

Abstract
Background
Mitophagy, a selective autophagy process, plays various roles in tumors. Prohibitin 2 (PHB2) is an inner-mitochondrial membrane protein that participates in parkin-induced mitophagy. However, the role of PHB2 in non-small cell lung carcinoma (NSCLC) has not been previously reported.
Material/Methods
PHB2 protein or PHB2-mRNA in NSCLC and paired normal tissues was determined by Western blot, qRT-PCR, and immunohistochemical staining. Cell proliferation was detected by CCK-8 assay. Cell migration was evaluated by wound healing and transwell migration assays. A 3D live-cell confocal system was used to monitor autophagic flux. Mitochondrial autolysosomes were observed by transmission electron microscopy (TEM). Finally, we performed JC-1 assay to measure mitochondrial membrane potential (MMP).
Results
The level of PHB2 was significantly increased in human NSCLC specimens compared to paired adjacent specimens. Inhibition of PHB2 expression attenuated mitophagy in A549 and H1299 cells, as indicated by decreased levels of LC3 II/I and parkin markers and increased level of p62 protein. Furthermore, the inhibition caused reduction in mitochondrial autolysosomes and autophagic flux, as shown by TEM and live-cell imaging, respectively. In addition, PHB2 inhibition caused a remarkable increase in MMP and suppressed the proliferation and migration of A549 and H1299 cells.
Conclusions
Our results suggest that downregulation of PHB2 reduced parkin-mediated mitophagy, which suppressed proliferation and migration of A549 and H1299 cells.
MeSH Keywords: Carcinoma, Non-Small-Cell Lung; Cell Migration Inhibition; Cell Proliferation; Membrane Proteins; Mitochondrial Degradation
Background
Lung carcinoma is a malignant tumor with the highest rates of cancer-related morbidity and mortality worldwide, with a global 5-year survival rate of less than 16% [1]. Due to recent developments in surgical technology and advances in precision medicine, the ability to treat this lung cancer have improved significantly [2–6]. Clinical types of lung carcinoma include non-small cell lung carcinoma (NSCLC) and small cell lung carcinoma (SCLC), with NSCLC being the most common, accounting for the vast majority of all lung cancer cases. At present, the treatment of NSCLC is a comprehensive treatment based on surgery. Despite the continuous development of surgical technology, the prognosis of NSCLC is still unsatisfactory [7]. Therefore, it is urgent to further study the molecular biological mechanism of NSCLC in order to provide new targets and ways to better utilize precision medicine and effectively control the incidence and reduce the mortality of lung cancer.
Autophagy is a self-cleaning process in cells subjected to adverse environmental stress such as hypoxia, infection, or deficiency in nutrition [8]. Through autophagosome formation, the aberrant proteins in the cells and damaged or excess organelles are packaged and fused by lysosomes, which then degrade them, thus maintaining a healthy intracellular environment [8,9]. This also includes mitophagy, which clears away dysfunctional mitochondria [10]. Mitophagy is a selective autophagy process specifically involved in degradation of damaged or redundant mitochondria in cells to maintain cellular homeostasis [11]. Recently, Wei et al. reported that prohibitin 2 (PHB2), a protein in the inner mitochondrial membrane (IMM), acts as a receptor for mitophagy [12]. PHB2 is involved in parkin-induced mitophagy and participates in autophagy by binding the autophagosomal membrane-associated protein LC3. Recent studies have shown that PHB2-mediated mitophagy is dependent on the parkin pathway, which is involved in various human diseases, including cancer [13–15]. In malignant tumors, mitophagy may provide protective effects to tumor cells under metabolic pressure or/and hypoxia due to high proliferation rates and selective pressure. Interestingly, it was reported that PHB2 is overexpressed in prostate carcinoma, liver carcinoma, pancreatic carcinoma, esophagus carcinoma, bladder carcinoma, and breast carcinoma compared with corresponding normal tissues [16–21]. Similarly, it has also been reported that PHB2 is a critical factor in the proliferation of melanoma cells, human rhabdomyosarcoma cells, and hematologic malignancies [22–24]. However, the role of PHB2 in NSCLC is still unclear.
