeled CK-PAMAM-PTX and Cys-PAMAM-PTX were prepared following the same procedure as the nonlabeled nanomedicines.
Preparation of Cy5.5-Labeled CK-PAMAM-PTX and Cys-PAMAM-PTX
PTX-COOH (10 mg, 0.01 mmol), PAMAM (10 mg, 0.0007 mmol), EDC·HCl (2 mg, 0.01 mmol), and NHS (1.2 mg, 0.01 mmol) were dissolved in 0.5 mL of anhydrous DMSO and stirred at room temperature for 2 h. The crude product was purified by precipitation from DMSO into diethyl ether/acetone (4:1, v/v) three times. The obtained product and 2 mg of Cy5.5-NHS (0.0028 mmol) were dissolved in 0.5 mL of anhydrous DMSO and stirred for 1 h. Then, Cy5.5-labeled CK-PAMAM-PTX and Cys-PAMAM-PTX were prepared following the same procedures as the FITC-labeled nanomedicines.
Preparation of CK-PAMAM
PAMAM (10 mg, 0.0007 mmol) and Mal-PEG-NHS (110 mg, 0.055 mmol, MW 2000) were dissolved in anhydrous DMSO and stirred overnight to give PEG-PAMAM, which was purified by dialysis against PBS (pH 6.8) for 2 h and then by ultrafiltration (Millipore Amicon Ultra-15 centrifugal filter tube, 3000 NMWL) to remove the impurities. After lyophilization of PEG-PAMAM, 50 mg of PEG-PAMAM-PTX and 5 mg of CRGDK (0.009 mmol) were dissolved in 0.5 mL of PBS (0.01 M, pH 7.4) and stirred at room temperature overnight. The crude product was purified by ultrafiltration to give the drug carrier CK-PAMAM.
Chemical Component Analysis of CK-PAMAM-PTX
The chemical components of CK-PAMAM-PTX were analyzed by measuring the residual reactants in each reaction step by HPLC. PTX-COOH (5 mg, 0.005 mmol) and PAMAM (5 mg, 0.0004 mmol) were dissolved in 0.2 mL of anhydrous DMSO in the presence of EDC·HCl (1 mg, 0.005 mmol) and NHS (0.6 mg, 0.005 mmol). After stirring for 2 h, the crude product was precipitated by dropping into 5 mL of a diethyl ether/acetone (4:1, v/v) solution, and the obtained precipitate was further purified by washing with diethyl ether/acetone (4:1, v/v) three times. All of the supernatants were collected after centrifugation for further HPLC analysis of the residual PTX-COOH. The obtained product was reacted with 45 mg of Mal-PEG-NHS (MW 2000, 0.022 mmol) in anhydrous DMSO and stirred overnight to give PEG-PAMAM-PTX, which was precipitated with diethyl ether/acetone (4:1, v/v), and the precipitate was dissolved in purified water and further purified by ultrafiltration (Millipore Amicon Ultra-15 centrifugal filter tube, 3000 NMWL) three times. The supernatant and filtrates were collected for further HPLC analysis of the residual PEG. After lyophilization of PEG-PAMAM-PTX, 25 mg of PEG-PAMAM-PTX and 2.5 mg of CRGDK (0.0004 mmol) were dissolved in 0.5 mL of PBS (0.01 M, pH 7.4) and stirred at room temperature overnight. The crude product was purified by ultrafiltration to give the NRP-1-targeted drug-delivery nanoparticle CK-PAMAM-PTX, and the filtrate was collected for HPLC analysis of CRGDK.
Chromatographic separation was performed on a Waters HPLC system with a C18 column (4.6 × 150 mm2, 5 μm, C18, Sunfire Technologies). The flow rate was set to 1 mL/min, and the detection wavelength was 228 nm. For the analysis of the residuals in the PTX and PEG solution, the mobile phase consisted of 1/1 double-distilled water/acetonitrile (HPLC grade, Anaour Chemicals Supply) and the retention times of PTX-COOH and NHS-PEG-Mal were 9.47 and 1.3–1.48 min, respectively. The concentrations of PTX-COOH, NHS-PEG-Mal, and CRGDK were determined by preestablished calibration curves. The amounts of the unreacted PTX-COOH, NHS-PEG-Mal, and CRGDK were calculated by multiplication of the calculated concentration and the volume of the supernatants or filtration solution. The loading of PTX-COOH, NHS-PEG-Mal, and CRGDK on PAMAM was calculated by subtracting the unreacted reagent from the starting reactant.
