

Graphical abstract
Keywords: RNA-seq, Metabolism, Galaxy, Sample stratification, TCGA
Abstract
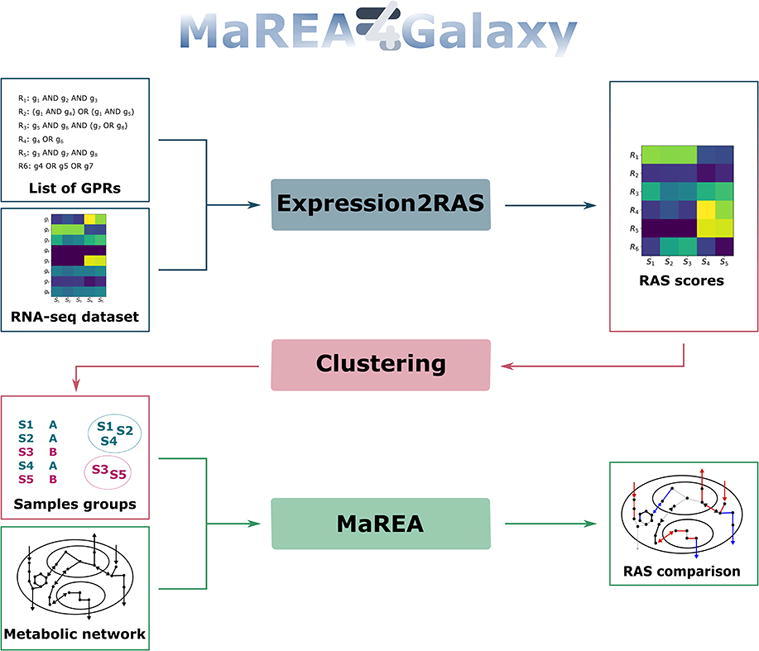
We present MaREA4Galaxy, a user-friendly tool that allows a user to characterize and to graphically compare groups of samples with different transcriptional regulation of metabolism, as estimated from cross-sectional RNA-seq data. The tool is available as plug-in for the widely-used Galaxy platform for comparative genomics and bioinformatics analyses. MaREA4Galaxy combines three modules. The Expression2RAS module, which, for each reaction of a specified set, computes a Reaction Activity Score (RAS) as a function of the expression level of genes encoding for the associated enzyme. The MaREA (Metabolic Reaction Enrichment Analysis) module that allows to highlight significant differences in reaction activities between specified groups of samples. The Clustering module which employs the RAS computed before as a metric for unsupervised clustering of samples into distinct metabolic subgroups; the Clustering tool provides different clustering techniques and implements standard methods to evaluate the goodness of the results.
1. Introduction
In the last recent years, life sciences have witnessed a renewed focus on phenotype level phenomena. Accordingly, there has been an increasing attention towards cellular metabolism, which is regarded as the ultimate level of phenotype, reflecting the response of biological systems to regulatory and environmental changes.
Alteration of metabolic processes plays a pivotal role in many pathologies, such as cancer, metabolic syndromes, neurodegenerative diseases [10], [16], as well as in aging processes [12].
Although quantification of metabolites has become more and more feasible [9], the difficulty of inferring changes in metabolic pathways based on metabolomics data [3] is pushing the need to understand metabolic alterations by leveraging gene expression data.
To this end, computational strategies are being proposed to integrate –omics data into metabolic networks [13], [18], [14]. Within this context, we have recently introduced the pipeline MaREA (Metabolic Reaction Enrichment Analysis) [8]. MaREA characterizes the metabolic disregulations that distinguish sets of individuals, by projecting RNA-seq data onto metabolic networks, without requiring explicit metabolic measurements.
MaREA computes a Reaction Activity Score (RAS) for each metabolic reaction and each sample/individual, based on the read count of the set of genes that encode the catalyzing enzyme(s). The scores are first used as features for cluster analysis and then to visualize the metabolic disregulations that distinguish the identified clusters, in a form understandable to life scientists.
In [8], we have demonstrated that MaREA can efficiently stratify cancer patients according to their metabolic activity, as proven by significantly different survival expectancy. Moreover, MaREA proved to be able to readily capture metabolic differences between two conditions, such as the properties that distinguish normal from tumor samples.
