

Significance Statement
In males with classic Fabry disease, the processes leading to the frequent outcome of ESKD are poorly understood. Mutations in the gene encoding α-galactosidase A leads to globotriaosylceramide accumulation in various cell types; in podocytes, this accumulation progresses with age. In this study of 55 males with classic Fabry disease genotype and/or phenotype, the authors found an increasing fraction of podocyte cytoplasm occupied by globotriaosylceramide, which plateaued at around the age of 27 years. At the same time, podocyte volume continued to rise, apparently at the expense of increasing podocyte stress (indicated by increasing podocyte foot process width) and podocyte loss. These changes associated with increasing urinary protein excretion, a strong prognosticator of adverse renal outcomes, and with reduction in GFR, indicating a need for early intervention before critical podocyte loss.
Keywords: Fabry disease, podocyte, chronic kidney disease, pathology
Visual Abstract

Abstract
Background
In males with classic Fabry disease, the processes leading to the frequent outcome of ESKD are poorly understood. Defects in the gene encoding α-galactosidase A lead to accumulation of globotriaosylceramide (GL3) in various cell types. In the glomerular podocytes, accumulation of GL3 progresses with age. Of concern, podocytes are relatively resistant to enzyme replacement therapy and are poorly replicating, with little ability to compensate for cell loss.
Methods
In this study of 55 males (mean age 27 years) with classic Fabry disease genotype and/or phenotype, we performed unbiased quantitative morphometric electron microscopic studies of biopsied kidney samples from patients and seven living transplant donors (to serve as controls). We extracted clinical information from medical records and clinical trial databases.
Results
Podocyte GL3 volume fraction (proportion of podocyte cytoplasm occupied by GL3) increased with age up to about age 27, suggesting that increasing podocyte GL3 volume fraction beyond a threshold may compromise survival of these cells. GL3 accumulation was associated with podocyte injury and loss, as evidenced by increased foot process width (a generally accepted structural marker of podocyte stress and injury) and with decreased podocyte number density per glomerular volume. Worsening podocyte structural parameters (increasing podocyte GL3 volume fraction and foot process width) was also associated with increasing urinary protein excretion—a strong prognosticator of adverse renal outcomes in Fabry disease—as well as with decreasing GFR.
Conclusions
Given the known association between podocyte loss and irreversible FSGS and global glomerulosclerosis, this study points to an important role for podocyte injury and loss in the progression of Fabry nephropathy and indicates a need for therapeutic intervention before critical podocyte loss occurs.
Fabry disease is caused by mutations in the GLA gene leading to deficiency in the lysosomal enzyme α-galactosidase A (α-Gal-A) and accumulation of its substrates, primarily globotriaosylceramide (GL3), in various cell types and organs.1 Especially in males with more severe α-Gal-A gene mutations and little or no residual α-Gal-A enzyme activity, Fabry disease often results in severe vital organ injury that manifests as strokes, cardiomyopathy, arrhythmias, renal failure, neuropathy, and premature death.2
Enzyme replacement therapy (ERT) may relatively quickly (within 5 months) eliminate microscopically detectable GL3 accumulation in endothelial cells in the skin, kidney, and heart, as well as in additional cell types such as glomerular mesangial cells and fibroblasts.3 However, some cell types such as glomerular podocytes, vascular smooth muscle cells, and cardiac myocytes are considerably more resistant to ERT.3,4 These are all complex, terminally differentiated, poorly replicating cells with great functional significance whose injury or loss can lead to severe organ dysfunction. Thus, although a long-term, randomized, placebo-controlled ERT trial reported reductions in serious clinical events,5 there are substantial residual risks despite ERT.6 Because podocytes play a crucial role in preserving nephron structure and function, they have little ability to replenish when lost.7 Podocyte dysfunction/loss is closely associated with proteinuria, the strongest available biomarker for predicting GFR loss in patients with Fabry disease.8 In addition, there is a large and ever-increasing body of human and animal research which is consistent with the concept that substantial podocyte loss is associated with FSGS and global glomerulosclerosis, important and irreversible lesions on the path to ESKD.9,10 In young patients with Fabry disease who are ERT naive, podocyte GL3 accumulation increases with age, whereas in glomerular endothelial and mesangial cells it does not.11 Given that the development of the clinical events in Fabry disease is highly age dependent,6,12,13 cells with progressive Fabry changes with increasing age (e.g., podocytes) are more likely to contribute to these events. This study focuses primarily on podocyte changes over a wide age range in male patients with untreated Fabry disease.
Methods
Patients
We studied male patients with Fabry disease who were treatment naive. Renal biopsies were performed as baselines for clinical trials, as clinical assessments before the initiation of ERT, or for diagnosis of clinical renal abnormalities. All patients provided written informed consent (or parental consent in the case of children). Kidney biopsies from seven living transplant donors obtained before organ removal were studied as controls. The research was performed in accordance with principles of the Declaration of Helsinki and was approved by the Institutional Review Boards of the Universities of Minnesota and Washington.
Clinical Information
Patients’ age at the biopsy, serum creatinine, urinary protein excretion rate (UPER; based on urine protein-creatinine ratio or 24-hour urine protein), Fabry-related symptoms, GLA mutation, α-Gal-A activity, and plasma GL3 values were extracted from medical records or from the clinical trial databases. Leukocyte and plasma α-Gal-A activity levels are strongly correlated.14 To make leukocyte and plasma α-Gal-A activity values measured in different laboratories and at different times comparable, we expressed α-Gal-A activity as a percentage of the lower limit of a given laboratory’s normal range (Table 1). All protein changes were checked with the MutationTaster software (http://www.mutationtaster.org/cgi-bin/MutationTaster/MutationTaster69.cgi). For mutation descriptions at the cDNA and protein levels, we followed the Human Genome Variation Society recommendations.15 GFR, where available, was measured by the plasma disappearance of iohexol, or was estimated based on serum creatinine values using the CKD Epidemiology Collaboration equation if age ≥1816 or the modified Schwartz equation if age <18 years.17 UPER was derived from timed urine collections or spot urine samples obtained close to the time of the biopsy.
Table 1.