In our study, we evaluated PHB2 expression in human tissues and cell lines of NSCLC, and studied the anti-tumor effect of inhibition of PHB2 in vitro. We also explored the specific mechanism underlying the anti-tumor effect of inhibition of PHB2. Our results may contribute to a new perspective for the treatment of NSCLC.
Material and Methods
NSCLC and adjacent normal tissue samples
Human NSCLC tissue samples and adjacent normal samples (confirmed by pathology) were obtained during surgery from 38 patients. These patients were consecutively admitted to the Second Affiliated Hospital of Harbin Medical University (Harbin, China) between November 2017 and July 2018. All these patients signed the written informed consent and the study was conducted under the supervision of Ethics and Scientific Committees of Harbin Medical University. No patient had received chemotherapy, radiotherapy, or immunotherapy before surgery. Table 1 shows the clinicopathological data of 38 patients.
Table 1.
The clinicopathological data of the 38 study participants.
| n | (%) | |
|---|---|---|
| Age (years) | ||
| ≥60 | 24 | (63.2) |
| <60 | 14 | (36.8) |
| Sex | ||
| Male | 23 | (60.5) |
| Female | 15 | (39.5) |
| Histology | ||
| SCC | 11 | (28.9) |
| AC | 27 | (71.1) |
| Stage | ||
| I | 23 | (60.5) |
| II | 4 | (10.5) |
| III | 10 | (26.3) |
| IV | 1 | (2.6) |
Cell culture and treatment
H1299, H460, A549, PC9, H1915, H2170, and human bronchial epithelial cells (HBE) were cultured as described previously [9]. Cells were transfected with si-PHB2 (RiboBio, Guangzhou, China), negative control, and PHB2-overexpressed plasmid (IbsBio, Shanghai, China) as described previously [9].
RNA isolation and real-time quantitative RT-PCR (qRT-PCR)
Total RNA was extracted as previously described [9]. The expressions of PHB2 mRNA were quantified by qRT-PCR, and GAPDH was used as the reference gene. Primers used for qRT-PCR were: PHB2 forward primer, 5′-CAGAAAATTGTGCAGGCCGA-3′; PHB2 reverse primer, 5′-GATGAGGCTGTCACTTCCCC-3′.
Cell proliferation
A549 and H1299 cells were used for our experiment. Overexpression of PHB2 was carried out by transfecting with lentivirus plasmid. Si-PHB2 was used to knock down PHB2 expression. Transfected cells were seeded as described previously [9]. Based on the manufacturer’s protocol, we used CCK-8 assay to evaluate cell proliferation. CCK-8 kits were purchased from Dojindo Biotechnology Co. (Kumamoto, Japan). Finally, OD value was recorded by a microplate reader at 450 nm.
Cell migration
To evaluate cell migration ability, wound healing assay was performed as described previously [9]. We observed cell migration into the scratches and took pictures under a microscope. In the transwell migration assay, transwell chambers were the same as described previously [9]. Cells were transfected for 48 h, then we seeded serum-free medium cultured cells suspension in the upper chamber. The lower chamber was prepared with addition of 500 μl culture medium with FBS (fetal bovine serum). Cells were incubated for 48 h at 37°C, then migrated cells were fixed as described previously [9]. We looked through a microscope (Olympus, Japan) to observe migrated cell numbers.
Western blotting
RIPA buffer was used to lyse transfected cells and human tissue samples as previously described, and protein concentrations were tested via BCA method (Beyotime, Shanghai, China) [25], using SDS-PAGE gel electrophoresis to separate proteins. After this, separated proteins were transferred to polyvinylidene fluoride (PVDF) membranes. Then, primary antibodies against PHB2 (Abcam, MA, USA), p62 (Cell Signaling Technology, Danvers, USA), LC3 II/I (Cell Signaling Technology, Danvers, USA), and parkin (Invitrogen, MA, USA) were added and cells were incubated at 4°C overnight. Protein expression was detected as described previously [9].