The MaREA pipeline is highly versatile and can be applied to virtually any study aiming at comparing the metabolism of samples in different conditions or experimental settings.
In [8], we released a MATLAB-based tool that implements the methodology. However, some technical barriers current limit the application of the pipeline: MATLAB is a proprietary software and many life scientists do not have the software licence; moreover, the tool is not web-based and, therefore, it requires local resources; also, most life scientists are not familiar with the MATLAB environment.
To overcome these limitations, we present here the freely available open source web-based tool MaREA4Galaxy which embeds the MaREA pipeline within the widely-used platform Galaxy [1].
Galaxy is a user-friendly web-based workflow system that allows biomedical researchers to use computational biology tools even without sophisticated computer science skills.
As compared to other user-friendly web-based metabolic network visualization tools, such as Escher [11] or Fame [2], which mainly focus on Flux Balance Analysis, MaREA4Galaxy specifically enables metabolic reaction enrichment analysis of gene expression data which may have been obtained directly within the Galaxy environment, for instance by using Galaxy tools to produce read counts from raw RNA-seq data. MaREA4Galaxy automatically recognizes most common gene nomenclature systems. It also enables cluster analysis of samples based on reaction activities, as well as on any other features. The cluster analysis module implements new functionalities, as compared to the previously released MATLAB-based MaREA tool. New clustering algorithms have been included, as well as new instruments for the evaluation of clustering goodness and for the selection of optimal number of clusters.
Moreover, MaREA4Galaxy inherits the benefits of Galaxy. In particular, Galaxy allows to build multi-step computational analyses. It allows users to upload their own data, as well as to interface with public databases, and enables researchers to perform the text manipulation required to properly format data for analysis without requiring advanced programming skills. Galaxy can be downloaded, customized and installed either locally or on a dedicated server. It also provides a comprehensive documentation.
In order to illustrate the functionalities of MaREA4Galaxy, we show a novel example on real data obtained from The Cancer Genome Atlas (TCGA) [17]. In particular, we perform an unsupervised cluster analysis of the gene expression of liver hepatocellular carcinoma tumors and we analyze the obtained clusters.
2. Implementation and availability
Following the recommendation by Galaxy’s core developers’ team, the back-end development of MaREA4Galaxy is based on Python and the front-end development on XML. The interaction between front-end and back-end is based on the template engine Cheetah. MaREA4Galaxy is built on top of the following libraries: lxml, svglib, reportlab, pandas, scipy, python-libsbml, matplotlib, numpy and scikit-learn for clustering analysis.
MaREA4Galaxy is stored in a versioned code archive in ToolShed, at: https://toolshed.g2.bx.psu.edu/repos/bimib/marea. ToolShed allows the administrators of the hundreds of public and private Galaxy servers worldwide to easily install MaREA4Galaxy, as well as any other Galaxy utility, into their instances.
Once installed, MaREA4Galaxy appears in the Galaxy toolbar (left bar) under the name MaREA (see Fig. 1 for an example). A demo of MaREA4Galaxy is available at:http://bimib.disco.unimib.it:5555.
Fig. 1.

Screenshot of the MaREA4Galaxy interface. The module for RAS computation is illustrated. In particular, the built-in (default) HMRcore GPR rules are chosen. In the ‘add dataset’ field there is the RNA-seq dataset which has been previously uploaded and that appears in green in the History panel on the right.
3. Functionalities and workflow
MaREA4Galaxy processes datasets stored in the history panel of Galaxy. These datasets can be uploaded directly from the user’s computer as structured text file by using, e.g., the Galaxy built-in tool Get Data, or obtained as output of intermediary analyses performed with other tools.
MaREA4Galaxy consists in three interconnected modules that may also work independently.
-
•
The RASs computation module (Expression2RAS).
-
•
The metabolic reaction enrichment analysis module (MaREA).
-
•
The cluster analysis module (Clustering).
As better detailed in the following sections, the Expression2RAS module computes a RAS for each reaction in each sample. The MaREA module allows to visualize on a metabolic map the metabolic reactions that are up- or down- regulated in different groups of samples either defined a priori or identified by the Clustering module. The Clustering module allows to identify sample subgroups (or clusters) that share similar metabolic properties, ideally by employing the RAS computed by the Expression2RAS module. Any other data can however be used as feature for unsupervised clustering of samples. The metabolic differences between the clusters thus obtained can, in turn, be analyzed with the MaREA module.