Clinical and renal functional characteristics in male patients with classic Fabry disease
| Case | Age (yr) | Protein Change | cDNA Mutation | Mutation Category | α-Gal-A Activity (%LNL) | Plasma GL3 | GFR | UPER | RAAS Blockade | Nonrenal Fabry Features |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 4 | p.(Tyr216Asp) | c.646T>G | Missense | NK | NK | 92 | NK | N | NK |
| 2 | 5 | p.(Arg112Cys) | c.334C>T | Missense | 0% | NK | 122 | 0.460 | N | None |
| 3 | 7 | p.(Met267Arg) | c.800T>G | Missense | 14% | 7.5 | 106 | 0.250 | N | CO, PN, GI |
| 4 | 11 | p.(His302Alafs*13) | c.903_904insG | Frameshift | 0% | NK | 121 | 0.150 | N | NK |
| 5 | 11 | NK | NK | NK | 20% | NK | NK | 0 | N | CO, AK |
| 6 | 12 | NK | NK | NK | 38% | NK | NK | 0.039 | N | CO, AK |
| 7 | 13 | NK | NK | NK | 0% | NK | 122 | 0.169 | N | NK |
| 8 | 15 | NK | NK | NK | 3% | NK | 93 | 0.082 | N | NK |
| 9 | 15 | NK | NK | NK | 0% | NK | 103 | 0.088 | N | NK |
| 10 | 15 | NK | NK | NK | 3% | NK | 134 | 0.079 | N | NK |
| 11a | 15 | NK | NK | NK | NK | NK | NK | 0.350 | N | CO, AK, PN |
| 12 | 16 | p.(Arg404del) | c.1212_1214delAAG | Deletion | 49% | 13.4 | 112 | 0.090 | N | CO, AK, PN, GI |
| 13 | 17 | p.(Arg227Ter) | c.679C>T | Nonsense | 2% | 17.1 | 190 | 0.229 | N | CO, AK, PN |
| 14 | 17 | p.(Asn272Lys) | c.816C>A | Missense | 2% | NK | 107 | 0.110 | N | PN, GI |
| 15 | 17 | NK | NK | NK | 0% | NK | 97 | 0.082 | N | NK |
| 16 | 17 | NK | NK | NK | 3% | NK | 127 | 0.069 | N | NK |
| 17 | 18 | p.(Met267Arg) | c.800T>G | Missense | 19% | 4.8 | 96 | 0.220 | N | CO, AK, PN, GI |
| 18 | 18 | p.(Arg227Ter) | c.679C>T | Nonsense | 2% | 19.3 | 156 | 0.167 | N | CO, AK, PN |
| 19 | 20 | NK | c.802–3_802-2delCA | Intronic deletion/ splicing | 3% | NK | 153 | 0.167 | N | CO, AK |
| 20 | 20 | p.(Arg220Lys) | c.658C>T | Nonsense | 2% | 7.7 | 70 | 0.211 | Y | CO, AK, PN |
| 21 | 20 | p.(Asn272Lys) | c.816C>A | Missense | <24% | 27.3 | 131 | 0.024 | N | CO, AK, PN, GI |
| 22 | 21 | p.(Arg227Ter) | c.679C>T | Nonsense | 2% | 15.8 | 130 | 0.102 | N | AK, PN, GI |
| 23 | 21 | p.(Arg227Ter) | c.679C>T | Nonsense | NK | 2 | 289 | 0.140 | N | CO, PN, GI |
| 24 | 23 | p.(Asn298Lysfs*2) | c.893_894insG | Insertion | 6% | 7 | 112 | 0.102 | N | CO, AK, GI |
| 25 | 23 | NK | NK | NK | 5% | 8.5 | 178 | NK | N | CO, AK, LVH, PN |
| 26 | 23 | p.(Gly132Arg) | c.394G>A | Missense | <24% | 9.9 | 88 | NK | N | CO, AK, |
| 27 | 23 | p.(Arg404del) | c.1212_1214delAAG | Deletion | 14% | 8.5 | 113 | 0.240 | N | CO, PN, GI |
| 28 | 23 | NK | NK | NK | 6% | 10.5 | 148 | NK | N | CO, AK, PN |
| 29 | 24 | NK | NK | NK | 4% | 20 | 126 | 0.140 | N | CO, AK, PN |
| 30 | 25 | NK | NK | NK | <24% | 13.9 | 146 | 0.402 | N | CO, AK, AR, PN, GI |
| 31 | 25 | NK | NK | NK | 6% | NK | 110 | NA | N | CO, AK |
| 32 | 25 | p.([Asp55Val; Gln57Leu]) | c.(164A>T; 170A>T) | Double missense | 0% | NK | 114 | 0.018 | NK | NK |
| 33 | 26 | p.(Trp204Ter) | c.612G>A | Nonsense | 6% | 9.9 | 96 | NK | N | CO, AK, PN, GI |
| 34 | 30 | p.(Arg404del) | c.1212_1214delAAG | Deletion | 28% | 10.2 | 86 | 0.220 | N | CO, AK, PN, GI |
| 35 | 31 | NK | c.639+4A>T | Splicing | 3% | 5.3 | 167 | 1.150 | N | CO |
| 36 | 33 | p.(Asp244Asn) | c.730G>A | Missense | NK | NK | 115 | 0.023 | NK | NK |
| 37 | 33 | p.(Val339Alafs*32) | c.1016_1026delTGTGGGAACGA | Deletion | 0% | 7.7 | 103 | 0.383 | N | CO, AK, PN |
| 38 | 34 | p.(Tyr216Cys) | c.647A>G | Missense | 0% | NK | 119 | 0.029 | NK | NK |
| 39 | 34 | NK | c.639+4A>T | Splicing | 3% | 29.5 | 137 | 0.297 | N | CO, AK, LVH, AR, MI, PN |
| 40 | 35 | p.(Ser102Glnfs*19) | c.304delC | Deletion | 3% | 16.9 | 102 | NK | Y | CO, AK, PN |
| 41 | 35 | p.(Gly144Val) | c.431G>T | Missense | 0% | NK | 105 | 0.009 | NK | NK |
| 42 | 36 | p.(Val281_Thr282delinsAla) | c.842_844delTAA | Deletion | <24% | 10.8 | 102 | 1.267 | N | CO, AK, PN |
| 43 | 37 | p.(Ser148Arg) | c.444T>G | Missense | 6% | 13.7 | 101 | 0.359 | N | CO, AK, PN |
| 44 | 38 | NK | NK | NK | <24% | 35.5 | 135 | 1.615 | N | CO, AK, PN |
| 45 | 38 | p.(Ser148Arg) | c.444T>G | Missense | 6% | 12 | 101 | NA | N | CO, AK, MI |
| 46 | 40 | p.(Asp153del) | c.457_459delGAC | Deletion | 0% | NK | 76 | 3.380 | Y | LVH |
| 47 | 40 | p.(Arg112Cys) | c.334C>T | Missense | NK | 4.8 | 79 | 0.230 | N | CO, AK, LVH |
| 48 | 40 | p.(Arg301Ter) | c.901C>T | Nonsense | 0% | 19.2 | 133 | 0.462 | N | CO, AK, GI |
| 49 | 45 | p.(Pro259Arg) | c.776C>G | Missense | 3% | NK | 104 | 0.027 | NK | NK |
| 50 | 45 | p.(Ala156Thr) | c.466G>A | Missense | NK | NK | 74 | 0.016 | NK | NK |
| 51 | 45 | p.(Leu243Phe) | c.729G>C | Missense | 0% | NK | 104 | 0.008 | NK | NK |