Immunofluorescent staining
After transfection for 24 h, we used 4% buffered paraformaldehyde in PBS to fix cells, and then blocked the cells in a solution of 1% BSA and 0.1% Triton-X (both in PBS) for 2 h at 37°C. Primary antibody against PHB2 (1: 150) was added, followed by incubation at 4°C for 12 h. Cells were washed 3 times with PBS, followed by incubation with secondary antibody for 1 h at room temperature. DAPI (1: 100) staining was then performed for 20 min to stain the nuclei. Immunofluorescence was assessed by laser confocal microscopy.
Live-cell imaging for autophagic flux
Transfected cells were infected with mRFP-GFP-LC3 adenovirus particles (Hanbio, Shanghai, China) for 24 h. Then, the images were taken using the VoX 3D live-cell confocal system [9].
Transmission electron microscopy (TEM)
Transfected cells were fixed in glutaraldehyde as described previously [9]. Sections were electron-stained before they were observed under an electron microscope (JEM-1200, Japan).
Immunohistochemical analysis
Human NSCLC and adjacent normal tissue samples were fixed as described previously [9], then incubated with primary antibodies against PHB2 at 4°C for 12 h. On the next day, all slides were incubated with secondary antibody followed by diaminobenzidine staining. All slides were then examined by light microscopy.
Measurement of the mitochondrial membrane potential with JC-1
MMP was detected by use of the JC-1 Assay Kit (Beyotime, Shanghai, China). After transfection for 24 h, A549 and H1299 cells were stained with JC-1 staining solution. Fluorescence intensity was measured]under a fluorescence microscope (FV300, Olympus, Japan) with single excitation (argon-ion 488 nm) and dual emission (shift from green [530 nm] to red [590 nm]) [26]. The ratio of green and red fluorescent intensities indicated the changes of MMP in the cells.
Statistical analysis
Data were assessed using GraphPad Prism 5.0 software. The t test was used for comparisons between 2 groups, and multiple groups were compared by one-way ANOVA. P values <0.05 were regarded as a significant.
Results
PHB2 was overexpressed in NSCLC
We evaluated PHB2 expression in NSCLC and adjacent normal tissues. As shown in Figure 1A, NSCLC tissues had more obviously upregulated PHB2 than matched (normal) tissues. Using qRT-PCR, the same trend was observed (Figure 1B). For further confirmation, we evaluated the expression of PHB2 in NSCLC by comparing with matched tissues by immunohistochemical staining. The results clearly showed that PHB2 expression was higher in NSCLC (Figure 1F). Next, we assessed the level of PHB2 in A549, H1299, H460, H1915, PC9, and H2170 cell lines and human bronchial epithelial (HBE) cell line. In comparison with HBE, the levels of PHB2 protein and mRNA were highest in A549 and H1299 cells (Figure 1C–1E). Thus, A549 and H1299 cells were selected for subsequent experiments.
Figure 1.

PHB2 expression in NSCLC. (A) PHB2 expression in human tissues was detected by Western blot (n=5). * P<0.05 vs. Normal. (B) mRNA expression of PHB2 in human tissues were measured by qRT-PCR (n=38). *** P<0.001 vs. normal. (C, D) PHB2 protein expression in H1299, H460, A549, PC9, H1915, H2170, and HBE cells were measured (n=5). *** P<0.01 vs. HBE. (E) The relative quantities of PHB2 mRNA in A549, H1299, and HBE cells were measured (n=5). ** P<0.01 vs. HBE, *** P<0.001 vs. HBE. (F) Expression of PHB2 protein in human NSCLC and paired normal tissues was detected by immunohistochemistry (n=5). Representative pictures are shown. Scale bar indicates 100 μm.