3.1. Computation of reaction activity scores
The Expression2RAS tool selects and extracts metabolic genes from a gene-expression dataset and, by solving Gene-Protein-Reaction (GPR) association rules, computes a Reaction Activity Score (RAS) for metabolic reactions of interest, as illustrated in [8]. The assumption is that enzyme isoforms contribute additively to the overall activity of a given reaction, whereas enzyme subunits limit its activity, by requiring all the components to be present for the reaction to be catalyzed [8].
3.1.1. Input
The Expression2RAS tool (Fig. 1) takes two main inputs: 1) the list of GPRs; 2) the normalized read count of genes from a given cross-sectional RNA-seq dataset, as, e.g., RPKM (Reads per Kilobase per Million mapped reads) or TPM (Transcripts Per Kilobase Million).
The first input is a representation of the metabolic model being studied and it is basically a dictionary (key-value data structure), which associates a set of genes to each metabolic reaction. Both reactions and genes must be defined by a unique identifier.
Boolean operators AND and OR define the relationship between genes and enzymes. The AND operator joins genes that encode for different subunits of the same enzyme, whereas the OR operator joins genes that encode for isoforms of the same enzyme.
For the user’s convenience, two human metabolic network models have been made directly available within the tool: HMRcore and Recon 2.2. HMRcore corresponds to the set of GPR rules included in the core model of central carbon metabolism introduced in [6] and was used and curated in [4], [7], [5], [8], whereas Recon 2.2 [15] is a genome-wide model encompassing virtually all reactions encoded in human metabolism. However, the user can also opt to upload any custom metabolic network model of her/his choice.
The ID of genes in the dataset must of course coincide with the ID used in the GPR rules. In case built-in GPRs are used, the following gene nomenclatures are automatically recognized: HUGO ID, Ensemble ID, HUGO symbol, Entrez ID.
In case of missing expression value, referred to as NaN (Not a Number), for a gene joined with an AND operator in a given GPR rule, the user can choose to solve the rule ‘A AND NaN’ as A, or to disregard it tout-court (i.e., treated as NaN).
3.1.2. Output
The tool simply returns as output a dataset reporting the RAS computed for each sample for each reaction in the chosen metabolic network. The RAS dataset is displayed in the History panel.
3.2. Metabolic reaction enrichment analysis
The MaREA tool statistically compares the RAS of user-defined groups of samples [8] and visualizes the identified differences.
According to the user’s preference the tool performs the following comparisons.
-
•
Pairwise comparison of each group against all other groups.
-
•
Comparison of each group against the rest of the samples.
-
•
Comparison of each group against a user-defined control group.
3.2.1. Input
The MaREA tool (Fig. 2) takes as main input the Reaction Activity Scores of each sample, as computed by the Expression2RAS module and, if given, the eventual partition of samples/patients into distinct classes.
Fig. 2.

Screenshot of the MaREA4Galaxy interface. The module for metabolic reaction enrichment analysis is illustrated. The input format option ‘RNAseq dataset of all samples + sample group specification’ has been selected and the best clustering obtained with the k-means algorithm in the History has been selected as sample group specification.
The input RAS dataset can be organized in two alternative ways: 1) two or more separate RAS datasets, each relative to a different set of samples/patients; 2) a unique RAS dataset for all samples/patients, plus a file that associates to each sample its affiliation to a set.
As (optional) input, the user may also supply a graphical map of the metabolic network for an efficient visualization of the analysis outputs. If the HMRcore model is chosen, the corresponding map is included within the tool and does have not to be uploaded. The metabolic map format is a svg file, reporting metabolites and products of each reaction linked with an arrow, whose ID matches the name of the reaction in the GPR file.
The following advanced options can also be displayed and specified.
-
•
The P-Value threshold, used for significance Kolmogorov-Smirnov (KS) test, to verify whether the distributions of RASs over the samples in two sets are significantly different.
-
•
The threshold of the fold-change between the average RAS of two groups. Among the reactions that pass the KS test, only fold-change values larger than the indicated threshold will be visualized on the output metabolic map.
