

SUMMARY
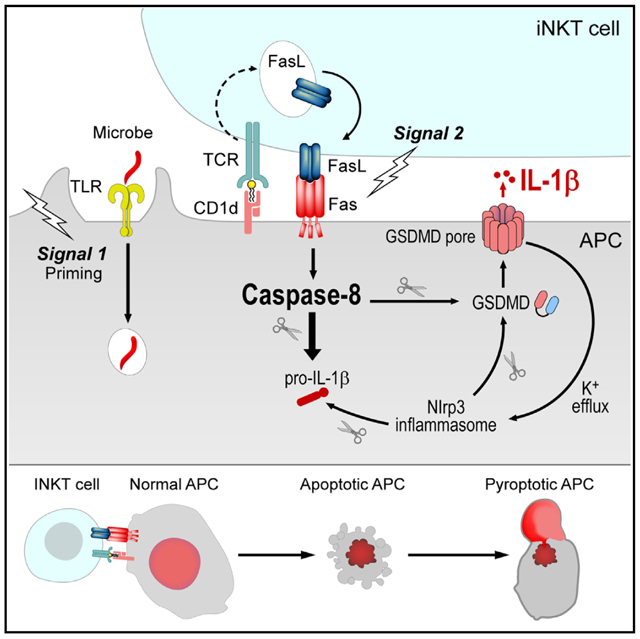
Interleukin-1β (IL-1β) is a key orchestrator of anti-microbial immunity whose secretion is typically dependent on activation of inflammasomes. However, many pathogens have evolved strategies to evade inflammasome activation. Here we describe an alternative, two-cell model for IL-1β release where invariant natural killer T (iNKT) cells use the death receptor pathway to instruct antigen-presenting cells to secrete IL-1β. Following cognate interactions with TLR-primed bone marrow-derived dendritic cells (BMDCs), iNKT cells rapidly translocate intracellular Fas ligand to the surface to engage Fas on BMDCs. Fas ligation activates a caspase-8-dependent signaling cascade in BMDCs that drives IL-1β release largely independent of inflammasomes. The apoptotic program initiated by Fas ligation rapidly transitions into a pyroptosis-like form of cell death mediated by gasdermin D. Together, our findings support a two-cell model for IL-1β secretion that may supersede inflammasome activation when cytosolic triggers fail.
Graphical Abstract
In Brief
How the immune system triggers IL-1β release during infection with inflammasome-evasive microbes is largely unknown. Donado et al. describe an alternative, caspase-8-dependent IL-1β secretion pathway where a second cell, the invariant natural killer T (iNKT) cell, can provide antigen-presenting cells with cell-extrinsic IL-1β release signals through FasL.
INTRODUCTION
Interleukin-1β (IL-1β) is a central mediator of inflammation that orchestrates essential immune responses against microbial invaders. It elicits widespread local and systemic effects that are intended to culminate in pathogen elimination, including leukocyte recruitment and activation, production of acute-phase reactants, and induction of fever (Dinarello, 2011). IL-1β is synthesized as an immature cytosolic precursor that requires cleavage to become biologically active. Its processing and secretion are largely regulated by inflammasomes, cytosolic multiprotein complexes that assemble following detection of microbial products, endogenous danger signals, or perturbations of cellular homeostasis in the cytosol of host cells (Latz et al., 2013; Martinon et al., 2002; Schroder and Tschopp, 2010). Most inflammasomes consist of a cytosolic sensor, the adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and the effector protease caspase-1. Following inflammasome assembly, active caspase-1 catalyzes proteolytic maturation of the leaderless pro-inflammatory cytokine IL-1β and the related IL-1 family member IL-18 (Broz and Dixit, 2016; Latz et al., 2013; Schroder and Tschopp, 2010). In addition, caspase-1 cleaves the pore-forming protein gasdermin D (GSDMD), generating an N-terminal fragment that forms pores in the plasma membrane (Broz and Dixit, 2016; Kayagaki et al., 2015; Shi et al., 2015). Although these pores can serve as conduits for the release of mature IL-1β (Evavold et al., 2018; Heilig et al., 2018; Kayagaki et al., 2015; Shi et al., 2015), they also trigger pyroptosis, a lytic form of cell death characterized by cell swelling, membrane rupture, and release of soluble cytosolic contents (Aglietti et al., 2016; Ding et al., 2016; Kayagaki et al., 2015; Liu et al., 2016; Sborgi et al., 2016; Shi et al., 2015).
Inflammasome activation is controlled by two signals. The first signal, termed priming, generally induces expression of an inflammasome sensor and the substrate pro-IL-1β. The priming signal can be provided through stimulation of various Toll-like receptors (TLRs) or by the cytokine tumor necrosis factor (TNF). This first step is necessary to render the cell responsive to a second signal, which can come in the form of diverse microbial, endogenous, and environmental stimuli that trigger inflammasome assembly and activation (Broz and Dixit, 2016; Latz et al., 2013; Schroder and Tschopp, 2010). Because of their cytosolic residence, inflammasome sensors are poised to detect the presence of invasive microbes that perturb the host cell cytosol. However, many pathogens have evolved strategies to inhibit or evade the inflammasomes. Microbial invaders can do this by repressing or structurally modifying ligands or by directly inhibiting inflammasome activation through specific virulence factors (Higa et al., 2013; Maltez and Miao, 2016; Shin and Brodsky, 2015; Stewart and Cookson, 2016; Ulland et al., 2015). Here we asked whether alternative mechanisms beyond inflammasome activation may mediate the processing and secretion of IL-1β during conditions where cell-intrinsic recognition of cytosolic triggers of inflammasomes is compromised.
Invariant natural killer T (iNKT) cells are innate T cells that play a major role in host defense against bacterial, fungal, and viral infections. They exist in a poised effector state that allows them to respond in synchrony with the innate immune response, performing distinct effector functions significantly more rapidly than adaptive, major histocompatibility complex (MHC)-restricted T cells. iNKT cells express evolutionarily conserved, semi-invariant αβ T cell receptors (TCRs) responsive to endogenous and microbial lipid antigens bound to the non-polymorphic, class I-like molecule CD1d. One of the functional hallmarks of iNKT cells is their remarkable ability to catalyze immune responses in large part by rapidly transactivating other leukocytes through cytokine production and contact-dependent mechanisms (Brennan et al., 2013). Given their role as cellular adjuvants during the early stages of infection, we hypothesized that iNKT cells might contribute to the production of IL-1β by antigen-presenting cells (APCs) and that this may be important in the recognition of pathogens that evade inflammasome activation from within the cytosol of host cells.
Here we describe a two-cell model for IL-1β release where iNKT cells instruct TLR-primed bone marrow-derived dendritic cells (BMDCs) to secrete IL-1β. Following cognate, CD1d-dependent recognition of primed BMDCs, we found that iNKT cells rapidly translocate pre-existing intracellular Fas ligand (FasL) to their surface and engage Fas on BMDCs. Fas ligation resulted in limited activation of the Nlrp3 inflammasome in BMDCs, but most of the IL-1β was released in an inflammasome-independent manner via caspase-8. Although Fas engagement initiated the apoptotic program in BMDCs, cells quickly transitioned to pyroptosis in a manner that was dependent on the pore-forming protein GSDMD. Importantly, we show that iNKT cells can employ FasL to elicit IL-1β release by BMDCs infected with the commensal microbe Bacteroides fragilis (B. fragilis) and the obligate intracellular pathogen Chlamydia trachomatis (C. trachomatis) under conditions where these organisms fail to activate inflammasomes in a cell-intrinsic manner. Thus, iNKT cells are poised to deliver IL-1β-secretion signals through FasL, and this two-cell mechanism for IL-1β release may be important against microbes that fail to activate the inflammasomes from within the host cell cytosol.
RESULTS
iNKT Cells Induce IL-1β Secretion by BMDCs through Cognate Interactions
Bi-directional cross-talk with APCs is central to the ability of iNKT cells to enhance immunity (Brennan et al., 2013). Thus, we hypothesized that iNKT cells might promote IL-1β release from APCs. To assess whether iNKT cells could trigger the release of IL-1β, we co-cultured iNKT cells with BMDCs that had been primed with the TLR2 agonist Pam3CSK4 (P3C). Although a priming signal alone proved to be insufficient to elicit secretion of IL-1β by BMDCs, addition of iNKT cells to P3C-primed BMDCs led to a significant release of IL-1β. Indeed, iNKT-driven IL-1β release was comparable to that induced by the Nlrp3 inflammasome activator monosodium urate (MSU) (Figure 1A). When P3C-primed BMDCs were loaded with a strong synthetic lipid agonist for the iNKT cell TCR, α-galactosylceramide (αGalCer), even higher levels of IL-1β were released (Figure 1B). Co-culture with iNKT cells resulted in accumulation of extracellular IL-1β over time (Figure 1C), suggesting that they induce gradual rather than the rapid, all-or-none release of IL-1β induced by the canonical Nlrp3 inflammasome agonist nigericin. Immunoblotting showed that iNKT cells induced secretion of the processed form of IL-1β by P3C-primed BMDCs, and its release coincided with depletion of intracellular pro-IL-1β (Figure 1D). To determine whether the release of IL-1β was dependent on cognate recognition of the lipid antigen-presenting molecule CD1d by iNKT cells, we used BMDCs from CD1d-deficient mice. Strikingly, co-culture of iNKT cells with P3C-primed BMDCs from mice deficient in CD1d resulted in the complete absence of extracellular IL-1β, whereas the ability of nigericin to elicit this cytokine was unaffected in CD1d-deficient BMDCs (Figure 1E). Together, these results demonstrate that cognate interactions between iNKT cells and BMDCs, whether in the presence of self or foreign lipid antigens, are sufficient to drive IL-1β release by TLR-primed BMDCs.
Figure 1. iNKT Cells Induce IL-1β Secretion by BMDCs in a CD1d-Dependent Manner.
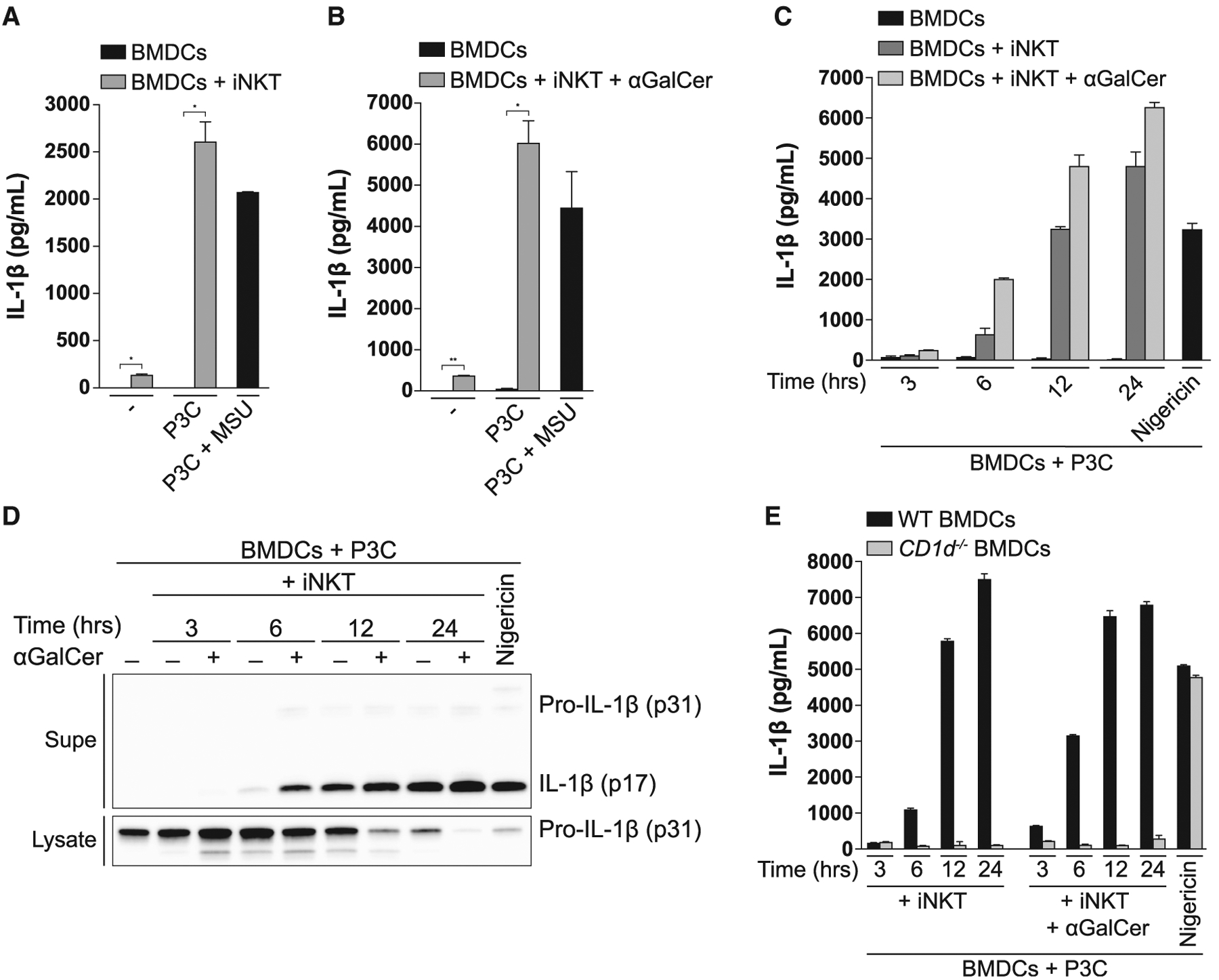
(A and B) BMDCs were left unprimed or primed with Pam3CSK4 (P3C; 0.5 μg/mL) for 24 h and subsequently co-cultured with iNKT cells for 24 h in the (A) absence or (B) presence of αGalCer (50 ng/mL). IL-1β release was quantified by ELISA. As a control, BMDCs were primed with P3C for 4 h, followed by 6-h stimulation with MSU (100 μg/mL). The same concentrations of P3C, αGalCer, and MSU were used throughout all experiments.
(C and D) P3C-primed BMDCs were cultured alone or with iNKT cells for the indicated times in the presence or absence of αGalCer. IL-1β release was quantified by ELISA (C), and cell-associated and extracellular IL-1β were analyzed by immunoblot (D). As a control, BMDCs were primed with P3C for 4 h, followed by 1-h stimulation with nigericin (10 μM).
(E) P3C-primed BMDCs from WT and CD1d−/− mice were co-cultured with iNKT cells for the indicated times in the absence or presence of αGalCer. IL-1β release was quantified by ELISA. P3C-primed BMDCs stimulated with nigericin were used as a control.
Error bars represent mean + SD of triplicate samples. Each panel is representative of at least three different experiments. *p < 0.05, **p < 0.01, as determined by Student’s unpaired t test. Supe, supernatant.
iNKT Cells Employ the Death Receptor Ligand FasL to Elicit IL-1β Release by TLR-Primed BMDCs
We next sought to determine the mechanism by which iNKT cells induced IL-1β release by BMDCs. We failed to detect secreted IL-1β when P3C-primed BMDCs were cultured with the cell-free supernatant of TCR-activated iNKT cells (Figure 2A), suggesting that iNKT cells employ a cell-surface molecule, rather than a soluble factor, to elicit IL-1β release. Based on how rapidly iNKT cells induced TLR-primed BMDCs to secrete IL-1β (Figures 1C and 1D), we reasoned that iNKT cells were likely to express the relevant molecule at resting state as part of their “innate-like” phenotype (Cohen et al., 2013; Kim et al., 2015). To identify a candidate molecule, we queried the ImmGen database (Heng et al., 2008) and compared the expression of all cell-surface molecules between iNKT cells and other αβ T cells. One of the most highly and differentially expressed surface molecules we found in iNKT cells was Fasl (Figure 2B). To determine whether iNKT cells employ FasL to elicit IL-1β release by BMDCs, we co-cultured iNKT cells and P3C-primed BMDCs in the presence of a FasL-blocking antibody (Figure 2C). Strikingly, FasL-blockade completely abrogated the ability of iNKT cells to induce IL-1β secretion by BMDCs. As observed with FasL blockade, iNKT cells were unable to elicit IL-1β release by Fas-deficient BMDCs (Figure 2D). Together, these results suggest that iNKT cells employ the death receptor ligand FasL to instruct TLR-primed BMDCs to secrete IL-1β.