PHB2 inhibition suppresses proliferation and migration in vitro
To explore the function of PHB2 in proliferation and migration in NSCLC, pcDNA3.1-PHB2 plasmid or si-PHB2 were transfected into NSCLC cells. Transfection efficiency was assessed by qRT-PCR and immunofluorescence staining (Figure 2A, 2F). We utilized CCK-8 assay to evaluate cell proliferation. As shown in Figure 2B, transfection with si-PHB2 remarkably suppressed cell proliferation in NSCLC cells. In addition, wound healing assay indicated that downregulation of PHB2 inhibited cell migration compared to their corresponding controls (Figure 2C, 2D). Similarly, transwell migration assay demonstrated that transfection with si-PHB2 led to suppressed NSCLC cell migration (Figure 2E). In summary, our results indicated that PHB2 inhibition suppresses proliferation and migration in vitro.
Figure 2.

Si-PHB2 suppressed proliferation and progression of H1299 and A549 cells. (A) qRT-PCR was performed to detect transfection efficiency (n=5). *** P<0.001 vs. Control. (B) Cell proliferation were measured with an CCK-8 assay (n=5). *** P<0.001 vs. Control. (C, D) Wound healing assay showed that si-PHB2 inhibited cell migration compared to their corresponding controls (n=5). *** P<0.001 vs. Control. (E) Invasion ability was measured by transwell migration assay (n=5). Representative pictures are shown. (F) Transfection efficiency was detected by immunofluorescence staining (n=5). Representative pictures are shown. Scale bar indicates 50 um.
PHB2 promotes proliferation and migration in vitro
Detection of overexpression efficiency was determined by qRT-PCR and immunofluorescence (Figure 3A, 3F). In CCK-8 assay, the cell proliferation rate of cells was obviously increased when cells were transfected with the plasmid (Figure 3B), and PHB2 overexpression accelerated wound healing (Figure 3C, 3D) and migration of A549 and H1299 cells (Figure 3E). In summary, our results indicated that PHB2 overexpression promotes proliferation and migration in vitro.
Figure 3.

PHB2 promoted proliferation and migration in H1299 and A549 cells. (A) Transfection efficiency was detected by qRT-PCR (n=5). ** P<0.01, *** P<0.001 vs. Control. (B) Cell proliferation were measured with an CCK-8 assay (n=5). *** P<0.001 vs. Control. (C, D) PHB2 overexpression accelerated wound healing compared to their corresponding controls (n=5). *** P<0.001 vs. Control. (E) Invasion ability was analyzed by transwell migration assay (n=5). Representative pictures are shown. (F) Transfection efficiency was detected by immunofluorescence staining (n=5). Representative pictures are shown. Scale bar indicates 50 μm.
PHB2 regulates mitophagy
To determine the underlying mechanism behind increased NSCLC cell progression due to PHB2, investigated the mitochondrial autophagy markers. Western blot results (Figure 4A, 4B) showed that the cells transfected with si-PHB2 had decreased SQSTM1/p62 degradation and LC3 II/I expression. Cells transfected with si-PHB2 expressed lower levels of parkin protein, but cells transfected with PHB2 plasmid had increased endogenous LC3 II/I and parkin levels as well as p62 degradation level (Figure 4C, 4D). Therefore, our data revealed that PHB2 promotes lysosome function and at least partially affects parkin-mediated mitophagy in NSCLC cells.
Figure 4.

PHB2 increased expression of mitochondrial autophagy markers in NSCLC cells. (A, B) After transfection with si-PHB2 for 48 h, Western blot analysis was used measure mitophagy-related protein levels in H1299 and A549 cells (n=5). Representative pictures are shown. (C, D) After transfection with PHB2 plasmid for 48 h, we performed Western blot analysis to detect mitophagy-related protein levels in H1299 and A549 cells (n=5). Representative pictures are shown. * P<0.05; ** P<0.01; *** P<0.001 vs. Control.