-
•
optional outputs to be displayed in the History panel.
The reader is referred to [8] for further theoretical aspects regarding the options above, whereas further technical details regarding formatting of input files are available in the help section in Galaxy.
3.2.2. Output
MaREA returns for each evaluated comparison, a collection output in the History including the following items.
-
•
A table reporting the fold-change between RASs and p-value of the Kolmogorov-Smirnov test.
-
•
The modified metabolic map (whenever supplied as input). Reactions up-regulated in the first class as compared to the second class are marked in red, whereas reactions down-regulated in the former are marked in blue. Thickness of arrows is proportional to the fold-change between the average RASs of the two classes. Non-Classified reactions, i.e., reactions without information about the corresponding gene-enzyme rule, are marked in black. Reactions that display a non-significant p-value or a RAS fold-change below the threshold are marked in gray color. The pdf of the map can be directly visualized within Galaxy. The user can also download the svg format of the map in order to apply changes.
-
•
A log file, reporting possible warning or error messages. Problems that prevent the pipeline’s functioning, such as wrong format of files, gene ID type not supported or duplicated IDs, insufficient number of classes, as well as minor problems, such as extra-columns, duplicated labels, missing gene values, or empty classes, are properly notified in detail.
3.3. Cluster analysis
The Clustering tool has been conceived to cluster gene expression data, by using the RAS scores computed by MaREA4Galaxy as features, given its efficacy in stratifying cancer patients according to metabolic phenotype, as demonstrated in [8]. However, it is suited to cluster observations in any dataset in which rows indicate different variables/features and columns different observations.
The Clustering tool implements three of the main existing algorithms to cluster data, namely: K-means, agglomerative clustering and DBSCAN (Density Based Spatial Clustering of Applications with Noise). Parameters and outputs of the tool are specific of each algorithm as briefly described in the following. A screenshot of this feature of the tool is reported in Fig. 3.
Fig. 3.

Screenshot of the MaREA4Galaxy interface. The module for cluster analysis is illustrated. The RAS computed by the MaREA tool have been selected as input dataset and K-means has been chosen as clustering method. 2 to 5 number of clusters will be tested. The elbow and silhouette plots will be generated.
3.3.1. K-means
Given that K-means clustering requires the number of clusters k to be set by the user, and that it is usually difficult to know the correct number of clusters a priori, the Clustering tool allows to evaluate different values of k and provides standard methods to estimate the goodness of each clustering in order to choose the best one. In particular, the elbow plot is generated, which allows to identify the “elbow” (the point of inflection on the curve) as the best candidate. The tool also generates a silhouette plot for each k, which reports the cohesion and the separation indexes of each element. The tool also computes the silhouette score of each element, as well as the average silhouette of each k, returning the k with the best (highest) silhouette. The user can specify the minimum and maximum number of clusters to be evaluated and whether elbow and dendrogram plots must be generated.
3.3.2. Agglomerative clustering
In the case of agglomerative clustering, the hierarchical output illustrated by the dendrogram facilitates the choice of the best clustering. The Clustering tool returns the set of clusters obtained when cutting the dendrograms at different points. The user can specify the minimum and maximum number of clusters to be tested and whether the dendrogram plot must be generated.
3.3.3. DBSCAN
The DBSCAN method automatically chooses the number of clusters, based on parameters that define when a region is to be considered dense. Custom parameters may be used, namely the maximum distance between two samples for one to be considered as in the neighborhood of the other and the number of samples in a neighborhood for a point to be considered as a core point.
4. Application example
To illustrate MaREA4Galaxy functioning, we show here an example of application. We analyze a liver hepatocellular carcinoma RNA-seq dataset taken from the TCGA pancancer study, including 372 patients. The original data is available at:http://download.cbioportal.org/blca_tcga.tar.gz.
The goal of our example is to identify patients’ cohorts with different metabolic features, without assuming any prior knowledge about the dataset. To this end, we first upload our dataset in the History panel of Galaxy by means of the Get Data tool. Based on the HMRcore metabolic map, we then compute the RAS for each reaction in each patient, by means of Expression2RAS tool, see Fig. 1.