Figure 2. iNKT Cells Employ the Death Receptor Ligand FasL to Instruct TLR-Primed BMDCs to Release IL-1β.
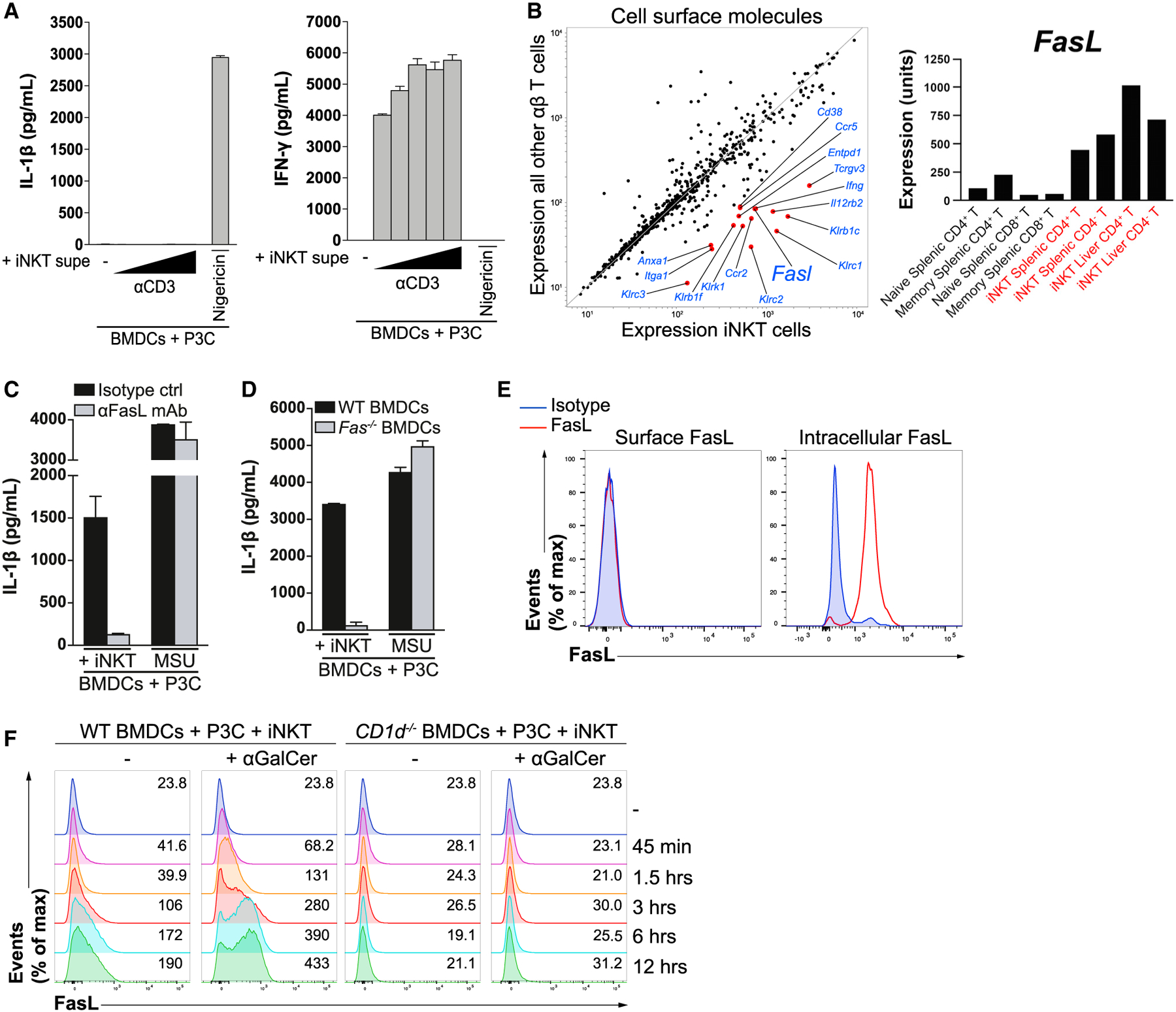
(A) iNKT cells were cultured in the presence of increasing concentrations of plate-bound anti-CD3 for 24 h, after which their cell-free Supes were used to stimulate P3C-primed BMDCs for 24 h. IL-1β (left) and interferon γ (IFN-γ; right) in the Supes were quantified by ELISA. Nigericin-stimulated BMDCs were used as a control.
(B) The ImmGen database was mined to compare the expression of cell-surface proteins (Gene Ontology term “cell surface,” GO0009986) expressed by iNKT cells versus other αβ T cells. The red dots represent genes expressed by iNKT cells with a fold change of greater than 5 compared with all other αβ T cells. Expression values comparing class means for specific T cell populations are shown in the right panel.
(C) P3C-primed BMDCs were co-cultured with iNKT cells for 24 h in the presence of a FasL-blocking antibody (aFasL mAb) or an isotype control. IL-1β release was quantified by ELISA. MSU-treated BMDCs were used as a control.
(D) P3C-primed BMDCs from WT and Fas−/− mice were co-cultured with iNKT cells for 24 h. IL-1β release was quantified by ELISA. MSU-treated BMDCs were used as a control.
(E) Resting iNKT cells were stained with a FasL-specific antibody or an isotype control and subsequently analyzed by flow cytometry. The left panel represents surface staining, whereas the right panel represents permeabilized iNKT cells.
(F) P3C-primed BMDCs from WT and CD1d−/− mice were co-cultured with CellTrace Violet-labeled-iNKT cells for the indicated times in the presence or absence of αGalCer, and the expression of FasL on the surface of iNKT cells was analyzed by flow cytometry. Numbers on the right of each histogram represent mean fluorescence intensity values.
Error bars represent mean + SD of triplicate samples. Each panel is representative of at least three different experiments.
GM-CSF-derived bone marrow cultures, widely referred to as BMDCs, serve as useful model APCs and consist of a heterogeneous population of monocyte-derived macrophages and conventional DC-like cells (Helft et al., 2015). It has been shown recently that monocyte-derived macrophages, the most abundant cell population found in GM-CSF-derived BM cultures, are the cellular source of IL-1β following classical inflammasome activation in these cultures (Erlich et al., 2019). To identify which cell type secretes IL-1β in response to Fas ligation, we sorted monocyte-derived macrophages (MDMs; CD11c+CD11bhi MHC-IIlo-intCD115+MerTK+) and DCs (GM-DCs; CD11c+ CD11bintMHC-IIhighCD115−CD135+) from GM-CSF-derived BM cultures using fluorescence-activated cell sorting (FACS) and stimulated them with recombinant hexameric FasL (rFasL) (Holler et al., 2003) after priming with P3C. In agreement with a prior report studying inflammasome activation (Erlich et al., 2019), we found that, within BMDC cultures, MDMs, but not GM-DCs, were responsible for IL-1β secretion following stimulation with rFasL or the Nlrp3 inflammasome agonist MSU (Figures S1A and S1B). It is likely that the inability of GM-DCs to secrete IL-1β in response to Fas ligation is due to their lack of expression of inflammasome components and pro-IL-1β, as reported previously (Erlich et al., 2019).
We next investigated the kinetics of FasL expression on the surface of iNKT cells. Although iNKT cells lacked detectable FasL on their surface, they expressed a large pool of intracellular FasL at steady state (Figure 2E). iNKT cells rapidly mobilized intracellular FasL to their surface upon co-culture with BMDCs, and this translocation was entirely dependent on CD1d (Figure 2F). The stronger TCR stimulus provided by αGalCer elicited even more rapid and pronounced expression of surface FasL by iNKT cells (Figure 2F). These data suggest that iNKT cells constitutively express intracellular FasL stores that are rapidly translocated to the surface upon engagement of their TCRs. iNKT cells are thus poised to engage Fas on TLR-primed BMDCs following cognate interactions, resulting in release of bioactive IL-1β.
Fas Ligation Triggers Inflammasome-Dependent and -Independent IL-1β Release via Caspase-8
Having demonstrated that iNKT cells employ FasL to transactivate an IL-1β secretion pathway in BMDCs, we next sought to determine the relevant signaling pathways downstream of Fas ligation. Among inflammasome sensors, Nlrp3 is the most promiscuous, being activated by a diverse array of stimuli ranging from microbial and sterile molecules to disturbances in cytosolic homeostasis (Broz and Dixit, 2016; Latz et al., 2013; Schroder and Tschopp, 2010; Swanson et al., 2019). Co-culture of iNKT cells with either Nlrp3- or caspase-1/11-deficient BMDCs resulted in a partial reduction in IL-1β release as detected by ELISA (Figure 3A) and immunoblot (Figure 3B). Although IL-1β release fell by approximately 50% at early time points, there was little effect on release of IL-1β over time (Figure S2A). Further, the partial dependence on the Nlrp3 inflammasome was similar in magnitude when αGalCer was added to the co-culture (Figures S2B and S2C), confirming that presentation of self or foreign lipid antigens results in activation of phenotypically equivalent pathways. We also failed to detect the active p20 subunit of caspase-1 in Nlrp3-deficient BMDCs, confirming that the Nlrp3 inflammasome is activated in response to Fas ligation (Figure S2D). These results suggest that, although iNKT cells can transactivate the Nlrp3 inflammasome in BMDCs, they also elicit activation of an inflammasome-independent pathway that has a larger contribution to IL-1β release.
Figure 3. Fas Ligation Triggers Inflammasome-Dependent and Independent IL-1β Release via Caspase-8.
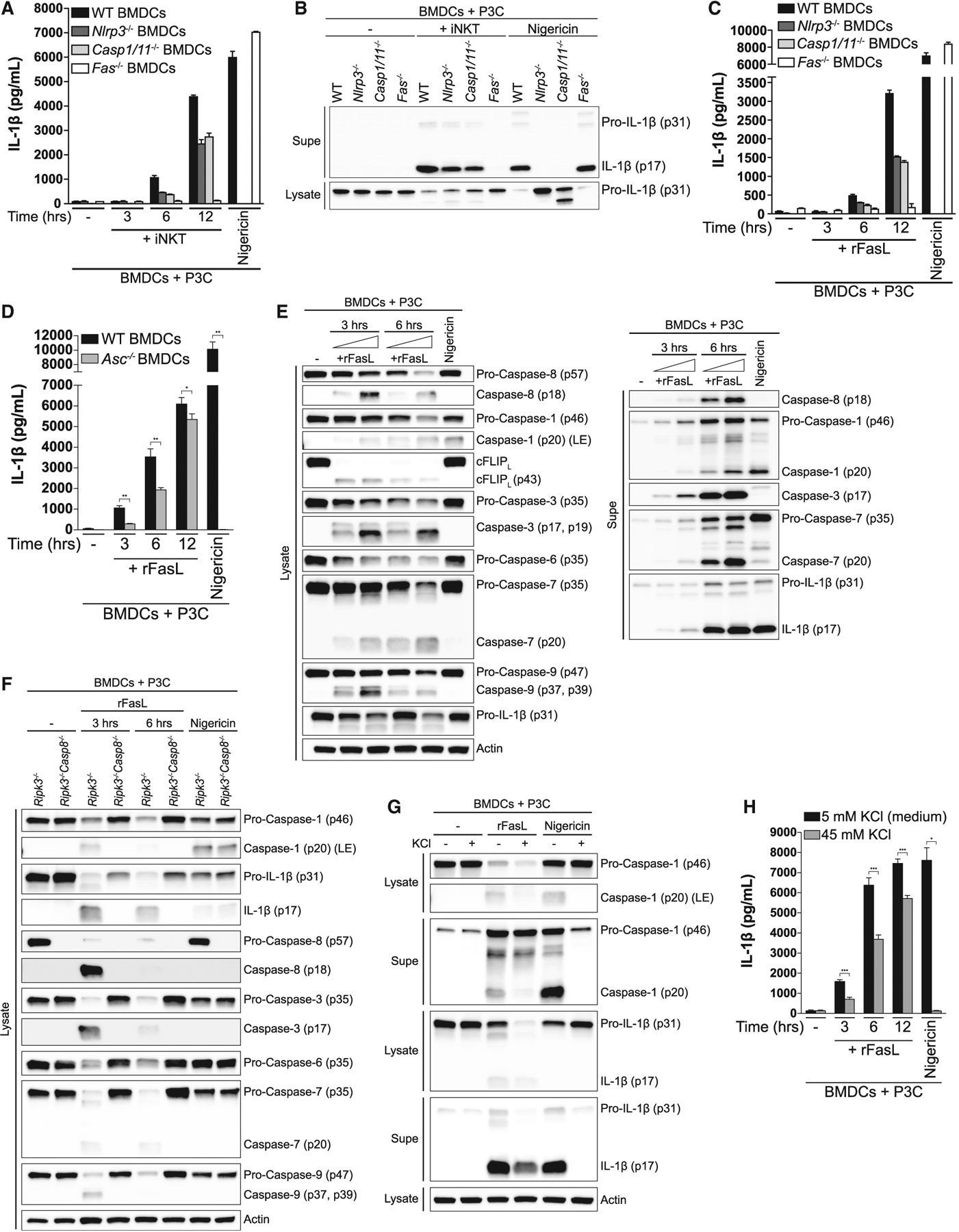
(A and B) P3C-primed BMDCs from WT, Nlrp3−/−, Caspase1/11−/− and Fas−/− mice were cultured alone or with iNKT cells for (A) the indicated times or (B) 12 h. IL-1β release was quantified by ELISA (A) and cell-associated (lysate) and extracellular (Supe) IL-1β were analyzed by immunoblot (B). Nigericin-treated BMDCs were used as a control.
(C and D) P3C-primed BMDCs from (C) WT, Nlrp3−/−, Caspase1/11−/−, and Fas−/− or (D) WT and Asc−/− mice were cultured alone or in the presence of recombinant, hexameric FasL (rFasL) for the indicated times. IL-1β release was quantified by ELISA.
(E) P3C-primed BMDCs were cultured in the presence of increasing concentrations of rFasL for the indicated times, and the cell lysates (left) and cell-free Supes (right) were probed for the indicated proteins by immunoblot.
(F) P3C-primed BMDCs from Ripk3−/− and Ripk3−/−Caspase-8−/− mice were cultured in the presence of rFasL for the indicated times, and the cell lysates were probed for the indicated proteins by immunoblot.
(G and H) P3C-primed BMDCs were treated with rFasL in the presence or absence of 45 mM KCl for (G) 6 h or (H) the indicated times. The cell lysates and cell-free Supes were probed for the indicated proteins by immunoblot (G) and IL-1β release was quantified by ELISA (H).
Error bars represent mean + SD of triplicate samples. Each panel is representative of at least three different experiments. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by Student’s unpaired t test. LE, long exposure. See also Figures S2 and S3.
