

Abstract
The available active surface area and the density of probes immobilized on this surface are responsible for achieving high specificity and sensitivity in electrochemical biosensors that detect biologically relevant molecules, including DNA. Here, we report the design of gold-coated, silicon micropillar-structured electrodes functionalized with modified poly-l-lysine (PLL) as an adhesion layer to concomitantly assess the increase in sensitivity with the increase of the electrochemical area and control over the probe density. By systematically reducing the center-to-center distance between the pillars (pitch), denser micropillar arrays were formed at the electrode, resulting in a larger sensing area. Azido-modified peptide nucleic acid (PNA) probes were click-reacted onto the electrode interface, exploiting PLL with appended oligo(ethylene glycol) (OEG) and dibenzocyclooctyne (DBCO) moieties (PLL-OEG-DBCO) for antifouling and probe binding properties, respectively. The selective electrochemical sandwich assay formation, composed of consecutive hybridization steps of the target complementary DNA (cDNA) and reporter DNA modified with the electroactive ferrocene functionality (rDNA-Fc), was monitored by quartz crystal microbalance. The DNA detection performance of micropillared electrodes with different pitches was evaluated by quantifying the cyclic voltammetric response of the surface-confined rDNA-Fc. By decrease of the pitch of the pillar array, the area of the electrode was enhanced by up to a factor 10.6. A comparison of the electrochemical data with the geometrical area of the pillared electrodes confirmed the validity of the increased sensitivity of the DNA detection by the design of the micropillar array.
Introduction
The invention of (bio)sensors represents a breakthrough for life sciences. The possibility of detecting specific bioanalytes rapidly and quantitatively has propelled the economic and scientific effort toward the design and production of functional biorecognition devices.1 Owing to their well-developed miniaturized fabrication processes, high signal-to-noise ratio, and real-time response, electrochemical biosensors are currently the gold standard for daily-life applications such as food/environmental control and point-of-care devices for the detection of biologically relevant molecules such as glucose,2 phenol,3 and drugs (e.g., doxorubicin/gentamicin,4 tobramycin5).
At the same time, the progress in genetics and genomics has led to the insight that biomolecules, such as proteins and DNA/RNA variants, can act as biomarkers providing valuable information for early diagnosis and monitoring of several types of tumors and genetic diseases.6−8 Due to the small sample volume needed for analysis, the possibility of working with body fluids, and the relatively high sensitivity,9 electrochemical DNA biosensors have gained popularity as clinical assay devices for diseases that can be detected by DNA biomarkers,10,11 such as Crohn’s disease, multiple sclerosis, cystic fibrosis, and cancer.12−14 Nevertheless, the low concentration of DNA biomarkers (up to 104 units/mL of plasma/serum)15,16 has fueled the development of signal amplification methods to achieve highly sensitive electrochemical detection.17 Strategies have been reported based on enzyme amplification,18−20 postmodification polymerization of conductive materials,21,22 as well as the use of electroactive reporter probes.23−25
For surface-confined electrochemical DNA biosensors, the intrinsic sensitivity depends strongly on the surface architecture, which affects the display of probes at the interface together with the probe density (which have been defined as crucial parameters in DNA detection),26−28 and the electrochemically active surface area of the biosensor.5,29,30 The use of three-dimensional (3D) structures such as polymer brushes,31 hydrogels,32 nanodisks,33 gold nanoparticles,34 nanoporous gold,35 and nanotextured microelectrodes36 has significantly improved the electrochemical device performance and, consequently, the detection limit of target DNA analytes compared to flat substrates. Similarly, conductive micro- and nanopillar-structured substrates have been reported (though not applied in DNA sensing) to provide a higher surface area than planar electrodes.37−40
Schröper et al. have demonstrated that the electroactive surface area for nanopillar-based devices is often lower than predicted due to the strong dependence on the diffusivity of the electroactive species compared to a flat substrate.41 On the other hand, microelectrodes and micropillar-structured substrates can steadily generate a higher signal,42,43 thus representing an alternative for improving the sensitivity of the electrochemical biosensor. The groups of Compton and del Campo showed with simulations the positive effect of reducing the center-to-center separation (pitch) of gold-coated micropillars and increasing their aspect ratio on the electrochemical performance in a cyclic voltammetry (CV) setup in the case of electroactive species in solution.44,45 An experimental demonstration showed a maximal signal enhancement for the peak current density of 1.6 and 6.2 times with respect to the projected surface area, using two different pillar geometries. In addition, del Campo et al. showed the importance of achieving full wetting of the gold-coated micropillar-structured substrates to achieve full penetration of the pillar array by the solution and thus an optimal electrochemical sensitivity of the device.44
Poly-l-lysine (PLL) polymers grafted with poly(ethylene glycol) (PEG) units (PLL-PEG) have been shown to increase the lubrication properties of both hydrophobic (poly(dimethylsiloxane), PDMS) and metal oxide substrates, due to the hydrophilic nature of the PEG chains and the high content of trapped water.46−48 In addition, modified PLL has been used to functionalize surfaces allowing fast and orthogonal biomolecule immobilization and to provide good antifouling properties.49,50 In particular, PLL grafted with oligo(ethylene glycol) (OEG) and maleimide (Mal) moieties has been exploited by us to control the probe density of both engineered peptide nucleic acid (PNA) and DNA probes for DNA recognition.27
Here, we report the design and use of gold-coated, micropillar-structured electrodes, with control over the micropillar pitch to increase the sensitivity, and modified with functionalized PLL to anchor the probes for electrochemical DNA detection. The positively charged PLL, grafted with OEG and dibenzocyclooctyne (DBCO) groups (PLL-OEG-DBCO),51 was self-assembled on the electrode surface to form a hydrophilic, orthogonally biofunctionalizable layer. An azido-PNA probe with a complementary sequence capable of detecting the KRAS gene16 was used to illustrate potential applications for tumor DNA detection. The choice of PNA as a probe for DNA detection was driven by the higher affinity and selectivity for complementary DNA (cDNA) compared to DNA probes52 and their resistance to enzymes present in biological fluids.53 In addition, the combination of modified PLL anchored with PNA probes has been proven to provide high hybridization efficiencies at elevated surface probe densities by the suppression of electrostatic repulsion occurring in DNA-probe devices.27 The electrochemical detection of cDNA by a sandwich assay was performed using a reporter probe DNA, complementary to the free 5′-end of the target cDNA, bearing an electroactive ferrocene moiety (rDNA-Fc). The signal generated by CV was evaluated as a function of the micropillar pitch, to establish the relationship between the signal gain and the increased surface area, thus providing an insight into the potential of the sensitivity gain reached by electrode microstructuring.