| 52 | 46 | NK | NK | NK | <24% | 25.3 | 154 | 0.209 | N | CO, AK, LVH, PN, GI |
| 53 | 52 | p.(Asp33Gly) | c.98A>G | Missense | 0% | NK | 83 | 0.016 | NK | NK |
| 54 | 53 | p.(Trp44Cys) | c.132G>T | Missense | NK | NK | 75 | 0.090 | Y | CO, AK, LVH, GI |
| 55 | 60 | p.(Asp322Glu) | c.966C>G | Missense | 1% | NK | NK | NK | NK | NK |
Plasma GL3 measred in μg/mL; GFR measured in ml/min per 1.73 m2; UPER, measured in mg/g if urine protein-creatinine ratio or g/d if 24 h collection. LNL, lower normal limit; RAAS, renin-angiotensin aldosterone system; NK, not known; N, no; CO, corneal opacity; PN, peripheral neuropathy; GI, gastrointestinal; AK, angiokeratoma; Y, yes; AR, arrhythmia; LVH, left ventricular hypertrophy; MI, myocardial infarction.
Fabry disease diagnosis confirmed by clinical history of periodic lower-extremity pain crises with and without fevers, angiokeratomas, cornea verticillata, mild proteinuria at age 14 yr, kidney biopsy findings consistent with Fabry nephropathy in the absence of lysosomotrophic medications, and a maternal uncle with acroparesthesias and cardiac disease who died prematurely at age 40 yr.
Renal Biopsy Studies
Sections (1 µm) of 2.5% glutaraldehyde-fixed, plastic-embedded tissues were stained with toluidine blue for identification of glomeruli.18 Random glomerular sections were prepared for stereologic studies as described elsewhere.11 Overlapping digital low-magnification (approximately 8000×) images of entire glomerular profiles were obtained using a JEOL 1010 electron microscope for masked review by two observers (B.N. and M.M.) to select two to five nonsclerosed glomeruli per biopsy with minimal or no artifacts for stereologic studies as described below. High-magnification (approximately 30,000×) images were obtained according to a systematic uniform random sampling protocol for estimation using point counting of the fraction of the volume (Vv) of podocyte cytoplasm occupied by GL3 inclusions (Vv[Inc/PC]), hereafter called “podocyte GL3 volume fraction” for simplicity (Figure 1).11 This was also done for glomerular endothelial (Vv[Inc/Endo]) and mesangial (Vv[Inc/Mes]) cells.11 Average volume of podocyte nuclei (VPCN) was estimated using the point-sampled intercept method19 with a modified sampling strategy to reduce the volume-weighted property of the method (Figure 1).20 This provides shape- and size-independent volume estimates. The fraction of the volume of podocytes occupied by podocyte nuclei (Vv[PCN/PC]) and fraction of the volume of the glomerulus occupied by podocytes (Vv[PC/glom]) were estimated using point counting. The average volume of podocytes was calculated as 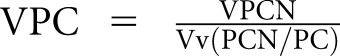 . The total volume of GL3 inclusions per podocyte (V[Inc/PC]), hereafter called “podocyte GL3 volume” for simplicity, was then calculated as VPC·Vv(Inc/PC). Number density of podocytes per glomerular volume (Nv[PC/glom]) was calculated as the fraction of the volume of the glomerulus occupied by podocytes divided by the average podocyte volume or Vv(PC/glom)/VPC.21 Podocyte average foot process width (FPW) was estimated as the reciprocal of slit-length density as previously described (Figure 1).11,22 All stereologic estimates were done by masked observers.
. The total volume of GL3 inclusions per podocyte (V[Inc/PC]), hereafter called “podocyte GL3 volume” for simplicity, was then calculated as VPC·Vv(Inc/PC). Number density of podocytes per glomerular volume (Nv[PC/glom]) was calculated as the fraction of the volume of the glomerulus occupied by podocytes divided by the average podocyte volume or Vv(PC/glom)/VPC.21 Podocyte average foot process width (FPW) was estimated as the reciprocal of slit-length density as previously described (Figure 1).11,22 All stereologic estimates were done by masked observers.
Figure 1.

Illustration of morphometric methods. (A) Systematic uniform random sampling of a glomerular profile for stereologic measurements from a patient with Fabry disease. Red boxes represent locations where higher-magnification images are obtained by transmission electron microscopy. Red arrows show the path of sampling. Yellow asterisk marks the box that is magnified in (B) (montage low magnification, about 8000×). (B) Magnified view of a portion of glomerular tuft with a superimposed point grid used for fractional volume estimation. (C) Higher magnification (approximately 30,000×) image with an unbiased counting frame superimposed for estimation of FPW based on the number of line intercepts with the glomerular basement membrane (yellow arrowheads) and number of slits (green arrows).