Drug Loading into CK-PAMAM-PTX
The amount of PTX in CK-PAMAM-PTX was also analyzed by spectrophotometry. A calibration curve of PTX was established by plotting the absorbance of PTX solutions at 233 nm versus concentration. The absorbance of PTX in CK-PAMAM-PTX was determined by subtracting the background absorbance of a sample of CK-PAMAM, and the PTX loading of CK-PAMAM-PTX was calculated according to the preestablished calibration curve.
In Vitro Release of PTX
CK-PAMAM-PTX (6 mg) was dispersed in 0.5 mL of pH 7.4 PBS or FBS and dialyzed against 20 mL of the corresponding release medium (containing 0.1% V/V Tween 80) at 37 °C. At predetermined time points, aliquots (0.5 mL) were withdrawn, and PTX was quantified with the HPLC method described above.
In Vitro Cell Uptake and Competition Experiments
MDA-MB-231 cells were seeded into 35 mm glass-bottom culture dishes and allowed to adhere overnight. Then, the cells were incubated with FITC-labeled CK-PAMAM-PTX or Cys-PAMAM-PTX in DMEM containing 10% FBS for 1 h or with 0.5 mg/mL free CRGDK for 0.5 h and then treated with FITC-labeled CK-PAMAM-CK for another 1 h. After washing with PBS, fixing with 4% paraformaldehyde, and staining with DAPI, the cells were examined by CLSM (Olympus, FV 1000).
The quantitative analysis of cellular uptake was performed by flow cytometry. MDA-MB-231 cells were seeded in a six-well plate and allowed to adhere overnight. After incubation with FITC-labeled CK-PAMAM-PTX and Cys-PAMAM-PTX for 1 h, the cells were trypsinized, washed, and detected with a flow cytometer.
In Vitro Cytotoxicity
MDA-MB-231 and L929 cells were seeded in 96-well plates at a density of 5000 cells per well. After incubation for 24 h, MDA-MB-231 cells were incubated with CK-PAMAM-PTX, Cys-PAMAM-PTX, and clinical Taxol at PTX concentrations from 0.8 to 25 μg/mL, and MDA-MB-231 and L929 cells were incubated with the empty CK-PAMAM carrier at concentrations from 12.5 to 400 μg/mL. After 24 h, 10 μL of CCK8 solution was added to each well followed by incubation for another 1 h. The absorbance was measured at 450 nm with a microplate reader.
In Vivo and Ex Vivo Fluorescence Imaging
All animal experiments were performed in compliance with the guidelines set by the Animal Care Committee of Southeast University. The mouse model was established by subcutaneously injecting MDA-MB-231 tumor cells (5 × 106 cells per mouse) into the proximal thigh region of female BALB/c nude mice. The tumor-bearing mice were divided into two groups (n = 3), which were intravenously injected with either Cy5.5-labeled CK-PAMAM-PTX or Cys-PAMAM-PTX through the tail vein. In vivo fluorescence imaging was performed with a fluorescence imaging system at scheduled time points after injection of CK-PAMAM-PTX or Cys-PAMAM-PTX. The T/N ratio of the in vivo fluorescence images was calculated using the following formula: T/N ratio = SIT/SIM, where SIT is the average signal intensity of the tumor and SIM is the average signal intensity of the contralateral thigh muscle. The signal intensity was determined using region of interest (ROI) functions of Maestro 2.10.0 software. The mice were sacrificed 24 h post injection of the nanoparticles. The tumor tissues and main organs were harvested to be imaged with the fluorescence imaging system.
Tumor Penetration
After in vivo fluorescence imaging, the tumor tissues were collected, processed routinely into paraffin, and sectioned at a thickness of 4 μm. After staining with DAPI, the sections were subjected to fluorescence microscopy analysis (Olympus, VS120). Fluorescence images of the full-scale tumor sections were taken with a fixed exposure time to show the micro-biodistribution of the nanoparticles within the tumors. The fluorescence profile was analyzed by ImageJ. To visualize the tumor blood vessels, we stained the tumor sections with a CD31 primary antibody and Alexa Fluor 488-conjugated secondary antibody.
In Vivo Tumor Inhibition
The mouse model was established by subcutaneously injecting MDA-MB-231 tumor cells (5 × 106 cells per mouse) into the armpit of female BALB/c nude mice. When the tumor reached a mean volume of 150 mm3, the mice were divided into five groups (n = 6) and were injected intravenously with different formulations of PTX (CK-PAMAM-PTX, Cys-PAMAM-PTX, and clinical Taxol), CK-PAMAM, and neat PBS (used as a negative control). The injection dose of all of the PTX formulations was normalized to be equivalent to 7.5 mg/kg PTX. The tumor volumes were measured every 2 days and calculated using the following formula: tumor volume = (width)2 × length/2. The body weights of the tested mice were also measured. Tumor growth inhibition (TGI) was calculated by the following equation
where V̅ is the average of the relative tumor volume.