Results and Discussion
Micropillar-Based Electrode Design and Probe Functionalization
To investigate the sensitivity gain in electrochemical biosensors with higher surface areas created by 3D microstructuring, we employed flat (control) and micropillar-structured, Au-coated Si substrates with four different pitches (19, 14, 10, and 8 μm). These were used in combination with a proof-of-principle, sandwich assay-mediated recognition of the complementary DNA (cDNA, the model analyte) and rDNA-Fc to assess the electrochemical performance. Figure 1a shows the schematic overview of the micropillar biorecognition interface to detect DNA. Smaller pitches result in a larger density of pillars and concomitantly higher electroactive surface area available for subsequent functionalization and DNA binding. Consequently, more modified PLL is adsorbed, and a higher density of DBCO moieties per projected electrode area is displayed on the surface. The probe density on the electrode surface is set by the grafting density of DBCO groups attached to the PLL,27,51 upon reacting the PNA-azide molecules to the DBCO groups by the strain-promoted azide–alkyne cycloaddition (SPAAC) click reaction (Figures 1b,c, and S1–S3).54 The target cDNA (43 nt) and the reporter rDNA-Fc (23 nt) molecules (Table S1) are consecutively hybridized to the surface. Such a sandwich-like assay avoids the necessity of DNA postmodification for introducing the ferrocene redox moiety after the successful dual-hybridization event.55 The main objective of this work was to observe the relationship between the response obtained from the electroactive rDNA-Fc specifically anchored at the interface and the surface area (enhanced by reducing the pillar pitch), thus demonstrating the signal amplification by micropillar-based electrodes in CV experiments.
Figure 1.

(a) Schematic representation illustrating the concept of increased electrochemical surface area by reducing the pitch of the electrode design to gain electrochemical sensitivity. (b) Overview of the chemical steps occurring at the PLL-OEG-DBCO-modified, gold-coated electrode interface, showing the sequential deposition of: the N3-PNA probe, cDNA, and rDNA-Fc. (c) Chemical structure of the PLL-OEG-DBCO used, together with the SPAAC reaction scheme between DBCO and azido-PNA (structures, sequences, and characterization of the PNA and DNA molecules are shown in Figures S1–S3 and Table S1).
Silicon substrates with hexagonal micropillar arrays with a height of 36.7 μm and a diameter of 4.0 μm were fabricated by photolithography and deep reactive-ion etching (DRIE) according to the procedure reported by Elbersen et al.56 (Figure S4). Figure 2a shows the micropillar array with a pitch of 8 μm, characterized by high-resolution scanning electron microscopy (HR-SEM; top view shown in Figure S5). The final electrode architecture was formed by sputtering a thin layer (∼200 nm) of gold on top of the substrate. Due to the sputter direction normal to the surface, the gold layer was thinner (∼50 nm on average) at the sides of the pillars. Nevertheless, a conformal and fully conductive layer was achieved (Figure 2b).
Figure 2.

HR-SEM images showing: (a) a tilted cross section of an 8 μm pitch micropillar-structured silicon substrate, with pillar height and diameter of 36.7 and 4 μm, respectively; (b) zoom-in image of a cross-sectioned Si micropillar (taken at approximately halfway the pillar) showing the conformal gold coating (bright) inside the scallops.
The PLL polymer (15–30 kDa) grafted with OEG and DBCO functionalities (Figure 1c) was synthesized adapting a previously reported procedure.27,51 The OEG and DBCO groups were covalently grafted to the PLL side chains in a one-step synthesis by N-hydroxysuccinimide (NHS) ester coupling (see Experimental Section). The mole fractions of appended groups in PLL-OEG-DBCO were determined by 1H NMR, which yielded percentages of 27.6 for the OEG and 3.0 for the DBCO moieties (Figure S6). The total grafting density of functionalized lysine side chains was kept below 35–40% to ensure strong adsorption to the surface.57,58 The azido-PNA probe was synthesized as previously reported.59
As a proof of concept, the surface functionalization processes of modified-PLL deposition, azido-PNA immobilization, and consecutive cDNA and rDNA-Fc hybridization steps were followed on a flat substrate by quartz crystal microbalance with dissipation (QCM-D) monitoring (Figure 3). Upon mounting a UV–ozone-activated gold chip in the QCM chamber, both the PLL-OEG-DBCO adsorption and azido-PNA steps showed a decrease of the resonance frequency (blue line, fifth overtone), which remained stable after washing with phosphate-buffered saline (PBS) (see Figure 3a). Consecutive injections of cDNA (43 nt) and rDNA-Fc (23 nt) solutions produced frequency shifts (Δf) of ∼16 and ∼12 Hz (average of two measures), respectively, demonstrating the recognition ability of the DNA-bioresponsive interface and the feasibility of the sandwich assay. These frequency shifts correspond, using the Sauerbrey equation,60 to cDNA and rDNA-Fc densities of 2.5 × 1012 and 3.4 × 1012 molecules/cm2, assuming that 80% of the mass is due to adsorbed water.61 The length difference between the DNA molecules and their hybridization can cause hydration changes and consequently an overestimation of the hybridization efficiency. By taking into account the different numbers of nucleotides between cDNA and rDNA-Fc, the hybridization efficiency was approximately 140% (not corrected for differences in hydration, as well as the mass and hydration of the Fc moiety), which is within the error for QCM monitoring, as already reported by the Knoll and Höök groups.62,63
Figure 3.

QCM-D time traces of: (a) the stepwise adsorption process comprising PLL-OEG-DBCO deposition, anchoring of azido-PNA, and the detection of the cDNA and rDNA-Fc; (b) control using ncDNA instead of cDNA; (c) control without cDNA; (d) control without azido-PNA. In all experiments, the concentrations were 0.5 mg/mL for the modified-PLL solutions and 0.5 μM for the azido-PNA, cDNA, rDNA-Fc, and ncDNA solutions. PBS (pH 7.4) washing steps (gray bars) were performed before and after every adsorption step. The fifth overtone was used for both Δf and ΔD.
The selectivity of the PNA-modified surface was investigated by controls, using a noncomplementary DNA sequence (ncDNA, 43 nt; Figure 3b) and by leaving out either the cDNA (Figure 3c) or the PNA probe anchoring steps (Figure 3d) in the sandwich assay. All controls showed a near or full absence of signal for the DNA and/or rDNA-Fc steps, after the rinsing step. Noteworthy, despite a Δf of approximately 2 Hz for the ncDNA (Figure 3b) and 0.5 Hz for the cDNA (in the absence of the azido-PNA step, Figure 3d, corresponding to a 3.1% of the total cDNA binding in the detection in Figure 3a), fouling was largely absent. In conclusion, a selective response of rDNA-Fc was obtained only in the case of PNA probe anchoring and the double hybridization sequence in the sandwich assay.
