

Abstract
KCC2 is a vital neuronal K+/Cl− co-transporter that is implicated in the etiology of numerous neurological diseases. In normal cells, KCC2 undergoes developmental dephosphorylation at Thr906 and Thr1007. We engineered mice with heterozygous phospho-mimetic mutations T906E and T1007E (KCC2E/+) to prevent the normal developmental dephosphorylation of these sites. Immature (postnatal day 15) but not juvenile (postnatal day 30) KCC2E/+ mice exhibited altered GABAergic inhibition, an increased glutamate/GABA synaptic ratio, and greater susceptibility to seizure. KCC2E/+ mice also had abnormal ultra-sonic vocalizations at postnatal days 10 to 12 and impaired social behavior at postnatal day 60. Postnatal bumetanide treatment restored network activity by postnatal day 15 but failed to restore social behavior even by postnatal day 60. Our data indicate that posttranslational KCC2 regulation controls the GABAergic developmental sequence in vivo, indicating that deregulation of KCC2 could be a risk factor for the emergence of neurological pathology.
INTRODUCTION
Neurodevelopmental disorders (NDDs), including specific epilepsy subtypes, schizophrenia, intellectual disability, and autism spectrum disorders (ASDs), exhibit a shared spectrum of pathological changes in neuronal morphology, synapse function, and network properties (1–3). A fundamental obstacle to developing novel therapies for NDDs is our limited knowledge of disease pathogenesis. Impaired function of the neuronal potassium/chloride extruder KCC2 has been implicated in the pathogenesis of multiple NDDs (4).
KCC2 (SLC12A5) is essential for establishing and maintaining the low intracellular chloride concentration ([Cl−]i) of mature mammalian neurons. The link between KCC2 and NDDs has been highlighted by several studies showing a decreased functional expression of KCC2 in human patients with epilepsy (5), schizophrenia (6, 7), and Rett syndrome (8), as well as in mouse models of Fragile X (9) and Rett syndromes (10, 11). However, due to the posttranslational regulation of KCC2, there is no simple relationship between the abundance of KCC2 protein and its activity as chloride extruder (12). Phosphorylation of KCC2 at specific residues strongly affects the ion transport activity of the transporter (13–18).
Among multiple KCC2 phosphorylation sites (13, 16–20), the dual (de)phosphorylation of threonine 906 and 1007 (Thr906/Thr1007) is a particularly potent regulator of KCC2 activity (13–15, 21–23). Thr906/Thr1007 becomes progressively dephosphorylated during neuronal development (14, 21). The phospho-mimetic threonine-to-glutamate (T906E/T1007E) mutations of KCC2 result in strong inhibition of the ion-transport activity in different cell lines and cultured neurons (15, 21, 24). Unlike wild-type KCC2, whose in vitro overexpression in immature neurons reduces [Cl−]i and produces hyperpolarizing shift of gamma-Aminobutyric acid (GABA) receptor type A (GABAAR) responses, the overexpression of KCC2 T906E/T1007E results in an increase in [Cl−]i, and a depolarizing shift of GABAAR responses (14, 15).
The importance of precise posttranslational KCC2 regulation has been corroborated by the identification of mutations that impair these processes in human patients with NDDs, including epilepsy (25–27). However, the pathophysiological consequences and mechanisms of impaired K-Cl co-transporters phosphorylation has only begun to be explored in vivo (23, 28, 29).
Here, we have generated a KCC2 transgenic mouse carrying the phospho-mimetic mutations at KCC2 Thr906 and Thr1007, which mimics constitutive phosphorylation and prevents normal developmental dephosphorylation at these sites (14, 15, 21). KCC2E/E homozygous mice die 4 to 12 hours after birth (30) , highlighting that precise phospho-regulation of these sites is essential for postnatal survival. In contrast, heterozygous (KCC2E/+) mice are viable and fertile. By studying these mice, we have found that heterozygous constitutive KCC2 Thr906/Thr1007 phosphorylation does not produce detectable changes in the overall abundance of KCC2, but significantly impairs the inhibitory strength of GABAergic neurotransmission and network properties during the first post-natal weeks of life. Moreover, KCC2E/+ mice exhibit increased seizure susceptibility and altered ultra-sonic vocalization at early ages [ponstnatal (P) days P2 to P15] and social interaction deficits at adult ages (2 months). Postnatal treatment with the NKCC1 blocker bumetanide in vivo normalized the increased seizure susceptibility and aberrant network properties of KCC2E/+ mice at early ages but failed to restore social behavior at adult ages. Together, these results provide the first in vivo evidence of the importance of KCC2 post-translational modification for normal CNS development, and implicate dysregulated KCC2 Thr906/Thr1007 phosphorylation as a potential mechanism in the pathogenesis of NDDs.
RESULTS
Heterozygous KCC2E/+ mice do not exhibit any obvious mutant phenotype.
To determine the in vivo significance of phosphorylation at the WNK/SPAK-kinase dependent KCC2 Thr906/Thr1007 phosphorylation motif (14, 15, 21), we generated mice harboring phospho-mimetic glutamic acid substitutions at these sites (T906E/T1007E) via homologous recombination (Fig. 1A). Homozygous KCC2E/E mice died within the first 4–12 hours after birth from apparent respiratory distress, but heterozygous KCC2E/+ mice were viable, fertile and survived through adulthood. Compared to KCC2+/+ littermates, the KCC2E/+ mice showed no difference in the time of eye opening or in weight gain from P0 to P15 (Fig. 1, B and C). Immunoprecipitation of KCC2 from hippocampal lysates using antibodies that recognize the native phosphorylated, but not mutated (T906E and T1007E) forms of KCC2 (31), showed a significant decrease in KCC2E/+ mice as compared to KCC2+/+ littermates (Fig. 1, D to F, and table S1). The abundance of total KCC2 in hippocampal lysates from KCC2+/+ mice increased progressively from P5 to P15 (Fig. 1, D and G, and table S1), in agreement with previous reports (32, 33). A similar developmental up-regulation of KCC2 abundance was observed in hippocampi from KCC2E/+ mice (Fig. 1, D and G, and table S1). No difference in total KCC2 abundance was seen between KCC2+/+ and KCC2E/+ mice at all studied time points (Fig. 1, D and G, and table S1), or KCC2 Ser940 phosphorylation (fig. S1, A and B, and table S1). In addition, no difference in the total abundance of NKCC1 or the NKCC1/KCC2 regulatory kinases WNK1, WNK3, SPAK, or OSR1 were noted in KCC2+/+ and KCC2E/+ mice (fig. S1, C and D, and table S1).
Figure 1. Characterization of KCC2E/+ phospho-mimetic mice.
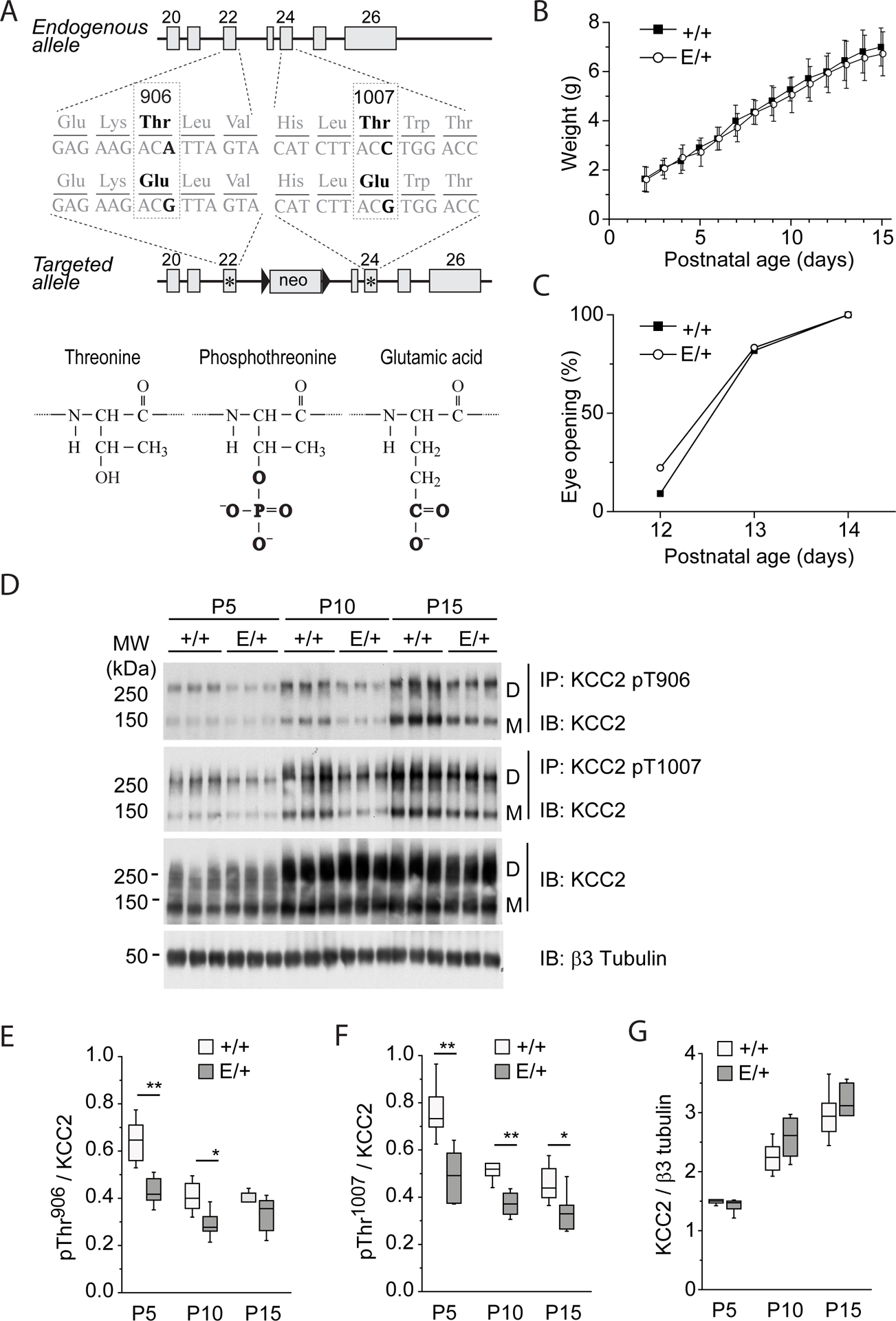
(A) Mutagenesis scheme. Thr906 and Thr1007 are located in exons 22 and 24, respectively, and are preserved in endogenous allele. T906E and T1007E mutations are located in the second targeted allele that contains a Neomycin selection cassette excised in vivo by Cre recombinase. (B) Weight gain by KCC2E/+ and KCC2+/+pups. The plot represents mean±SD of values obtained from 11 KCC2+/+ pups [4 litters] and 18 KCC2E/+ pups [4 litters]. Two-way ANOVA analysis revealed no statistically significant difference between KCC2E/+ and KCC2+/+pups (P=0.18, F=1.84) and strong difference of weight during development (P=1.1E-8, F=205.39). (C) Age dependence of eye opening, an external sign of normal CNS maturation in vertebrates (69), in KCC2E/+ and KCC2+/+pups. The plot represents percentage of animals with open eyes (N=11 KCC2+/+ pups [4 litters] and 18 KCC2E/+ pups [4 litters]). The Chi-square analysis revealed no statistically significant difference between KCC2E/+ and KCC2+/+pups (P=0.83). (D) Abundance of KCC2 in KCC2E/+ and KCC2+/+pups. Hippocampal lysates from KCC2E/+ and KCC2+/+ at the indicated time pointspoints were subjected to immuno-precipitation (IP) with the indicated phosphorylation site-specific antibodies recognizing pThr906- or pThr1007- KCC2, and the immuno-precipitated monomer (“M”) and dimer (“D”) products were detected with the pan-KCC2 antibody (IB). The same antibody was used for detection of total KCC2 protein abundance in the same input material. An antibody recognizing neuron specific β3 tubulin was employed to normalize total protein amounts for sample loading. Each condition illustrates migration of extracts from 3 mice. (E to G) Developmental abundance of immunoprecipitated phosphorylated forms of Thr906 (E), Thr1007 (F) and total KCC2 (G). Data are from N=6 mice per condition from 4 different litters. * P <0.05 and ** P <0.01 indicate statistical difference between KCC2+/+ and KCC2E/+ littermates for pairwais groups, Mann-Whitney test. The Kruskal–Wallis test applied for analysis of P5 to P15 samples in G revealed significant age-dependent increase of total KCC2 both in KCC2+/+ (P=6.3E-4) and KCC2E/+ (P=7.6E-4) littermates. For details on statistical analyses, see table S1.