PHB2 changes MMP and reinforces mitophagy in vitro
To further verify that PHB2 promotes mitophagy, changes of MMP were detected in vitro. After transfection with si-PHB2 or PHB2 plasmid for 48 h, JC-1 assay was used to measure MMP. The findings showed that MMP was decreased in tumor cells treated with PHB2 plasmid. In contrast, tumor cell MMP was more stable after si-PHB2 treatment (Figure 5A). The transfected cells were infected as described previously to detect autophagic flux [9]. H1299 and A549 cells transfected with PHB2 plasmid shown increased level of autophagic flux. In contrast, H1299 and A549 cells transfected with si-PHB2 shown decreased levels of autophagic flux (Figure 5B, 5C), and TEM showed that PHB2 overexpression increased the number of mitochondrial autophagosomes (Figure 5D). The above results proved that PHB2 could change MMP and reinforce mitophagy in NSCLC cells.
Figure 5.

PHB2 changes MMP and reinforces mitophagy in NSCLC cells. (A) MMP was determined by JC-1 assay. The red and green fluorescence indicates changes in MMP quantified using Image-pro plus software (n=5). Representative pictures are shown. (B, C) Representative images of autophagic flux (autophagosomes: yellow, autolysosomes: red, n=3). Representative pictures are shown. Scale bar indicates 10 μm. *** P<0.001. (D) Representative images of electronic micrographs. Scale bar indicates 500 nm. The arrows (red) depict mitochondrial autophagosomes, and cell nuclei are represented by N (n=5, magnification 10 000×).
Discussion
In our study, the first discovery was that PHB2 was obviously higher in NSCLC tissues and in A549 and H1299 cells compared with normal counterparts. Secondly, upregulation of PHB2 facilitated detrimental proliferation and migration in vitro. Thirdly, we found that PHB2 enhanced parkin-mediated mitophagy in NSCLC cells. Our findings are the first to reveal the clinical potential of PHB2 in NSCLC.
In malignant tumors, autophagy can give tumor cells a survival advantage under the selective pressure which comes from high proliferation rates, hypoxia, and therapeutic interventions [27]. Damaged or dysfunctional mitochondria are swallowed into autophagosomes, which degrades them through the autophagy pathway, a process known as mitophagy. Mitophagy, a selective autophagic removal of mitochondria, is a conserved cellular procedure vital for maintaining cellular homeostasis. The accumulation of damaged mitochondria by dysregulated mitophagy is crucial for carcinogenesis and tumor progression [28]. Mitophagy is like a switch regulating metabolism, reducing the number of mitochondria and inhibiting the activity of mitochondrial network structure, which can directly reduce the amount of glucose required for mitochondrial oxidative phosphorylation then transfer glucose out and use it in glycolysis or other anabolic pathways to provide more energy for tumor cells (Figure 6). When mitochondrial dysfunction occurs, cancer cells phagocytose their own mitochondria through mitophagy, reducing the production of destructive reactive oxygen species (ROS), while providing nutrients to tumor cells to support cell division [29]. The absence of mitophagy increases oxidative stress and inhibits tumor cell growth. On one hand, several proteins that help with mitophagy, such as FUNDC1, parkin, BNIP3, and BNIP3L/NIX, have been proved to be upregulated in cancers. On the other hand, p62/SQSTM1 is downregulated in cancers to protect cancer cells [30,31]. Prior research on mitophagy has concentrated on the function of the outer mitochondrial membrane (OMM) [32]. When mitochondria are damaged, the stable of PINK1, a mitochondrial kinase, causes parkin to be recruited to damaged mitochondria, triggering mitophagy. After this, these ubiquitinated OMM proteins in combination with autophagy receptors promote autophagosome formation through the LC3-interacting region (LIR) domain, triggering mitophagy [33]. Recently, Wei et al. reported that the autophagosome LC3 II protein directly binds to the IMM protein PHB2 to capture the dysfunctional mitochondria during mitophagy [12]. This shows that the IMM protein PHB2 is an extremely important mitophagy receptor. In addition, Yan et al. found that overexpression of PHB2 in HeLa cells promoted the translocation of LC3 II protein to mitochondria, and overexpression of PHB2 in HeLa cells with parkin gene could promote the level of mitophagy, while cells lacking the parkin gene did not show these effects [34]. In our study, we provided solid evidence that overexpression of PHB2 resulted in upregulation of parkin and LC3 II/I protein level expression and downregulation of p62 expression in vitro, thereby improving the mitophagy level of tumor cells. Therefore, inhibition of mitophagy activity by decreasing the expression of PHB2 in NSCLC cells will likely be a new mechanism for treating NSCLC.