In addition, to demonstrate a good wettability of the micropillar-structured substrates by means of modified PLL, contact angle goniometry was performed on three model substrates, two with a micropillar array having pitches of 19 and 8 μm and a flat substrate. The low values reported in Table S2 indicate good wetting, in agreement with the presence of the adsorbed PLL-OEG-DBCO and the increased hydrophilicity in the micropillar area even after 24 h. Overall, these results indicate the feasibility of probe anchoring and the performance of the sandwich assay in a micropillar array without wetting problems.
Performance of Micropillar Electrodes in Electrochemical DNA Detection
The micropillar-structured electrodes used in this work consist of two parts, namely, the pillared section, where the micropillars are positioned, and a flat area surrounding the pillar array, as schematically displayed in Figure 4. The projected areas of the flat and pillared sections (PAf and PAp) are 0.19 and 0.25 cm2, respectively (thus, the total projected surface area PAtot = 0.44 cm2). Upon the introduction of the micropillars in the electrode design, a theoretical surface area enhancement factor (SE) (= Atot/PAtot, where Atot is the total geometric electrode area) is defined. When taking into account only the micropillared section of the electrode, the geometric surface area of that section (Ap) is expected to increase with a factor 1 + 2/3·√3·π·dp·hp/p2 (i.e., the theoretical surface enhancement factor for the pillared section, SEp; see Table S3), where dp is the pillar diameter, hp is the pillar height, and p is the pitch of the hexagonal micropillar array (see the Electrochemical Analysis section in the SI and Figure S7).
Figure 4.
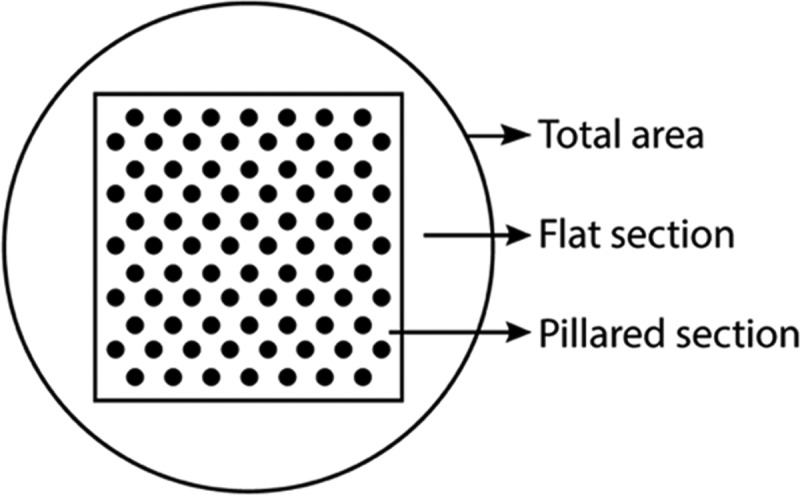
Schematic top-view representation of the whole projected micropillar-structured electrode area (PAtot = 0.44 cm2), showing the micropillar (pillared section, PAp = 0.25 cm2) and the surrounding flat (flat section, PAf = 0.19 cm2) area.
The increase of the surface area by the micropillars was experimentally assessed using CV. The electrochemically active area of two gold-coated micropillar-structured substrates (19 and 8 μm pitch) and of a flat sample was determined using a 0.1 M solution of H2SO4.41 Qualitatively, the areas of both the oxidation and reduction peaks were significantly higher for the micropillar-structured substrates compared to the flat one (Figure S8). By integrating the reduction peaks, the total electroactive surface areas (Atot,exp) were 0.57 cm2 for the flat electrode and 1.26 and 3.68 cm2 for the micropillar-structured ones (with pitches of 19 and 8 μm, respectively, Table S4). For the flat sample, the observed area is 1.29 times higher than the geometric area, which is attributed to substrate roughness introduced by the gold sputtering process. For the pillared samples, however, the experimental areas were approximately a factor 1.5 higher than the expected geometric surface areas. This higher increase for the pillared samples is attributed to the combined roughness effects caused by scallop formation, the roughness of the Si surface (both introduced by the DRIE etching process), and the Au sputtering process. For this reason, two different roughness factors were defined, one for the flat section (sf = 1.29, Electrochemical Analysis in the SI) and one for the pillared section (sp = 1.48; see Figure S9). The Atot,exp values were found to be in good agreement with the geometric surface area calculations after including the roughness factors, with a maximum error <8%. The experimental surface enhancement factors for the total area (flat + pillared sections) were then calculated by the ratio of the experimental surface area of a micropillared electrode and that of the flat one, amounting to 2.21 and 6.46 for the 19 and 8 μm pitch, respectively. When viewing the area increase effect of the pillared section only, surface enhancement factors of 3.14 and 10.6 were found, showing a clear contribution of the pillars and a decreasing pitch on the expected electrochemical signal amplification (see Electrochemical Analysis in the SI).
The relationship between the electrode surface area and the DNA sensitivity was quantified by CV experiments using the DNA sandwich detection scheme (Figure 1b). Micropillar-structured electrodes with pitches of 19, 14, 10, and 8 μm and a flat substrate, preincubated with PLL-OEG-DBCO and azido-PNA probe solutions, were covered with a cDNA solution followed by rDNA-Fc deposition to perform the CV measurements after the sandwich assay hybridization (Figure S10). The dependence of the total charge involved in the redox process (Q) due to the surface-anchored electroactive Fc moiety, which is related to the peak area in the CV, was evaluated as a function of the scan rate, and the results are presented in Figure S11. The constant values of Q vs scan rate indicate that the electron transfer processes occurred at the interface as expected for surface-confined species. Control experiments performed by exploiting ncDNA (Figure S12a) or by omitting one step of the sandwich assay (azido-PNA anchoring or cDNA hybridization, Figure S12b,c) showed the absence of physisorbed electroactive material, which is attributed to the retained antifouling properties of the self-assembled modified PLL at the electrode interface, and thus confirms the specificity of the sandwich assay.