To further investigate the molecular events that occur downstream of Fas ligation in BMDCs, we used rFasL (Holler et al., 2003). The addition of rFasL to P3C-primed BMDCs resulted in robust release of IL-1β that was qualitatively equivalent to that induced by iNKT cells (Figure 3C). We also detected the active, cleaved form of caspase-1 in the supernatants of TLR-primed BMDCs treated with rFasL (Figure 3E), indicating that, like iNKT cells, rFasL was able to induce inflammasome activation. Further, we observed a similar reduction in IL-1β release in ASC-deficient BMDCs compared with wild-type (WT) BMDCs (Figure 3D), as observed in Nlrp3- and caspase-1/11-deficient BMDCs (Figure 3C).
Fas ligation elicits formation of a death-inducing signaling complex (DISC) that includes the adaptor protein Fas-associated death domain (FADD) and caspase-8, the initiator caspase of the extrinsic apoptotic pathway. Engagement of Fas triggers recruitment of FADD to the intracellular region of Fas, allowing caspase-8 to bind to Fas-associated FADD. This results in recruitment of additional caspase-8 molecules, leading to caspase-8 oligomerization and subsequent self-cleavage-induced activation. Active caspase-8 in turn cleaves and activates the executioner caspases-3, −6, and −7, which go on to cleave a number of substrates to drive the cellular changes characteristic of apoptosis (Green and Llambi, 2015; Strasser et al., 2009). As expected, addition of rFasL to P3C-primed BMDCs resulted in robust cleavage of caspase-8 into the active p18 fragment (Figure 3E). Likewise, Fas ligation also resulted in cleavage of the executioner caspases-3, −6, and −7 into their active forms (Figure 3E).
Next we tested whether caspase-8 is required for FasL-induced IL-1β cleavage by BMDCs. Genetic ablation of caspase-8 results in embryonic lethality in mice because of uncontrolled necroptosis, a form of programmed necrosis that requires the kinase activity of receptor-interacting protein kinases 1 and 3 (RIPK1 and RIPK3, respectively) and is executed by the pseudokinase mixed lineage kinase-like (MLKL) (Galluzzi et al., 2017; Silke et al., 2015). Thus, to find out whether caspase-8 is required for FasL-induced IL-1β processing, we assessed the effect of loss of caspase-8 in Ripk3-deficient BMDCs. Before doing so, we confirmed that RIPK3 and MLKL are not required for IL-1β release following Fas crosslinking (Figure S3A). Caspase-8-deficient BMDCs were unable to process IL-1β following Fas ligation (Figure 3F). Further, in the absence of caspase-8, we failed to detect the active, cleaved forms of caspase-1; the executioner caspase-3, −6, and −7; and caspase-9, the initiator caspase of the intrinsic apoptotic pathway (Figure 3F). Thus, caspase-8 orchestrates the major events occurring downstream of Fas ligation; it directly cleaves the majority of pro-IL-1β, amplifies its processing and release by driving activation of the Nlrp3 inflammasome, and triggers activation of all pro-apoptotic caspases.
We next asked whether potassium efflux, a common trigger of the Nlrp3 inflammasome (Muñoz-Planillo et al., 2013), drives inflammasome activation downstream of Fas ligation. Consistent with published data, nigericin-driven caspase-1 cleavage and IL-1β processing and release were completely inhibited when potassium efflux was blocked (Figure 3G). In contrast, although raising the concentration of extracellular potassium completely inhibited FasL-induced caspase-1 processing (Figure 3G), it only partially blocked IL-1β cleavage and secretion (Figures 3G and 3H). The reduction in IL-1β release was similar to that observed in inflammasome-deficient BMDCs stimulated with rFasL (Figures 3C and 3D). Thus, Fas ligation appears to induce potassium efflux via caspase-8 to trigger activation of the Nlrp3 inflammasome.
Caspase-8 oligomerizes at the DISC, where it can form homodimers or heterodimers with cellular FLICE/caspase-8-like inhibitory protein (cFLIP), a caspase-8-like protein that lacks proteolytic activity. Caspase-8 homodimers become catalytically active following autoproteolytic cleavage, and these active homodimers drive apoptosis downstream of death receptor ligation. On the other hand, heterodimerization with cFLIP confers enzymatic activity to the heterodimer even in the absence of proteolytic processing of caspase-8, and these caspase-8:cFLIP heterodimers inhibit necroptosis by cleaving RIPK1 and RIPK3 (Boatright et al., 2004; Green and Llambi, 2015; Oberst et al., 2011; Pop et al., 2011; Tummers and Green, 2017). Moreover, cFLIP itself is a substrate for active caspase-8, which can cleave it when engaged in homodimers or heterodimers with cFLIP (Micheau et al., 2002). We observed complete cleavage of the long isoform of cFLIP (cFLIPL) into the p43 form (Figure 3E), suggesting that the entire pool of intracellular cFLIP in P3C-primed BMDCs is cleaved by caspase-8 homodimers or capase-8/cFLIP heterodimers following Fas ligation. We next asked whether proteolytic processing of caspase-8 is required for IL-1β release downstream of Fas ligation and whether this mechanism is driven by caspase-8 homodimers or caspase-8:cFLIP heterodimers. To this end, we used BMDCs from a caspase-8 mutant knockin mouse (Casp8DA/DA) in which autoproteolytic processing is prevented by replacing aspartate 387 with an alanine (Philip et al., 2016). We found that BMDCs that are incapable of undergoing caspase-8 processing were significantly impaired in their ability to release IL-1β following Fas ligation by iNKT cells (Figure S3B), suggesting that FasL-induced IL-1β release requires caspase-8 self-processing and is most efficiently driven by caspase-8 homodimers. Together, our results suggest that Fas ligation deploys caspase-8 to elicit activation of inflammasome-dependent and -independent pathways for release of IL-1β.
iNKT Cells Instruct BMDCs to Secrete IL-1β following Infection with Microbes that Fail to Activate the Inflammasome
We next asked whether iNKT cells can trigger IL-1β secretion by BMDCs in situations where microbes fail to activate the inflammasome via cell-intrinsic mechanisms. The intestinal microbe B. fragilis has been reported to lack the ability to induce significant IL-1β secretion by macrophages in a cell-autonomous setting (Chen et al., 2016). Similarly, we found that infection with live B. fragilis failed to induce substantial IL-1β secretion by BMDCs (Figure 4A). In contrast, co-culture of iNKT cells with BMDCs that had been infected with B. fragilis resulted in a significant dose-dependent increase in the amount of secreted IL-1β. Addition of a FasL-blocking antibody to the culture completely abrogated the ability of iNKT cells to elicit secretion of IL-1β by B. fragilis-infected BMDCs. Next we asked whether iNKT cells could also elicit IL-1β release by BMDCs infected with the obligate intracellular pathogen C. trachomatis, a microorganism known to cause significant immunopathology. Although infection with C. trachomatis induced weak IL-1β release by BMDCs, addition of iNKT cells resulted in robust secretion of IL-1β by BMDCs (Figure 4B). As observed for B. fragilis, the release of IL-1β induced by iNKT cells was entirely dependent on FasL. These results suggest that iNKT cells have the capacity to fill a gap in the recognition of microbes that fail to activate the inflammasome from within the cytosol of host cells.
Figure 4. iNKT Cells Induce IL-1β Secretion by BMDCs during Infection with Microbes that Fail to Elicit Strong IL-1β Release in a Cell-Intrinsic Manner.
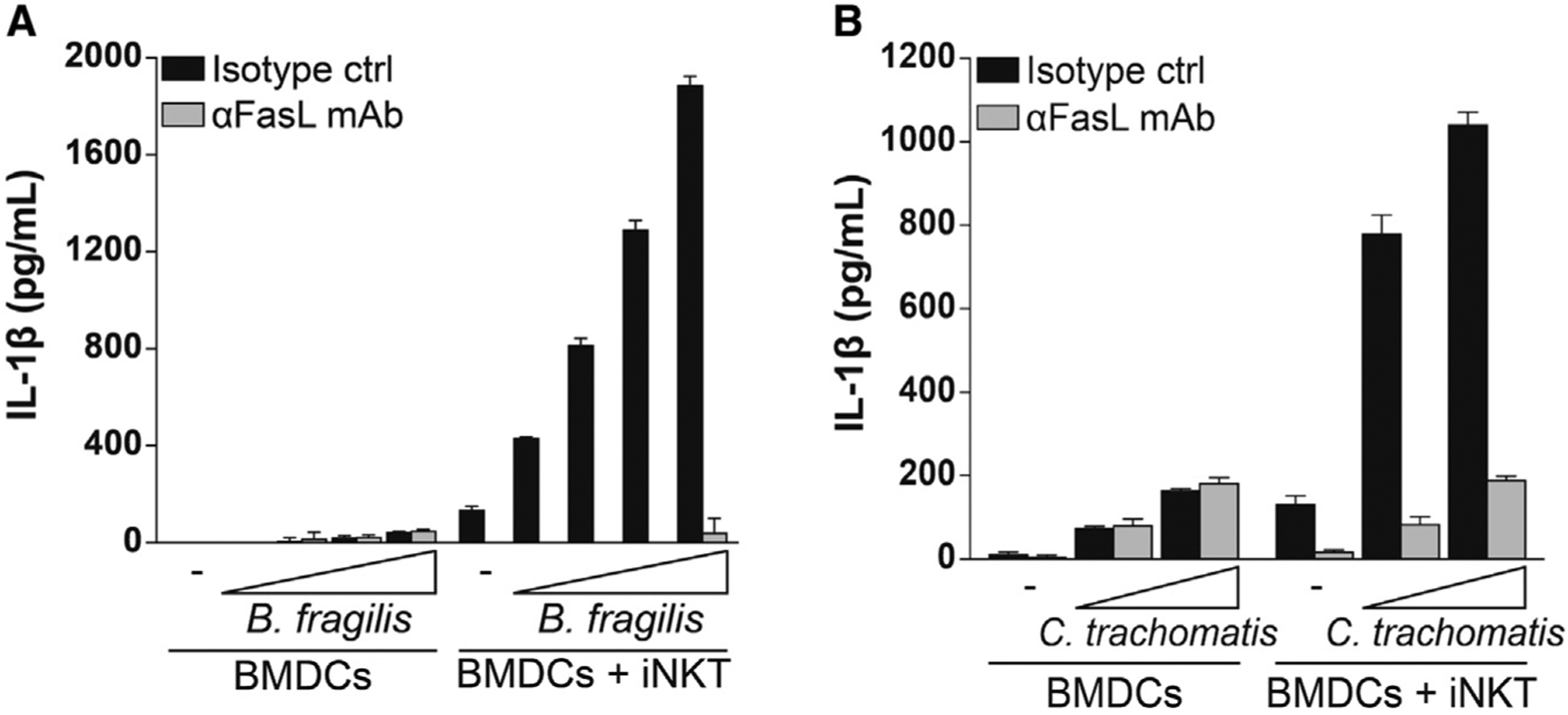
BMDCs were infected with (A) B. fragilis or (B) C. trachomatis at increasing MOIs, washed, and cultured alone or with iNKT cells for 24 h in the presence of a FasL-blocking antibody (αFasL mAb) or an isotype control. IL-1β release was quantified by ELISA. Error bars represent mean + SD of triplicate samples; panels are representative of three and two independent experiments, respectively.
Fas Ligation Drives the Transition from Apoptosis to Necrosis in BMDCs
Ligation of the death receptor Fas drives extrinsic apoptosis, classically a form of non-inflammatory cell death that is morphologically and biochemically distinct from necrotic cell death pathways like pyroptosis. Some of the features of apoptotic cell death include cytoplasmic shrinkage, phosphatidylserine exposure on the outer leaflet of the plasma membrane, chromatin condensation, membrane blebbing, and formation of apoptotic bodies (Galluzzi et al., 2018; Nagata, 2018). In contrast, pyroptosis is characterized by cell swelling and plasma membrane rupture, which are dependent on formation of plasma membrane pores by members of the gasdermin family of proteins (Kovacs and Miao, 2017; Shi et al., 2017). The consequent release of intracellular contents and proinflammatory molecules defines pyroptosis as an inflammatory form of cell death. To begin to characterize cell death following co-culture of iNKT cells with P3C-primed BMDCs, we stained the cells with the phosphatidylserine-binding protein annexin V and the membrane-impermeant DNA-intercalating dye Sytox Green and analyzed the cells by flow cytometry. Cells that become single-positive for annexin V are considered to be undergoing early apoptosis, whereas cells that also internalize Sytox Green have become membrane permeable and are deemed necrotic. Although a fraction of BMDCs co-cultured with iNKT cells became positive only for annexin V, a substantial percentage also stained positive with Sytox Green at later time points (Figure 5A). A stronger TCR stimulus provided by αGalCer resulted in more pronounced death of BMDCs, characterized by increases in the annexin V single-positive and annexin V/Sytox Green-double positive populations (Figure S4A). Following Fas crosslinking using rFasL, P3C-primed BMDCs showed flow cytometry staining patterns comparable with those observed with iNKT cell co-culture (Figure 5B). We next measured the release of cytosolic lactate dehydrogenase (LDH) into the medium as an indication of loss of plasma membrane integrity, a feature of necrotic cell death. We detected a significant amount of LDH in the supernatants of P3C-primed BMDCs following Fas crosslinking, and the appearance of extracellular LDH correlated with the release of IL-1β (Figure 5C). Together, these results suggest that Fas ligation drives cell death that exhibits features of both apoptosis and necrosis in TLR-primed BMDCs.
Figure 5. Fas Ligation Drives the Transition from Apoptosis to Necrosis in BMDCs.
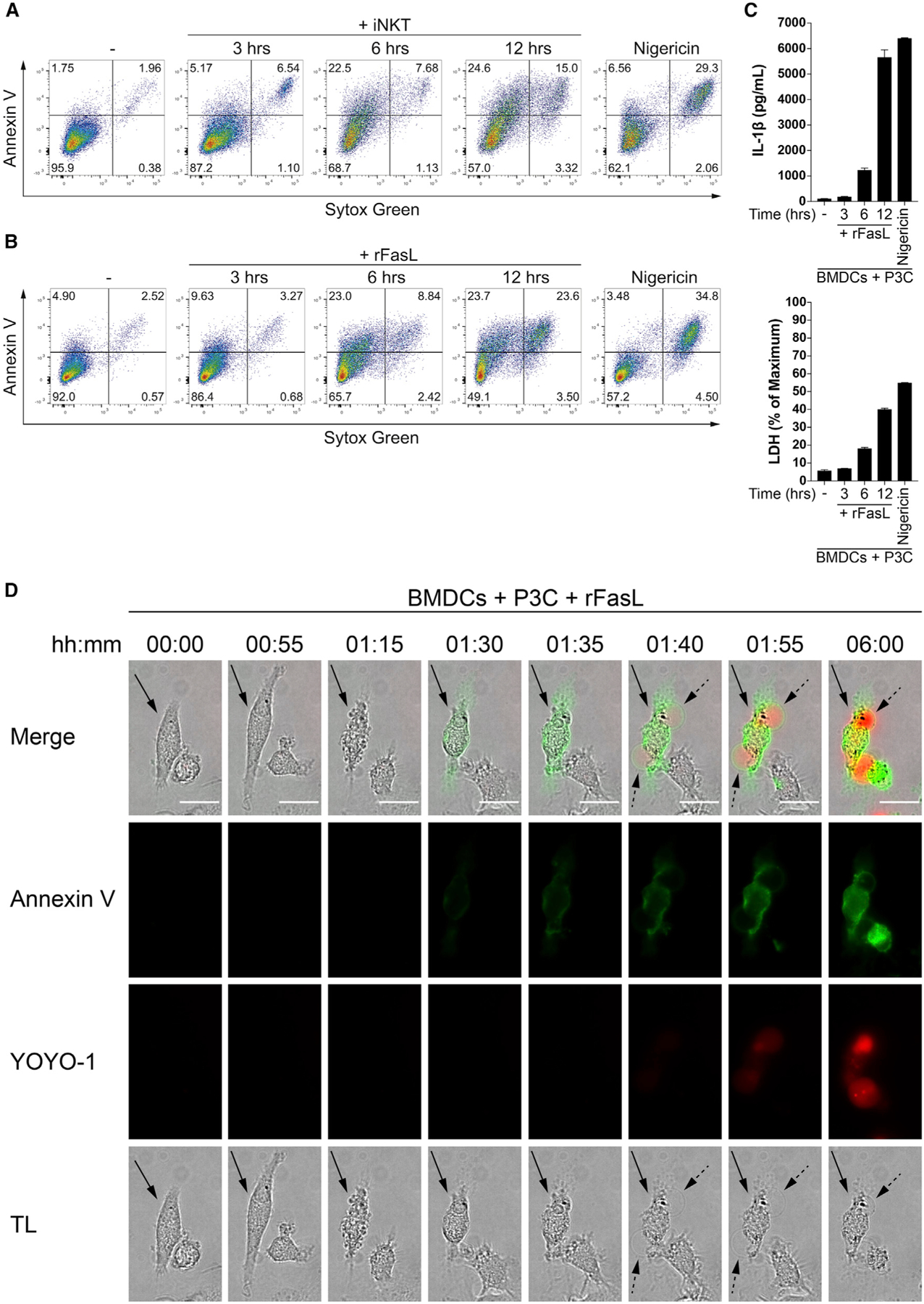
(A and B) P3C-primed BMDCs were cultured alone or with (A) CellTrace Violet-labeled iNKT cells or (B) rFasL. After the indicated times, the cells were harvested, labeled with Sytox Green and annexin V, and the extent of early-apoptotic and necrotic BMDCs was analyzed by gating on CellTrace Violet-negative, CD11c-positive BMDCs. Nigericin-treated BMDCs were used as a control.