Statistical Analyses
Statistica 13.0 (Statsoft, Inc.) software was used. Parametric or nonparametric tests were used based on the variable characteristics and distribution. Comparison of variables in patients with Fabry disease and normal controls was done using the t test or Kolmogorov–Smirnov test. Relationships between variables were evaluated using Pearson correlation. Multiple regression analysis was performed to identify factors associated with podocyte loss and GFR. Piecewise linear regression analysis was performed to study nonlinear relationships. P≤0.05 was considered statistically significant.
Results
Patient Characteristics
We aimed to study podocyte injury in kidney biopsies from male patients with ERT-naive Fabry disease with the classic genotype/phenotype. A total of 67 males with ERT-naive Fabry disease with a median age of 26 (range, 4–60) years were initially considered for enrollment. The phenotype of the disease (classic versus late onset/cardiac variant) could be defined in 60 patients based on the available literature for known mutations and/or clinical findings; 48 (80%) had classic and 12 (20%) had late-onset/cardiac phenotypes of Fabry disease. The available information was insufficient for reliable classification in seven patients. Clinical parameters (age, GFR, UPER) and Fabry-specific structural data including GL3 inclusion volume fraction in podocytes and mesangial cells were not statistically different between these seven patients and the patients with classic phenotype, and inclusion volume fraction in endothelial cells was greater in these seven patients with unknown phenotype compared with the patients with the classic phenotype (Supplemental Table 1). We therefore combined these two groups for all subsequent analyses (n=55) and excluded the subjects with the late-onset/cardiac variant from the study. Subjects’ characteristics are listed in Table 1.
Relationships between GL3 Accumulation, Podocyte Injury, and Loss with Age
The scatterplot of age versus podocyte GL3 volume fraction suggested that the relationship between these two parameters may be best explained through two regression lines with a slope change somewhere between ages 20 and 30 years (Figure 2A). Piecewise linear regression analysis showed that 63% of podocyte GL3 volume fraction can be explained by age, with the breakpoint at age 27±13 years (i.e., podocyte GL3 volume fraction correlated directly with age in patients <27 years [R=0.62, P=0.0001], but did not increase with age thereafter; Figure 2A). This suggests that, beyond a threshold, increasing podocyte GL3 volume fraction may compromise the survival of these cells. To further examine this possibility, we determined the relationship between GL3 inclusion volume fraction in podocyte profiles with a visible nucleus and the size of that podocyte profile as an indicator of podocyte size in 193 podocytes from seven randomly selected biopsies. There was a statistically significant, direct correlation between podocyte profile area and GL3 inclusion volume fraction in each biopsy that was consistent with larger podocytes having a greater fraction of cytoplasm filled with GL3; this suggests that the rate of GL3 accumulation in these cells exceeds their rate of cellular enlargement (Supplemental Table 2). Furthermore, the plot of pooled data from these biopsies showed an initial linear relationship between GL3 inclusion volume fraction and podocyte profile area followed by a plateau (Figure 2B). This supports a threshold for GL3 inclusion volume fraction and/or podocyte size beyond which podocyte survival is compromised (Figure 2B).
Figure 2.

Relationships between podocyte structural parameters and age. (A) Relationship between the podocyte inclusion volume fraction (Vv[Inc/PC]) and age. Regression line shows an exponential model with plateau. (B) Relationship between podocyte profile area and Vv(Inc/PC). (C) Inverse relationship between the fractional volume of podocytes per glomerulus (Vv[PC/glom]) and age (r=−0.54, P=0.0001). (D) Relationship between number density of podocytes per glomerulus (Nv[PC/glom]) and age. Solid lines represent regression lines. Dashed lines represent 95% confidence interval.
Because we observed a different pattern of relationship between fractional volume of inclusions per podocyte and age in patients <27 versus those ≥27 years, we also separately analyzed the data in each of these age groups. In contrast to podocyte GL3 volume fraction which (as noted above) reached a plateau at about 27 years of age, both mean podocyte volume (VPC) and the podocyte GL3 volume (V[Inc/PC]) correlated directly with age in patients <27 years (R=0.50; P=0.017 and R=0.59; P=0.004, respectively) and >27 years (R=0.55; P=0.026 and R=0.51; P=0.046, respectively). Thus, although the podocyte GL3 volume fraction did not appear to increase beyond a certain level, podocyte cell volume and total GL3 content per podocyte continued to increase with increasing age. Comparison of a subset of patients with Fabry disease (n=20) with similar ages to the living kidney donors showed that mean podocyte cell volume in patients with Fabry disease was approximately fourfold greater than these normal controls (Supplemental Figure 1).
Although podocytes were enlarged in those with Fabry disease, there was progressive decline in the fraction of glomerular volume occupied by podocytes (Vv[PC/glom]) with age (R=−0.57; P<0.001; Figure 2C). Likewise, by simple linear regression analysis, number density of podocytes per glomerular volume (Nv[PC/glom]) declined with age (R=−0.47, P=0.033; Figure 2D), confirming podocyte loss with increasing age. Inverse correlations between age and the fraction of glomerular volume occupied by podocytes and between age and podocyte number density were present both in patients who were younger and who were older than 27 years of age (Figures 2C and 2D). Moreover, in 33 subjects where values for both plasma or leukocyte α-Gal-A activity and podocyte number density were available, there was a direct relationship between α-Gal-A activity and podocyte number density (r=0.46, P=0.007), consistent with a relationship between α-Gal-A deficiency and podocyte loss.
To identify factors associated with podocyte loss, multiple regression analysis was performed with podocyte number density (Nv[PC/glom]) as the dependent variable and age, podocyte GL3 volume, and GL3 inclusion volume fraction per podocyte, endothelial cell, and mesangial cell as predictor variables. Because podocyte GL3 volume and mean podocyte volume were highly correlated and showed redundancy with a tolerance of >0.01, mean podocyte volume was not included in the model. The model explained 40% (adjusted R2=0.40, P=0.003) of podocyte number density variance and podocyte GL3 volume was the only independent predictor of podocyte number density (P=0.0004), consistent with a strong negative effect of podocyte GL3 accumulation on podocyte survival in Fabry disease. Importantly, the addition of α-Gal-A activity substantially improved the model, where now 61% (adjusted R2=0.61, P=0.0005) of podocyte number density was explained, with both podocyte GL3 volume (P<0.001) and α-Gal-A activity (P=0.001) being independent predictors. Mean podocyte FPW, a generally accepted structural marker of podocyte stress and injury, was approximately 1.5-fold greater in patients with Fabry disease compared with normal controls (P=0.004; Supplemental Figure 2). Also, FPW correlated with both podocyte GL3 volume fraction (R=0.40, P=0.004) and podocyte GL3 volume (R=0.36, P=0.03).