Statistical Analysis
Student’s t-test was used to assess the statistical significance of differences, and P values of less than 0.05 were considered statistically significant.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 81571789, 81602728, and 81671796), Science Fund for Creative Research Groups of the National Natural Science Foundation of China (61821002), and Clinical Innovation Center of Medical Imaging and Interventional Radiology (No. YXZXA2016005).
Author Contributions
The paper was written through contributions of all the authors. All the authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Wilhelm S.; Tavares A. J.; Dai Q.; Ohta S.; Audet J.; Dvorak H. F.; Chan W. C. W. Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater. 2016, 1, 16014 10.1038/natrevmats.2016.14. [DOI] [Google Scholar]
- Wang X.; Wang Y.; Chen Z. G.; Shin D. M. Advances of Cancer Therapy by Nanotechnology. Cancer Res. Treat. 2009, 41, 1–11. 10.4143/crt.2009.41.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin C. H.; Rubin K.; Pietras K.; stman A. Ö. A High interstitial fluid pressure - an obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- Khawar I. A.; Kim J. H.; Kuh H. J. Improving drug delivery to solid tumors: priming the tumor microenvironment.. J. Controlled Release 2015, 201, 78–79. 10.1016/j.jconrel.2014.12.018. [DOI] [PubMed] [Google Scholar]
- Ji J.; Ma F.; Zhang H.; Liu F.; He J.; Li W.; Xie T.; Zhong D.; Zhang T.; Tian M.; Zhang H.; Santos H. A.; Zhou M. Light-Activatable Assembled Nanoparticles to Improve Tumor Penetration and Eradicate Metastasis in Triple Negative Breast Cancer. Adv. Funct. Mater. 2018, 28, 1801738 10.1002/adfm.201801738. [DOI] [Google Scholar]
- Raza F.; Zafar H.; You X.; Khan A.; Wu J.; Ge L. Cancer nanomedicine: focus on recent developments and self-assembled peptide nanocarriers. J. Mater. Chem. B 2019, 7, 7639–7655. 10.1039/C9TB01842E. [DOI] [PubMed] [Google Scholar]
- Raza F.; Zhu Y.; Chen L.; You X.; Zhang J.; Khan A.; Khan M. W.; Hasnat M.; Zafar H.; Wu J.; Ge L. Paclitaxel-loaded pH responsive hydrogel based on self-assembled peptides for tumor targeting. Biomater. Sci. 2019, 7, 2023–2036. 10.1039/C9BM00139E. [DOI] [PubMed] [Google Scholar]
- Raza F.; Zafar H.; Zhu Y.; Ren Y.; Ullah A.; Khan A. U.; He X.; Han H.; Aquib M.; Boakye-Yiadom K. O.; Ge L. A Review on Recent Advances in Stabilizing Peptides/Proteins upon Fabrication in Hydrogels from Biodegradable Polymers. Pharmaceutics 2018, 10, 16 10.3390/pharmaceutics10010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspel N.; Zanuy D.; Nussinov R.; Teesalu T.; Ruoslahti E.; Aleman C. Binding of a C-End Rule Peptide to the Neuropilin-1 Receptor: A Molecular Modeling Approach. Biochemistry 2011, 50, 1755–1762. 10.1021/bi101662j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielenberg D. R.; Pettaway C. A.; Takashima S.; Klagsbrun M. Neuropilins in neoplasms: expression, regulation, and function. Exp. Cell Res. 2006, 312, 584–593. 10.1016/j.yexcr.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Lambert S.; Bouttier M.; Vassy R.; Seigneuret M.; Petrow-Sadowski C.; Janvier S.; Heveker N.; Ruscetti F. W.; Perret G.; Jones K. S.; Pique C. HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF(165). Blood 2009, 113, 5176–5185. 10.1182/blood-2008-04-150342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberici L.; Lise R.; Kazuki N. S.; Lilach A.; Venkata R. K.; Tambet T.; Claudio B.; Catia T.; Rizzardi G. P.; Ruoslahti E. De Novo Design of a Tumor-Penetrating Peptide. Cancer Res. 2013, 73, 804–812. 10.1158/0008-5472.CAN-12-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Huang H.; Wang D.; Lu H.; Chen J.; Chai Z.; Yao S. Q.; Hu Y. Light-Triggered PEGylation/dePEGylation of the Nanocarriers for Enhanced Tumor Penetration. Nano Lett. 2019, 19, 3671–3675. 10.1021/acs.nanolett.9b00737. [DOI] [PubMed] [Google Scholar]