(C) P3C-primed BMDCs were cultured alone or in the presence of rFasL for the indicated times. Top: IL-1β release was quantified by ELISA. Bottom: LDH in the extracellular medium was monitored by an enzymatic assay. Nigericin-treated BMDCs were used as a control.
(D) P3C-primed BMDCs were stimulated with rFasL, and morphological changes (transmitted light, TL), annexin V (green), and YOYO-1 (red) staining were assessed by live imaging over 6 h. Images were captured every 5 min, and representative time points are shown. The arrow indicates a representative cell. Dotted arrows point at necrotic plasma membrane balloons. The scale bars represent 10 μm.
Error bars represent mean + SD of triplicate samples. Each panel is representative of at least three different experiments. See also Figure S4.
We considered the possibility that apoptotic cell death and necrosis could be occurring sequentially or, alternatively, that they could occur in exclusive cell populations. To distinguish these possibilities, we used a live microscopy assay to discriminate apoptotic cells from necrotic cells based on three criteria. First, we tracked whether cells underwent shrinkage or swelling, morphological hallmarks of apoptotic and necrotic cells, respectively. Second, we evaluated whether cells externalized phosphatidylserine and, thus, bound annexin V on the outer leaflet of their plasma membrane, as expected of cells undergoing apoptosis. Third, we monitored loss of plasma membrane integrity, a feature of necrosis (Galluzzi et al., 2018), as assessed by staining with the membrane-impermeable, nucleic acid-binding dye YOYO-1. Fas ligation rapidly induced cytoplasmic shrinkage and membrane blebbing, both features of apoptosis (Figures 5D, S4B, and S4C; Videos S1 and S2). BMDCs then proceeded to stain with annexin V, and this was rapidly followed by swelling and lysis, as evidenced by formation of large plasma membrane balloons that are characteristic of necrosis. Although the majority of BMDCs exhibited a brief lag between annexin V staining and lysis (~80%), about 20% of BMDCs progressed so rapidly from an apoptotic to a necrotic morphotype that our 5-min imaging intervals failed to capture annexin V staining of the plasma membrane preceding cell lysis (Figure S4C and S4D). The lack of a substantial lag between annexin V and YOYO-1 staining suggested that BMDCs initiated but did not complete the classic apoptotic program. Further, annexin V staining was not limited to the outer leaflet of the plasma membrane. Following membrane permeabilization, annexin V increasingly stained the cytoplasm of cells, and this occurred concomitantly with incorporation of YOYO-1.
Following Fas crosslinking, we also noticed that the YOYO-1 staining spread from the nucleus to the cytoplasm and plasma membrane balloons as BMDCs adopted a pyroptotic-like morphotype (Figures 5D, S4B, S4C, S5A, and S5B). Apoptotic and pyroptotic cells have been reported to undergo chromatin condensation (pyknosis), but although the nuclei of apoptotic cells then experience membrane rupture and DNA fragmentation (karyorrhexis), the nuclei of pyroptotic cells remain intact (Galluzzi et al., 2018; Miao et al., 2011). Thus, we reasoned that classical pyroptosis is characterized by confinement of nuclear DNA in the nucleus, but if it is preceded by apoptosis, then DNA may diffuse into the cytoplasm and pyroptotic balloons. To test this hypothesis, we labeled P3C-primed BMDCs with the fluorescent, cell-permeable DNA probe silicon-rhodamine Hoechst (SiR DNA) and tracked the nuclear DNA over time. After addition of rFasL, P3C-primed BMDCs underwent substantial nuclear condensation concurrently with membrane blebbing and cytoplasmic shrinkage. When the cells transitioned to necrosis, as characterized by formation of large plasma membrane balloons and YOYO-1 staining, the nuclear DNA diffused into the cytoplasm and plasma membrane balloons (Figures 6A, 6C, S5A, and S5B; Video S3). This observation was in stark contrast to BMDCs stimulated to undergo canonical pyroptosis using nigericin (Figures 6B and 6C), where the nucleus underwent condensation but the SiR-DNA staining persisted as a single large speck over time, suggesting that the nuclear DNA remained confined to the nucleus. Thus, it is likely that fragmented nuclear DNA leaks into the cytoplasm and necrotic plasma membrane balloons when apoptosis is converted into necrosis, a feature not apparent in cells that undergo classical pyroptosis. Indeed, TUNEL staining confirmed that only BMDCs treated with rFasL, but not those stimulated with nigericin, experienced significant DNA fragmentation (Figure S5C). Together, the data suggest that Fas ligation drives the rapid transition from early apoptosis to necrosis in TLR-primed BMDCs.
Figure 6. Nuclear DNA Diffuses to the Cytosol and Plasma Membrane Balloons in BMDCs When Necrosis Is Preceded by Apoptosis.
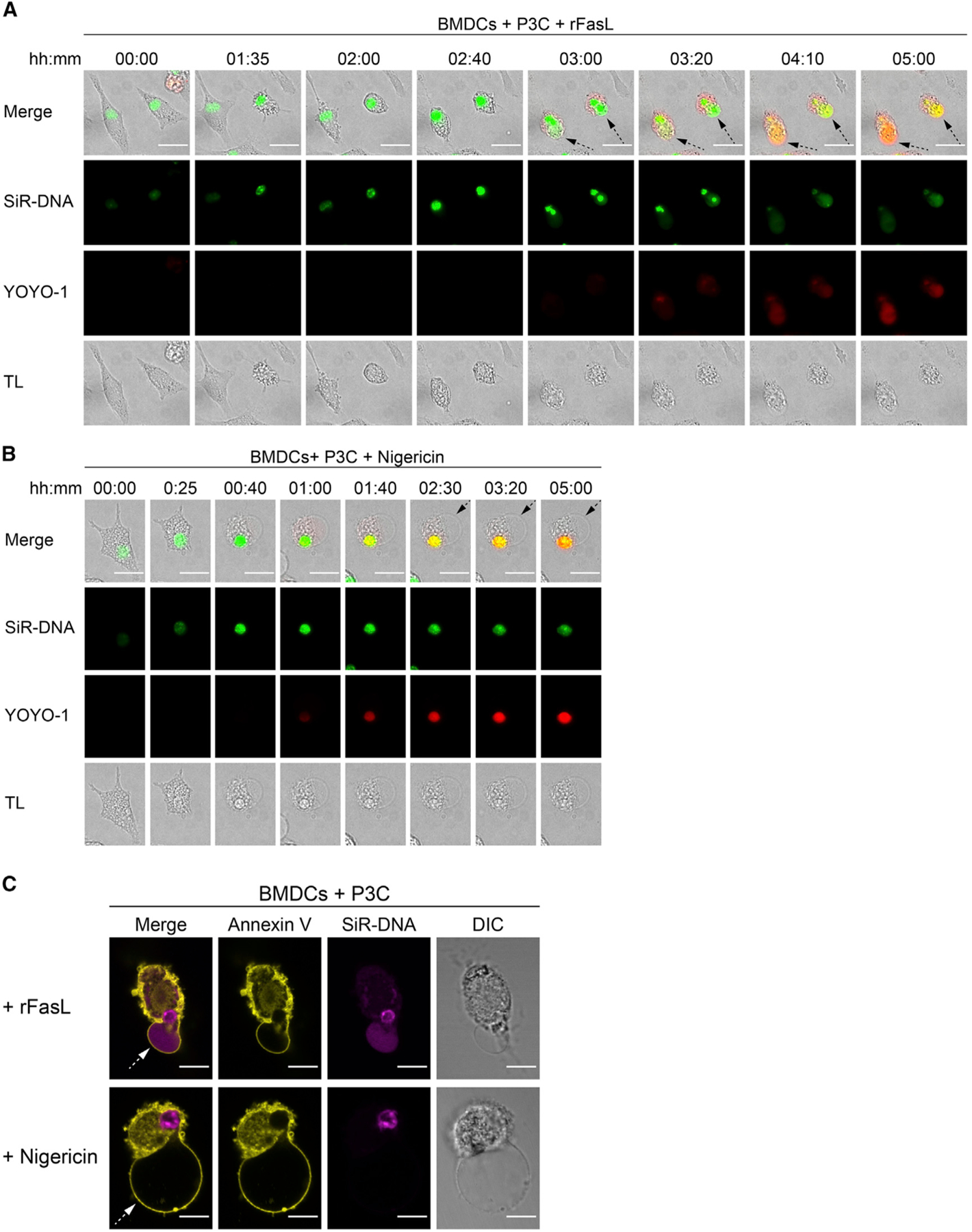
(A and B) P3C-primed BMDCs were labeled with SiR-DNA and stimulated with rFasL (A) or nigericin (B). Morphological changes (TL), nuclear DNA (SiR-DNA, green), and YOYO-1 (red) staining were monitored by live imaging over 6 h. Images were captured every 5 min, and representative time points are shown. Dotted arrows point at necrotic plasma membrane balloons. The scale bars represent 10 μm.
(C) P3C-primed BMDCs were labeled with SiR-DNA and stimulated with (top) rFasL or (bottom) nigericin in the presence of annexin V. Morphological changes (TL), nuclear DNA (SiR-DNA, magenta), and annexin V (yellow) staining were assessed after 6 h of stimulation. Dotted arrows point at necrotic plasma membrane balloons. The scale bars represent 10 μm.
Panels are representative of at least three different experiments. See also Figure S5.
Fas Ligation Drives Pyroptosis and IL-1β Release by TLR-Primed BMDCs through the Pore-Forming Protein GSDMD
Following assembly of the Nlrp3 inflammasome, active caspase-1 not only processes pro-IL-1β but also cleaves the pore-forming protein GSDMD. The cleaved N-terminal domain of GSDMD interacts with the plasma membrane and oligomerizes, forming pores that serve as conduits for the release of IL-1β (Evavold et al., 2018; Heilig et al., 2018) and ultimately cause the cell to swell and lyse (Hagar et al., 2013; Liu et al., 2016; Shi et al., 2015). Given that FasL-induced IL-1β release by TLR-primed BMDCs was partially dependent on the Nlrp3 inflammasome (Figures 3A–3D), we next tested whether GSDMD contributed to IL-1β release and the progression to pyroptosis observed following Fas ligation. Consistent with published data, we found that nigericin-induced IL-1β release and pyroptosis were completely dependent on GSDMD. In contrast, the ability of GSDMD-deficient BMDCs to release IL-1β (Figure 7A) and become pyroptotic (Figure 7B) following Fas ligation was lost early on but was progressively regained over time. Analysis of GSDMD cleavage by immunoblot confirmed that Fas ligation resulted in processing of GSDMD into the pore-forming N-terminal fragment (Figure 7C). Fas ligation also resulted in generation of a GSDMD fragment of about 20 kDa (GSDMD inactive). The appearance of this 20-kDa band is consistent with previous reports showing that caspase-3 and −7 can inactivate GSDMD by cleaving it at aspartate 87 (Taabazuing et al., 2017), suggesting that this fragment may be the result of combined cleavage between caspase-1 and caspase-3 or −7. The abundance of this inactive fragment may partly explain the limited role of the Nlrp3 inflammasome and GSDMD in the late phase of FasL-induced pyroptosis and IL-1β release by TLR-primed BMDCs.
Figure 7. Fas Ligation Drives Pyroptosis and IL-1β release by TLR-primed BMDCs through the Pore-Forming Protein GSDMD.
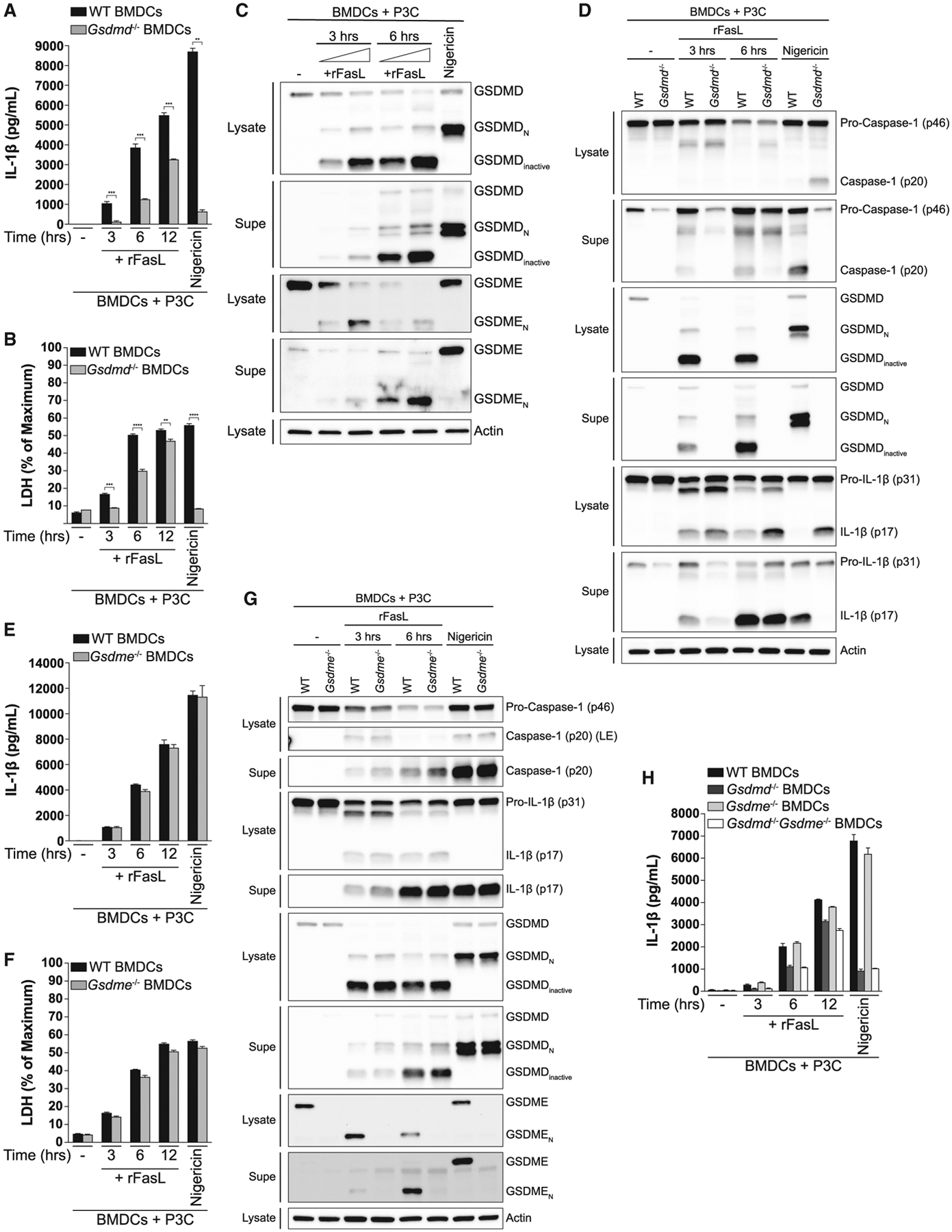
(A and B) P3C-primed BMDCs from WT and Gsdmd−/− mice were stimulated with rFasL for the indicated times. IL-1β release was quantified by ELISA (A), and LDH in the extracellular medium (B) was monitored by an enzymatic assay. Nigericin-treated BMDCs were used as a control.