Relationships between Podocyte Structural Parameters and Renal Function
By simple linear regression analysis, UPER correlated with increasing podocyte GL3 volume fraction (R=0.44, P=0.003) and FPW (R=0.41, P=0.007) (Figure 3, A and B). Using simple linear regression analysis, the fraction of glomerular volume occupied by podocytes (R=0.24, P=0.09), mean podocyte volume (R=0.30, P=0.07), and podocyte GL3 volume (R=0.30, P=0.08) showed trends of relationship with GFR. Using multiple regression analysis with a tolerance >0.01, 13% of GFR variance (adjusted R2=0.13, P=0.03) was explained by the fraction of glomerular volume occupied by podocytes, FPW, and podocyte GL3 volume fraction, where the fraction of glomerular volume occupied by podocytes and FPW were independent predictors. Addition of age, podocyte GL3 volume, and average podocyte volume or podocyte number density made the model statistically insignificant.
Figure 3.

Relationships between podocyte structural parameters and urinary protein excretion, and podocyte loss (A) Relationship between UPER (logarithmic scale, in mg/g if urine protein-creatinine ratio or g/d if 24-hour collection) and podocyte inclusion volume fraction (Vv[Inc/PC]; R=0.44, P=0.003). (B) Relationship between UPER (logarithmic scale) and podocyte FPW (R=0.41, P=0.007). (C) Relationship between number density of podocytes per glomerular volume (Nv[PC/glom]) and podocyte total inclusion volume. Regression line shows an exponential model with two-phase decay. Solid line represents regression lines. Dashed lines represent 95% confidence interval.
The scatterplot of podocyte number density versus podocyte GL3 volume showed that the relationship between these two parameters follows an initial steep downward slope followed by a milder slope, consistent with a two-phase exponential decay function (Figure 3C). Piecewise linear regression analysis identified podocyte GL3 volume of 2009 µm3 as the breakpoint with maximum slope shift. Patient and biopsy characteristics in relation to this breakpoint are listed in Table 2. Aside from podocyte number density, podocyte GL3 volume, and average podocyte volume, the other clinical and structural parameters studied were not statistically different in patients with biopsies with podocyte GL3 volume above or below the breakpoint. Patients with podocyte GL3 volume greater than the breakpoint showed an inverse correlation between age and podocyte number density (r=−0.70, P=0.008), and direct correlations between age and podocyte GL3 volume (r=0.57, P=0.04) and mean podocyte volume (r=0.67, P=0.01). Also, in patients with podocyte GL3 volume greater than the breakpoint, UPER correlated inversely with podocyte number density (r=−0.64, P=0.03) and directly with podocyte volume (r=0.79, P=0.002), whereas FPW correlated inversely only with podocyte number density (r=−0.74, P=0.04). Subjects with podocyte GL3 volume less than or equal to the breakpoint showed no statistically significant relationship between age or UPER and podocyte number density, podocyte GL3 volume, or podocyte volume. Although there was a direct relationship between UPER and the podocyte GL3 fraction (r=0.60, P=0.009), this was not found in subjects with podocyte GL3 volume greater than the breakpoint.
Table 2.
Clinical and renal structural characteristics of male “classic” patients with Fabry disease whose total volume of GL3 inclusions per podocyte (V[Inc/PC]) is below or above the breakpoint (2009 µm3) determined by piecewise linear regression analysis of relationship between the volume fraction of GL3 inclusions per podocyte (Vv[Inc/PC]) and number density of podocytes per glomerular volume (Nv[PC/glom])
| Characteristic | V(Inc/PC)≤Breakpoint (n=21) | V(Inc/PC)>Breakpoint (n=13) | P Value |
|---|---|---|---|
| Age, yr | 30±11 | 28±11 | 0.64 |
| GFR, ml/min/1.73m2 | 110±25 | 124±30 | 0.15 |
| UPERa | 0.14±0.14 | 0.39±0.54 | 0.16 |
| α-Gal-A activityb | 0.07%±0.13% | 0.07%±0.06% | 0.98 |
| Vv(PC/glom) | 0.31±0.05 | 0.35±0.03 | 0.05 |
| Nv(PC/glom) | 0.000157±0.000082 | 0.0000477±0.000022 | 0.0003 |
| Vv(Inc/PC) | 0.41±0.06 | 0.43±0.06 | 0.47 |
| V(Inc/PC), µm3 | 990±486 | 3514±1415 | —c |
| VPC, µm3 | 2460±1327 | 8326±3238 | <0.001 |
| FPW, nm | 703±200 | 795±271 | 0.28 |
| Vv(Inc/Endo) | 0.13±0.04 | 0.11±0.03 | 0.27 |
| Vv(Inc/Mes) | 0.06±0.03 | 0.05±0.04 | 0.40 |
Data for this analysis was available for 34 patients. V(Inc/PC), podocyte GL3 volume; average total volume of GL3 inclusions per podocyte.
Measured in mg/g if urine protein-creatinine ratio or g/d if 24-h collection.
Percentage lower limit of normal range.
Different by design.