The DNA sensitivity enhancement was assessed by evaluating the dependence of Q (from Figure S11) on the pitch p of the micropillar-structured electrodes. Figure 5 shows a linear increase of Qvs 1/p2, confirming the effect of the pillar architecture on the detected signal. In this graph, the intercept with the y-axis indicates the flat sample. The linearity of the fit demonstrates not only the absence of diffusion effects but also the uniformity of the detected rDNA-Fc and cDNA. Consequently, assuming both hybridization steps to be 100% efficient, the surface coverage Γ could be derived from the slope (see the Electrochemical Analysis section in the SI), providing a value of (9.0 ± 0.2) × 10–12 mol/cm2 or (5.4 ± 0.1) × 1012 rDNA-Fc moieties/cm2, which matches well the results obtained from QCM as described above. When using the semiempirical method described in our recent publication,27 using PLL-appended maleimide reactive groups to bind PNA probes applied to a flat substrate, a density of 3.7 × 1012 molecules/cm2 was expected, which compares well with the value observed here. Thus, these results validate the modified-PLL approach to control the density of probe molecules at both flat and micropillar-structured substrates.
Figure 5.

Dependence of the total charge Q involved in the surface-confined redox process of the Fc-covered electrodes (0.1 M NaClO4 electrolyte) resulting from the sandwich assay, as a function of 1/p2 (where p is the pitch). Datapoints correspond to the average values of Q for the flat and 19, 14, 10, and 8 μm pitch samples, derived from the experiments shown in Figure S10. The concentrations of the species used for the DNA binding scheme were 1.0 mg/mL for modified PLL, 0.5 μM for azido-PNA, cDNA, and rDNA-Fc in PBS (pH 7.4). The equation of the linear fitting is y = 117.10 × (±3.80) + 0.25 (±0.04).
A comparison of the two extremes in Figure 5, corresponding to the flat and the 8 μm pitch micropillar-structured substrates, shows that the signal enhancement due to the occurrence of the pillar array was approximately a factor of 7.0. The reported value agrees with the experimental surface enhancement factors found by the sulfuric acid measurements, confirming that the increase of the electroactive surface area is the key factor in improving the performance of surface-based electrochemical DNA sensors. When comparing only the pillared section of the micropillar array sample to the flat sample, an enhancement factor of 10.6 was deduced. Overall, these data confirm that the 3D electrode interface design, in correlation with the probe density control, can rationally define the gain in sensitivity for electrochemical DNA detection, showing how the combinations of substrate and biosensing layer can determine the outcome of the recognition device.
Conclusions
In summary, we have presented the potential gain in sensitivity for the electrochemical DNA detection by the application of azido-PNA probes clicked to a PLL-OEG-DBCO adhesion layer adsorbed on micropillar-structured substrates at various pitches. Compared to flat substrates, the densely structured micropillar arrays allowed up to 1 order of magnitude larger electrochemical active surface areas that can accommodate comparatively more PNA probes and consequently more target DNA to be detected by the final sandwich assay. The total amount of redox charges from the hybridized reporter rDNA-Fc probe scaled linearly with the electrochemically active surface area, defined by the pitch, indicating the surface-confined electron transfer and similar probe densities between substrates. Overall, this proof-of-concept micropillar-structured electrode design, combined with the surface functionalization approach of modified PLL, increased the total sensitivity by more than 1 order of magnitude.
Micropillar-structured electrodes improve the electrochemical detection of, among others, DNA molecules in buffer and clinical samples exploiting the third dimension by enhancing the detection area and, thus, the sensitivity. The proposed design gives room for further customizing the physical (substrate) and chemical (adhesion layer) characteristics of the biorecognition surface. Consequently, the geometrical parameters and the probe density can be varied to maximize the sensitivity. By eliminating the flat section of the current electrode, a fully covered micropillar electrode might further improve the amplification factor. Moreover, different etching recipes could produce taller pillars. As an example, the hypothetical use of 125 μm long pillars, as exploited by del Campo, together with halving both the pitch and pillar diameter, would have generated a 70 times higher electrochemical signal compared to a flat substrate. Other contributions can be obtained from the surface roughness and the probe density, by enhancement of the grafting density of appended groups at the PLL, which could result in a final signal enhancement factor of 2–3 orders of magnitude. The occurrence of diffusion limitations, as well as electrostatic and steric repulsion of the incoming DNA, needs to be investigated. However, these effects might be reduced by integrating such 3D architectures in microfluidic devices. All in all, the advantages of probing more sensing surface, the possible extension to other detection systems, together with the orthogonal control of the biorecognition interface at the molecular level, define the potential directions for producing label-free signal amplification at electrochemical, optical, and gravimetric biosensing devices.
Experimental Section
Materials
Poly-l-lysine hydrobromide (MW = 15–30 kDa by viscosity), NaClO4, deuterated water, and tablets for a 10 mM PBS solution (pH 7.4) were obtained from Sigma-Aldrich. H2SO4 (95%) was purchased from VWR Chemical, and HCl was obtained from SelectiPur. Methyl-OEG4-NHS ester was obtained from Thermo Fisher Scientific, while DBCO-OEG4-NHS was obtained from Click Chemistry Tools. The membrane for dialysis (Spectra/Por; 6–8 kDa cutoff; diameter, 6.4 mm) was purchased from Spectrum Labs, Greece. cDNA (complementary to KRAS sequence: 43 nt, 5′-ATG ACTGAATATAAACTTGTGGTAGTTGGAGCTGGTGGCGTAG-3′) and ncDNA (42 nt, 5′-CTACGCCACCTCAACCTA CGCCACCTCCACCTACGCCACCTC-3′) were purchased from Eurofins Genomics and used as received. The ferrocene-labeled DNA (23 nt; MW, 7487 g/mol; 5′-ACCACAAGTTTATATTCAGTCAT-Fc-3′) was acquired from Biomers.net GmbH. Gold QCM chips (with a fundamental frequency of 5 MHz) were purchased from Biolin Scientific. Silicon p++ wafers (⟨100⟩-oriented, one-side polished, 525 ± 25 μm substrate thickness, 0.01–0.025 Ω cm resistivity) were obtained from Okmetic Finland, while the positive Olin 907-17 photoresist was obtained from Arch Chemicals. The PNA probe was synthesized using a previously described procedure.59
Synthesis and Quantification of PLL-OEG-DBCO-Grafted Percentages
The synthesis of PLL-OEG-DBCO and the quantification of the mole fractions of OEG and DBCO grafted to the PLL backbone were performed using previously reported procedures.27,51,57 Briefly, 10 mg/mL of PLL HBr was dissolved in PBS 7.4, and stoichiometric amounts of methyl-OEG4-NHS and DBCO-OEG4-NHS (both dissolved in DMSO at a concentration of 250 mM) were added under vigorous stirring. After 4 h, the solution was dialyzed with a dialysis membrane (molecular cutoff, 6–8 kDa) against decreasing concentrations of PBS in Milli-Q water, until a full 24 h cycle in Milli-Q water. The final solution was freeze-dried overnight. The obtained product was analyzed by NMR and stored at −20 °C in Milli-Q water. The quantification of the mole fractions is reported in the Supporting Information.