- Wei T.; Liu J.; Ma H.; Cheng Q.; Huang Y.; Zhao J.; Huo S.; Xue X.; Liang Z.; Liang X. J. Functionalized Nanoscale Micelles Improve Drug Delivery for Cancer Therapy in Vitro and in Vivo. Nano Lett. 2013, 13, 2528–2534. 10.1021/nl400586t. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; You X.; Guan S.; Huang H.; Wang L.; Zhang J.; Wu J. Poly(Ferulic Acid) with an Anticancer Effect as a Drug Nanocarrier for Enhanced Colon Cancer Therapy. Adv. Funct. Mater. 2019, 29, 1808646. [Google Scholar]
- Tang L.; Gabrielson N. P.; Uckun F. M.; Fan T. M.; Cheng J. Size-Dependent Tumor Penetration and in Vivo Efficacy of Monodisperse Drug-Silica Nanoconjugates. Mol. Pharmaceutics 2013, 10, 883–892. 10.1021/mp300684a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco E.; Shen H.; Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xun L.; Si J.; Zhang Qi.; Huang Q.; Danxia G.; Yang H.; Chen X.; Shen Y.; Sui M. Functionalized Nanoparticles Efficiently Enhancing the Targeted Delivery, Tumor Penetration, and Anticancer Activity of 7-Ethyl-10-Hydroxycamptothecin. Adv. Healthcare Mater. 2018, 7, 1701140. 10.1002/adhm.201701140. [DOI] [PubMed] [Google Scholar]
- Min H.; Huang-Fu M. Y.; Guo W. W.; Guo N. N.; Chen J.; Liu H. N.; Xie Z. Q.; Lin M. T.; Wei Q. C.; Gao J. Q. MMP-2-Sensitive HA End-Conjugated Poly(amidoamine) Dendrimers via Click Reaction To Enhance Drug Penetration into Solid Tumor. ACS Appl. Mater. Interfaces 2017, 9, 42459–42470. 10.1021/acsami.7b10098. [DOI] [PubMed] [Google Scholar]
- Li H. J.; Du J. Z.; Liu J.; Du X. J.; Shen S.; Zhu Y. H.; Wang X. Y.; Ye X.; Nie S.; Wang J. Smart Superstructures with Ultrahigh pH-Sensitivity for Targeting Acidic Tumor Microenvironment: Instantaneous Size Switching and Improved Tumor Penetration. ACS Nano 2016, 10, 6753–6761. 10.1021/acsnano.6b02326. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Park E. J.; Na D. H. Recent progress in dendrimer-based nanomedicine development. Arch. Pharm. Res. 2018, 41, 571–582. 10.1007/s12272-018-1008-4. [DOI] [PubMed] [Google Scholar]
- Wang K.; Zhang X.; Liu Y.; Liu C.; Jiang B.; Jiang Y. Tumor penetrability and anti-angiogenesis using iRGD-mediated delivery of doxorubicin-polymer conjugates. Biomaterials 2014, 35, 8735–8747. 10.1016/j.biomaterials.2014.06.042. [DOI] [PubMed] [Google Scholar]
- Yu S.; Zhang Y.; Wang X.; Zhen X.; Zhang Z.; Wu W.; Jiang X. Synthesis of Paclitaxel-Conjugated β-Cyclodextrin Polyrotaxane and Its Antitumor Activity. Angew. Chem., Int. Ed. 2013, 52, 7272–7277. 10.1002/ange.201301397. [DOI] [PubMed] [Google Scholar]
- Cao J.; Ge R.; Zhang M.; Xia J.; Han S.; Lu W.; Liang Y.; Zhang T.; Sun Y. A triple modality BSA-coated dendritic nanoplatform for NIR imaging, enhanced tumor penetration and anticancer therapy. Nanoscale 2018, 10, 9021–9037. 10.1039/C7NR09552J. [DOI] [PubMed] [Google Scholar]
- Deutsch H. M.; Glinski J. A.; Hernandez M.; Haugwitz R. D.; Narayanan V. L.; Suffness M.; Zalkow L. H. Synthesis of Congeners and Prodrugs. 3. Water-soluble Prodrugs of Taxol with Potent Antitumor Activity. J. Med. Chem. 1989, 32, 788–792. 10.1021/jm00124a011. [DOI] [PubMed] [Google Scholar]