Discussion
This is the first study that addresses detailed structural changes in podocytes in a relatively large number of patients with Fabry disease. The podocyte23 is among a group of terminally differentiated, relatively poorly replicating but important cell types that also includes vascular smooth muscle cells and cardiac myocytes,24 all of which have relatively poor responses to ERT.3,25 Severe damage to these cell types can have serious clinical consequences including, for vascular smooth muscle cells, downstream ischemia and tissue infarction (e.g., strokes, cardiac arrhythmias, myocardial fibrosis, renal interstitial fibrosis, and global glomerulosclerosis) and, for cardiac myocytes, cardiomyopathy-related cardiac failure.26 The podocyte is critical to the maintenance of glomerular permselectivity27 and podocyte damage is associated with proteinuria which, in untreated males with Fabry disease, is a very powerful predictor of progressive GFR loss8 as well as a predictor of the failure of long-term ERT to prevent further GFR loss.6 As noted above, podocyte loss cannot be easily compensated for by podocyte regeneration and, if sufficiently severe, leads to irreversible global glomerulosclerosis and the spiral of CKD progression related to reductions in the number of functioning nephrons.28
We have previously shown that podocyte GL3 volume fraction (i.e., the fraction of podocyte cytoplasm filled with GL3 inclusions) increases in patients with Fabry disease with classic GLA mutations who were <19 years.11 Our hypothesis regarding the relationships between aging, GL3 accumulation, and podocyte loss is based on the findings from this study which involved a larger patient cohort across a wider age span and is summarized in Figure 4. We found that the increase in podocyte GL3 volume fraction with increasing age continues until about the age of 25–30 years. Thereafter there is a plateau in this relationship, suggesting that podocyte viability is compromised by greater proportions of podocyte cytoplasm filled with GL3 inclusions. The direct relationship between podocyte GL3 volume fraction and podocyte profile area clearly shows that podocyte GL3 accumulation cannot be adequately compensated for by podocyte enlargement; if this were the case, podocyte GL3 volume fraction would have remained constant while podocytes enlarged. The direct relationships between FPW and podocyte GL3 volume fraction is indicative of increasing podocyte stress and injury with increasing GL3 accumulation; this is supported by the direct relationships between podocyte GL3 volume fraction and UPER. These results also support the validity of our sampling and measuring methodologies in that our structural results from three glomeruli are reflective of the permselectivity properties of all approximately 2 million glomeruli.
Figure 4.

Visual depiction of proposed relationships between age and podocyte parameters in Fabry patients and normal controls. Relationships between aging and podocyte GL3 volume (dotted line), podocyte GL3 volume fraction (dashed line), and podocyte loss in Fabry disease (black bold line). The gray line represents physiologic podocyte loss with aging. Initially, the rate of GL3 accumulation is greater than the rate of podocyte enlargement, this leading to increasing podocyte GL3 volume fraction with increasing age up to 25–30 years of age. Thereafter, podocyte GL3 volume fraction plateaus while GL3 accumulation continues in parallel with podocyte enlargement, and this is associated with podocyte loss from aging aggravated by additional podocyte loss from Fabry disease.
Importantly, this podocyte injury was associated with podocyte loss. Although mean podocyte volume was increased in these male patients with classic Fabry disease, in some instances to 300%–400% above the upper limit of normal, the fraction of glomerular volume occupied by podocytes actually decreased with increasing age. These findings are most consistent with decreasing numbers of podocytes per glomerulus. Although increased number of podocytes in the urine (i.e., podocyturia) has been shown in Fabry disease,29–31 this study is, in fact, the first to directly document podocyte loss in biopsies from patients with Fabry disease. Also important was the finding of an inverse relationship between both podocyte size and GL3 content and number density of podocytes per glomerulus. Podocytes undergo compensatory hypertrophy as a result of glomerular enlargement and/or reduced number of podocytes. Surpassing the capacity of podocytes for hypertrophy leads to podocyte loss and segmental glomerular sclerosis.32,33 Several mechanisms have been proposed for explaining podocyte injury in Fabry disease.34–36 Regardless of the injury process, the strong inverse relationship found between podocyte GL3 volume and podocyte number density supports clinical relevance of using quantitative measures of podocyte GL3 volume as an indicator of response to Fabry-specific treatments.20,37,38 Importantly, the relationship between podocyte GL3 content and podocyte number density followed an initial phase with a steep slope and a later phase with a milder slope, with a breakpoint at the transition between these two phases. Although the proposed value for this breakpoint in this study needs to be confirmed in studies with a larger number of biopsy samples, a few important points can be derived from the pattern of the relationship between these two parameters. Biopsies with greater GL3 content per podocyte showed more prominent podocyte loss and were also associated with more progressive podocyte GL3 accumulation as well as with increasing podocyte loss with aging. Although these cross-sectional observations suggest that quantitative assessment of podocyte GL3 volume may be of prognostic value and may help identify patients with more severe renal phenotypes, it will be important to confirm such conclusions in longitudinal studies.
As this study strongly suggests, the process of podocyte loss begins relatively early in Fabry disease, arguing for earlier institution of treatment, certainly before significant proteinuria develops. It will also be important to conduct longitudinal studies to document if a given treatment can lead to amelioration of podocyte loss. Nevertheless, a beneficial effect of earlier institution of ERT on podocyte survival would not be surprising because, beyond a certain point in the process of podocyte loss, glomerular scarring is the regular outcome28 and, with higher levels of proteinuria, GFR loss continues despite the institution of ERT.6 Our findings also lend support for implementing kidney biopsies in the ascertainment of the severity of renal injury and timing of therapeutic strategies in males with Fabry disease. Whereas a baseline biopsy before initiation of treatment will provide valuable information, performing follow-up biopsies may be even more informative in assessment of the efficacy of treatment on the injury to podocytes and other kidney structures.20,39,40 The situation for females is more complex because their podocyte involvement and injury is also affected by mosaicism resulting from random X-inactivation.41
This study has some limitations. The cross-sectional nature of this study limits our ability to draw firm conclusions regarding prognostic significance of the podocyte injury findings. The available α-Gal-A activity values included plasma or leukocyte values measured in different laboratories over a wide time span required a normalization process in an attempt to derive comparative values. Also, these observations in untreated males with classic Fabry disease should not be generalized to patients with atypical forms of Fabry disease, to females, or to patients treated with ERT or other forms of treatment.
In summary, podocyte GL3 accumulation in Fabry disease is associated with podocyte injury which progresses with age and this is associated with proteinuria and podocyte loss.