Figure 6.
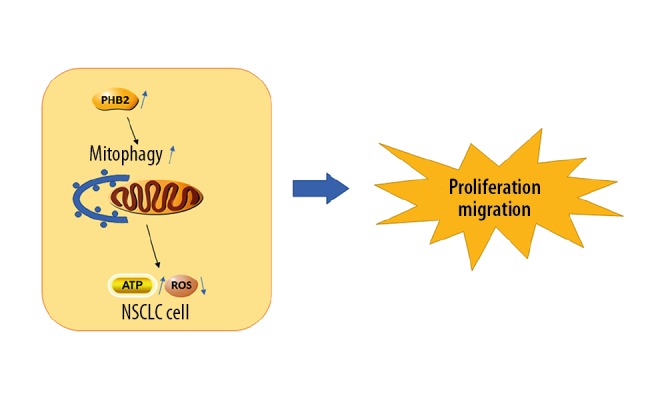
Schematic diagram: Mitophagy promotes the growth of tumor cells by reducing the production of ROS and providing energy to support tumor cells by swallowing mitochondria.
Studies have shown that PHB2 is overexpressed in esophageal, gastric, colorectal, prostate, breast, bladder, papillary thyroid and liver carcinoma [13,18,19,35]. For the first time, our research discovered that PHB2 was remarkably higher in human NSCLC tissues and in cancer cell lines than in normal counterparts. However, the mechanism of PHB2 overexpression and its role in NSCLC are unclear. Mitochondrial damage causes mitochondrial depolarization. Depolarized mitochondria express PINK1 on the outer membrane, resulting in recruitment of parkin, followed by ubiquitination and degradation of OMM proteins. After the destruction of the OMM by a proteasome-dependent mechanism, the mitochondrial inner membrane protein PHB2 is exposed, and its LIR is prone to interact with LC3 II. At the site of mitochondrial outer membrane damage, LC3 II binds to PHB2, ultimately leading to mitophagy. We confirmed that PHB2 can increase autophagy flux and PHB2 regulates the level of parkin-mediated mitophagy and then affects the number of autolysosomes in tumor cells. Overexpression of PHB2 significantly enhanced the destruction of MMP and increased the expression of mitophagy-related proteins in tumor cells. We also found that downregulation of PHB2 remarkably decreased tumor cells proliferation and migration and that decreased PHB2 remarkably attenuated proliferation and migration in NSCLC by inhibiting parkin-mediated mitophagy. In summary, our data suggest that inhibition of PHB2 expression can significantly suppress NSCLC proliferation and migration in vitro by inhibiting parkin-mediated mitophagy. However, more work is necessary to confirm the effect of PHB2 on NSCLC and to elucidate the underlying mechanism.
Conclusions
We found that PHB2 was obviously higher in human NSCLC tissues and in NSCLC cell lines (H1299 and A549) when compared with normal counterparts. Furthermore, our data revealed that si-PHB2 exerts anti-tumor effects by regulating parkin-mediated mitophagy, which may be a potential treatment for NSCLC.
Footnotes
Conflict of interest
None.