(C and D) P3C-primed BMDCs from WT (C) or WT and Gsdmd−/− (D) mice were stimulated with rFasL for the indicated times, and the cell lysates and cell-free Supes were probed for the indicated proteins by immunoblot.
(E–G) P3C-primed BMDCs from WT and Gsdme−/− mice were treated with rFasL for the indicated times. IL-1β release was quantified by ELISA (E), LDH in the extracellular medium was monitored by an enzymatic assay (F), and the cell lysates and cell-free Supes were probed for the indicated proteins by immunoblot (G).
(H) P3C-primed BMDCs from WT, Gsdmd−/−, Gsdme−/−, and Gsdmd−/−Gsdme−/− mice were stimulated with rFasL for the indicated times. IL-1β release was quantified by ELISA.
Error bars represent mean + SD. Each panel is representative of at least three different experiments. **p < 0.01, ***p < 0.001, ****p < 0.0001, as determined by Student’s unpaired t test. See also Figure S6.
Given that loss of GSDMD appeared to have a more profound effect on IL-1β release and cell death than loss of the inflammasome components Nlrp3, caspase-1/11, or ASC (Figures 7A, 7B, and 3A–3D), we asked whether an alternative, inflammasome-independent pathway may be contributing to GSDMD cleavage following Fas ligation. Nlrp3 and caspase-1/11-deficient BMDCs were indeed still capable of cleaving GSDMD into its pore-forming fragment, albeit to a lesser extent than WT BMDCs (Figure S6A). Because caspase-8 has recently been shown to cleave GSDMD in the context of Yersinia infection (Orning et al., 2018; Sarhan et al., 2018), we asked whether caspase-8 was responsible for the inflammasome-independent processing of GSDMD downstream of Fas ligation. Although caspase-8-deficient BMDCs were unaffected in their ability to cleave GSDMD following nigericin-induced Nlrp3 inflammasome activation, they were completely unable to process GSDMD in response to Fas ligation (Figure S6B).
Following our observation that caspase-8 can induce GSDMD cleavage independent of the inflammasome, we hypothesized that GSDMD pores might mediate activation of the Nlrp3 inflammasome downstream of Fas ligation. Indeed, caspase-1 cleavage was significantly impaired in GSDMD-deficient BMDCs following Fas ligation (Figure 7D). Together, our data demonstrate that caspase-8 is the main driver of IL-1β release downstream of Fas ligation and that it does so by directly cleaving IL-1β and GSDMD. The resulting GSDMD-mediated pore formation likely leads to potassium efflux, activating the Nlrp3 inflammasome and amplifying IL-1β release.
In addition to GSDMD, pyroptosis has been reported to occur in response to chemotherapy drugs via caspase-3-mediated cleavage of gasdermin E (GSDME), another gasdermin family member with pore-forming activity (Wang et al., 2017). Moreover, the N-terminal fragment of GSDME has been shown to mediate breakdown of the plasma membrane during the late stages of apoptosis, driving the transition from apoptosis to secondary necrosis (Rogers et al., 2017). Because Fas ligation resulted in robust activation of caspase-3 (Figure 3E), we next asked whether GSDME was involved in FasL-induced IL-1β release and pyroptosis. Although GSDME was rapidly processed into its pore-forming N-terminal fragment (Figure 7C), the ability of GSDME-deficient BMDCs to process and secrete IL-1β (Figures 7E and 7G) or release LDH (Figure 7F) in response to Fas ligation remained intact. Further, analysis of caspase-1 and GSDMD cleavage revealed that GSDME did not play a role in FasL-induced inflammasome activation (Figure 7G). We also considered the possibility that a role of GSDME was obscured by the effect of GSDMD. To address this, we assessed IL-1β release in GSDMD−/− GSMDE−/− double-knockout BMDCs but found no additional defect other than that observed in GSDMD single-deficient BMDCs (Figure 7H). These results suggest that the pore-forming activity of GSDMD, but not GSDME, drives IL-1β release and the transition from apoptosis to pyroptosis in TLR-primed BMDCs following Fas ligation.
We next sought to determine whether a two-cell model for IL-1β release could also be operative in humans. Human iNKT cells have been reported previously to induce IL-1β secretion by peripheral blood monocytes (Felley et al., 2016), although the mechanism through which they do this was not elucidated. We cultured P3C-primed human MDMs with iNKT cells in the presence of αGalCer. As in the mouse system, a priming signal was insufficient to elicit release of IL-1β by BMDCs, but addition of iNKT cells triggered IL-1β secretion to an extent comparable with that induced by nigericin (Figure S6C). Addition of rFasL to P3C-primed MDMs also resulted in significant release of IL-1β, and this effect was blocked by a FasL-neutralizing antibody (Figure S6D). Although rFasL was able to drive IL-1β release, we failed to observe inhibition of IL-1β release when two distinct FasL-blocking antibodies were individually added to the MDM-iNKT cell co-culture (data not shown). Thus, it appears that, although human MDMs can be triggered to secrete IL-1β by FasL (Figure S6D) and are also responsive to iNKT cells in co-culture (Figure S6C), human iNKT cells may employ additional molecules that can drive this pathway.
DISCUSSION
During infection, processing and secretion of IL-1β have mainly been thought to hinge on inflammasome activation following recognition of microbes in the host cell cytosol. However, pathogenic microorganisms employ a number of strategies to suppress or evade this cell-autonomous mechanism of inflammasome activation. Further, there are several examples of infections in which IL-1β plays an important protective role independent of caspase-1 or the inflammasomes, including infection with C. trachomatis (Bellocchio et al., 2004; Chen et al., 2016; Lu et al., 2000), Pseudomonas aeruginosa (Karmakar et al., 2012), and Mycobacterium tuberculosis (Dorhoi et al., 2012; Juffermans et al., 2000; McElvania Tekippe et al., 2010). These observations suggest that there are important mechanisms of IL-1β release in addition to the inflammasomes. The ability of iNKT cells to instruct BMDCs to process and secrete IL-1β represents an important mechanism by which the immune system could overcome microbial inflammasome evasion. Although cell-intrinsic cytosolic sensing by inflammasomes is undoubtedly important, perhaps it is not surprising that additional pathways to promote IL-1β release have evolved in the complex multicellular immune systems of mammals.
iNKT cells, “cellular adjuvants” of the immune system, are characterized by a number of features that endow them with the ability to respond to infection in an innate-like manner. They reside in a poised effector state and can respond within minutes to hours of being activated. Their ability to rapidly exert their effector functions is partly due to their constitutive expression of preformed cytokine-encoding mRNAs, a feature that does not require prior recognition of foreign antigen (Bendelac et al., 2007; Brennan et al., 2013; Cohen et al., 2013; Gutierrez-Arcelus et al., 2019; Kohlgruber et al., 2016; Liu et al., 2016). Our studies revealed that iNKT cells express FasL transcripts and protein at steady state and that they rapidly translocate pre-existing intracellular FasL stores to their surface following cognate interactions with TLR-primed BMDCs. These pre-existing FasL stores and the ability to rapidly deploy FasL to elicit IL-1β release by TLR-primed BMDCs are further evidence of the innate-like functional role of iNKT cells that distinguishes them from adaptive, MHC-restricted T cells.
iNKT cells recognize distinct endogenous and exogenous lipid antigens through a hotspot of invariant, germline-encoded residues in their TCR, a property that allows iNKT cells to respond en masse, essentially as a clonal population (Scott-Browne et al., 2007). This mode of recognition is very similar to how pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs) (Chuenchor et al., 2014). Based on our findings, we propose that the iNKT cell TCR can function as a surrogate PRR that may complement sensing of microbes that fail to elicit strong inflammasome activation from within the cytosol of an APC. We hypothesize that this alternative, two-cell mechanism of IL-1β release may permit specific recognition of microorganisms that have high pathogenic potential but fail to activate inflammasomes in a cell-autonomous manner. It is also conceivable that iNKT cells may trigger release of IL-1β by APCs that have been exposed to commensal microbes that otherwise do not activate the inflammasomes. This may occur in the liver, which captures gut commensal bacteria that enter the bloodstream during intestinal pathology (Balmer et al., 2014) and where iNKT cells make up to 30% of the lymphocyte compartment (Brennan et al., 2013). Both of these theories are supported by our findings that iNKT cells can elicit IL-1β release by BMDCs exposed to the obligate intracellular pathogen C. trachomatis (Figure 4B) or the gut commensal B. fragilis (Figure 4A). Because iNKT cells can be activated by bacteria, viruses, fungi, and even sterile inflammation, the mechanisms defined in this report provide a pathway for IL-1β release across a range of inflammatory situations.
Although iNKT cells can be activated through direct recognition of microbial lipids presented on CD1d, it is now appreciated that they may be even more frequently activated by mammalian self-lipid antigens induced in host cells by TLR agonists or infections, including microbes that do not harbor cognate lipid antigens (Brennan et al., 2011; Brigl et al., 2011; Salio et al., 2007). This provides a nearly universal mechanism of iNKT cell activation by mammalian cells that allows them to respond to a vast array of microorganisms with the simple requirement that a microbe engages a TLR on the APC. Importantly, here we found that iNKT cells can instruct TLR-primed BMDCs to secrete IL-1β in response to endogenous or foreign lipid antigens presented on CD1d. Thus, our studies suggest that any microorganism that stimulates a TLR on an APC could elicit activation of this two-cell pathway for IL-1β release.
We observed that iNKT cell-driven IL-1β release is mediated entirely through ligation of Fas on APCs. Fas ligation has been reported previously to drive secretion of IL-1β through caspase-8 independent of the Nlrp3 inflammasome (Bossaller et al., 2012; Miwa et al., 1998; Uchiyama et al., 2013). In contrast to these studies, we found that, although caspase-8 directly cleaves the majority of IL-1β secreted in response to Fas ligation (Figures 3B and 3F), it also cleaves GSDMD (Figures S6A and S6B) to induce Nlrp3 inflammasome activation. GSDMD-mediated pore formation then leads to potassium efflux, resulting in activation of the Nlrp3 inflammasome (Figures 3G and 3H) as a mechanism to amplify IL-1β release.
Apoptosis has long been regarded as an immunologically silent form of cell death. Classically, this feature is achieved through organized dismantling of the cell into apoptotic bodies that are rapidly taken up and cleared by phagocytes. As this process unfolds, the paradigm has remained that the plasma membrane preserves its integrity, preventing putative damage-associated molecular patterns (DAMPs) from leaking into the extracellular space and causing inflammation (Galluzzi et al., 2018). Nonetheless, several studies have shown that apoptosis is not always silent and can induce potent inflammatory responses in vitro and in vivo (Bossaller et al., 2012; Faouzi et al., 2001; Feltham et al., 2017). Secondary necrosis has been thought to occur when cells undergo the complete apoptotic program but are not scavenged by phagocytes. In contrast, we found that TLR-primed BMDCs initiate apoptosis, exhibiting cytoplasmic shrinkage and membrane blebbing that very rapidly progressed to cell swelling and lysis. Further, annexin V staining revealed that BMDCs did not always externalize phosphatidylserine prior to undergoing swelling, as would be expected during classical apoptosis, whereas cells that exposed phosphatidylserine did so for merely minutes before lysing. This suggests that TLR-primed BMDCs initiate the apoptotic program in response to Fas ligation but then rapidly switch to pyroptosis through the action of the pore-forming protein GSDMD. Of note, although we found that GSDMD plays a major role in FasL-induced IL-1β release and cell death, GSDMD-deficient BMDCs were still capable of releasing IL-1β and undergoing necrosis at late time points (Figures 7A and 7B). Given our observation that GSDME does not play a role in FasL-induced IL-1β or cell death (Figures 7E and 7F), it is conceivable that other pore-forming proteins are activated late during execution of apoptosis and that these can compensate for the lack of GSDMD. Taken together, our findings demonstrate that iNKT cells can co-opt the extrinsic apoptotic pathway to instruct TLR-primed BMDCs to undergo an inflammatory form of cell death characterized by release of bioactive IL-1β, a fascinating repurposing of signaling modules that evolved for cell death and cytoplasmic danger sensing.
In summary, we described a two-cell model for IL-1β release where iNKT cells employ FasL to instruct TLR-primed BMDCs to process and secrete IL-1β. This is made possible by the ability of iNKT cells to rapidly translocate pre-existing intracellular FasL stores to their surface following cognate interactions. Therefore, iNKT cells are positioned to rapidly instruct APCs to secrete IL-1β, and they can do so following infection with any microorganism that stimulates a PRR on APCs (Figure S7). This mechanism may also be at play in the setting of non-infectious or sterile inflammation, where FasL-expressing T cells may drive the autoimmune pathology associated with dysregulated IL-1β release. We show that, in addition to “ancient” cell-autonomous cytosolic sensing by inflammasomes, mammals have evolved a multi-cellular, cell-extrinsic pathway for IL-1β release that combines signaling components of the inflammasomes and the extrinsic cell death pathways. Our study provides a mandate to examine the likely possibility that other innate-like T cells, including γδ T cells, mucosa-associated invariant T cells, and even adaptive effector T cells, can also co-opt the death receptor pathway to provide the innate immune system with cell-extrinsic IL-1β secretion signals.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Michael B. Brenner (mbrenner@research.bwh.harvard.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Wild-type C57BL/6J, Caspase1/11−/− (Kuida et al., 1995), Nlrp3−/− (Kovarova et al., 2012), Fas−/−, and CD1d−/− (Exley et al., 2003) mice were purchased from Jackson Laboratories and were housed under specific pathogen-free conditions at the Center for Comparative Medicine at Brigham and Women’s Hospital (BWH). All procedures were approved by the BWH Institutional Animal Care and Use Committee. Mice were euthanized by CO2 asphyxiation followed by cervical dislocation in accordance to the BWH IACUC guidelines. Bone marrow from the following mice were kindly provided by the following investigators: Caspase-8DA/DA (Philip et al., 2016) was provided by Dr. Igor Brodsky, Gsdmd−/− (Kambara et al., 2018) was provided by Dr. Hongbo Luo, Ripk3−/− (Newton et al., 2004) and Mlkl−/− (Murphy et al., 2013) were provided by Dr. Ben Croker, Ripk3−/−Casp8−/− (Kaiser et al., 2011) was provided by Dr. Edward Mocarski, Gsdme−/− and Gsdmd−/−Gsdme−/− (Chen et al., 2019) were provided by Dr. Petr Broz, and Asc−/− (Ozören et al., 2006) was provided by Dr. Gabriel Nuñez.