We then switch to the Clustering tool and we select as input dataset the RASs computed before. As a proof of principle, we choose K-means as clustering algorithm. We test a number of clusters k from 2 to 5, and we indicate that we want to generate both elbow and silhouette plots for each tested k (see a screenshot of the tool in Fig. 3).
The execution of the tool returns the clustering output for each value of k, and indicates as best clustering the one that maximizes the average silhouette coefficient. In this case, the tools returns as the best clustering. The generated elbow and silhouette plots are reported in Fig. 4. It can be noticed that, although we have identified a good stratification of liver cancer patients, by qualitative observation of the elbow plot one may also choose as best clustering.
Fig. 4.

Evaluation of clustering goodness by MaREA4Galaxy. Left panel: elbow plot generated by the Clustering module, showing an elbow for . Right panel: silhouette plot generated by the Clustering module for , which has been returned as best clustering according to the average silhouette score reported in the plot’s title.
Finally, we can use the MaREA tool to promptly analyze the differences between the two patients’ cohorts. As shown in the screenshot in Fig. 2, we select this time the input format option ‘RNAseq of all samples +sample group specification’ and we select the best clustering output obtained with Clustering as sample group specification file. We flag the option of generating the pdf map and once we execute the tool we obtain the map reported in Fig. 5.
Fig. 5.

Example of metabolic map generated by MaREA4Galaxy. In the example, red arrows indicate reactions up-regulated, whereas blue arrows reactions down-regulated, in a subgroup of liver hepatocellular carcinoma patients. Black arrows refer to reactions without information about the corresponding gene-enzyme rule. Dashed gray arrows refer to non significant disregulations according Kolmogorov-Smirnov test with p-value 0.01. Solid gray arrows refer to reactions with a variation lower then 20%. As output maps are provided as vector graphics (in svg/pdf file formats), they can be zoomed-in at will.
Although a few reactions significantly differ between the two cluster, an expert who is familiar with this classical representation of central carbon metabolism can immediately notice (Fig. 5) the main metabolic features that distinguish the two patients cohorts.
For example, in the first group, the upper glycolytic pathway is up-regulated, whereas lower glycolysis, upstream of lactate production, is down-regulated. Lactate production from pyruvate is instead up-regulated, indicating that pyruvate production derives from alternative routes, such as from the amino acid serine derived from glucose. The reactions that go from serine to pyruvate are indeed up-regulated. Other differences involve the utilization of glutamine and synthesis of amino acids derived from glutamine, such as asparagine and aspartate, as well as production of putrescine and urea in the urea cycle.
5. Conclusion
We have shown how with a few intuitive steps, without the need to set technical parameters, and in a very short time, MaREA4Galaxy enables to uncover and characterize the differences in metabolic activity observed in different sample subgroups, as in the case of cancer patients.
Being empowered by the well-known open and web-based platform Galaxy for performing accessible, reproducible, and transparent bioinformatics science, MaREA4Galaxy can support many life scientists who may have little knowledge of computational methods for analyzing the metabolic variability underlying gene-expression datasets, no matter whether collected in their labs or available in public databases, thus paving the way to tackle metabolic plasticity and heterogeneity.
As an example, we have shown a novel application of the MaREA pipeline to liver hepatocellular carcinoma and we have identified two groups with well distinct metabolic features. Investigating the implications of these differences is out of the scope of this work. However, it would be interesting to analyze whether the two groups of patients differ in other aspects, such as their prognosis, (epi) genomic makeup or regulation of signaling pathways.
A better understanding of the fundamental causes of metabolic heterogeneity is important for personalised treatment of the many diseases involving metabolic alterations, as well as for targeted nutrition recommendations and intervention the field of personalized nutrition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Chiara Damiani: Conceptualization, Investigation, Supervision, Writing - original draft, Writing - review & editing. Lorenzo Rovida: Software. Davide Maspero: Conceptualization, Data curation. Irene Sala: Software. Luca Rosato: Software. Marzia Di Filippo: Visualization. Dario Pescini: Visualization. Alex Graudenzi: Writing - review & editing. Marco Antoniotti: Writing - review & editing. Giancarlo Mauri: Writing - review & editing, Funding acquisition.
Acknowledgments
The institutional financial support to SYSBIO – within the Italian Roadmap for ESFRI Research Infrastructures – is gratefully acknowledged. CD and GM received funding from FLAG-ERA grant ITFoC.