KCC2E/+ CA3 pyramidal neurons exhibit a delay in the GABAergic developmental sequence.
Phospho-mimetic KCC2 T906E/T1007E mutation impairs Cl− extrusion capacity in different transient overexpression models (14, 15). To test whether constitutive genomic T906E/T1007E mutations also alter neuronal Cl− homeostasis, we measured the driving force of GABAA channels (DFGABA) in CA3 pyramidal neurons of acute hippocampal slices from KCC2+/+ and KCC2E/+ mice using non-invasive cell-attached single-GABAA channel recordings (9, 34). In agreement with previous reports (9, 35), in slices prepared from P7–9 KCC2+/+ mice, the DFGABA was positive in most recorded neurons (Fig. 2B and table S2). Onwards from P12–14 in KCC2+/+ mice, DFGABA shifted towards less depolarizing and even hyperpolarizing values (Fig. 2, A and B, and table S2). In contrast, in CA3 pyramidal neurons from KCC2E/+ mice, DFGABA remained depolarized until P17–20 (Fig. 2, A and B, and table S2). Notably, the hyperpolarizing shift in KCC2E/+ mice occurred at P30 (Fig. 2B). There was, however, no significant difference in the resting membrane potential of KCC2E/+ and KCC2+/+ CA3 pyramidal neurons at P12–14 and P17–20 (Fig. 2C, fig. S2, and table S2). These data show that the developmental hyperpolarizing GABA shift is delayed in the CA3 pyramidal neurons of KCC2E/+ mice.
Figure 2. Delayed depolarizing shift of GABA transmission in KCC2E/+ CA3 neurons.
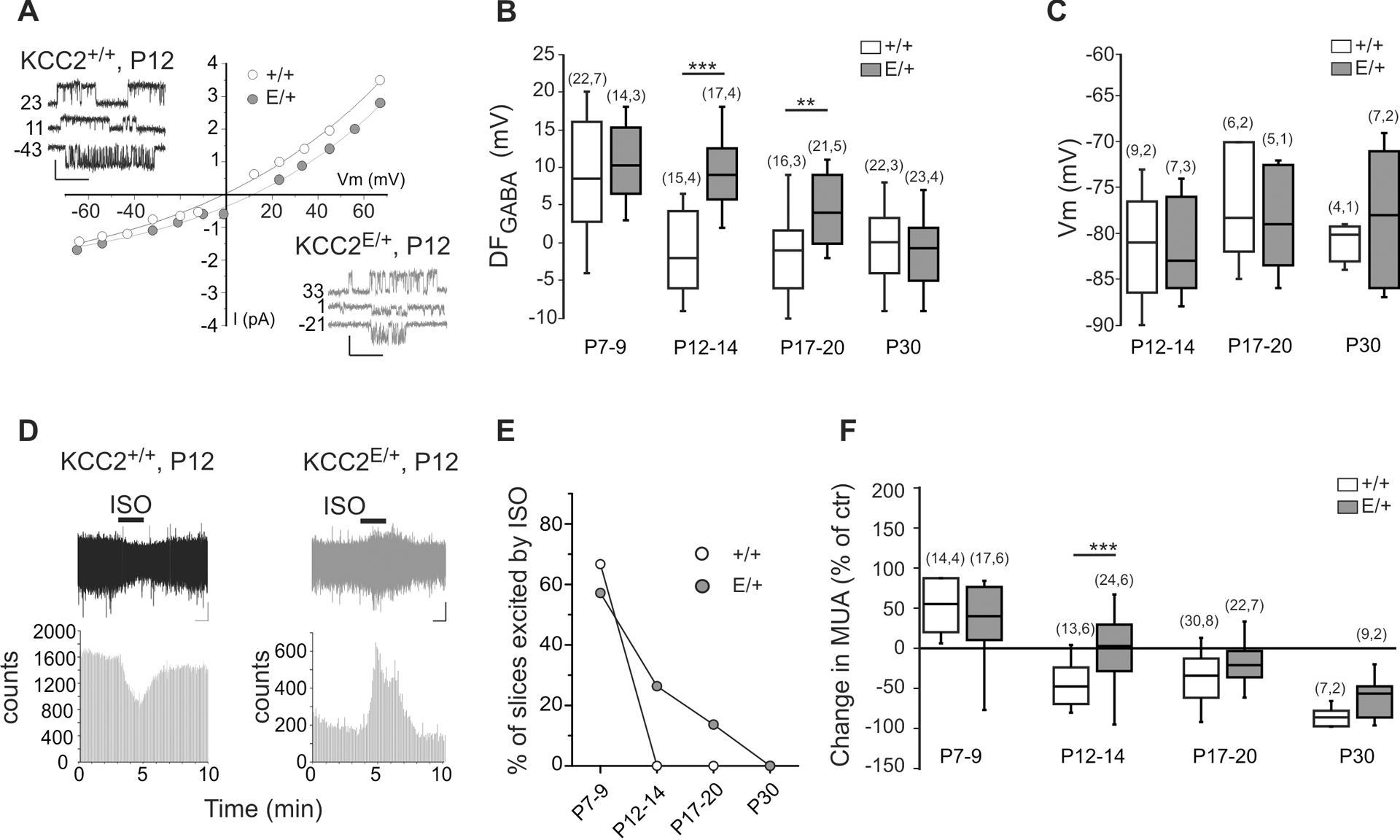
Representative example of I-V curves and traces at different membrane potentials of single GABAA channel currents recorded from CA3 neurons at P12. DFGABA was determined as the intercept of the I-V curve with the x-axis. Scale values: 1 s, 1 pA. B) Boxplots of DFGABA at different age frames. **P<0.01; ***P<0.001, Mann-Whitney test. Numbers in parenthesis indicate the number of recorded neurons (first value) and number of animals (value after comma). C) Boxplots of resting membrane potential (Vm) in CA3 neurons of indicated age frames analyzed using single NMDA-channel recordings, shown in fig. S2. Mann-Whitney test revealed no difference between KCC2+/+ and KCC2E/+ mice (N indication same as in (B)). For details, see table S2. D) Traces illustrate the representative field potential recordings from CA3 neurons of inhibitory (left plot) and excitatory (right plot) actions of isoguvacine applied as indicated with horizontal bar. Scale values: 1 min, 20 μV. The histograms show the quantification of spike frequencies in illustrated traces. E) Percentage of slices showing an increase of MUA in response to isoguvacine. Chi-square test was used to compare KCC2E/+ and KCC2E/+ mice (P=1.0E-5). F) Boxplot of the relative change of isoguvacine-dependent MUA frequency at different age frames. ***P<0.001, Mann-Whitney test, numbers in parenthesis indicate the number of recorded slices (first value) and number of animals (value after comma). Data in (E and F) were obtained from the same set of slices. For statistical analyses details, see table S2.
We next examined the emergence of functional GABAergic inhibition in the developing KCC2+/+ and KCC2E/+ hippocampus. We performed non-invasive extracellular recordings of MUA (36, 37) in acute hippocampal slices from KCC2+/+ and KCC2E/+ littermates, and investigated the effect of bath application of the GABAAR agonist isoguvacine (10μM) on the firing of the CA3 pyramidal neurons from P6 to P30. Consistent with previous results (36), in wild-type KCC2+/+ mice, isoguvacine induced an increase of firing of CA3 pyramidal neurons in 67% of P7–9 slices (Fig. 2E). Starting from P12 no firing increase was observed in response to isoguvacine (Fig. 2, D and E). In KCC2E/+ mice, the isoguvacine-induced increase of firing persisted until P17-P20 (Fig. 2, D and E). Averaging the overall effect of isoguvacine on the firing of CA3 pyramidal neurons confirmed the delayed hyperpolarizing shift of GABA action in KCC2E/+ mice (Fig. 2F and table S2).
Altered glutamate/GABA balance in juvenile KCC2E/+ CA3 pyramidal neurons.
A loss of equilibrium in glutamatergic and GABAergic synaptic drive has been observed in several NDDs associated with a delayed developmental GABA shift (9, 10). We therefore performed whole cell recordings of CA3 pyramidal neurons in acute hippocampal slices at P15–20 and P30 to measure spontaneous GABAAR-mediated and glutamate-mediated postsynaptic currents (sGABA-PSCs and sGlut-PSCs) with a low Cl− pipette solution (Fig. 3, A to F, and table S3). At P15–20, we found that the frequency of sGlut-PSCs (Fig. 3B), but not their amplitude (Fig. 3E), was increased in KCC2E/+ CA3 pyramidal neurons compared to KCC2+/+ littermates. In contrast, the frequencies of sGABA-PSCs were decreased in KCC2E/+ CA3 pyramidal neurons (Fig. 3C), with no change in amplitude (Fig. 3F). At P30, whole cell recordings revealed no significant difference in the frequency or amplitude of sGlut-PSCs and sGABA-PSCs (Fig. 3, B to F). Calculation of the ratio of sGlut-PSCs and sGABA-PSCs frequencies (hereafter referred to as excitatory-inhibitory (E/I) balance) recorded from the same CA3 pyramidal neurons at P15–20 showed a significant increase in KCC2E/+ neurons compared to KCC2+/+ neurons (Fig. 3D). There was, however, no difference in the E/I balance at P30–35 (Fig. 3D and table S3). These data show that the E/I balance is impaired in CA3 neurons of P15-P20 KCC2E/+ mice, coinciding with the depolarizing DFGABA.