1H NMR of PLL-OEG-DBCO (400 MHz D2O) δ [ppm] = 1.26–1.56 (lysine γ-CH2), 1.61–1.82 (lysine β, δ-CH2), 2.48 (ethylene glycol CH2 from both OEG and DBCO coupled, −CH2–C(=O)–NH), 2.96 (free lysine, H2N–CH2), 3.14 (ethylene glycol CH2 of coupled lysine from both OEG and DBCO, C(=O)–NH–CH2−), 3.35 (OEG methoxy, −O–CH3), 3.59–3.77 (ethylene glycol from both OEG and DBCO, CH2–O−), 4.27 (lysine backbone, NH–CH–C(O)−), 7.22–7.69 (DBCO from coupled DBCO, CArH).
Micropillar-Structured Substrate Fabrication
Micropillar-structured electrodes were fabricated according to a reported procedure.56 In summary, a positive photoresist (Olin 907-17) was deposited on a p++ silicon substrate followed by photolithography to create a circular patterned photoresist (5 × 5 mm2) with spacing between the circles varying between 8 and 19 μm (Figure S4 step A). Micropillar-structured substrates were formed via DRIE (SPTS Pegasus, etching rate of ∼ 10 μm/min, 20 °C) until the desired micropillar height was achieved (Figure S4 step B). The created substrates were then cleaned in O2/CF4 plasma (Tepla 360) for 30 min and in a solution of HCl, H2O2, and H2O (1:1:5 ratio, 70 °C) for 15 min to strip the fluorocarbon residues and photoresist from the substrates. Prior to gold sputtering, the silicon-modified substrates were cleaned for 10 min in HNO3 and 30 s in HF to remove the silicon dioxide layer. Immediately after the HF step, a gold layer was sputtered (TCOathy) conformally over the entire substrate at 10–2 mbar and 50 W for 1800 s (Figure S4 step C).
Quartz Crystal Microbalance (QCM)
Gold-coated (50 nm, QSX301) QCM-D chips from LOT-Quantum were cleaned for 5 min in a basic piranha solution (H2O/NH4OH/H2O2 in ratio 5:1:1) at 70 °C for 5 min and then washed extensively with Milli-Q water and EtOH. After drying under nitrogen flow and oxidized with UV–ozone (BioForce chamber, Nanosciences) for 15 min, the chips were mounted in the chambers and a flow rate of 80 μL/min was used for all of the steps. QCM-D measurements were performed using a Q-Sense E4 4-channel quartz crystal microbalance with a peristaltic pump (Biolin Scientific), monitoring the fifth fundamental overtone. All experiments were performed in a PBS solution (10 mM, pH 7.4) at 22 °C. The Δf’s for cDNA and rDNA-Fc are averaged from two measurements.
Scanning Electron Microscopy
Micropillar-structured substrates were visualized using HR-SEM (FEI Sirion HR-SEM) at an acceleration voltage of 10 kV. The cross-sectional image was taken after cutting the micropillar-structured electrode with a diamond cut pen and sonication in ethanol for 30 min.
Cyclic Voltammetry Experiments
Gold-coated CV chips (flat and micropillar-structured substrates) were cleaned for 30 s in a piranha solution, washed extensively with water and EtOH, and dried with nitrogen. The experiments for the determination of the active surface area were performed using a 0.1 M H2SO4 solution as the electrolyte, at a scan rate of 100 mV/s.41 The reduction peak area was used to determine the active electrochemical surface area via the theoretical charge density value of 448 μC/cm2 for gold surfaces.41 The theoretical surface area of the substrate used as the working electrode is 0.44 cm2 due to the O-ring, which used to have a conformal contact between the electrochemical cell and the substrate.
In the case of the electrochemical DNA detection by sandwich assay for both flat and micropillar-structured substrates, the gold chips were immersed in a solution consisting of PLL-OEG(27.6)-DBCO(3.0) (1 mg/mL, PBS pH 7.4) for 60 min, after activation by UV–ozone for 15 min. Then, PBS (pH 7.4) solutions containing azide-PNA (0.5 μM), cDNA (0.5 μM), and rDNA (0.5 μM) were consecutively deposited on top of the functionalized PLL-OEG-DBCO substrate for 4 and 1 h for each step of hybridization, under gentle shaking. After each deposition, a rinsing step with Milli-Q water followed by a drying step with N2 was performed. Alternatively, a solution of ncDNA (0.5 μM in PBS 7.4, for 1 h) was used for the selectivity experiment. All of the CV experiments were performed varying the scan rate between 10 and 200 mV/s in fresh 0.1 M NaClO4 as the electrolyte (degassed for 5 min).
Electrochemical measurements were performed in a three-electrode setup (custom-built glass electrochemical cell) with a platinum disk as the counter electrode, a red rod reference electrode (Ag/AgCl, saturated KCl solution, Radiometer Analytical), and the gold substrate as the working electrode (theoretical surface area of 0.44 cm2). Data analysis was done using CHI760D software (CH Instruments, Inc. Austin) and the methodology reported in the Electrochemical Analysis section in the SI. The CV experiments were repeated twice.
Acknowledgments
The Horizon 2020 Health project “ULTRAPLACAD” (no. 633937) and the Weijerhorst project are acknowledged for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.langmuir.0c00144.