Footnotes
Supplementary data associated with this article can be found, in the online version, athttps://doi.org/10.1016/j.csbj.2020.04.008.
Supplementary data
The following are the Supplementary data to this article:
References
- 1.Afgan E., Baker D., Van den Beek M., Blankenberg D., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Eberhard C. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucl Acids Res. 2016;44(W1):W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boele J., Olivier B.G., Teusink B. Fame, the flux analysis and modeling environment. BMC Syst Biol. 2012;6(1):8. doi: 10.1186/1752-0509-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damiani C., Colombo R., Di Filippo M., Pescini D., Mauri G. Italian Workshop on Artificial Life and Evolutionary Computation. Springer; 2016. Linking alterations in metabolic fluxes with shifts in metabolite levels by means of kinetic modeling; pp. 138–148. [Google Scholar]
- 4.Damiani C., Di Filippo M., Pescini D., Maspero D., Colombo R., Mauri G. popFBA: tackling intratumour heterogeneity with Flux Balance Analysis. Bioinformatics. 2017;33(14):i311–i318. doi: 10.1093/bioinformatics/btx251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Damiani C., Maspero D., Di Filippo M., Colombo R., Pescini D., Graudenzi A. Integration of single-cell rna-seq data into metabolic models to characterize tumour cell populations. PLOS Computat Biol. 2018;15(2) doi: 10.1371/journal.pcbi.1006733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Filippo M., Colombo R., Damiani C., Pescini D., Gaglio D., Vanoni M., Alberghina L., Mauri G. Zooming-in on cancer metabolic rewiring with tissue specific constraint-based models. Computat Biol Chem. 2016;62:60–69. doi: 10.1016/j.compbiolchem.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Graudenzi A, Maspero D, Damiani C. Modeling spatio-temporal dynamics of metabolic networks with cellular automata and constraint-based methods. In: Cellular Automata. ACRI 2018. Lecture Notes in Computer Science. vol. 11115. Springer, Cham; 2018, p. 16–29.
- 8.Graudenzi A., Maspero D., Di Filippo M., Gnugnoli M., Isella C., Mauri G., Medico E., Antoniotti M., Damiani C. Integration of transcriptomic data and metabolic networks in cancer samples reveals highly significant prognostic power. J Biomed Inform. 2018;87:37–149. doi: 10.1016/j.jbi.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 9.Holmes E., Wilson I.D., Nicholson J.K. Metabolic phenotyping in health and disease. Cell. 2008;134(5):714–717. doi: 10.1016/j.cell.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 10.Hotamisligil G.S. Inflammation and metabolic disorders. Nature. 2006;444(7121):860. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 11.King Z.A., Dräger A., Ebrahim A., Sonnenschein N., Lewis N.E., Palsson B.O. Escher: a web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLoS Comput Biol. 2015;11(8) doi: 10.1371/journal.pcbi.1004321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.López-Otín C., Galluzzi L., Freije J.M., Madeo F., Kroemer G. Metabolic control of longevity. Cell. 2016;166(4):802–821. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 13.Machado D., Herrgård M. Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism. PLoS Comput Biol. 2014;10(4) doi: 10.1371/journal.pcbi.1003580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Opdam S., Richelle A., Kellman B., Li S., Zielinski D.C., Lewis N.E. A systematic evaluation of methods for tailoring genome-scale metabolic models. Cell Syst. 2017;4(3):318–329. doi: 10.1016/j.cels.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swainston N., Smallbone K., Hefzi H., Dobson P.D., Brewer J., Hanscho M., Zielinski D.C., Ang K.S., Gardiner N.J., Gutierrez J.M. Recon 2.2: from reconstruction to model of human metabolism. Metabolomics. 2016;12(7):1–7. doi: 10.1007/s11306-016-1051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward P.S., Thompson C.B. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M., Network C.G.A.R. The cancer genome atlas pan-cancer analysis project. Nature Genet. 2013;45(10):1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yizhak K., Chaneton B., Gottlieb E., Ruppin E. Modeling cancer metabolism on a genome scale. Mol Syst Biol. 2015;11(6):817. doi: 10.15252/msb.20145307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.