Figure 3. The KCC2E/+ P15 but not P30 mice show an altered glutamate/GABA balance.

A) Representative whole cell recordings of brain slices spontaneous post-synaptic currents (holding potential =−45mV) in 2 week-old KCC2+/+ and KCC2E/+ CA3 pyramidal neurons. Scale values: 500 ms, 50 pA. B to F) Boxplots of frequencies (B,C) and amplitudes (E,F) of the spontaneous glutamatergic (B and E) and GABAergic (C and F) post-synaptic currents, and their ratios (D), recorded from 2 and 4 week-old KCC2+/+ and KCC2E/+ CA3 pyramidal neurons. Numbers in parenthesis indicate the number of cells recorded and mice used. *P<0.05, ***P<0.001, Mann-Whitney test (see table S3 for details on statistical analyses).
Increased seizure susceptibility in juvenile KCC2E/+ mice.
Genetic studies have shown an association between KCC2 dysfunction and different types of human epilepsy syndromes (5, 38). To determine whether impaired KCC2 Thr906/Thr1007 phosphorylation impacts seizure susceptibility, pairs of immature and juvenile (P15 and P30) KCC2+/+ or KCC2E/+ mice were placed in a hermetic chamber containing two compartments separated using holey wall (hereafter termed compartments a and b), thereby ensuring simultaneous exposure of both animal groups to the convulsant agent flurothyl (2,2,2-trifluoroethyl ether) (see the associated Methods for further details). Flurothyl induced severe tonic-clonic seizures with a loss of posture and jumping in both KCC2+/+ and KCC2E/+ mice (stage 4 in Fig. 4A). As a control, when both compartments included P15 wild-type KCC2+/+ animals, the latencies of seizure appearance in both animals were similar (Fig. 4B). When compartment a included P15 wild-type KCC2+/+ mice and compartment b contained their heterozygous KCC2E/+ littermates, the population of seizure latency values of heterozygous KCC2E/+ mice was significantly shorter as compared to their wild-type KCC2+/+ littermates (Fig. 4B, fig. S3, and table S4). In contrast, 30 day-old KCC2+/+ and KCC2E/+ mice had no difference in seizure latency (Fig. 4C and table S4). These results show KCC2E/+ mice have increased seizure susceptibility at P15, coinciding with their depolarizing DFGABA and increased neuronal network activity.
Figure 4. KCC2E/+ P15 but not P30 mice show an increased onset of flurothyl-induced seizures.
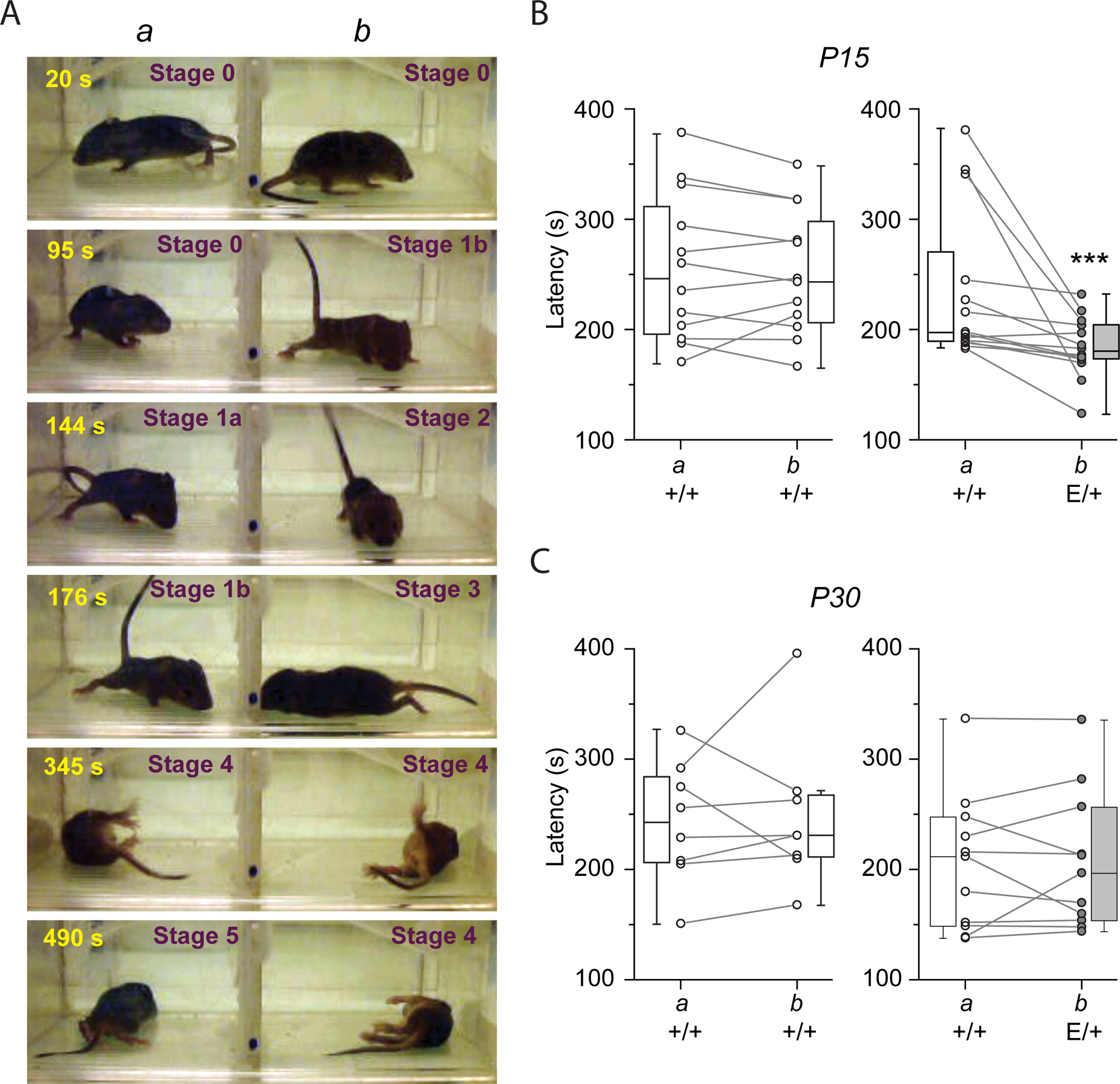
A) Photos illustrating different stages of the responses in P15 mice to flurothyl. The number in the upper left corner of each image indicates the time elapsed after beginning of flurothyl injection. Because the latencies of the effects of flurothyl depend on large number of parameters (see Methods) two mice of same or different phenotypes were placed in compartments “a” and “b” of transparent, ventilated but hermetically-sealed cage in which flurothyl was injected. In the illustrated experiment, compartments a and b housed KCC2+/+ and KCC2E/+ mice, respectively. The first behavioral response to flurothyl was animal immobility (Stage 0) followed by rigid posture (Stage 1a) and tail extension (Stage 1b). The stages 0 and 1 were observed in 100% of P15 mice. After Stage 1a, ~50% of animals exhibited brief (1–10s) period of tranquility with one-two irregular low-amplitude body jiggles (Stage 2) and/or 1st short seizure (1–10s) episodes (Stage 3) followed by severe long-lasting (10–30 s) generalized (tonic-clonic) seizures (Stages 4). After tonic-clonic seziures, mice returned to four-limb posture and remain immobile for 10–30 min (Stage 5). N=at least 8 pairs of mice per group. B, C) Latency of onset of tonic-clonic seizures of P15 (B) and P30 (C) KCC2+/+ and KCC2E/+ mice placed in compartments a and b as indicated. Connected points indicate pairs of animals. ***P<0.001, Wilcoxon Paired test, N=12 pairs for left plot (B), 14 pairs for right plot (B), 8 pairs for left plot (C), and 11 pairs for right plot (C) (see fig. S3 and table S3 for more details on statistical analysis).
Altered vocalization and social interactions in KCC2E/+ mice.
An increase in seizure susceptibility is a co-morbidity frequently associated with NDDs (39). In addition, alterations in DFGABA and deficiency in synaptic activity were reported in different animal models of NDD (9, 10, 40). We therefore assessed several neuro-behavioral tests in KCC2+/+ and KCC2E/+ mice, including ultrasonic vocalization (USVs) (alteration of communication) (41), open field (anxiety and hyperactivity) (42), three-chamber test (sociability) (43), and grooming splash tests (depression) (44).
USVs test were assessed in P2 to P12 mice. In KCC2+/+ mice, the number of calls varied at different postnatal days and showed a typical ontogenetic profile (45) (Fig. 5A). KCC2E/+pups compared to KCC2+/+ animals showed a statistically significant different ontogenetic profile (Fig. 5A and table S5). In P2 to P8 KCC2E/+ mice, the number of ultrasonic calls was similar to KCC2+/+ animals but at P10 and P12 the number of calls was higher (Fig. 5A).
Figure 5. KCC2 T906E/T1007E impairs newborn communication and adult social-behavior.

A) Number of calls in responses to separation from mother during 3 min sessions at P2-P12. Data are mean ± SD. Two-way ANOVA test revealed difference between KCC2+/+ and KCC2E/+ mice (P=0.003) and during development (P=1.8E-6). **P<0.01 between KCC2+/+ and KCC2E/+, post-hoc Tukey test, table S5. Numbers in table indicate the number of studied pups (first value) and litters (value after comma). B-C) Time spent by KCC2+/+ (B) and KCC2E/+ (C) mice in the stranger chamber, empty chamber, and inbetween (middle). Upper panels illustrate heat maps of mice movements in the sociability tests. White circles show location of plastic cup-like cage containing stranger (s) or empty (e). Collumns illustrate the mean ± SD of the time spent by tested mice in the indicated compartments. ns= non-significant (P=0.1), ***P<0.001, one-way ANOVA test (N = 15 for +/+ and N = 14 for E/+), table S5.