Chemical structures of the PNA and DNA linkers; 1H NMR spectra of PLL-OEG-DBCO; contact angle goniometry values; illustration of the geometrical micropillar electrode formation and theoretical model based on its geometry; CV data acquisition, selectivity experiments, and data treatment; and surface probe density calculation (PDF)
Author Contributions
∥ J.M. and R.W.K. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Kawamura A.; Miyata T.. Biosensors. In Biomaterials Nanoarchitectonics; Ebara M., Ed.; Elsevier, 2016; pp 157–176. [Google Scholar]
- Wang J. Electrochemical Glucose Biosensors. Chem. Rev. 2008, 108, 814–825. 10.1021/cr068123a. [DOI] [PubMed] [Google Scholar]
- Liu F.; Piao Y.; Choi J. S.; Seo T. S. Three-Dimensional Graphene Micropillar Based Electrochemical Sensor for Phenol Detection. Biosens. Bioelectron. 2013, 50, 387–392. 10.1016/j.bios.2013.06.055. [DOI] [PubMed] [Google Scholar]
- Arroyo-Currás N.; Somerson J.; Vieira P. A.; Ploense K. L.; Kippin T. E.; Plaxco K. W. Real-Time Measurement of Small Molecules Directly in Awake, Ambulatory Animals. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 645–650. 10.1073/pnas.1613458114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo-Currás N.; Scida K.; Ploense K. L.; Kippin T. E.; Plaxco K. W. High Surface Area Electrodes Generated via Electrochemical Roughening Improve the Signaling of Electrochemical Aptamer-Based Biosensors. Anal. Chem. 2017, 89, 12185–12191. 10.1021/acs.analchem.7b02830. [DOI] [PubMed] [Google Scholar]
- Rifai N.; Gillette M. A.; Carr S. A. Protein Biomarker Discovery and Validation: the Long and Uncertain Path to Clinical Utility. Nat. Biotechnol. 2006, 24, 971–983. 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- Dawson S.; Tsui D.; Murtaza M.; Biggs H.; Rueda O. M.; Chin S.; Dunning M. J.; Gale D.; Forshew T.; Mahler-Araujo B.; Rajan S.; Humphray S.; Becq J.; Halsall D.; Wallis M.; Bentley D.; Caldas C.; Rosenfeld N. Analysis of Circulating Tumor DNA to Monitor Metastatic Breast Cancer. N. Engl. J. Med. 2013, 368, 1199–1209. 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- Yong E. Cancer Biomarkers: Written in Blood. Nature 2014, 511, 524–526. 10.1038/511524a. [DOI] [PubMed] [Google Scholar]
- Grieshaber D.; MacKenzie R.; Vörös J.; Reimhult E. Electrochemical Biosensors - Sensor Principles and Architectures. Sensors 2008, 8, 1400–1458. 10.3390/s80314000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond T. G.; Hill M. G.; Barton J. K. Electrochemical DNA Sensors. Nat. Biotechnol. 2003, 21, 1192–1199. 10.1038/nbt873. [DOI] [PubMed] [Google Scholar]
- Ricci F.; Plaxco K. W. E-DNA Sensors for Convenient, Label-Free Electrochemical Detection of Hybridization. Microchim. Acta 2008, 163, 149–155. 10.1007/s00604-008-0015-4. [DOI] [Google Scholar]
- Das J.; Ivanov I.; Montermini L.; Rak J.; Sargent E. H.; Kelley S. O. An Electrochemical Clamp Assay for Direct, Rapid Analysis of Circulating Nucleic Acids in Serum. Nat. Chem. 2015, 7, 569–575. 10.1038/nchem.2270. [DOI] [PubMed] [Google Scholar]
- Grody W. W.; Cutting G. R.; Klinger K. W.; Richards C. S.; Watson M. S.; Desnick R. J. Laboratory Standards and Guidelines for Population-Based Cystic Fibrosis Carrier Screening. Genet. Med. 2001, 3, 149–154. 10.1097/00125817-200103000-00010. [DOI] [PubMed] [Google Scholar]
- Maurano M.; Humbert R.; Rynes E.; Thurman R.; Haugen E.; Wang H.; Reynolds A.; Sandstrom R.; Qu H.; Brody J.; Shafer A.; Neri F.; Lee K.; Kutyavin T.; Stehling-Sun S.; Johnson A.; Canfield T.; Giste E.; Diegel M.; Bates D.; Hansen R.; Neph S.; Sabo P.; Heimfeld S.; Raubitschek A.; Ziegler S.; Cotsapas C.; Sotoodehnia N.; Glass I.; Sunyaev S.; Kaul R.; Stamatoyannopoulos J. A. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellassai N.; Spoto G. Biosensors for Liquid Biopsy: Circulating Nucleic Acids to Diagnose and Treat Cancer. Anal. Bioanal. Chem. 2016, 408, 7255–7264. 10.1007/s00216-016-9806-3. [DOI] [PubMed] [Google Scholar]
- Thierry A.; Mouliere F.; El Messaoudi S.; Mollevi C.; Lopez-Crapex E.; Rolet F.; Gillet B.; Gongora C.; Dechelotte P.; Robert B.; Del Rio M.; Lamy P.; Bibeau F.; Nouaille M.; Loriot V.; Jarrousse A.; Molina F.; Mathonnet M.; Pezet D.; Ychou M. Clinical Validation of the Detection of KRAS and BRAF Mutations from Circulating Tumor DNA. Nat. Med. 2014, 20, 430–435. 10.1038/nm.3511. [DOI] [PubMed] [Google Scholar]
- Nguyen A. H.; Sim S. J. Nanoplasmonic Biosensor: Detection and Amplification of Dual Bio-Signatures of Circulating Tumor DNA. Biosens. Bioelectron. 2015, 67, 443–449. 10.1016/j.bios.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Campbell C. N.; Gal D.; Cristler N.; Banditrat C.; Heller A. Enzyme-Amplified Amperometric Sandwich Test for RNA and DNA. Anal. Chem. 2002, 74, 158–162. 10.1021/ac015602v. [DOI] [PubMed] [Google Scholar]
- Patolsky F.; Lichtenstein A.; Willner I. Electronic Transduction of DNA Sensing Processes on Surfaces: Amplification of DNA Detection and Analysis of Single-Base Mismatches by Tagged Liposomes. J. Am. Chem. Soc. 2001, 123, 5194–5205. 10.1021/ja0036256. [DOI] [PubMed] [Google Scholar]
- Wang J.; Kawde A. N.; Musameh M.; Rivas G. Dual Enzyme Electrochemical Coding for Detecting DNA Hybridization. Analyst 2002, 127, 1279–1282. 10.1039/b207424a. [DOI] [PubMed] [Google Scholar]
- Yang I. V.; Thorp H. H. Modification of Indium Tin Oxide Electrodes with Repeat Polynucleotides: Electrochemical Detection of Trinucleotide Repeat Expansion. Anal. Chem. 2001, 73, 5316–5322. 10.1021/ac010062n. [DOI] [PubMed] [Google Scholar]