Bacterial strains and growth
B. fragilis (NCTC 9343, a kind gift from Dr. Laurie Comstock) was grown anaerobically at 37°C in Brain Heart Infusion broth (Anaerobe Systems). The culture’s optical density at 600 nm was measured to determine the colony-forming units. C. trachomatis serovar D (UW-3/Cx; ATCC, kindly provided by Dr. Michael Starnbach) was propagated within McCoy cells grown in RPMI 1640 (Thermo Fisher) supplemented with 10% FBS, 2 mM L-glutamine, 0.1 mM nonessential amino acids and 1 mM sodium pyruvate. Cells were lysed with sterile glass beads followed by sonication to release bacteria from the inclusion. Elementary bodies (EBs) were purified by density gradient centrifugation, aliquoted and stored at −80°C in medium containing 220 mM sucrose, 15 mM sodium phosphate, 4 mM potassium phosphate and 5 mM L-glutamic acid. Aliquots were thawed immediately before use. Inclusion forming units (IFUs) of thawed EBs were tittered on McCoy cell monolayers.
Human samples
All human samples were used with approval from the Brigham and Women’s Hospital Institutional Review Board. As a source of human peripheral blood mononuclear cells (PBMCs), leukoreduction filters were obtained from the Brigham and Women’s Hospital Kraft Family Blood Donor Center. Human iNKT cells were derived from healthy donor peripheral blood as previously described (Brigl et al., 2006).
METHOD DETAILS
Preparation of murine bone marrow-derived dendritic cells (BMDCs)
To generate BMDCs, the femoral and tibial bones of 6-15-week old female mice were flushed with RPMI media (Thermo Fisher) and the bone marrow suspension was passed through a 40 μm cell strainer. The filtered bone marrow cells were depleted of red blood cells (RBCs) following a 1-minute treatment with ACK lysing buffer (Thermo Fisher) on ice. 4×106 bone marrow cells per untreated 10-cm dish were cultured in 10 mL of IMDM medium (Thermo Fisher) supplemented with 10% heat-inactivated fetal bovine serum (Gemini Bio-Products), 2 mM L-glutamine, 100 U/ml penicillin / 100 μg/ml streptomycin, 25 mM HEPES, 50 μM β-mercaptoethanol (all Thermo Fisher), and 20 ng/ml of recombinant murine GM-CSF (Peprotech) and kept in a humidified incubator at 37°C with 5% CO2. An additional 10 mL of media was added to plates on day 4 of differentiation. After 7 days of differentiation, the non-adherent cells in the culture supernatant and loosely adherent cells harvested by gentle washing with 2 mM EDTA (Thermo Fisher) in HBSS (Thermo Fisher) were pooled and used as the starting source of cells for most experiments. For all microscopy-based experiments, the CD11c+ fraction was enriched to 99% purity by incubating the starting population with CD11c microbeads (Miltenyi Biotec) and passing them sequentially through two LS columns (Miltenyi Biotec).
Preparation of human monocyte-derived macrophages (MDMs)
PBMCs were isolated using Ficoll-Paque PLUS (GE Lifesciences) according to the manufacturer’s instructions. To prepare MDMs, CD14+ cells were isolated using CD14 microbeads (Miltenyi Biotec). CD14+ cells were plated in 6-well plates at 3×106 cells per well in 3 mL of RPMI medium (Thermo Fisher) supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin / 100 μg/ml streptomycin, 25 mM HEPES, 50 μM β-mercaptoethanol, and 100 ng/ml of recombinant human GM-CSF (Peprotech) and kept in a humidified incubator at 37°C with 5% CO2. An additional 3 mL of media was added to wells on day 3 of differentiation. After 6 days of differentiation, the non-adherent cells in the culture supernatant and the loosely adherent cells lifted by gentle use of a cell scraper were pooled and used as the starting source of cells for experiments.
Generation of iNKT cell lines
Murine iNKT cells were enriched from the spleens of female C57BL/6J mice as previously described (Chiba et al., 2009). Briefly, splenocytes were harvested from female C57BL/6J mice by pressing spleens through a 40-μm cell strainer, followed by 1-minute treatment with ACK lysis buffer on ice to deplete RBCs. T cells were negatively selected using the Pan T Isolation Kit II following the manufacturer’s protocol (Miltenyi Biotec). iNKT cells were labeled with APC-conjugated CD1d tetramer loaded with PBS-57 lipid antigen (National Institutes of Health Tetramer Core Facility) and iNKT cells were enriched using anti-APC beads (Miltenyi Biotec). To prepare APCs for expansion of iNKT cells, CD11c+ splenocytes were enriched by pressing spleens of female C57BL/6J mice through a 40-mm cell strainer followed by digestion in DMEM (Thermo Fisher) containing 10% FBS, collagenase type IV (Worthington Biochemical, 1 mg/ml), and DNase1 (Sigma, 20 μg/ml) for 30 minutes at 37°C. Splenocytes were depleted of RBCs following a 1-minute treatment with ACK lysis buffer on ice. CD11c+ splenocytes were positively selected using CD11c microbeads following the manufacturer’s protocol. CD11c+ splenocytes were irradiated, plated at 2×105 cells per well in 24 well plates and were loaded with αGalCer (50 ng/ml) for 6 hours. 2×106 freshly isolated iNKT cells were added per well in 24-well plates (1:10 ratio of CD11c+ splenocytes to iNKT cells) in complete RPMI medium containing 10% FBS. Two days later, IL-2 (Peprotech, 0.5 ng/ml) and IL-7 (Peprotech, 10 ng/ml) were added to the media. 21 days following the start of the expansion, iNKT cells were enriched to 99% purity by FACS-sorting using APC-conjugated CD1d tetramer loaded with PBS-57 lipid antigen (NIH Tetramer Core Facility, 1:500) and an antibody against the TCR β chain (Biolegend, 1:200). iNKT cells were then stimulated with freshly-isolated CD11c+ splenocytes as described above and were frozen 21 days later for use in experiments. The human iNKT cell clone J24L.17 was previously generated from healthy donor peripheral blood as described (Brigl et al., 2006). Briefly, iNKT cells sorted using APC-conjugated CD1d tetramer loaded with PBS-57 lipid antigen (1:500) were cultured at 37°C with 5% CO2 in RPMI culture medium containing 10% FBS, 2% human AB serum, 1% penicillin and streptomycin, and 1% L-glutamine in the presence of irradiated allogeneic PBMCs and PHA. After 5–10 days of culture, 200 U/ml recombinant human IL-2 (Peprotech) was added to the medium.
Cell stimulation
Where indicated, BMDCs were primed with P3C (Invivogen, 0.5 μg/ml) prior to addition of inflammasome agonists, iNKT cells, or recombinant, hexameric FasL (rFasL or MegaFasL, Adipogen, 0.1 μg/ml). The following stimulation conditions apply for ELISAs, LDH release assays, and flow cytometry-based assays. For inflammasome activation controls, BMDCs were primed for 4 hours and treated with monosodium urate (MSU) crystals (Invivogen, 100 μg/ml) for 6 hours or nigericin (Invivogen, 10 μM) for 1 hour. BMDCs were primed for 24 hours in the presence or absence of αGalCer (generously provided by Dr. Gurdyal Besra, 50 ng/ml) prior to addition of iNKT cells or rFasL. BMDCs were routinely plated at 5×104 cells per well in 96-well plates for ELISAs, LDH release assays, and flow cytometry-based assays. For immunoblots, BMDCs were plated in 6-well plates at 1.25×106 cells per well. For microscopy, BMDCs were plated in glass-bottom 96-well plates at 2×104 cells per well. A 4:1 ratio of iNKT cells to BMDCs was used for co-culture assays. For experiments with human cells, MDMs were primed with P3C (2 μg/ml) for 24 hours in the presence or absence of αGalCer (50 ng/ml) prior to addition of iNKT cells, rFasL, or nigericin. MDMs were cultured in the presence of iNKT cells or rFasL for 24 hours, and were stimulated with nigericin for 2 hours. The human iNKT cell line J24L.17 has been previously described (Brigl et al., 2006) and was used for the co-culture experiments. 5×104 MDMs and 2×105 human iNKT cells were used per condition for co-culture experiments. Antibody-mediated neutralization experiments were performed in the presence of a FasL-blocking antibody (clones MFL3 and NOK-1 for murine and human studies, respectively, both from Biolegend) or an isotype control (clones HTK888 and MOPC-21 for murine and human studies, respectively, both from Biolegend) at 10 ug/ml. All stimulations were performed in complete RPMI medium containing 10% FBS.
ELISAs and LDH release assays
At the indicated time points, cell-free supernatants were harvested by spinning the plates twice at 400 × g for 5 minutes, serially, while transferring to new plates. Quantification of secreted murine IL-1β and IFNγ was performed using ELISA kits from Thermo Fisher according to the manufacturer’s protocol. For LDH release assays, fresh cell-free supernatants were assayed at the indicated time points using the Pierce LDH cytotoxicity assay kit based on the manufacturer’s instructions. LDH release is depicted as percentage of maximum death (cells treated with 0.1% Triton X-100). The percentage of LDH was calculated as follows: LDH (% of Maximum): (sample LDH – background LDH) / (max LDH – background LDH in the presence of lysis buffer)
Flow Cytometric Analysis of FasL Expression
iNKT cells were labeled with 0.5 μM CellTrace Violet (Thermo Fisher) in HBSS for 30 minutes at 37°C. They were spun down at 300 × g for 10 minutes and any remaining CellTrace Violet was quenched by incubating the iNKT cells in complete RPMI for 5 minutes. iNKT cells were co-cultured with unlabeled BMDCs in 96-well plates at a ratio of 4:1 and the cells were harvested by gentle pipetting after the indicated time points. Cells were washed 2X in PBS and dead cell exclusion was performed by staining cells in PBS with Zombie Green live/dead dye (Biolegend, 1:500) for 30 minutes on ice. Following two washes in FACS buffer, cells were incubated with an Fc receptor-blocking antibody (16-0161-86, Thermo Fisher) for 15 minutes on ice. For assessment of surface FasL expression, cells were stained with a PE-conjugated antibody against FasL (MFL3, Thermo Fisher, 1:50) for 30 minutes at room temperature. During the last 15 minutes of the incubation, an antibody against PE (PE001, Biolegend, 1:25) was added to amplify the signal of the PE-conjugated anti-FasL antibody. Cells were washed twice in FACS buffer and were immediately analyzed by flow cytometry using an LSRFortessa (BD Biosciences). To assess the expression of intracellular (or whole cell) FasL, iNKT cells were stained with APC-conjugated PBS-57 CD1d tetramer, fixed, and permeabilized using the eBioscience intracellular fixation and permeabilization kit (Thermo Fisher) according to the manufacturer’s instructions, followed by staining with PE-conjugated anti-FasL (MFL3, Thermo Fisher, 1:50). The mean fluorescence intensity values reflecting FasL expression on iNKT cells were quantified using FlowJo software (BD Biosciences) by gating on the live, CellTrace Violet-positive cells (for cell surface FasL) or by gating on the live, PBS-57 CD1d tetramer positive cells (for intracellular or whole-cell staining).
Immunoblotting
For co-culture experiments, BMDCs were primed and co-cultured with iNKT cells in complete RPMI medium containing 10% FBS. Cell-free supernatants were transferred to 2 mL Eppendorf Safe-Lock tubes and extracellular IL-1β was immunoprecipitated by rotating the tubes overnight at 4°C in the presence of 0.5 mg of biotinylated polyclonal goat anti-mouse IL-1β (BAF-401, R&D) and 200 μg of Dynabeads MyOne Streptavidin T1 magnetic beads (Thermo Fisher). For experiments were BMDCs were stimulated with rFasL (and corresponding untreated and nigericin-treated controls), BMDCs were washed 3x with ice-cold PBS after priming, switched to serum-free Opti-MEM media, and the cell-free supernatants were precipitated with 10% Trichloroacetic acid (TCA, Sigma) after the indicated time points. Cell lysates were prepared by lysing cells in RIPA buffer supplemented with cOmplete protease inhibitor cocktail (Roche), and the concentrations of the lysates were quantified using the Pierce BCA protein assay kit (Thermo Fisher). Cell lysates, supernatants, and immunoprecipitated IL-1β were separated by SDS-PAGE on 12% Mini-PROTEAN TGX or Criterion TGX gels (Bio-Rad) and transferred onto 0.2 mm PVDF membranes using a Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blocked with 5% nonfat dry milk containing 0.1% Tween 20 in TBST for 1 hour at room temperature, and then incubated with primary antibodies overnight at 4C unless noted otherwise. The following were the primary antibodies used: Actin (Sigma, A5441, 1:10000), IL-1β (R&D, AF-401, 1:800), IL-1β (Genetex, GTX74034, 1:1000, used to probe immunoprecipitated IL-1β), caspase-1 (Adipogen, AG-20B-0042-C100, 1:1000), caspase-8 (Enzo, ALX-804-447-C100, 1:1000), cleaved caspase-8 (Cell Signaling, 8592, 1:1000), caspase-3 (Cell Signaling, 9662, 1:1000), cleaved caspase-3 (Cell Signaling, 9661, 1:1000), FLIP (Cell Signaling, 56343, 1:1000), caspase-6 (Cell Signaling, 9762, 1:1000), caspase-7 (Cell Signaling, 12827, 1:1000), caspase-9 (Cell Signaling, 9508, 1:1000), GSDMD (Abcam, ab209845, 1:1000), GSDME (Abcam, ab215191, 1:1000, incubation performed at room temperature for 1.5 hours). Membranes were incubated with the following horseradish peroxidase-conjugated secondary antibodies at a 1:10000 dilution for 1 hour at room temperature: goat anti-mouse IgG (115-035-146), donkey anti-rabbit IgG (711-035-152), bovine anti-goat IgG (805-035-180), and donkey anti-rat IgG (712-035-150), all from Jackson Immunoresearch. Membranes were developed using Clarity Western ECL (Bio-Rad) and imaged using a ChemiDoc Touch MP (Bio-Rad).
Flow Cytometric Quantification of Cell Death
To distinguish early apoptosis from necrosis, BMDCs were stimulated on non-treated 96-well plates. After the indicated time points, the supernatants were removed and the cells were incubated in 5 mM EDTA in HBSS for 5 minutes at 37°C. BMDCs were harvested from the plate by gentle pipetting, transferred to 96-well V-bottom plates, and washed twice in FACS buffer (Fluorobrite DMEM supplemented with 5% FBS, 25 mM HEPES, and 2 mM EDTA). BMDCs were incubated with an Fc receptor-blocking antibody (16-0161-86, Thermo Fisher) for 15 minutes prior to staining with antibodies against CD11c (N418, Thermo Fisher, 1:300) and MHC-II (M5/114.15.2, Thermo Fisher, 1:400) for 30 minutes on ice. Cells were then washed twice, once in FACS buffer followed by another wash in annexin V binding buffer (Biolegend). BMDCs were then stained with annexin V-APC (Biolegend, 1:50) and Sytox Green (Thermo Fisher, 50 nM) in 100 μL for 5 minutes on ice after which they were diluted to 300 μL with annexin V binding buffer. CD11c+ BMDCs staining positive or negative for annexin V and Sytox Green were immediately analyzed immediately by flow cytometry using an LSRFortessa and the percentages of differently labeled cells were calculated using FlowJo software. For co-culture experiments, iNKT cells were labeled with 0.5 μM CellTrace Violet prior to co-culture with BMDCs, and the CellTrace Violet-negative, CD11c+ BMDCs were gated on for the flow cytometric analysis of cell death.