The three-chamber test that was performed on P60 mice using widely explored paradigm (46) revealed difference in social behavior of KCC2+/+ and KCC2E/+ mice; while the wild type KCC2+/+ mice spent ~35% more time to exploring the compartment with the stranger than the empty compartment (Fig. 5B), the KCC2E/+ mice showed no interest to stranger and the difference in the times spent in stranger-containing compartments and empty compartments was less pronounced and statistically not significant at 0.05 level (Fig. 5C). The three-chamber test allows determination of social novelty and exploration behavior. Both KCC2+/+ and KCC2E/+ mice spent more time to exploring the novel stranger compartment than the familiar stranger compartment (fig. S4A), and there were no difference between KCC2+/+ and KCC2E/+ mice in the number of entries in the 3 different chambers compartment (fig. S4B and table S5). Two other behavioral tests that are used to determine anxiety behavior, locomotion, and depression revealed no difference in the behavior of KCC2+/+ and KCC2E/+ mice (fig. S5 and table S5). These data show that KCC2 T906E/T1007E impaired communication and sociability behavior.
Bumetanide restores altered glutamate/GABA balance and seizure susceptibility, but not vocalization and social interactions, in KCC2E/+ mice.
Prenatal (9) or adult (40, 47) administration of the NKCC1 inhibitor bumetanide has been shown to alleviate symptoms in rat and mice animal models of NDDs, presumably due to rescuing of depolarizing action of GABA (1). We therefore examined whether bumetanide could restore neuronal network dysfunction and correct behavioral alterations in KCC2E/+ mice. To this aim, KCC2E/+ mice received daily intraperitoneal (i.p.) bumetanide (0.2mg/kg) or DMSO (sham) injection from P6 to P15, a period during which the hyperpolarizing shift of GABA responses occurs in KCC2+/+ mice but failed in KCC2E/+ mice. We found that the bumetanide treatment fully restored the E/I balance in KCC2E/+ mice (Fig. 6A). The sGlut-PSCs to sGABA-PSCs ratio was similar between the naïve and bumetanide-treated KCC2+/+ mice but significantly lower in bumetanide-treated KCC2E/+ mice compared to sham KCC2E/+ mice (table S6). Next, to determine whether the rescue effect of bumetanide relies on the restoration of the GABA hyperpolarizing shift or on the reorganization of neuronal circuits, we assessed the effect of acute bumetanide treatment in vitro. 3h treatment of P15 hippocampal slices with bumetanide (10μM) fully restored the sGlut-PSCs to sGABA-PSCs ratio of CA3 pyramidal neurons (Fig. 6A).
Figure 6. Bumetanide treatment restores the Glutamate/GABA balance in KCC2E/+ mice.
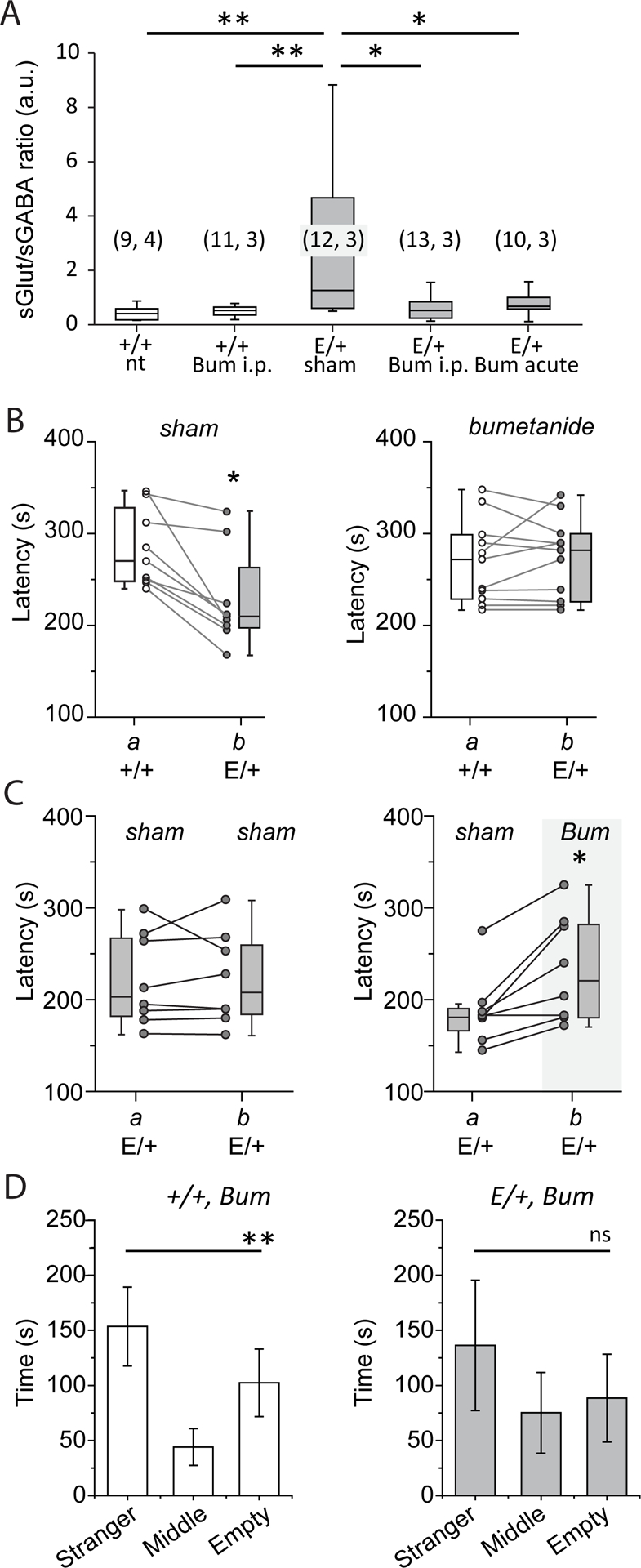
A) Box plot of the frequency ratio of spontaneous glutamatergic to GABAergic postsynaptic currents recorded at P17-P20 from CA3 pyramidal neurons of brain slices prepared from KCC2+/+ (“+/+”) or KCC2E/+ (“E/+”) mice that obtained i.p. daily injection (from P6 to P15) of bumetanide (bum i.p.) or DMSO (sham). “nt” indicates values in non-treated mice. “Bum acute” shows values from slices that were prepared from non-treated KCC2E/+ mice and pre-incubated after dissection during 3 h with bumetanide (10 μM). *P<0.05, **P<0.05, Mann-Whitney test. Numbers in parenthesis indicate the number of cells recorded from the number of mice. B) Latency of the onset of tonic-clonic seizures of KCC2+/+ and KCC2E/+ mice in compartments a and b and treated daily (i.p. injections from P6 to P15) with DMSO (sham, left plot, N = 9 pairs) or bumetanide (right plot, N = 11 pairs). The connected points indicate pairs of animals. **P<0.01, Wilcoxon paired test. C) KCC2E/+ mice treated as in (B) with bumetanide (bum, compartment b, right plot) exhibit longer latencies of seizures onset compared to DMSO-treated mice (compartment a, left plot), (N = 8 pairs). Left plot shows control experiments when a and b compartments contained KCC2E/+ mice treated with DMSO (sham), (N = 8 pairs). The connected points indicate pairs of animals. *P<0.05, Wilcoxon Paired test. D) Social behavior of KCC2+/+ (N=10) and KCC2E/+ (N=7) mice treated with bumetanide from P6 to P15 and studied at P60. ns= non-significant, **P<0.01, one-way ANOVA test).
Chronic bumetanide administration from P6 to P15 in P15 KCC2E/+ mice fully restored the latency of flurothyl-induced tonic-clonic seizures as compared with KCC2+/+ littermates (Fig. 6B, right plot, and table S6). The observed effect was specific for bumetanide, as KCC2E/+ animals treated from P6 to P15 with vehicle only (DMSO) showed shorter latencies of seizures induction (Fig. 6B, left plot), similar to those observed above (Fig. 4B). Consistent with above observation, P15 KCC2E/+ mice treated with bumetanide showed significantly longer latencies of seizure responses as compared to their KCC2E/+ littermates treated with vehicle (Fig. 6C, right plot), while vehicle-treated KCC2E/+ mice placed into two compartments showed similar seizure latencies (Fig. 6C).
In contrast, chronic bumetanide treatment from P6 to P15 did not normalize the social behavior deficits observed in KCC2E/+ mice tested at P60. KCC2E/+ mice treated with bumetanide spent as much time exploring the empty compartment and the compartment with strangers (Fig. 6D and table S6) as did untreated KCC2E/+ mice (Fig. 5D). As control experiments, KCC2+/+ mice were treated with bumetanide from P6 to P15. Like KCC2+/+mice, bumetanide-treated KCC2+/+ mice spent more time to explore stranger compartment than empty compartment (Fig. 6D).
DISCUSSION
Our multidisciplinary approach that includes biochemistry, electrophysiology, and neurobehavior in Thr906/Thr1007 phospho-mimetic KCC2E/+ mice revealed several, previously unknown observations that illustrate the importance of KCC2 Thr906/Thr1007 phosphorylation for post-natal brain physiology and neurodevelopment. We have shown that P15, but not P30, KCC2E/+ mice exhibit an alteration in strength of GABAergic inhibition and an enhanced excitatory/inhibitory (E/I) ratio, two characteristic electrophysiological signatures associated with multiple NDDs (1, 48, 49). Furthermore, behavioral tests show phospho-mimetic KCC2 Thr906/Thr1007 mutation lead to increased ultra-sonic vocalization during the two first post-natal weeks, higher susceptibility for seizure generation at P15, and impaired social interaction in P60 mice, three characteristic neurobehaviors associated with multiple NDDs (2, 41, 50). Post-natal treatment with the NKCC1 blocker bumetanide restored E/I balance and normalized seizure thresholds but failed to restore the social interaction impairments. These data highlight the functional importance of KCC2’s post-translational control in CNS development, and suggest impairment of this mechanism may contribute to the pathogenesis of certain symptoms associated with multiple different NDDs.
By performing electrophysiological recordings in acute hippocampal slices, we showed that the developmental maturation of Cl−-dependent GABAergic neurotransmission is delayed in KCC2E/+ mice. At P12-P15, DFGABA is shifted towards negative values in KCC2+/+ mice, but remained positive in KCC2E/+ mice. Likewise, at P12, isoguvacine reduced the firing of CA3 pyramidal cell in KCC2+/+ mice, but enhanced the firing in KCC2E/+ mice. These results corroborate previous in vitro studies showing that phospho-mimetic mutations of KCC2 Thr906/Thr1007 down-regulate transporter activity, leading to an increase in [Cl−]i, and consequently, reduced inhibitory strength of GABA (14). Interestingly, the developmental action of GABA in KCC2E/+ mice was not abolished but delayed; at P30, the DFGABA and effect of isoguvacine were similar in KCC2+/+ and KCC2E/+ animals. This could be explained by compensation of the KCC2 T906E/T1007E allele by the developmental up-regulation in KCC2 abundance and simultaneous Thr906/Thr1007 de-phosphorylation in the wild type KCC2 allele. Together, these results indicate that post-translational modification of KCC2 is a determining factor in the post-natal emergence of functional GABAergic inhibition.