Source of support: The National Natural Science Foundation of China (81773735)
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Xu H, Zhang L. Dissection of incomplete fissure using electrocautery is a safe and acceptable method for thoracoscopic right upper lobectomy. Thorac Cardiovasc Surg. 2019;67:227–31. doi: 10.1055/s-0038-1645880. [DOI] [PubMed] [Google Scholar]
- 3.Xu H, Zhang L. The bronchus first and vessels simultaneously stapled technique: A safe and simple method for video-assisted right upper lobe lobectomy. Thorac Cardiovasc Surg. 2019;67:131–36. doi: 10.1055/s-0037-1620276. [DOI] [PubMed] [Google Scholar]
- 4.Su X, Wan Y, Xie L, et al. Expression of SUMO1P3 compared with SUMO1 is an independent predictor of patient outcome in lung adenocarcinoma. Med Sci Monit. 2019;25:6691–701. doi: 10.12659/MSM.916887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan H, Li J, Wang J, Hu Z, et al. Long non-coding RNAs (lncRNAs) tumor-suppressive role of lncRNA on chromosome 8p12 (TSLNC8) inhibits tumor metastasis and promotes apoptosis by regulating interleukin 6 (IL-6)/signal transducer and activator of transcription 3 (STAT3)/hypoxia-inducible factor 1-alpha (HIF-1alpha) signaling pathway in non-small cell lung cancer. Med Sci Monit. 2019;25:7624–33. doi: 10.12659/MSM.917565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tian LJ, Wu YP, Wang D, et al. Upregulation of long noncoding RNA (lncRNA) X-inactive specific transcript (XIST) is associated with cisplatin resistance in non-small cell lung cancer (NSCLC) by downregulating microRNA-144-3p. Med Sci Monit. 2019;25:8095–104. doi: 10.12659/MSM.916075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang J, Ding Z, Luo Q, Xu W. Cancer cell-derived exosomes promote cell proliferation and inhibit cell apoptosis of both normal lung fibroblasts and non-small cell lung cancer cell through delivering alpha-smooth muscle actin. Am J Transl Res. 2019;11:1711–23. [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Y, Rucker EB, 3rd, Zhou BP. Autophagy regulation in the development and treatment of breast cancer. Acta Biochim Biophys Sin (Shanghai) 2016;48:60–74. doi: 10.1093/abbs/gmv119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin C, Zhang H, Liu X, et al. Downregulated MCOLN1 attenuates progression of non-small-cell lung cancer by inhibiting lysosome-autophagy. Cancer Manag Res. 2019;11:8607–17. doi: 10.2147/CMAR.S216538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wrighton KH. Metabolism: Mitophagy turns beige adipocytes white. Nat Rev Mol Cell Biol. 2016;17:607. doi: 10.1038/nrm.2016.125. [DOI] [PubMed] [Google Scholar]
- 11.Georgakopoulos ND, Wells G, Campanella M. The pharmacological regulation of cellular mitophagy. Nat Chem Biol. 2017;13:136–46. doi: 10.1038/nchembio.2287. [DOI] [PubMed] [Google Scholar]
- 12.Wei Y, Chiang WC, Sumpter R, Jr, et al. Prohibitin 2 is an Inner Mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–38.e10. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Artal-Sanz M, Tavernarakis N. Prohibitin and mitochondrial biology. Trends Endocrinol Metab. 2009;20:394–401. doi: 10.1016/j.tem.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 14.Merkwirth C, Langer T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 15.Theiss AL, Sitaraman SV. The role and therapeutic potential of prohibitin in disease. Biochim Biophys Acta. 2011;1813:1137–43. doi: 10.1016/j.bbamcr.2011.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai XW, Yu WW, Yu W, et al. Tissue-based quantitative proteomics to screen and identify the potential biomarkers for early recurrence/metastasis of esophageal squamous cell carcinoma. Cancer Med. 2018;7:2504–17. doi: 10.1002/cam4.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chowdhury I, Garcia-Barrio M, Harp D, et al. The emerging roles of prohibitins in folliculogenesis. Front Biosci (Elite Ed) 2012;4:690–99. doi: 10.2741/410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen Y, Gao Y, Yuan H, et al. Prohibitin-2 negatively regulates AKT2 expression to promote prostate cancer cell migration. Int J Mol Med. 2018;41:1147–55. doi: 10.3892/ijmm.2017.3307. [DOI] [PubMed] [Google Scholar]