Live Imaging of Cell Death
Dead cells were depleted from BMDCs using a dead cell removal kit (Miltenyi Biotec) and CD11c+ BMDCs were subsequently enriched to 99% purity by incubating the live fraction with CD11c microbeads following the manufacturer’s instructions. CD11c+ BMDCs were plated on 96-well glass bottom plates (Greiner Bio-One) coated with Poly-D-Lysine (Sigma, 0.5 mg/ml in 0.1 M borate buffer) in complete Fluorobrite DMEM (Thermo Fisher) supplemented with 2.5 mM CaCl (Sigma). CD11c+ 2 BMDCs were primed for 6 or 24 hours prior to stimulation with nigericin or rFasL, respectively. To visualize phosphatidylserine exposure and membrane permeabilization by time-lapse microscopy, Annexin V-AF647 (Thermo Fisher) or Annexin V-AF488 (Thermo Fisher) were added into the media at 1:50 and YOYO-1 (Thermo Fisher) was added at 250 nM for the duration of the stimulation. To track the nuclear DNA over time, the live CD11c+ BMDCs were enriched as mentioned above and were labeled with 0.25 μM SiR-DNA (Cytoskeleton, Inc) for 6 hours prior to stimulation with nigericin or rFasL. Cells were washed three times with complete Fluorobrite DMEM media and YOYO-1 was added at 250 nM for the duration of the stimulation. Cells were then stimulated with nigericin or rFasL and were immediately visualized by time-lapse widefield microscopy. Images were acquired every 5 minutes for 4 – 6 hours.
Time-lapse widefield microscopy was performed using a Molecular Devices ImageXpress Micro Confocal that was maintained at 37C and 5% CO2 for the duration of the stimulation. Images were acquired using a 40X Plan Apo objective and analyzed using MetaXpress software. A Zeiss LSM 800 confocal microscope equipped with a 63X Plan Apo oil objective and an incubation chamber maintained at 37C and 5% CO2 was also used. Images were analyzed using Zen 2.3 software (Zeiss). Final image analysis was performed using ImageJ software (NIH).
TUNEL staining
BMDCs were plated on 96-well glass bottom, Poly-D-Lysine-coated plates (MatTek) coated with Poly-D-Lysine in complete RPMI media. BMDCs were primed for 6 and 24 hours prior to stimulation with nigericin and rFasL, respectively. Following 1 or 6 hour stimulation with nigericin or rFasL, respectively, the supernatant was removed and the cells were immediately fixed with 4% paraformaldehyde for 30 minutes at room temperature. The fixative was then removed, and the cells were permeabilized (0.25% Triton X-100 in PBS) for 20 minutes at room temperature. After two washes with deionized water, the cells were stained with a Click-iT Plus TUNEL Assay kit (Thermo Fisher) according to the manufacturer’s instructions. Following completion of TUNEL staining, the cells were stained with ActinGreen (Thermo Fisher) as suggested by the manufacturer. The cells were then stained with 5 mg/ml Hoechst 33342 (Thermo Fisher) for 15 minutes at room temperature. After two PBS washes, the cells were mounted with SlowFade Diamond Antifade Mountant (Thermo Fisher). The samples were imaged using a Zeiss LSM 800 confocal microscope equipped with a 20X Plan Apo oil objective. Images were analyzed using Zen 2.3 software. Final image analysis was performed using ImageJ software.
Cell Sorting
BMDCs differentiated from the bone marrow of five C57BL/6J mice were pooled and sorted on a BD FACS Aria (BD Biosciences). Briefly, BMDCs were harvested, pooled, incubated with Fc receptor-blocking antibody (16-0161-86, Thermo Fisher) for 15 minutes on ice, and the CD11c+ BMDCs were enriched using CD11c microbeads following the manufacturer’s instructions. To sort GM-DCs and GM-Macs, the CD11c-enriched BMDCs were stained with antibodies against CD11c (N418, Thermo Fisher, 1:300), CD11b (M1/70, Thermo Fisher, 1:300) MHC-II (M5/114.15.2, Thermo Fisher, 1:400), CD115 (AFS98, Biolegend, 1:100), MerTK (DS5MMER, Thermo Fisher, 1:100), and CD135 (A2F10, Biolegend, 1:50). GM-Macs were sorted as CD11c+CD11bhiMHCIIlo-intCD115+MerTK+ while GM-DCs were sorted as CD11c+CD11bintMHCIIhiCD115−CD135+ (see also Figure S1). Dead cells were excluded with PI.
Bacterial Infections
BMDCs were plated in 96-well plates at 5×104 cells per well and were incubated with log-phase B. fragilis (NCTC 9343) at multiplicities of infection (MOIs) of 10, 20, 40, and 80. Plates were spun at 300 × g for 5 minutes and incubated for 3 hours at 37°C to allow uptake of B. fragilis by BMDCs. The cells were then washed 2x with complete RPMI and were allowed to rest for 21 hours to complete 24 hours of exposure to priming stimulus provided by B. fragilis. For infections with C. trachomatis serovar D (UW-3/Cx; ATCC), BMDCs were plated as described above and infected by adding C. trachomatis at inclusion forming units (IFUs) of 1.5 and 3 and spinning the plates at 37°C for 1 hour. BMDCs were then incubated at 37°C for another 3 hours. Following 4 hours of infection, BMDCs were washed 3x with complete RPMI and were allowed to rest for 20 hours to complete 24 hours of exposure to priming stimulus provided by C. trachomatis. 24 hours after BMDCs were first exposed to B. fragilis or C. trachomatis, a monoclonal antibody that blocks FasL (MFL3, Biolegend, 20 μg/ml) or an isotype control (HTK888, Biolegend, 20 μg/ml) was added to BMDCs, followed by addition of iNKT cells at a 1:1 ratio. Plates were spun at 300 × g for 5 minutes and were incubated at 37°C 24 hours prior to analysis of the cell-free supernatants by ELISA.
ImmGen expression data
Expression data for iNKT and T cell populations is publicly available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE15907 or http://www.immgen.org.
To identify surface-expressed proteins, population mean expression data was first filtered to include genes within Gene Ontology (http://geneontology.org/) term “cell surface” GO0009986 with an expression cutoff of greater than 120 units (Cohen et al., 2013). Multiplot Studio was used to generate the expression-expression plot in Figure 2B (GenePattern, Broad Institute).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± standard deviation (SD) as indicated in the figure legends and were analyzed in GraphPad Prism 7. Statistical analyses were performed in R using Student’s unpaired t test as indicated in the legends. Significance is depicted with asterisks on graphs as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat polyclonal anti-IL-1β | R&D Systems | Cat#AF-401-NA; RRID:AB_416684 |
| Rabbit polyclonal anti-IL-1β | Genetex | Cat#GTX74034; RRID:AB_378141 |
| Mouse monoclonal anti-Caspase-1 (clone Casper-1) | Adipogen | Cat#AG-20B-0042-C100; RRID:AB_2755041 |
| Rat monoclonal anti-Caspase-8 (clone 1G12) | Enzo Life Sciences | Cat#ALX-804-447-C100; RRID:AB_2050952 |
| Rabbit monoclonal anti-cleaved Caspase-8 (clone D5B2) | Cell Signaling Technology | Cat#8592; RRID:AB_10891784 |
| Rabbit polyclonal anti-Caspase-3 | Cell Signaling Technology | Cat#9662; RRID:AB_331439 |
| Rabbit polyclonal anti-cleaved Caspase-3 | Cell Signaling Technology | Cat#9661; RRID:AB_2341188 |
| Rabbit monoclonal anti-FLIP (clone D5J1E) | Cell Signaling Technology | Cat#56343; RRID:AB_2799508 |
| Rabbit polyclonal anti-Caspase-6 | Cell Signaling Technology | Cat#9762; RRID:AB_10829240 |
| Rabbit monoclonal anti-Caspase-7 (clone D2Q3L) | Cell Signaling Technology | Cat#12827; RRID:AB_2687912 |
| Mouse monoclonal anti-Caspase-9 (clone C9) | Cell Signaling Technology | Cat#9508; RRID:AB_2068620 |
| Rabbit monoclonal anti-GSDMD (clone EPR19828) | Abeam | Cat#ab209845; RRID:AB_2783550 |
| Rabbit monoclonal anti-DFNA5/GSDME (clone EPR19859) | Abeam | Cat#ab215191; RRID:AB_2737000 |
| Mouse monoclonal anti-actin (clone AC-15) | Sigma-Aldrich | Cat#A5441; RRID:AB_476744 |
| Anti-mouse CD11c (clone N418) | Thermo Fisher | Cat#MCD11c05; RRID:AB_10373550 |
| Anti-mouse I-A/I-E (clone M5/114.15.2) | Thermo Fisher | Cat#46-5321-82; RRID:AB_1834439 |
| Anti-mouse CD11b (clone M1/70) | Thermo Fisher | Cat#11-0112-82; RRID:AB_464935 |
| Anti-mouse CD115 (clone AFS98) | Thermo Fisher | Cat#17-1152-82; RRID:AB_1210789 |
| Anti-mouse CD135 (clone A2F10) | Thermo Fisher | Cat#12-1351-82; RRID:AB_465859 |
| Anti-mouse MerTK (clone DS5MMER) | Thermo Fisher | Cat#25-5751-82; RRID:AB_2573466 |
| Anti-mouse TCR β chain (clone H57-597) | Biolegend | Cat#109229; RRID:AB_10933263 |
| Anti-mouse CD3 (clone 145-2C11) | Biolegend | Cat#100340; RRID:AB_11149115 |
| Anti-mouse FasL (clone MFL3) | Biolegend | Cat#106606; RRID:AB_313279 |
| Anti-phycoerythrin antibody (clone PE001) | Biolegend | Cat#408102; RRID:AB_2168924 |
| Anti-mouse FasL blocking antibody (clone MFL3) | Biolegend | Cat#106612; RRID:AB_2813954 |
| Armenian Hamster IgG isotype control antibody | Biolegend | Cat#400940; RRID:AB_11203529 |
| Anti-human FasL blocking antibody (clone NOK-1) | Biolegend | Cat#306415; RRID:AB_2810458 |
| Mouse IgG1, κ isotype control antibody (clone MOPC-21) | Biolegend | Cat#400165; RRID:AB_11150399 |
| Biotinylated polyclonal goat anti-mouse IL-1β | R&D Systems | Cat#BAF401; RRID:AB_356450 |
| Goat anti-mouse IgG (H+L) | Jackson ImmunoResearch | Cat#115-035-146; RRID:AB_2307392 |
| Donkey anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat#711-035-152; RRID:AB_10015282 |
| Donkey anti-Rat IgG (H+L) | Jackson ImmunoResearch | Cat#712-035-150; RRID:AB_2340638 |
| Bovine anti-Goat IgG (H+L) | Jackson ImmunoResearch | Cat#805-035-180; RRID:AB_2340874 |
| Rat anti-mouse CD16/CD32 (clone 93) | Thermo Fisher | Cat#16-0161-86; RRID:AB_468900 |
| Bacterial and Virus Strains | ||
| Bacteroidis fragilis (strain NCTC 9343) | Laboratory of Dr. Laurie Comstock | ATCC 25285 |
| Chlamydia trachomatis (strain D/UW-3/Cx) | Laboratory of Dr. Michael Starnbach | ATCC VR-85 |
| Biological Samples | ||
| Human peripheral blood mononuclear cells | Brigham and Women’s Hospital Kraft Family Blood Donor Center | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant, multimeric FasL (MegaFasL) | Adipogen | Cat#AG-40B-0130-C010 |
| Pam3CSK4 | Invivogen | Cat#tlrl-pms |
| Nigericin | Invivogen | Cat#tlrl-nig |
| MSU Crystals | Invivogen | Cat#tlrl-msu |
| Alpha Galactosylceramide | Dr. Gurdyal Besra | N/A |
| Recombinant murine GM-CSF | Peprotech | Cat#315-03 |
| Recombinant human GM-CSF | Peprotech | Cat#300-03 |
| Recombinant murine IL-2 | Peprotech | Cat#212-12 |
| Recombinant murine IL-7 | Peprotech | Cat#217-17 |
| Recombinant human IL-2 | Peprotech | Cat#200-02 |
| Mouse PBS-57 CD1d-APC tetramer | NIH Tetramer Core Facility | N/A |
| Collagenase, type 4 | Worthington Biochemical | Cat#LS004186 |
| DNase 1 | Sigma-Aldrich | Cat#DN25 |
| ACK lysing buffer | Thermo Fisher | Cat#A1049201 |
| SYTOX Green | Thermo Fisher | Cat#S34860 |
| YOYO-1 | Thermo Fisher | Cat#Y3601 |
| Annexin V Alexa Fluor 488 | Thermo Fisher | Cat#A13201 |
| Annexin V Alexa Fluor 647 | Thermo Fisher | Cat#A23204 |
| Annexin V APC | Biolegend | Cat#640941 |
| SiR-DNA | Cytoskeleton, Inc | Cat#CY-SC007 |
| FluoroBrite DMEM | Thermo Fisher | Cat#A1896702 |
| Opti-MEM | Thermo Fisher | Cat#11058021 |
| Poly-D-Lysine | Sigma-Aldrich | Cat#A-003-E |
| Glycine | Sigma-Aldrich | Cat#G7126 |
| CellTrace Violet | Thermo Fisher | Cat#C34557 |
| Zombie Green Fixable Viability Dye | Biolegend | Cat#423112 |
| Dynabeads MyOne Streptavidin T1 | Thermo Fisher | Cat#65601 |
| Trichloroacetic acid | Sigma-Aldrich | Cat#91228 |
| ActinGreen-488 | Thermo Fisher | Cat#R37110 |
| Hoechst 33342 | Thermo Fisher | Cat#H3570 |
| SlowFade Diamond Antifade Mountant | Thermo Fisher | Cat#S36963 |
| Brain Heart Infusion broth | Anaerobe Systems | Cat#AS-872 |
| Ficoll-Paque PLUS | GE Lifesciences | Cat#17144003 |
| Critical Commercial Assays | ||
| Mouse IL-1β ELISA kit | Thermo Fisher | Cat#88-7013-77; RRID:AB_2574944 |
| Mouse IFNγ ELISA kit | Thermo Fisher | Cat#88-7314-77; RRID:AB_2575068 |
| Human IL-1β ELISA kit | Thermo Fisher | Cat#88-7261-88; RRID:AB_2575054 |
| Pierce™ LDH cytotoxicity assay | Thermo Fisher | Cat#88954 |
| Anti-mouse CD11c microbeads, ultrapure | Miltenyi Biotec | Cat#130-108-338 |
| Mouse Pan T Cell Isolation Kit II | Miltenyi Biotec | Cat#130-095-130 |
| Anti-APC microbeads | Miltenyi Biotec | Cat#130-090-855 |
| Anti-human CD14 microbeads, ultrapure | Miltenyi Biotec | Cat#130-118-906 |
| Pierce BCA Protein Assay | Thermo Fisher | Cat#23227 |
| eBioscience Intracellular Fixation & Permeabilization Buffer Set | Thermo Fisher | Cat#88-8824-00 |
| Click-iT Plus TUNEL Assay, Alexa Fluor 647 dye | ThermoFisher | Cat#C10619 |
| Deposited Data | ||
| ImmGen Consortium | Heng et al., 2008 | https://www.immgen.org |
| Experimental Models: Cell Lines | ||
| Primary murine splenic iNKT cell line | This paper | N/A |
| Primary human iNKT cell clone J24L.17 | Brigl et al., 2006 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Fas−/−: B6.MRL-Faslpr/J | Jackson Laboratories | JAX stock #000482 |
| Mouse: Caspase1/11−/−: B6N.129S2-Casp1tm1Flv/J | Jackson Laboratories | JAX stock #016621 |
| Mouse: CD1d−/− | Exley et al., 2003 | N/A |
| Mouse: Caspase-8DA/DA | Philip et al., 2016 | N/A |
| Mouse: Gsdmd−/− | Kambara et al., 2018 | N/A |
| Mouse: Ripk3−/− | Newton et al., 2004 | N/A |
| Mouse: Mlkl−/− | Murphy et al., 2013 | N/A |
| Mouse: Ripk3−/−Casp8−/− | Kaiser et al., 2011 | N/A |
| Mouse: Gsdme−/−: C57BL/6N-Gsdmeem1Fsha/J | Jackson Laboratories | JAX stock #032411 |
| Mouse: Gsdmd−/−Gsdme−/− | Chen et al., 2019 | N/A |
| Mouse: Asc−/− | Ozören et al., 2006 | N/A |
| Mouse: WT: C57BL/6NJ (control for Gsdme−/− mice) | Jackson Laboratories | JAX stock #005304 |
| Mouse: WT: C57BL/6 | Jackson Laboratories | JAX stock #000664 |
| Mouse: Nlrp3−/−: B6.129S6-Nlrp3tm1Bhk/J | Jackson Laboratories | JAX stock #021302 |
| Software and Algorithms | ||
| FlowJo (v10.5.3) | BD Biosciences | https://www.flowjo.com/ |
| R (V3.3.2) | R Foundation for Statistical Computing | http://www.R-project.org |
| Fiji / ImageJ version 2.0.0 | Fiji contributors / ImageJ developers | https://imagej.net/Fiji |
| Zen (v2.3) | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| MetaXpress (v6.0) | Molecular Devices | https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metaxpress |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/ |
| Multiplot Studio v2 | GenePattern, Broad Institute | https://www.genepattern.org/ |
| Other | ||
| 96-well plate | No. 1.5 Coverslip | 5 mm Glass Diameter | Poly-D-Lysine Coated | MatTek Corporation | Cat#P96GC-1.5-5-F |
| Greiner Bio-One SensoPlate 96-Well Glass Bottom Microplates | Greiner Bio-One | Cat#655892 |
Highlights.