In addition to causing a reduction in the inhibitory strength of GABA, the T906E/T1007E phospho-mimetic mutations of KCC2 caused a transient enhancement of network excitability and the susceptibility to generate seizures. All three parameters (DFGABA, the E/I ratio, and seizure susceptibility) are altered at P15, but not P30, indicating a correlation between the polarity of GABA, an increase in neuronal network activity, and the sensitivity to epileptogenic agents. Furthermore, chronic post-natal treatment with bumetanide rescued the E/I ratio and alleviated the sensitivity to epileptogenic agents. Interestingly, the same rescue of E/I ratio was observed after acute bumetanide application to hippocampal slices. Thus, the increase in network excitability and seizure susceptibility in KCC2E/+ mice likely results from delayed depolarizing action of GABA and not from abnormal developmental sequences or reorganization of neuronal networks. Our findings also corroborate and extend recent work showing that genetic mutation of Thr 906/1007 to alanine (Ala), the inverse of our phospho-mimetic model that mimics constitutive dephosphorylation, enhances KCC2 activity and limits the onset and severity of seizures in homozygous mice (23).
We have shown that KCC2E/+ mice exhibit behavioral alterations, including an increase in the number of ultrasonic calls emitted by P10 and P12 isolated pups, and reduced social interactions in P60 mice, two key symptoms of ASDs. There was no difference in tests that evaluated anxiety, locomotion, and depression, although detailed investigations are needed to confirm these results. Previous work has implicated intrinsic KCC2 malfunction or dysregulation in different epilepsy subtypes (25, 51–54), and multiple NDDs such as Schizophrenia, ASDs, Rett syndrome (7, 9, 10, 27, 55, 56); The present work is the first study demonstrating that the post-translational KCC2 control could be a risk factor in the NDDs etiology. Our findings extend several recent genetic studies that have identified mutation-linked modifications of the post-translational control of KCC2 in human neurological disorders (25, 26, 54).
Impaired GABAergic neurotransmission and network activity dysregulation are hallmarks of NDD (1, 48, 49). Impaired Cl- homeostasis and excitatory GABA activity have been reported in adult mice model of Rett syndrome (10), Down syndrome (40) and Huntington’s disease (47). In these models, bumetanide treatment in adults restored the hyperpolarizing action of GABA and alleviated the symptoms. In contrast, in our study, the alterations of social interaction are present while GABA has shifted toward hyperpolarizing direction. Thus, the KCC2E/+ mice is a valuable model to study the patho-physiological importance of post-natal Cl- modification and neuronal network activity.
Previous studies have suggested dysregulation of the post-natal GABAergic sequence contributes to the pathogenesis of several neurological disorders by impairing neuronal network formation (9, 35, 57, 58). Consistent with this, hippocampal neurons of P0-P30 age Fragile X and Valproate mouse models of ASD exhibit increased post-natal Cl− and neuronal network activity (9, 57, 58). In these models, peri-natal treatment with bumetanide restored the GABAergic post-natal sequence (9) and adult behavior alterations (58). In our experiments, the P6-P15 post-natal treatment of KCC2E/+ animals with bumetanide restored neuronal network activity at P15-P20, but failed to rescue compromised social behavior at P60. Taken together, these data indicate that the post-natal alteration of GABAergic transmission and network functioning at least during the period from P6 to P15 is not implicated in social behavior modification and alteration of this phenomenon in KCC2E/+ mice involves other KCC2-dependent mechanisms. One putative mechanism is the requirement of “critical period” of KCC2-dependent changes in ion homeostasis In relation to this suggestion, previous work have shown that bumetanide treatment from E15 to P7 decreased cortical AMPA miniature currents, while treatment from E15 to E19 or P7 to P14 had no effect (59). The second possibility is that contrary to KCC2E/+ monogenic model, all cited above works that studied bumetanide treatment, were performed on multifactorial models involving long-lasting changes of large number of genes and signaling pathways (50, 60, 61) as well as reported changes of neuronal Cl− homeostasis in brain slices from juvenile (P30, (9)) or young adult (P60, (40, 47)) animals. Finally, the described bumetanide-resistant impairment of social interaction in KCC2E/+ mice might involve bumetanide/chloride homeostasis independent long lasting changes of the interaction with one of few out of multiple recently identified KCC2 partners (62).
When this work was in revision, it had been published a paper involving analysis of the behavior of two different mice lines with modified KCC2 phosphorylation (63). Authors convincingly showed that the homozygous S940A knockin mice with increased neuronal [Cl−]i during P10-P19 period has significantly reduced preference for social interaction, whereas homozygous T906A/T1007A mice with decreased neuronal [Cl−]i showed increased sociability compared to wild-type mice. These results are in perfect agreement with our finding showing that partial inactivation of KCC2 promotes reduction of sociability. Unfortunately authors did not reported yet whether mechanisms of altered/enhanced sociability of KCC2-mutated mice rely on KCC2 activity/[Cl−]i or involve other complementary pathways.
Recent clinical studies revealed that heterozygous mutations in human KCC2 are associated with multiple neurological disorders featuring impaired Cl− homeostasis including several forms of epilepsy (25, 26, 54, 64), ASD, and schizophrenia (27). How these mutations affect KCC2 activity and/or expression in humans and whether these mutations act in combination with other risk factors is presently unknown. In vitro experiments performed on cell cultures expressing KCC2 harboring human mutations suggest that at least some of these mutations impair critical post-translational modifications of KCC2 that alter its activity (25, 26). Our study supports these genetic findings by showing that impaired regulation of KCC2 phosphorylation in even one allele can directly contribute to the formation of pathology relevant for multiple NDDs. These KCC2 T906E(A)/T1007E(A) transgenic animals thus represent valuable models to dissect the importance and effects of regulated KCC2 phosphorylation in vivo, and serve as genetic proof that drug development targeting these sites is a compelling strategy to powerfully modulate KCC2 activity.
MATERIALS AND METHODS
Animals
The study was performed on KCC2E/+ and KCC2+/+ mixed background SV129/C57bl6-J mice. Animals were housed in a temperature-controlled environment with a 12-h light/dark cycle and free access to water and food. All procedures were in accordance with the European Communities Council Directive (86/609/EEC).
Production of KCC2 double point mutant targeted ES cell clones
Linearized targeting vector was transfected into 129Sv ES cells (genOway, Lyon, France) according to genOway’s electroporation procedures. PCR, Southern blot and sequence analysis of G-418 resistant ES clones revealed 2 clones as carrying the recombined locus. PCR over the 5’ end of the targeted locus was performed with a forward primer hybridizing upstream of the 5’ homology arm (5’- ATAGCGTTGGCTACCCGTGATATTGC-3’) and a reverse primer hybridizing within the Neomycin cassette (5’ AGGCTAGGCACAGGCTACATCCACAC-3’). Two Southern blot assays, assessing the correct recombination event at the 5’ end and at the 3’ end of the KCC2 locus, were performed. These assays are based on the use of an internal and of an external probe, respectively. Finally, the integrity of the point mutations was confirmed by sequence analysis.
Generation of chimeric mice and breeding scheme
Recombined ES cell clones were microinjected into C57BL/6 blastocysts, and gave rise to male chimeras with a significant ES cell contribution. These chimeras were bred with C57BL/6J mice expressing Cre-recombinase, to produce the KCC2 double point mutant heterozygous line devoid of the Neomycin cassette. For each line, F1 genotyping was performed by PCR and Southern blot. PCR primers hybridizing upstream (5’- GTGGTTCGCCTATGGGATCTGCTACTC-3’) and downstream (5’- AGACAAGGGTTCATGTAACAGACTCGCC-3’) of the Neomycin cassette allowed identification of the KCC2 endogenous, double point mutant allele harboring the Neomycin cassette, and double point mutant devoid of the Neomycin cassette (298-bp, 1946-bp and 387-bp, respectively). The Southern blot hybridized with an external probe allowed identification of the wild-type allele (14.1-kb) and the double point mutant allele (4.6-kb).
Genotyping
DNA extraction was performed with KAPA Mouse Genotyping Kit (Sigma-Aldrich). 88μl of water, 10μl of KAPA express extract buffer and 2μl of KAPA express extract enzyme was mixed and placed on the Eppendorf containing a small piece of biopsy. Lysis was performed on thermocycler during 10 min at 75°C for lysis and 5 min at 95°C for enzyme inactivation. The mix reaction for PCR was composed of 14.5μl H2O, 5μl 5X buffer (Promega), 1.5μl MgCl2 (25mM, Promega), 1.25μl primers, 0.25μl dNTP (25mM), 0.25μl GoTaq polymerase (Promega) for 1μl of lysis product. The primers were following: 93598cof-KKA1; AGA CAA GGG TTC ATG TAA CAG ACT CGC C and 93599cof-KKA1; GTG GTT CGC CTA TGG GAT CTG CTA CTC.
Hippocampal slice preparation and electrophysiological recordings
Brains were removed and immersed into ice-cold (2–4°C) artificial cerebrospinal fluid (ACSF) with the following composition (in mM): 126 NaCl, 3.5 KCl, 2 CaCl2, 1.3 MgCl2, 1.2 NaH2PO4, 25 NaHCO3 and 11 glucose, pH 7.4 equilibrated with 95% O2 and 5% CO2. Hippocampal slices (400 μm thick) were cut with a vibrating microtome (Leica VT 1000s, Germany) in ice cold oxygenated choline-replaced ACSF and were allowed to recover at least 90 min in ACSF at room (25°C) temperature. Slices were then transferred to a submerged recording chamber perfused with oxygenated (95% O2 and 5% CO2) ACSF (3 ml/min) at 34°C.
Whole-cell patch clamp recordings were performed from P15-P20 CA3 pyramidal neurons in voltage-clamp mode using an Axopatch 200B (Axon Instrument, USA). To record the spontaneous synaptic activity, the glass recording electrodes (4–7 MΩ) were filled with a solution containing (in mM): 100 KGluconate, 13 KCl, 10 HEPES, 1.1 EGTA, 0.1 CaCl2, 4 MgATP and 0.3 NaGTP. The pH of the intracellular solution was adjusted to 7.2 and the osmolality to 280 mOsmol l−1. The access resistance ranged between 15 to 30 MΩ. With this solution, the GABAA receptor-mediated postsynaptic current (GABAA-PSCs) reversed at −70mV. GABA-PSCs and glutamate mediated synaptic current (Glut-PSCs) were recorded at a holding potential of −45mV. At this potential, GABA-PSCs are outwards and Glut-PSCs are inwards. All recordings were performed using Axoscope software version 8.1 (Axon Instruments) and analyzed offline with Mini Analysis Program version 6.0 (Synaptosoft). For the acute bumetanide treatment, before recording, slices were incubated during 3h in ACSF containing 10μM of bumetanide.