Disclosures
Dr. Mauer is a recipient of investigator-initiated Sanofi Genzyme research grants. His laboratory performs kidney/skin biopsy laboratory studies for Sanofi Genzyme. He is also a consultant to Sanofi Genzyme for clinical trial design. He is a speaker at Sanofi Genzyme educational meetings. These interests have been reviewed and managed by the University of Minnesota in accordance with its conflict of interest policies. Dr. Mauer is also a consultant to and his laboratory performs kidney biopsy studies for Amicus Therapeutics and he is a consultant to Freeline Therapeutics. Dr. Mauer also reports other grants from Amicus Therapeutics, grants from National Institutes of Health, and Sanofi Genzyme, during the conduct of the study, Consultant fees from Freeline Therapeutics, and Sanofi Genzyme, outside the submitted work. Dr. Najafian is a recipient of investigator-initiated grants from Amicus Therapeutics and Sanofi Genzyme; has research contracts with Avrobio and Sanofi Genzyme; and is a consultant to Amicus Therapeutics, Freeline Therapeutics, and Sanofi Genzyme. Dr. Oliveira has received financial support for research from Sanofi Genzyme, honoraria for lecturing from Sanofi Genzyme and Shire, and honoraria for consultancies and advisory board membership from Sanofi Genzyme. Dr. Svarstad has received speakers’ fees and travel support from Amicus Therapeutics, Sanofi Genzyme, and Shire; and has participated in Amicus Therapeutics and Sanofi Genzyme Advisory Boards. Dr. Svarstad also reports personal fees from Amicus Therapeutics, grants from Sanofi Genzyme, personal fees from Sanofi Genzyme, and personal fees from Takeda Shire, outside the submitted work. Dr. Tøndel reports other support from Amicus, Freeline Therapeutics, Protalix, Sanofi Genzyme, and Shire during the conduct of the study. Dr. Tøndel has also received speakers’ fees and travel support from Amicus Therapeutics, Sanofi Genzyme, and Shire; and has participated in studies supported by Freeline Therapeutics, Protalix, Sanofi Genzyme, and Shire.
Funding
Sanofi Genzyme provided funding in the form of investigator-initiated grants on this subject. This study was supported by the National Center for Advancing Translational Sciences Rare Diseases Clinical Research Network grant U54NS065768, which is part National Institutes of Health Lysosomal Disease Network.
Supplementary Material
Acknowledgments
We thank Professor Chester Whitley from University of Minnesota for introducing our study to his patients and for providing clinical information of those who were enrolled. We thank Professor Robert Desnick and Ms. Yonina Loskove for providing correct and updated nomenclature of some of the mutations. We thank Professor Raphael Schiffmann from Baylor Scott & White Research Institute for assistance with normal laboratory ranges for α-Gal-A activity values for some of the subjects in this study. We greatly appreciate Dr. Alexey Sokolovskiy, Frida Meiers, Karen Zaruba, Ann Palmer, and Zour Yang for their excellent technical assistance with the electron microscopy studies and morphometric analyses as well as Cathy Bagne for her clinical coordinator role.
We thank Sanofi Genzyme and Amicus Therapeutics for providing us with research kidney biopsies from patients with Fabry who participated in prior clinical trials.
Dr. Najafian and Dr. Mauer designed the study and contributed equally to the writing of the manuscript. Dr. Najafian performed all of the statistical analyses. Dr. Tondel, Dr. Svarstad, Dr. Gubler, and Dr. Oliveira contributed renal biopsies and clinical data for this study and critically reviewed and edited the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019050497/-/DCSupplemental.
Supplemental Table 1. Comparison of clinical and Fabry specific renal structural data in Fabry patients with known “classic” phenotype and those with unknown phenotype.
Supplemental Table 2. Relationships between Vv(Inc/PC) and podocyte profile area in individual biopsies.
Supplemental Figure 1. Comparison of podocyte volume in male Fabry patients and healthy controls.
Supplemental Figure 2. Comparison of foot process width in Fabry and healthy control subjects.
References
- 1.Desnick RJ, Allen KY, Desnick SJ, Raman MK, Bernlohr RW, Krivit W: Fabry’s disease: Enzymatic diagnosis of hemizygotes and heterozygotes. Alpha-galactosidase activities in plasma, serum, urine, and leukocytes. J Lab Clin Med 81: 157–171, 1973 [PubMed] [Google Scholar]
- 2.MacDermot KD, Holmes A, Miners AH: Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet 38: 750–760, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thurberg BL, Rennke H, Colvin RB, Dikman S, Gordon RE, Collins AB, et al.: Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int 62: 1933–1946, 2002 [DOI] [PubMed] [Google Scholar]
- 4.Germain DP, Waldek S, Banikazemi M, Bushinsky DA, Charrow J, Desnick RJ, et al.: Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol 18: 1547–1557, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, et al.; Fabry Disease Clinical Trial Study Group: Agalsidase-beta therapy for advanced Fabry disease: A randomized trial. Ann Intern Med 146: 77–86, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Warnock DG, Ortiz A, Mauer M, Linthorst GE, Oliveira JP, Serra AL, et al.; Fabry Registry: Renal outcomes of agalsidase beta treatment for Fabry disease: Role of proteinuria and timing of treatment initiation. Nephrol Dial Transplant 27: 1042–1049, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liapis H, Romagnani P, Anders HJ: New insights into the pathology of podocyte loss: Mitotic catastrophe. Am J Pathol 183: 1364–1374, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wanner C, Oliveira JP, Ortiz A, Mauer M, Germain DP, Linthorst GE, et al.: Prognostic indicators of renal disease progression in adults with Fabry disease: Natural history data from the Fabry Registry. Clin J Am Soc Nephrol 5: 2220–2228, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kriz W: The pathogenesis of ‘classic’ focal segmental glomerulosclerosis-lessons from rat models. Nephrol Dial Transplant 18[Suppl 6]: vi39–vi44, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Kriz W, LeHir M: Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int 67: 404–419, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Najafian B, Svarstad E, Bostad L, Gubler MC, Tøndel C, Whitley C, et al.: Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int 79: 663–670, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel V, O’Mahony C, Hughes D, Rahman MS, Coats C, Murphy E, et al.: Clinical and genetic predictors of major cardiac events in patients with Anderson-Fabry Disease. Heart 101: 961–966, 2015 [DOI] [PubMed] [Google Scholar]