- 19.Sievers C, Billig G, Gottschalk K, Rudel T. Prohibitins are required for cancer cell proliferation and adhesion. PLoS One. 2010;5:e12735. doi: 10.1371/journal.pone.0012735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshimaru T, Komatsu M, Matsuo T, et al. Targeting BIG3-PHB2 interaction to overcome tamoxifen resistance in breast cancer cells. Nat Commun. 2013;4:2443. doi: 10.1038/ncomms3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yurugi H, Rajalingam K. A role for prohibitin in mast cell activation: location matters. Sci Signal. 2013;6:pe29. doi: 10.1126/scisignal.2004646. [DOI] [PubMed] [Google Scholar]
- 22.Djehal A, Krayem M, Najem A, et al. Targeting prohibitin with small molecules to promote melanogenesis and apoptosis in melanoma cells. Eur J Med Chem. 2018;155:880–88. doi: 10.1016/j.ejmech.2018.06.052. [DOI] [PubMed] [Google Scholar]
- 23.Ross JA, Robles-Escajeda E, Oaxaca DM, et al. The prohibitin protein complex promotes mitochondrial stabilization and cell survival in hematologic malignancies. Oncotarget. 2017;8:65445–56. doi: 10.18632/oncotarget.18920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Z, Ai H, Li K, et al. Prohibitin 2 localizes in nucleolus to regulate ribosomal RNA transcription and facilitate cell proliferation in RD cells. Sci Rep. 2018;8:1479. doi: 10.1038/s41598-018-19917-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu C, Liu X, Bai X, et al. MiR-519d suppresses breast cancer tumorigenesis and metastasis via targeting MMP3. Int J Biol Sci. 2018;14:228–36. doi: 10.7150/ijbs.22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun L, Zhao M, Yu XJ, et al. Cardioprotection by acetylcholine: a novel mechanism via mitochondrial biogenesis and function involving the PGC-1alpha pathway. J Cell Physiol. 2013;228:1238–48. doi: 10.1002/jcp.24277. [DOI] [PubMed] [Google Scholar]
- 27.Yang B, Ding L, Yao H, et al. A metal-organic framework (MOF) fenton nanoagent-enabled nanocatalytic cancer therapy in synergy with autophagy inhibition. Adv Mater. 2020;32:e1907152. doi: 10.1002/adma.201907152. [DOI] [PubMed] [Google Scholar]
- 28.Panigrahi DP, Praharaj PP, Bhol CS, et al. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin Cancer Biol. :2019. doi: 10.1016/j.semcancer.2019.07.015. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 29.Humpton TJ, Alagesan B, DeNicola GM, et al. Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 2019;9:1268–87. doi: 10.1158/2159-8290.CD-18-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drake LE, Springer MZ, Poole LP, et al. Expanding perspectives on the significance of mitophagy in cancer. Semin Cancer Biol. 2017;47:110–24. doi: 10.1016/j.semcancer.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamacher-Brady A, Brady NR. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci. 2016;73:775–95. doi: 10.1007/s00018-015-2087-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bingol B, Sheng M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic Biol Med. 2016;100:210–22. doi: 10.1016/j.freeradbiomed.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 33.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–73. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan C, Gong L, Chen L, et al. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy. 2019;16:419–34. doi: 10.1080/15548627.2019.1628520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elderwish S, Audebrand A, Nebigil CG, Désaubry L. Discovery of 3,3′-pyrrolidinyl-spirooxindoles as cardioprotectant prohibitin ligands. Eur J Med Chem. 2020;186:111859. doi: 10.1016/j.ejmech.2019.111859. [DOI] [PubMed] [Google Scholar]