Single GABAA and N-methyl-D-aspartate (NMDA) channel recordings were performed from P7 to P30 visually identified hippocampal CA3 pyramidal cells in cell-attached configuration using Axopatch-200A amplifier and pCLAMP acquisition software (Axon Instruments, Union City, CA). Data were low-pass filtered at 2 kHz and acquired at 10 kHz. The glass recording electrodes (4–7 MΩ) were filled with a solution containing (in mM) : (1) for recordings of single GABAA channels: GABA 0.01, NaCl 120, KCl 5, TEA-Cl 20, 4-aminopyridine 5, CaCl2 0.1, MgCl2 10, glucose 10, Hepes-NaOH 10 (9, 34); (2) for recordings of single NMDA channels: nominally Mg2+ free ACSF with NMDA (10 μM) and glycine (1 μM) (34). The pH of pipette solutions was adjusted to 7.2 and the osmolality to 280 mOsmol l−1. Both single GABAA and single NMDA channel currents were recorded in voltage clamp mode at different membrane potentials (from −80mV to 80mV for GABAA and from −120 to 40 mV for NMDA) in order to visualize outwardly and inwardly directed single channel currents. Analysis of currents trough single channels and I-V curves were performed using Clampfit 9.2 (Axon Instruments) as described by (34).
Sample size for each group reported in electrophysiological experiments was at least 5 neurons from at least 3 independent experiments.
Extracellular recording of the local field potentials (LFPs) were performed from P5-P20 CA3 region of hippocampus. Extracellular 50μm tungsten electrodes (California Fine Wire) were placed in the pyramidal cell layer to record the Multi Unit Activity (MUA). The signals were amplified using a DAM80i amplifier, digitized with an Axon Digidata 1550B, recorded with Axoscope software version 8.1 (Axon instruments) and analyzed offline with Mini Analysis Program version 6.0 (Synaptosoft). The frequency of MUA was analyzed before, during and after 2 min of application of isoguvacine (10μM). In order to collate the effects across experiments, we counted the proportion of slices that showed more that 20% MUA increase from the pre-drug period as excitatory, more than 20% decrease as inhibitory. The changes within range of −20% to + 20% were considered to be ‘no response’.
Antibodies for Western blot
The following antibodies were raised in sheep and affinity-purified on the appropriate antigen by the Division of Signal Transduction Therapy Unit at the University of Dundee: KCC2A phospho-Thr 906 (SAYTYER(T)LMMEQRSRR [residues 975 – 989 of human KCC3A] corresponding to SAYTYEK(T)LVMEQRSQI [residues 899 – 915 of human KCC2A]) (Catalog number: S959C); KCC2A phospho-Thr 1007 (CYQEKVHM(T)WTKDKYM [residues 1032 – 1046 of human KCC3A] corresponding to TDPEKVHL(T)WTKDKSVA [residues 998 – 1014 of human KCC2A]) (Catalog number: S961C); KCC2 phospho-Ser940 (Catalog number: NBP2–29513). Pan KCC2 total antibody (residues 932–1043 of human KCC2) was purchased from NeuroMab (Catalog number: 73–013). Anti-β-Tubulin III (neuronal) antibody was purchased from Sigma-Aldrich (Catalog number: T8578). Secondary antibodies coupled to horseradish peroxidase used for immunoblotting were obtained from Pierce. IgG used in control immunoprecipitation experiments was affinity-purified from pre-immune serum using Protein G-Sepharose.
Buffers for Western Blots
Buffer A contained 50 mM Tris/HCl, pH7.5 and 0.1mM EGTA. Lysis buffer was 50 mM Tris/HCl, pH 7.5, 1 mM EGTA, 1 mM EDTA, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1% (w/v) Triton-100, 0.27 M sucrose, 0.1% (v/v) 2-mercaptoethanol, and protease inhibitors (complete protease inhibitor cocktail tablets, Roche, 1 tablet per 50 mL). TBS-Tween buffer (TTBS) was Tris/HCl, pH 7.5, 0.15 M NaCl and 0.2% (v/v) Tween-20. SDS sample buffer was 1X NuPAGE LDS sample buffer (Invitrogen), containing 1% (v/v) 2-mercaptoethanol. Protein concentrations were determined following centrifugation of the lysate at 16,000 x g at 4°C for 20 minutes using the Bradford method with bovine serum albumin as the standard.
Immunoprecipitation with phosphorylation site-specific antibodies
KCCs phosphorylated at the KCC2 Thr906 and Thr1007 equivalent residue were immunoprecipitated from clarified hippocampal and cortical culture lysates (centrifuged at 16,000 x g at 4°C for 20 minutes) using phosphorylation site-specific antibody coupled to protein G–Sepharose as described (31). The phosphorylation site-specific antibody was coupled with protein-G–Sepharose at a ratio of 1 mg of antibody per 1 mL of beads in the presence of 20 μg/mL of lysate to which the corresponding non-phosphorylated peptide had been added. 2 mg of clarified cell lysate were incubated with 15 μg of antibody conjugated to 15 μL of protein-G–Sepharosefor 2 hours at 4°C with gentle agitation. Beads were washed three times with 1 mL of lysis buffer containing 0.15 M NaCl and twice with 1 mL of buffer A. Bound proteins were eluted with 1X LDS sample buffer.
Immunoblotting
Hippocampi tissue lysates (15 μg) in SDS sample buffer were subjected to electrophoresis on polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were incubated for 30 min with TTBS containing 5% (w/v) skim milk. The membranes were then immunoblotted in 5% (w/v) skim milk in TTBS with the indicated primary antibodies overnight at 4°C. Antibodies prepared in sheep were used at a concentration of 1–2 μg/ml. The incubation with phosphorylation site-specific sheep antibodies was performed with the addition of 10 μg/mL of the nonphosphorylated peptide antigen used to raise the antibody. The blots were then washed six times with TTBS and incubated for 1 hour at room temperature with secondary HRP-conjugated antibodies diluted 5000-fold in 5% (w/v) skim milk in TTBS. After repeating the washing steps, the signal was detected with the enhanced chemiluminescence reagent. Immunoblots were developed using a film automatic processor (SRX-101; Konica Minolta Medical) and films were scanned with a 600-dpi resolution on a scanner (PowerLook 1000; UMAX). Figures were generated using Photoshop and Illustrator (Adobe). The relative intensities of immunoblot bands were determined by densitometry with ImageJ software.
Behavior assessments
All behavioral procedures were carried between 8:00 and 17:00 h under dim light conditions. Male animals were moved to the testing room in their home cages 30 min prior to test beginning. The same animals have been used for all adulthood behavioral tests, starting at 6-week old. All experiments and analysis were done in blind genotyping by only one experimenter.
To induce vocalization, pups were isolated individually from their mother at P2–4-8–10-12. They were placed into an isolation box (23×28×18 cm) located inside a sound attenuating isolation cubicle (54×57×41 cm; Coulbourn Instruments, Allentown, PA, USA). An ultrasound microphone sensitive to frequencies of 10 to 250 kHz (Avisoft UltraSoundGate Condenser microphone capsule CM16/CMPA, Avisoft Bioacoustics, Berlin, Germany) was placed in the roof of the box. Vocalizations were recorded for 3 minutes using the Avisoft Recorder software (version 4.2) with a sampling rate of 250 kHz in 16 bit format. Recordings were transferred to SASLab Pro (version 5.2; AvisoftBioacoustics) and a fast Fourier transformation was conducted (512 FFT-length, 100% frame, Hamming window and 75%time window overlap, cut off frequencies high pass 20Khz) before analyzing the number of calls emitted by mice.
For the open field test, the mice were individually placed in the square field (38.5 × 38.5cm; Noldus, Netherlands). Recording began 10 seconds after the mouse was placed inside the apparatus for a 10 minutes trial. Behaviors were recorded by a video camera fixed above the apparatus and analyzed using the Ethovision 11.5 software (Noldus, Netherlands). After each trial, the open field was cleaned with a solution containing 70% of ethanol. Anxiety and locomotor-like behaviors were analyzed using three parameters: the time spent in the center (12.8cm × 12.8cm), the number of entries in the center and the distance travelled.
For the social interaction tests, we performed the 3-chamber test as previously described (46). The test was conducted in an apparatus (59 × 39.5cm; Noldus, Netherlands) divided in a central empty compartment (19.5 × 39.5 cm) and two side-compartments (19.5 × 39.5 cm) containing a plastic cup-like cage for the strangers. The strangers were of the same sex and age as the tested mice and were habituated during 15min (one habituation per day) to the plastic cup-like cage 4 days prior to the beginning of the test. The mouse was recorded during 4 consecutive trials of 5 min. Trial 1, habituation: the tested mouse was placed in the empty compartment with the access to the other compartments closed. Trial 2, sociability testing: a first stranger was placed in a plastic cup-like cage in one of the two-side compartments and the tested mouse could explore freely all the compartments. Trial 3, post-test: the tested mouse could explore freely its environment and be habituated for a longer time to the stranger 1 (familiar stranger). Trial 4, social novelty testing: the second stranger (novel stranger) was placed in the second plastic cup-like cage in the opposite side-compartment. For each tested mouse, the side-compartments where the strangers were placed were alternated to avoid any side preferences. All trials were recorded by a video camera placed above the apparatus by using the Ethovision 11.5 software (Noldus, Netherlands) and the time spent in each chamber was manually analyzed. Trials where the mice have returned less than once time to the chamber are removed.
The splash test was performed as previously described (Moretti et al; 2015). Mice were sprayed in the dorsal coat with a 10% sucrose solution. The viscosity of this solution dirties the mice and initiates the grooming behavior. After being sprayed, mice were individually placed in a plexiglass cylinder (15 × 45cm; Form X.L., France) and their behaviors were recorded for 5 minutes. Self-care and motivational behaviors were manually analyzed using three parameters: the duration of grooming, the latency to start the first grooming, and the frequency of grooming events. A grooming event was defined as at least one episode of any category of grooming (paw licking, head wash, body groom, leg licking, tail/genital licking).