- 13.Ortiz A, Abiose A, Bichet DG, Cabrera G, Charrow J, Germain DP, et al.: Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: Data from the Fabry Registry. J Med Genet 53: 495–502, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daitx VV, Mezzalira J, Goldim MP, Coelho JC: Comparison between alpha-galactosidase A activity in blood samples collected on filter paper, leukocytes and plasma. Clin Biochem 45: 1233–1238, 2012 [DOI] [PubMed] [Google Scholar]
- 15.den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al.: HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 37: 564–569, 2016 [DOI] [PubMed] [Google Scholar]
- 16.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, et al.; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration): A new equation to estimate glomerular filtration rate [published correction appears in Ann Intern Med 155: 408, 2011]. Ann Intern Med 150: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clermont MJ, Brion LP, Schwartz GJ: Reliability of plasma creatinine measurement in infants and children. Clin Pediatr (Phila) 25: 569–572, 1986 [DOI] [PubMed] [Google Scholar]
- 18.Fogo AB, Bostad L, Svarstad E, Cook WJ, Moll S, Barbey F, et al.; all members of the International Study Group of Fabry Nephropathy (ISGFN): Scoring system for renal pathology in Fabry disease: Report of the International Study Group of Fabry Nephropathy (ISGFN). Nephrol Dial Transplant 25: 2168–2177, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gundersen HJ, Jensen EB: Stereological estimation of the volume-weighted mean volume of arbitrary particles observed on random sections. J Microsc 138: 127–142, 1985 [DOI] [PubMed] [Google Scholar]
- 20.Najafian B, Tøndel C, Svarstad E, Sokolovkiy A, Smith K, Mauer M: One year of enzyme replacement therapy reduces globotriaosylceramide inclusions in podocytes in male adult patients with fabry disease. PLoS One 11: e0152812, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Looker HC, Mauer M, Saulnier PJ, Harder JL, Nair V, Boustany-Kari CM, et al.: Changes in albuminuria but not GFR are associated with early changes in kidney structure in type 2 diabetes. J Am Soc Nephrol 30: 1049–1059, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toyoda M, Najafian B, Kim Y, Caramori ML, Mauer M: Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes 56: 2155–2160, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Eng DG, Sunseri MW, Kaverina NV, Roeder SS, Pippin JW, Shankland SJ: Glomerular parietal epithelial cells contribute to adult podocyte regeneration in experimental focal segmental glomerulosclerosis. Kidney Int 88: 999–1012, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kresh JY, Chopra A: Intercellular and extracellular mechanotransduction in cardiac myocytes. Pflugers Arch 462: 75–87, 2011 [DOI] [PubMed] [Google Scholar]
- 25.Schiffmann R, Rapkiewicz A, Abu-Asab M, Ries M, Askari H, Tsokos M, et al.: Pathological findings in a patient with Fabry disease who died after 2.5 years of enzyme replacement. Virchows Arch 448: 337–343, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Germain DP: Fabry disease. Orphanet J Rare Dis 5: 30, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott RP, Quaggin SE: Review series: The cell biology of renal filtration. J Cell Biol 209: 199–210, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodgin JB, Bitzer M, Wickman L, Afshinnia F, Wang SQ, O’Connor C, et al.: Glomerular aging and focal global glomerulosclerosis: A podometric perspective. J Am Soc Nephrol 26: 3162–3178, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fall B, Scott CR, Mauer M, Shankland S, Pippin J, Jefferson JA, et al.: Urinary podocyte loss is increased in patients with fabry disease and correlates with clinical severity of fabry nephropathy. PLoS One 11: e0168346, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trimarchi H, Canzonieri R, Schiel A, Politei J, Stern A, Andrews J, et al.: Podocyturia is significantly elevated in untreated vs treated Fabry adult patients. J Nephrol 29: 791–797, 2016 [DOI] [PubMed] [Google Scholar]
- 31.Liern M, Collazo A, Valencia M, Fainboin A, Isse L, Costales-Collaguazo C, et al.: Podocyturia in paediatric patients with Fabry disease. Nefrologia 39: 177–183, 2019 [DOI] [PubMed] [Google Scholar]
- 32.Wiggins JE, Goyal M, Sanden SK, Wharram BL, Shedden KA, Misek DE, et al.: Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: Prevention by calorie restriction. J Am Soc Nephrol 16: 2953–2966, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Nishizono R, Kikuchi M, Wang SQ, Chowdhury M, Nair V, Hartman J, et al.: FSGS as an adaptive response to growth-induced podocyte stress. J Am Soc Nephrol 28: 2931–2945, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez-Niño MD, Carpio D, Sanz AB, Ruiz-Ortega M, Mezzano S, Ortiz A: Lyso-Gb3 activates Notch1 in human podocytes. Hum Mol Genet 24: 5720–5732, 2015 [DOI] [PubMed] [Google Scholar]
- 35.Ravarotto V, Simioni F, Carraro G, Bertoldi G, Pagnin E, Calò LA: Oxidative stress and cardiovascular-renal damage in fabry disease: Is there room for a pathophysiological involvement? J Clin Med 7: 409, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eikrem Ø, Skrunes R, Tøndel C, Leh S, Houge G, Svarstad E, et al.: Pathomechanisms of renal Fabry disease. Cell Tissue Res 369: 53–62, 2017 [DOI] [PubMed] [Google Scholar]
- 37.Ramaswami U, Bichet DG, Clarke LA, Dostalova G, Fainboim A, Fellgiebel A, et al.: Low-dose agalsidase beta treatment in male pediatric patients with Fabry disease: A 5-year randomized controlled trial. Mol Genet Metab 127: 86–94, 2019 [DOI] [PubMed] [Google Scholar]
- 38.Mauer M, Sokolovskiy A, Barth JA, Castelli JP, Williams HN, Benjamin ER, et al.: Reduction of podocyte globotriaosylceramide content in adult male patients with Fabry disease with amenable GLA mutations following 6 months of migalastat treatment. J Med Genet 54: 781–786, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skrunes R, Tøndel C, Leh S, Larsen KK, Houge G, Davidsen ES, et al.: Long-term dose-dependent agalsidase effects on kidney histology in fabry disease. Clin J Am Soc Nephrol 12: 1470–1479, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skrunes R, Svarstad E, Kampevold Larsen K, Leh S, Tøndel C: Reaccumulation of globotriaosylceramide in podocytes after agalsidase dose reduction in young Fabry patients. Nephrol Dial Transplant 32: 807–813, 2017 [DOI] [PubMed] [Google Scholar]
- 41.Mauer M, Glynn E, Svarstad E, Tøndel C, Gubler MC, West M, et al.: Mosaicism of podocyte involvement is related to podocyte injury in females with Fabry disease. PLoS One 9: e112188, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.