- Hu Q.; Kong J.; Han D.; Niu L.; Zhang X. Electrochemical DNA Biosensing via Electrochemically Controlled Reversible Addition-Fragmentation Chain Transfer Polymerization. ACS Sens. 2019, 4, 235–241. 10.1021/acssensors.8b01357. [DOI] [PubMed] [Google Scholar]
- Pheeney C. G.; Barton J. K. DNA Electrochemistry with Tethered Methylene Blue. Langmuir 2012, 28, 7063–7070. 10.1021/la300566x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M. U.; Nahar S.; Safavieh M.; Zourob M. Real-Time Electrochemical Detection of Pathogen DNA Using Electrostatic Interaction of a Redox Probe. Analyst 2013, 138, 907–915. 10.1039/C2AN36153A. [DOI] [PubMed] [Google Scholar]
- Hu Q.; Deng X.; Yu X.; Kong J.; Zhang X. One-Step Conjugation of Aminoferrocene to Phosphate Groups as Electroactive Probes for Electrochemical Detection of Sequence-Specific DNA. Biosens. Bioelectron. 2015, 65, 71–77. 10.1016/j.bios.2014.10.015. [DOI] [PubMed] [Google Scholar]
- Biagiotti V.; Porchetta A.; Desiderati S.; Plaxco K. W.; Palleschi G.; Ricci F. Probe Accessibility Effects on the Performance of Electrochemical Biosensors Employing DNA Monolayers. Anal. Bioanal. Chem. 2012, 402, 413–421. 10.1007/s00216-011-5361-0. [DOI] [PubMed] [Google Scholar]
- Movilli J.; Rozzi A.; Ricciardi R.; Corradini R.; Huskens J. Control of Probe Density at DNA Biosensor Surfaces Using Poly(l-Lysine) with Appended Reactive Groups. Bioconjug. Chem. 2018, 29, 4110–4118. 10.1021/acs.bioconjchem.8b00733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z. G.; Lai R. Y. Effect of Signaling Probe Conformation on Sensor Performance of a Displacement-Based Electrochemical DNA Sensor. Anal. Chem. 2013, 85, 3340–3346. 10.1021/ac3037987. [DOI] [PubMed] [Google Scholar]
- Suni I. I. Impedance Methods for Electrochemical Sensors Using Nanomaterials. TrAC, Trends Anal. Chem. 2008, 27, 604–611. 10.1016/j.trac.2008.03.012. [DOI] [Google Scholar]
- Wang X.; Smirnov S. Label-Free DNA Sensor Based on Surface Charge Modulated Ionic Conductance. ACS Nano 2009, 3, 1004–1010. 10.1021/nn900113x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry O. Y. F.; Kirwan S.; Debela A. M.; O’Sullivan C. K. Electrochemical Genosensor Based on Three-Dimensional DNA Polymer Brushes Monolayers. Electrochem. commun. 2011, 13, 1155–1158. 10.1016/j.elecom.2011.09.010. [DOI] [Google Scholar]
- Kowalczyk A.; Fau M.; Karbarz M.; Donten M.; Stojek Z.; Nowicka A. M. Hydrogel with Chains Functionalized with Carboxyl Groups as Universal 3D Platform in DNA Biosensors. Biosens. Bioelectron. 2014, 54, 222–228. 10.1016/j.bios.2013.11.017. [DOI] [PubMed] [Google Scholar]
- Kugel V.; Ji H. Nanopillars for Sensing. J. Nanosci. Nanotechnol. 2014, 14, 6469–6477. 10.1166/jnn.2014.9346. [DOI] [PubMed] [Google Scholar]
- Saeed A. A.; Sánchez J. L. A.; O’Sullivan C. K.; Abbas M. N. DNA Biosensors Based on Gold Nanoparticles-Modified Graphene Oxide for the Detection of Breast Cancer Biomarkers for Early Diagnosis. Bioelectrochemistry 2017, 118, 91–99. 10.1016/j.bioelechem.2017.07.002. [DOI] [PubMed] [Google Scholar]
- Daggumati P.; Matharu Z.; Seker E. Effect of Nanoporous Gold Thin Film Morphology on Electrochemical DNA Sensing. Anal. Chem. 2015, 87, 8149–8156. 10.1021/acs.analchem.5b00846. [DOI] [PubMed] [Google Scholar]
- Soleymani L.; Fang Z.; Lam B.; Bin X.; Vasilyeva E.; Ross A. J.; Sargent E. H.; Kelley S. O. Hierarchical Nanotextured Microelectrodes Overcome the Molecular Transport Barrier to Achieve Rapid, Direct Bacterial Detection. ACS Nano 2011, 5, 3360–3366. 10.1021/nn200586s. [DOI] [PubMed] [Google Scholar]
- Prehn R.; Cortina-Puig M.; Muñoz F. X. A Non-Enzymatic Glucose Sensor Based on the Use of Gold Micropillar Array Electrodes. J. Electrochem. Soc. 2012, 159, F134–F139. 10.1149/2.018206jes. [DOI] [Google Scholar]
- Anandan V.; Rao Y. L.; Zhang G. Nanopillar Array Structures for Enhancing Biosensing Performance. Int. J. Nanomed. 2006, 1, 73–79. 10.2147/nano.2006.1.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Kim D. S.; Yoon J. H.; Hong S. B.; Jeong S. W.; Yoo D. E.; Lee T. J.; Lee S. J.; Lee K. G.; Choi B. G. Nanopillar Films with Polyoxometalate-Doped Polyaniline for Electrochemical Detection of Hydrogen Peroxide. Analyst 2016, 141, 1319–1324. 10.1039/C5AN02134K. [DOI] [PubMed] [Google Scholar]
- Penmatsa V.; Kim T.; Beidaghi M.; Kawarada H.; Gu L.; Wang Z.; Wang C. Three-Dimensional Graphene Nanosheet Encrusted Carbon Micropillar Arrays for Electrochemical Sensing. Nanoscale 2012, 4, 3673–3678. 10.1039/c2nr30161j. [DOI] [PubMed] [Google Scholar]
- Schröper F.; Brüggemann D.; Mourzina Y.; Wolfrum B.; Offenhäusser A.; Mayer D. Analyzing the Electroactive Surface of Gold Nanopillars by Electrochemical Methods for Electrode Miniaturization. Electrochim. Acta 2008, 53, 6265–6272. 10.1016/j.electacta.2008.03.068. [DOI] [Google Scholar]
- Davies T. J.; Ward-Jones S.; Banks C. E.; del Campo J.; Mas R.; Muñoz F. X.; Compton R. G. The Cyclic and Linear Sweep Voltammetry of Regular Arrays of Microdisc Electrodes: Fitting of Experimental Data. J. Electroanal. Chem. 2005, 585, 51–62. 10.1016/j.jelechem.2005.07.021. [DOI] [Google Scholar]
- Numthuam S.; Kakegawa T.; Anada T.; Khademhosseini A.; Suzuki H.; Fukuda J. Synergistic Effects of Micro/Nano Modifications on Electrodes for Microfluidic Electrochemical ELISA. Sens. Actuators, B 2011, 156, 637–644. 10.1016/j.snb.2011.02.010. [DOI] [Google Scholar]