Infected APCs may fail to activate the inflammasome and release IL-1β
A second cell (e.g., iNKT cell) can employ FasL to rescue IL-1β production by APCs
Caspase-8 is the main driver of IL-1β release downstream of Fas ligation
Caspase-8 directly cleaves GSDMD, converting apoptosis to pyroptosis
ACKNOWLEDGMENTS
We thank Warren Alexander (Mlkl−/−), Hongbo Luo (Gsdmd−/−), Edward Mocarski (Ripk3−/− and Ripk3−/−Casp8−/−), Petr Broz (Gsdme−/− and Gsdmd−/−Gsdme−/−), Igor Brodsky (Caspase-8DA/DA), and Gabriel Nuñez (Asc−/−) for providing bone marrow from valuable mice. We thank Michael Starnbach and Eric Dumas for providing C. trachomatis serovar D (UW-3/Cx; ATCC) and Laurie Comstock for providing B. fragilis (NCTC 9343). This work was supported by NIH grants R21 AI44139, R01 AI44139, and R01 AI063428 (to M.B.B.); K08 AI102945 (to P.J.B.); institutional training grants T32 HL007627 and K08 AR075850 (to D.P.S.); and R01 HL124209 and funding from the American Asthma Foundation, Alex’s Lemonade Stand Foundation for Childhood Cancer, and the V Foundation for Cancer Research (to B.A.C.). We thank Jonathan C. Kagan for helpful discussions. The following reagent was obtained through the NIH Tetramer Core Facility: APC-conjugated CD1d tetramer loaded with PBS-57 lipid antigen.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.03.030.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, and Dueber EC (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 113, 7858–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer ML, Slack E, de Gottardi A, Lawson MA, Hapfelmeier S, Miele L, Grieco A, Van Vlierberghe H, Fahrner R, Patuto N, et al. (2014). The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci. Transl. Med 6, 237ra66. [DOI] [PubMed] [Google Scholar]
- Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, Vecchi A, Mantovani A, Levitz SM, and Romani L (2004). The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J. Immunol 172, 3059–3069. [DOI] [PubMed] [Google Scholar]
- Bendelac A, Savage PB, and Teyton L (2007). The biology of NKT cells. Annu. Rev. Immunol 25, 297–336. [DOI] [PubMed] [Google Scholar]
- Boatright KM, Deis C, Denault JB, Sutherlin DP, and Salvesen GS (2004). Activation of caspases-8 and −10 by FLIP(L). Biochem. J 382, 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossaller L, Chiang PI, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VA, Mocarski ES, Subramanian D, Green DR, Silverman N, et al. (2012). Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol 189, 5508–5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PJ, Tatituri RV, Brigl M, Kim EY, Tuli A, Sanderson JP, Gadola SD, Hsu FF, Besra GS, and Brenner MB (2011). Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat. Immunol 12, 1202–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PJ, Brigl M, and Brenner MB (2013). Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol 13, 101–117. [DOI] [PubMed] [Google Scholar]
- Brigl M, van den Elzen P, Chen X, Meyers JH, Wu D, Wong C-H, Reddington F, Illarianov PA, Besra GS, Brenner MB, and Gumperz JE (2006). Conserved and heterogeneous lipid antigen specificities of CD1d-restricted NKT cell receptors. J. Immunol 176, 3625–3634. [DOI] [PubMed] [Google Scholar]
- Brigl M, Tatituri RV, Watts GF, Bhowruth V, Leadbetter EA, Barton N, Cohen NR, Hsu FF, Besra GS, and Brenner MB (2011). Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J. Exp. Med 208, 1163–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, and Dixit VM (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol 16, 407–420. [DOI] [PubMed] [Google Scholar]
- Chen K, Shanmugam NK, Pazos MA, Hurley BP, and Cherayil BJ (2016). Commensal Bacteria-Induced Inflammasome Activation in Mouse and Human Macrophages Is Dependent on Potassium Efflux but Does Not Require Phagocytosis or Bacterial Viability. PLoS ONE 11, e0160937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P, and Broz P (2019). Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba A, Cohen N, Brigl M, Brennan PJ, Besra GS, and Brenner MB (2009). Rapid and reliable generation of invariant natural killer T-cell lines in vitro. Immunology 128, 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuenchor W, Jin T, Ravilious G, and Xiao TS (2014). Structures of pattern recognition receptors reveal molecular mechanisms of autoinhibition, ligand recognition and oligomerization. Curr. Opin. Immunol 26, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen NR, Brennan PJ, Shay T, Watts GF, Brigl M, Kang J, and Brenner MB; ImmGen Project Consortium (2013). Shared and distinct transcriptional programs underlie the hybrid nature of iNKT cells. Nat. Immunol 14, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA (2011). A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur. J. Immunol 41, 1203–1217. [DOI] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. [DOI] [PubMed] [Google Scholar]
- Dorhoi A, Nouailles G, Jörg S, Hagens K, Heinemann E, Pradl L, Oberbeck-Müller D, Duque-Correa MA, Reece ST, Ruland J, et al. (2012). Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is un-coupled from susceptibility to active tuberculosis. Eur. J. Immunol 42, 374–384. [DOI] [PubMed] [Google Scholar]
- Erlich Z, Shlomovitz I, Edry-Botzer L, Cohen H, Frank D, Wang H, Lew AM, Lawlor KE, Zhan Y, Vince JE, and Gerlic M (2019). Macrophages, rather than DCs, are responsible for inflammasome activity in the GM-CSF BMDC model. Nat. Immunol 20, 397–406. [DOI] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, and Kagan JC (2018). The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley MA, Bigley NJ, Cheng O, Shaulov A, Tahir SM, Carter QL, Garcia J, Wang C, Patten K, Stills HF, et al. (2003). Innate immune response to encephalomyocarditis virus infection mediated by CD1d. Immunology 110, 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faouzi S, Burckhardt BE, Hanson JC, Campe CB, Schrum LW, Rippe RA, and Maher JJ (2001). Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-kappa B-independent, caspase-3-dependent pathway. J. Biol. Chem 276, 49077–49082. [DOI] [PubMed] [Google Scholar]
- Felley LE, Sharma A, Theisen E, Romero-Masters JC, Sauer JD, and Gumperz JE (2016). Human Invariant NKT Cells Induce IL-1β Secretion by Peripheral Blood Monocytes via a P2X7-Independent Pathway. J. Immunol 197, 2455–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltham R, Vince JE, and Lawlor KE (2017). Caspase-8: not so silently deadly. Clin. Transl. Immunology 6, e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Chan FK, and Kroemer G (2017). Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol 12, 103–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. (2018). Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, and Llambi F (2015). Cell Death Signaling. Cold Spring Harb. Perspect. Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Arcelus M, Teslovich N, Mola AR, Polidoro RB, Nathan A, Kim H, Hannes S, Slowikowski K, Watts GFM, Korsunsky I, et al. (2019). Lymphocyte innateness defined by transcriptional states reflects a balance between proliferation and effector functions. Nat. Commun 10, 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, and Miao EA (2013). Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, and Broz P (2018). The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol 48, 584–592. [DOI] [PubMed] [Google Scholar]
- Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, and Reis e Sousa C (2015). GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c(+)MHCII(+) Macrophages and Dendritic Cells. Immunity 42, 1197–1211. [DOI] [PubMed] [Google Scholar]
- Heng TS, and Painter MW; Immunological Genome Project Consortium (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol 9, 1091–1094. [DOI] [PubMed] [Google Scholar]
- Higa N, Toma C, Nohara T, Nakasone N, Takaesu G, and Suzuki T (2013). Lose the battle to win the war: bacterial strategies for evading host inflammasome activation. Trends Microbiol 21, 342–349. [DOI] [PubMed] [Google Scholar]
- Holler N, Tardivel A, Kovacsovics-Bankowski M, Hertig S, Gaide O, Martinon F, Tinel A, Deperthes D, Calderara S, Schulthess T, et al. (2003). Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol. Cell. Biol 23, 1428–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juffermans NP, Florquin S, Camoglio L, Verbon A, Kolk AH, Speelman P, van Deventer SJ, and van Der Poll T (2000). Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J. Infect. Dis 182, 902–908. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, and Mocarski ES (2011). RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, Zhao L, Zhou S, Yu H, Zhou W, et al. (2018). Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep 22, 2924–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar M, Sun Y, Hise AG, Rietsch A, and Pearlman E (2012). Cutting edge: IL-1β processing during Pseudomonas aeruginosa infection is mediated by neutrophil serine proteases and is independent of NLRC4 and caspase-1. J. Immunol 189, 4231–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Kim EY, Lynch L, Brennan PJ, Cohen NR, and Brenner MB (2015). The transcriptional programs of iNKT cells. Semin. Immunol 27, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlgruber AC, Donado CA, LaMarche NM, Brenner MB, and Brennan PJ (2016). Activation strategies for invariant natural killer T cells. Immunogenetics 68, 649–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs SB, and Miao EA (2017). Gasdermins: Effectors of Pyroptosis. Trends Cell Biol 27, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z, Lommatzsch SE, Huang MT, Ting JP, and Koller BH (2012). NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J. Immunol 189, 2006–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, and Flavell RA (1995). Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 267, 2000–2003. [DOI] [PubMed] [Google Scholar]
- Latz E, Xiao TS, and Stutz A (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Shen C, and Brunham RC (2000). Chlamydia trachomatis infection of epithelial cells induces the activation of caspase-1 and release of mature IL-18. J. Immunol 165, 1463–1469. [DOI] [PubMed] [Google Scholar]
- Maltez VI, and Miao EA (2016). Reassessing the Evolutionary Importance of Inflammasomes. J. Immunol 196, 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, and Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. [DOI] [PubMed] [Google Scholar]
- McElvania Tekippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M, Braunstein M, and Ting JP (2010). Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS ONE 5, e12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Rajan JV, and Aderem A (2011). Caspase-1-induced pyroptotic cell death. Immunol. Rev 243, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, Thome M, Schneider P, Holler N, Tschopp J, Nicholson DW, Briand C, and Grütter MG (2002). The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem 277, 45162–45171. [DOI] [PubMed] [Google Scholar]
- Miwa K, Asano M, Horai R, Iwakura Y, Nagata S, and Suda T (1998). Caspase 1-independent IL-1βeta release and inflammation induced by the apoptosis inducer Fas ligand. Nat. Med 4, 1287–1292. [DOI] [PubMed] [Google Scholar]
- Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, and Núñez G (2013). K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. [DOI] [PubMed] [Google Scholar]
- Nagata S (2018). Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol 36, 489–517. [DOI] [PubMed] [Google Scholar]
- Newton K, Sun X, and Dixit VM (2004). Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol 24, 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, and Green DR (2011). Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al. (2018). Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozören N, Masumoto J, Franchi L, Kanneganti T-D, Body-Malapel M, Ertürk I, Jagirdar R, Zhu L, Inohara N, Bertin J, et al. (2006). Distinct roles of TLR2 and the adaptor ASC in IL-1β/IL-18 secretion in response to Listeria monocytogenes. J. Immunol 176, 4337–4342. [DOI] [PubMed] [Google Scholar]
- Philip NH, DeLaney A, Peterson LW, Santos-Marrero M, Grier JT, Sun Y, Wynosky-Dolfi MA, Zwack EE, Hu B, Olsen TM, et al. (2016). Activity of Uncleaved Caspase-8 Controls Anti-bacterial Immune Defense and TLR-Induced Cytokine Production Independent of Cell Death. PLoS Pathog 12, e1005910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, and Salvesen GS (2011). FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem. J 433, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, and Alnemri ES (2017). Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun 8, 14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salio M, Speak AO, Shepherd D, Polzella P, Illarionov PA, Veerapen N, Besra GS, Platt FM, and Cerundolo V (2007). Modulation of human natural killer T cell ligands on TLR-mediated antigen-presenting cell activation. Proc. Natl. Acad. Sci. USA 104, 20490–20495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, and Poltorak A (2018). Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 115, E10888–E10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P, and Hiller S (2016). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35, 1766–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, and Tschopp J (2010). The inflammasomes. Cell 140, 821–832. [DOI] [PubMed] [Google Scholar]
- Scott-Browne JP, Matsuda JL, Mallevaey T, White J, Borg NA, McCluskey J, Rossjohn J, Kappler J, Marrack P, and Gapin L (2007). Germline-encoded recognition of diverse glycolipids by natural killer T cells. Nat. Immunol 8, 1105–1113. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Shi J, Gao W, and Shao F (2017). Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci 42, 245–254. [DOI] [PubMed] [Google Scholar]
- Shin S, and Brodsky IE (2015). The inflammasome: Learning from bacterial evasion strategies. Semin. Immunol 27, 102–110. [DOI] [PubMed] [Google Scholar]
- Silke J, Rickard JA, and Gerlic M (2015). The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol 16, 689–697. [DOI] [PubMed] [Google Scholar]
- Stewart MK, and Cookson BT (2016). Evasion and interference: intracellular pathogens modulate caspase-dependent inflammatory responses. Nat. Rev. Microbiol 14, 346–359. [DOI] [PubMed] [Google Scholar]
- Strasser A, Jost PJ, and Nagata S (2009). The many roles of FAS receptor signaling in the immune system. Immunity 30, 180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson KV, Deng M, and Ting JP (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol 19, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taabazuing CY, Okondo MC, and Bachovchin DA (2017). Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol 24, 507–514.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummers B, and Green DR (2017). Caspase-8: regulating life and death. Immunol. Rev 277, 76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama R, Yonehara S, and Tsutsui H (2013). Fas-mediated inflammatory response in Listeria monocytogenes infection. J. Immunol 190, 4245–4254. [DOI] [PubMed] [Google Scholar]
- Ulland TK, Ferguson PJ, and Sutterwala FS (2015). Evasion of inflammasome activation by microbial pathogens. J. Clin. Invest 125, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, and Shao F (2017). Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.