To test seizure susceptibility, we used convulsant agent flurothyl (2,2,2-trifluoroethyl ether, Sigma) that is widely used to study epilepsy in different animal models (66, 67). The advantage of the flurothyl, as compared to other pro-convulsive agents is that it could be used to induce epilepsy-like activity in juvenile P0-P30 rats and mice (66, 68). Because the latencies of the effects of flurothyl depend on large number of parameters including atmospheric pressure, humidity, temperature, movements of the air in experimental chamber, animal weight and age (68) all experiments were performed using pairs of littermate males (P15 and P30) that were placed in transparent, ventilated but hermetically-sealed, cages, into which was delivered the epileptic agent, flurothyl. The progressive injection of the flurothyl into the cage produced a stereotypical behavioral manifestation of limbic seizure episodes that varied, depending on the animal age and genotype. At P15 these episodes commenced in all studied animals with forelimb and/or tail extension, rigid posture (stage 1) followed by period of tranquility with one-two irregular low-amplitude body jiggles(stage 2), one to three brief (1–2s) myoclonic jerks (stage 3), severe tonic-clonic seizures (stage 4) lasting 5–20 s that ended with falling and immobility of the animal (stage 5) (Fig. 4A). At P30 seizure episodes started from animal immobility and ended by severe tonic-clonic seizures (stage 4). The intermediate stages (rigid posture, tranquility period, jiggles and jerks) were absent or not clearly detectable at this age. 10 s after beginning of tonic-clonic seizures in more resistant animal from each pair, the injection of flurothyl was discontinued and cage was inhalated with fresh air. 2–5 min after stopping of the exposure to flurothyl most of mice returned to their 4-limb horizontal position, but remained immobile during 5–10 min. Thereafter all mice started moving and exploring the cage, although their moving activity was not scored.
Flurothyl was progressively injected into the cage using nano-pump (Harvard apparatus) and homogeneously distributed using mini-ventilator incorporated into the chamber. Behavioral responses were recorded using a video camera. The latency of tonic-clonic seizures was determined post hoc as the time of the first body convulsion in continuous series of tonic-clonic seizures.
Statistical analysis
Statistical analyses were conducted with OriginPro 9.0.0. Shapiro-Wilk normality test and Brown-Forsythe test of equal variance were used to determine the normality of distribution. A P<0.05 was considered significant for these and all subsequent tests. For data displaying normal distribution and equal variance one-way or two-way (as indicated) ANOVA and the post hoc Tukey test were used for multiple comparisons between groups. In the text and figures, the normally distributed data are represented as mean and SD, where SD is a standard deviation reflecting data variability. For data displaying non-normal distribution, unequal variance or data with number of studied animals lower than 7 per group, Kruskal-Wallis test was used for comparing three or more independent groups, Mann–Whitney U-test was used for comparison between 2 independent groups and Wilcoxon matched pairs test was employed to compare paired data. The Chi-square test was used to determine the significance of the difference between the frequencies of event occurrence. The data showing not normal distribution are illustrated as individual points and/or as boxplots. For the boxplots, the box extends from the first (Q1) to third (Q3) quartiles. The line inside the box represents the median. The whiskers define the outermost data point that falls within upper inner and lower inner Quartile fences [Q1–1.5(IQR)] and [Q3–1.5(IQR)], respectively.
Supplementary Material
Figure S1: KCC2 related signaling in KCC2E/+ phospho-mimetic mice.
Figure S2: Similar resting membrane potential in P13 CA3 neurons of acute slices from KCC2E/+ and KCC2+/+ mice.
Figure S3: Extended comparison procedure to verify statistical significance of the seizure susceptibility difference between KCC2+/+ and KCC2E/+ mice.
Figure S4: Mutations T906E/T1007E do not modify social novelty and exploration behaviors.
Figure S5: Results of additional behavior tests in KCC2+/+ and KCC2E/+ mice.
Table S1: Statistical differences among the samples illustrated in Fig. 1 and fig. S1.
Table S2: Statistical differences among the samples illustrated in Fig. 2.
Table S3: Statistical differences among the samples illustrated in Fig. 3.
Table S4: Statistical differences among the samples illustrated in Fig. 4 and fig. S3.
Table S5: Statistical differences among the samples in behavior experiments illustrated in Fig. 5 and figs. S4 and S5.
Table S6: Statistical differences among the samples illustrated in Fig. 6.
Acknowledgments:
We express our gratitude to the Neurochlore (www.neurochlore.fr) team (Drs Diana Ferrari, Morgane Chiesa and Sanaz Eftekhari) for their help in the mouse neurobehaviour studies; Mrs Marie Kurz for help in animal care; and Mrs Aurélie Montheil and Mrs Francesca Bader for genotyping. Dr. Marat Minlebaev for “statistics review” of the manuscript. Funding: French Foundation of Epilepsy Research (FFRE) for L.I.P. , French Ministry of Education (MRT) for L.I.P., Kahle T.K. for L.I.P., Ministry of Education and Research of Russian Federation (6.2313.2017/4.6 and Top-100) to I.K., Simons Foundation, March of Dimes Foundation, and NIH 4K12NS080223–05 (KTK).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES :
- 1.Deidda G, Bozarth IF, Cancedda L, Modulation of GABAergic transmission in development and neurodevelopmental disorders: investigating physiology and pathology to gain therapeutic perspectives. Front. Cell. Neurosci 8, 119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pasciuto E, Borrie SC, Kanellopoulos AK, Santos AR, Cappuyns E, D’Andrea L, Pacini L, Bagni C, Autism Spectrum Disorders: Translating human deficits into mouse behavior. Neurobiol. Learn. Mem 124, 71–87 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Cattane N, Richetto J, Cattaneo A, Prenatal exposure to environmental insults and enhanced risk of developing Schizophrenia and Autism Spectrum Disorder: focus on biological pathways and epigenetic mechanisms. Neurosci. Biobehav. Rev (2018), doi: 10.1016/j.neubiorev.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Schulte JT, Wierenga CJ, Bruining H, Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev 90, 260–271 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Kahle KT, Khanna AR, Duan J, Staley KJ, Delpire E, Poduri A, The KCC2 Cotransporter and Human Epilepsy: Getting Excited About Inhibition. Neuroscientist. 22, 555–562 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Hyde TM, Lipska BK, Ali T, V Mathew S, Law AJ, Metitiri OE, Straub RE, Ye T, Colantuoni C, Herman MM, Bigelow LB, Weinberger DR, Kleinman JE, Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci 31, 11088–95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sullivan CR, Funk AJ, Shan D, Haroutunian V, McCullumsmith RE, Decreased Chloride Channel Expression in the Dorsolateral Prefrontal Cortex in Schizophrenia. PLoS One. 10, e0123158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duarte ST, Armstrong J, Roche A, Ortez C, Pérez A, del M. O’Callaghan M, Pereira A, Sanmartí F, Ormazábal A, Artuch R, Pineda M, García-Cazorla A, Abnormal Expression of Cerebrospinal Fluid Cation Chloride Cotransporters in Patients with Rett Syndrome. PLoS One. 8 (2013), doi: 10.1371/journal.pone.0068851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, Khalilov I, Tsintsadze V, Brouchoud C, Chazal G, Lemonnier E, Lozovaya N, Burnashev N, Ben-Ari Y, Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. 343, 675–679 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Banerjee A, Rikhye RV, Breton-Provencher V, Tang X, Li C, Li K, Runyan CA, Fu Z, Jaenisch R, Sur M, Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc. Natl. Acad. Sci 113, E7287–E7296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Khoury R, Panayotis N, Matagne V, Ghata A, Villard L, Roux J-C, GABA and Glutamate Pathways Are Spatially and Developmentally Affected in the Brain of Mecp2-Deficient Mice. PLoS One. 9, e92169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B, Kaila K, Moss SJ, Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 36, 726–37 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee HHC, Walker JA, Williams JR, Goodier RJ, Payne JA, Moss SJ, Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem 282, 29777–84 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Friedel P, Kahle KT, Zhang J, Hertz NT, Pisella LI, Buhler E, Schaller F, Duan J, Khanna AR, Bishop PN, Shokat KM, Medina I, WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci. Signal 8, 23–26 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Inoue K, Furukawa T, Kumada T, Yamada J, Wang T, Inoue R, Fukuda A, Taurine inhibits K+-Cl- cotransporter KCC2 to regulate embryonic Cl- homeostasis via with-no-lysine (WNK) protein kinase signaling pathway. J. Biol. Chem 287, 20839–50 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee HHC, Jurd R, Moss SJ, Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell. Neurosci 45, 173–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber M, Hartmann A-M, Beyer T, Ripperger A, Nothwang HG, A novel regulatory locus of phosphorylation in the C terminus of the potassium chloride cotransporter KCC2 that interferes with N-ethylmaleimide or staurosporine-mediated activation. J. Biol. Chem 289, 18668–79 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cordshagen A, Busch W, Winklhofer M, Nothwang HG, Hartmann A-M, Phospho-regulation of the intracellular termini of K-Cl cotransporter 2 (KCC2) enables flexible control of its activity. J. Biol. Chem 293, 16984–16993 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Payne JA, Stevenson TJ, Donaldson LF, Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J. Biol. Chem 271, 16245–52 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Agez M, Schultz P, Medina I, Baker DJ, Burnham MP, Cardarelli RA, Conway LC, Garnier K, Geschwindner S, Gunnarsson A, McCall EJ, Frechard A, Audebert S, Deeb TZ, Moss SJ, Brandon NJ, Wang Q, Dekker N, Jawhari A, Molecular architecture of potassium chloride co-transporter KCC2. Sci. Rep 7, 16452 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rinehart J, Maksimova YD, Tanis JE, Stone KL, Caleb A, Zhang J, Risinger M, Pan W, Wu D, Christopher M, Forbush B, Joiner CH, Gulcicek EE, Gallagher PG, a Hodson C, Colangelo CM, Lifton RP, Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 138, 525–36 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HHC, Deeb TZ, Walker JA, Davies PA, Moss SJ, NMDA receptor activity downregulates KCC2 resulting in depolarizing GABA(A) receptor-mediated currents. Nat. Neurosci 14, 736–43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore YE, Deeb TZ, Chadchankar H, Brandon NJ, Moss SJ, Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl. Acad. Sci, 201810134 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedel P, Bregestovski P, Medina I, Improved method for efficient imaging of intracellular Cl(−) with Cl-Sensor using conventional fluorescence setup. Front. Mol. Neurosci 6, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kahle KTKT, Merner NDND, Friedel P, Silayeva L, Liang B, Khanna A, Shang Y, Lachance-Touchette P, Bourassa C, Levert A, Dion PAPA, Walcott B, Spiegelman D, Dionne-Laporte A, Hodgkinson A, Awadalla P, Nikbakht H, Majewski J, Cossette P, Deeb TZTZ, Moss SJSJ, Medina I, Rouleau GAGA, Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 15, 766–74 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stödberg