- Sánchez-Molas D.; Esquivel J. P.; Sabaté N.; Muñoz F. X.; del Campo F. J. High Aspect-Ratio, Fully Conducting Gold Micropillar Array Electrodes: Silicon Micromachining and Electrochemical Characterization. J. Phys. Chem. C 2012, 116, 18831–18846. 10.1021/jp305339k. [DOI] [Google Scholar]
- Prehn R.; Abad L.; Sánchez-Molas D.; Duch M.; Sabaté N.; del Campo F. J.; Muñoz F. X.; Compton R. G. Microfabrication and Characterization of Cylinder Micropillar Array Electrodes. J. Electroanal. Chem. 2011, 662, 361–370. 10.1016/j.jelechem.2011.09.002. [DOI] [Google Scholar]
- Lee S.; Spencer N. D. Adsorption Properties of Poly(l-Lysine)-Graft-Poly(Ethylene Glycol) (PLL-g-PEG) at a Hydrophobic Interface: Influence of Tribological Stress, pH, Salt Concentration, and Polymer Molecular Weight. Langmuir 2008, 24, 9479–9488. 10.1021/la801200h. [DOI] [PubMed] [Google Scholar]
- Lee S.; Mu M.; Ratoi-Salagean M.; Vörös J.; De Paul S. M.; Spikes H. A.; Textor M.; Spencer N. D.; et al. Boundary Lubrication of Oxide Surfaces by Poly(l-Lysine)-g-Poly(Ethylene Glycol) (PLL-g-PEG) in Aqueous Media. Tribol Lett. 2003, 15, 231–239. 10.1023/A:1024861119372. [DOI] [Google Scholar]
- Müller M.; Lee S.; Spikes H. A.; Spencer N. D. The Influence of Molecular Architecture on the Macroscopic Lubrication Properties of the Brush-like Co-Polyelectrolyte Poly(l-Lysine)-g-Poly(Ethylene Glycol) (PLL-g-PEG) Adsorbed on Oxide Surfaces. Tribol. Lett. 2003, 15, 395–405. 10.1023/B:TRIL.0000003063.98583.bb. [DOI] [Google Scholar]
- Huang N.; Vörös J.; De Paul S. M.; Textor M.; Spencer N. D. Biotin-Derivatized Poly(l-Lysine)-g-Poly(Ethylene Glycol): A Novel Polymeric Interface for Bioaffinity Sensing. Langmuir 2002, 18, 220–230. 10.1021/la010913m. [DOI] [Google Scholar]
- Pasche S.; De Paul S. M.; Vörös J.; Spencer N. D.; Textor M. Poly(l-Lysine)-Graft-Poly(Ethylene Glycol) Assembled Monolayers on Niobium Oxide Surfaces: A Quantitative Study of the Influence of Polymer Interfacial Architecture on Resistance to Protein Adsorption by ToF-SIMS and in Situ OWLS. Langmuir 2003, 19, 9216–9225. 10.1021/la034111y. [DOI] [Google Scholar]
- Di Iorio D.; Marti A.; Koeman S.; Huskens J. Clickable Poly-l-Lysine for the Formation of Biorecognition Surfaces. RSC Adv. 2019, 9, 35608–35613. 10.1039/C9RA08714A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti M. C.; Schwarz F. P. Thermal Stability of PNA/DNA and DNA/DNA Duplexes by Differential Scanning Calorimetry. Nucleic Acids Res. 1999, 27, 4801–4806. 10.1093/nar/27.24.4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demidov V. V.; Potaman V. N.; Frank-Kamenetskil M. D.; Egholm M.; Buchard O.; Sönnichsen S. H.; Nlelsen P. E. Stability of Peptide Nucleic Acids in Human Serum and Cellular Extracts. Biochem. Pharmacol. 1994, 48, 1310–1313. 10.1016/0006-2952(94)90171-6. [DOI] [PubMed] [Google Scholar]
- Kuzmin A.; Poloukhtine A.; Wolfert M. A.; Popik V. V. Surface Functionalization Using Catalyst-Free Azide-Alkyne Cycloaddition. Bioconjugate Chem. 2010, 21, 2076–2085. 10.1021/bc100306u. [DOI] [PubMed] [Google Scholar]
- Umek R.; Lin S.; Vielmetter J.; Terbrueggen R.; Irvine B.; Yu C.; Kayyem J.; Yowanto H.; Blackburn G.; Farkas D.; Chen Y. Electronic Detection of Nucleic Acids: A Versatile Platform for Molecular Diagnostics. J. Mol. Diagn. 2001, 3, 74–84. 10.1016/S1525-1578(10)60655-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbersen R.; Tiggelaar R. M.; Milbrat A.; Mul G.; Gardeniers H.; Huskens J. Controlled Doping Methods for Radial p/n Junctions in Silicon. Adv. Energy Mater. 2015, 5, 1401745 10.1002/aenm.201401745. [DOI] [Google Scholar]
- Duan X.; Mu L.; Sawtelle S. D.; Rajan N. K.; Han Z.; Wang Y.; Qu H.; Reed M. A. Functionalized Polyelectrolytes Assembling on Nano-BioFETs for Biosensing Applications. Adv. Funct. Mater. 2015, 25, 2279–2286. 10.1002/adfm.201500002. [DOI] [Google Scholar]
- Han Z.; Wang Y.; Duan X. Biofunctional Polyelectrolytes Assembling on Biosensors – A Versatile Surface Coating Method for Protein Detections. Anal. Chim. Acta 2017, 964, 170–177. 10.1016/j.aca.2017.01.051. [DOI] [PubMed] [Google Scholar]
- Veerbeek J.; Steen R.; Vijselaar W.; Rurup W. F.; Korom S.; Rozzi A.; Corradini R.; Segerink L.; Huskens J. Selective Functionalization with PNA of Silicon Nanowires on Silicon Oxide Substrates. Langmuir 2018, 34, 11395–11404. 10.1021/acs.langmuir.8b02401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauerbrey G. Verwendung von Schwingquarzen Zur Wägung Dünner Schichten Und Zur Mikrowägung. Z. Phys. 1959, 155, 206–222. 10.1007/BF01337937. [DOI] [Google Scholar]
- Höök F.; Ray a.; Nordén B.; Kasemo B. Characterization of PNA and DNA Immobilization and Subsequent Hybridization with DNA Using Acoustic-Shear-Wave Attenuation Measurements. Langmuir 2001, 17, 8305–8312. 10.1021/la0107704. [DOI] [Google Scholar]
- Su X.; Wu Y. J.; Knoll W. Comparison of Surface Plasmon Resonance Spectroscopy and Quartz Crystal Microbalance Techniques for Studying DNA Assembly and Hybridization. Biosens. Bioelectron. 2005, 21, 719–726. 10.1016/j.bios.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Larsson C.; Rodahl M.; Höök F. Characterization of DNA Immobilization and Subsequent Hybridization on a 2D Arrangement of Streptavidin on a Biotin-Modified Lipid Bilayer Supported on SiO2. Anal. Chem. 2003, 75, 5080–5087. 10.1021/ac034269n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.