T, McTague A, Ruiz AJ, Hirata H, Zhen J, Long P, Farabella I, Meyer E, Kawahara A, Vassallo G, Stivaros SM, Bjursell MK, Stranneheim H, Tigerschiöld S, Persson B, Bangash I, Das K, Hughes D, Lesko N, Lundeberg J, Scott RC, Poduri A, Scheffer IE, Smith H, Gissen P, Schorge S, Reith MEA, Topf M, Kullmann DM, Harvey RJ, Wedell A, Kurian MA, Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat. Commun 6, 8038 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merner ND, Chandler MR, Bourassa C, Liang B, Khanna AR, Dion P, Rouleau GA, Kahle KT, Regulatory domain or CpG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front. Cell. Neurosci 9, 386 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silayeva L, Deeb TZ, Hines RM, Kelley MR, Munoz MB, Lee HHC, Brandon NJ, Dunlop J, Maguire J, Davies PA, Moss SJ, KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. U. S. A (2015), doi: 10.1073/pnas.1415126112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kahle KT, Flores B, Bharucha-Goebel D, Zhang J, Donkervoort S, Hegde M, Begum G, Duran D, Liang B, Sun D, Bönnemann CG, Delpire E, Peripheral motor neuropathy is associated with defective kinase regulation of the KCC3 cotransporter. Sci. Signal 9, ra77–ra77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe M, Zhang J, Mansuri MS, Duan J, Karimy JK, Delpire E, Alper SL, Lifton RP, Fukuda A, Kahle KT, Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal 12, eaaw9315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Los Heros P, Alessi DR, Gourlay R, Campbell DG, Deak M, Macartney TJ, Kahle KT, Zhang J, The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl- co-transporters. Biochem. J 458, 559–573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Stein V, Hermans-Borgmeyer I, Jentsch TJ, a Hübner C, Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J. Comp. Neurol 468, 57–64 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Lu J, Karadsheh M, Delpire E, Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J. Neurobiol 39, 558–68 (1999). [PubMed] [Google Scholar]
- 34.Tyzio R, Ivanov A, Bernard C, Holmes GL, Ben-Ari Y, Khazipov R, Membrane potential of CA3 hippocampal pyramidal cells during postnatal development. J. Neurophysiol 90, 2964–72 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Tyzio R, Cossart R, Khalilov I, Minlebaev M, a Hübner C, Represa A, Ben-Ari Y, Khazipov R, Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science. 314, 1788–92 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Ben-Ari Y, Gaiarsa J-L, Tyzio R, Khazipov R, GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev 87, 1215–84 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Khazipov R, Khalilov I, Tyzio R, Morozova E, Ben-Ari Y, Holmes GL, Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur. J. Neurosci 19, 590–600 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Moore YE, Kelley MR, Brandon NJ, Deeb TZ, Moss SJ, Seizing Control of KCC2: A New Therapeutic Target for Epilepsy. Trends Neurosci. 40, 555–571 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Agrawal N, Govender S, Epilepsy and neuropsychiatric comorbidities. Adv. Psychiatr. Treat 17, 44–53 (2011). [Google Scholar]
- 40.Deidda G, Parrini M, Naskar S, Bozarth IF, Contestabile A, Cancedda L, Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med 21, 318–26 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Scattoni ML, Crawley J, Ricceri L, Ultrasonic vocalizations: A tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci. Biobehav. Rev 33, 508–515 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belzung C, Griebel G, Measuring normal and pathological anxiety-like behaviour in mice: a review. Behav. Brain Res 125, 141–9 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Silverman JL, Yang M, Lord C, Crawley JN, Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci 11, 490–502 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X-D, Yang G, Bai Y, Feng Y-P, Li H, The behavioral study on the interactive aggravation between pruritus and depression. Brain Behav. 8, e00964 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiaderkiewicz J, Głowacka M, Marta G, Barski J-J, Ultrasonic vocalizations (USV) in the three standard laboratory mouse strains. Developmental analysis. Acta Neurobiol. Exp. (Wars) 73, 557–563 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G, Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 472, 437–442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dargaei Z, Bang JY, Mahadevan V, Khademullah CS, Bedard S, Parfitt GM, Kim JC, Woodin MA, Restoring GABAergic inhibition rescues memory deficits in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci 115, E1618–E1626 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cellot G, Cherubini E, GABAergic Signaling as Therapeutic Target for Autism Spectrum Disorders. Front. Pediatr 2, 70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rubenstein JLR, Merzenich MM, Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes, Brain Behav. 2, 255–267 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bey AL, Jiang Y, in Current Protocols in Pharmacology (John Wiley & Sons, Inc., Hoboken, NJ, USA, 2014; http://www.ncbi.nlm.nih.gov/pubmed/25181011), vol. 66, pp. 5.66.1–5.66.26.25181011 [Google Scholar]
- 51.Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C, Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci 27, 9866–73 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Talos DM, Sun H, Kosaras B, Joseph A, Folkerth RD, Poduri A, Madsen JR, Black PM, Jensen FE, Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann. Neurol 71, 539–51 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palma E, Amici M, Sobrero F, Spinelli G, Di Angelantonio S, Ragozzino D, Mascia A, Scoppetta C, Esposito V, Miledi R, Eusebi F, Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc. Natl. Acad. Sci. U. S. A 103, 8465–8468 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Puskarjov M, Seja P, Heron SE, Williams TC, Ahmad F, Iona X, Oliver KL, Grinton BE, Vutskits L, Scheffer IE, Petrou S, Blaesse P, Dibbens LM, Berkovic SF, Kaila K, A variant of KCC2 from patients with febrile seizures impairs neuronal Cl- extrusion and dendritic spine formation. EMBO Rep. 15, 723–729 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arion D, Lewis DA, Altered expression of regulators of the cortical chloride transporters NKCC1 and KCC2 in schizophrenia. Arch. Gen. Psychiatry 68, 21–31 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang X, Kim J, Zhou L, Wengert E, Zhang L, Wu Z, Carromeu C, Muotri AR, Marchetto MCN, Gage FH, Chen G, KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. U. S. A 113, 751–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He Q, Nomura T, Xu J, Contractor A, The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci 34, 446–50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eftekhari S, Shahrokhi A, Tsintsadze V, Nardou R, Brouchoud C, Conesa M, Burnashev N, Ferrari DC, Ben-Ari Y, Response to Comment on “Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring.” Science. 346, 176–176 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Wang DD, Kriegstein AR, Blocking Early GABA Depolarization with Bumetanide Results in Permanent Alterations in Cortical Circuits and Sensorimotor Gating Deficits. Cereb. Cortex 21, 574–587 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Fu S-C, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, Carracedo A, Chahrour MH, Chiocchetti AG, Coon H, Crawford EL, Crooks L, Curran SR, Dawson G, Duketis E, Fernandez BA, Gallagher L, Geller E, Guter SJ, Sean Hill R, Ionita-Laza I, Jimenez Gonzalez P, Kilpinen H, Klauck SM, Kolevzon A, Lee I, Lei J, Lehtimäki T, Lin C-F, Ma’ayan A, Marshall CR, McInnes AL, Neale B, Owen MJ, Ozaki N, Parellada M, Parr JR, Purcell S, Puura K, Rajagopalan D, Rehnström K, Reichenberg A, Sabo A, Sachse M, Sanders SJ, Schafer C, Schulte-Rüther M, Skuse D, Stevens C, Szatmari P, Tammimies K, Valladares O, Voran A, Wang L-S, Weiss LA, Jeremy Willsey A, Yu TW, Yuen RKC, Cook EH, Freitag CM, Gill M, Hultman CM, Lehner T, Palotie A, Schellenberg GD, Sklar P, State MW, Sutcliffe JS, Walsh CA, Scherer SW, Zwick ME, Barrett JC, Cutler DJ, Roeder K, Devlin B, Daly MJ, Buxbaum JD, Devlin B, Daly MJ, Buxbaum JD, Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 515, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ehrhart F, Coort SLM, Cirillo E, Smeets E, Evelo CT, Curfs LMG, Rett syndrome – biological pathways leading from MECP2 to disorder phenotypes. Orphanet J. Rare Dis 11, 158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahadevan V, Khademullah CS, Dargaei Z, Chevrier J, Uvarov P, Kwan J, Bagshaw RD, Pawson T, Emili A, De Koninck Y, Anggono V, Airaksinen M, Woodin MA, Native KCC2 interactome reveals PACSIN1 as a critical regulator of synaptic inhibition. Elife. 6 (2017), doi: 10.7554/eLife.28270.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moore YE, Conway LC, Wobst HJ, Brandon NJ, Deeb TZ, Moss SJ, Developmental Regulation of KCC2 Phosphorylation Has Long-Term Impacts on Cognitive Function. Front. Mol. Neurosci 12, 173 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saitsu H, Watanabe M, Akita T, Ohba C, Sugai K, Ong WP, Shiraishi H, Yuasa S, Matsumoto H, Beng KT, Saitoh S, Miyatake S, Nakashima M, Miyake N, Kato M, Fukuda A, Matsumoto N, Impaired neuronal KCC2 function by biallelic SLC12A5 mutations in migrating focal seizures and severe developmental delay. Sci. Rep 6, 30072 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lemonnier E, Villeneuve N, Sonie S, Serret S, Rosier A, Roue M, Brosset P, Viellard M, Ernoux DB, Rondeau S, Thummler S, Ravel D, Ben-Ari Y, Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 7, e1056–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Villeneuve N, Ben-Ari Y, Holmes GL, Gaiarsa JL, Neonatal seizures induced persistent changes in intrinsic properties of CA1 rat hippocampal cells. Ann. Neurol 47, 729–38 (2000). [PubMed] [Google Scholar]
- 67.Velíšek L, Shang E, Velíšková J, Chachua T, Macchiarulo S, Maglakelidze G, Wolgemuth DJ, Greenberg DA, GABAergic neuron deficit as an idiopathic generalized epilepsy mechanism: the role of BRD2 haploinsufficiency in juvenile myoclonic epilepsy. PLoS One. 6, e23656 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Velíšková J, Shakarjian MP, Velíšek L, in Models of Seizures and Epilepsy (Academic Press, 2017; https://www.sciencedirect.com/science/article/pii/B9780128040669000353), pp. 491–512. [Google Scholar]
- 69.Stromland K, Pinazo-Durán MD, Ophthalmic involvement in the fetal alcohol syndrome: clinical and animal model studies. Alcohol Alcohol. 37, 2–8 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: KCC2 related signaling in KCC2E/+ phospho-mimetic mice.
Figure S2: Similar resting membrane potential in P13 CA3 neurons of acute slices from KCC2E/+ and KCC2+/+ mice.
Figure S3: Extended comparison procedure to verify statistical significance of the seizure susceptibility difference between KCC2+/+ and KCC2E/+ mice.
Figure S4: Mutations T906E/T1007E do not modify social novelty and exploration behaviors.
Figure S5: Results of additional behavior tests in KCC2+/+ and KCC2E/+ mice.
Table S1: Statistical differences among the samples illustrated in Fig. 1 and fig. S1.
Table S2: Statistical differences among the samples illustrated in Fig. 2.
Table S3: Statistical differences among the samples illustrated in Fig. 3.
Table S4: Statistical differences among the samples illustrated in Fig. 4 and fig. S3.
Table S5: Statistical differences among the samples in behavior experiments illustrated in Fig. 5 and figs. S4 and S5.
Table S6: Statistical differences among the samples illustrated in Fig. 6.