

Abstract
Background
Haemophilia is a genetic disorder characterized by spontaneous or provoked, often uncontrolled, bleeding into joints, muscles and other soft tissues. Current methods of treatment are expensive, challenging and involve regular administration of clotting factors. Gene therapy for haemophilia is a curative treatment modality currently under investigation. This is an update of a published Cochrane Review.
Objectives
To evaluate the safety and efficacy of gene therapy for treating people with haemophilia A or B.
Search methods
We searched the Cochrane Cystic Fibrosis & Genetic Disorders Group's Coagulopathies Trials Register, compiled from electronic database searches and handsearching of journals and conference abstract books. We also searched the reference lists of relevant articles and reviews.
Date of last search: 17 April 2020.
Selection criteria
Eligible trials include randomised or quasi‐randomised clinical trials, including controlled clinical trials comparing gene therapy (with or without standard treatment) with standard treatment (factor replacement) or other 'curative' treatment such as stem cell transplantation for individuals with haemophilia A or B of all ages who do not have inhibitors to factor VIII or IX.
Data collection and analysis
No trials of gene therapy for haemophilia matching the inclusion criteria were identified.
Main results
No trials of gene therapy for haemophilia matching the inclusion criteria were identified.
Authors' conclusions
No randomised or quasi‐randomised clinical trials of gene therapy for haemophilia were identified. Thus, we are unable to determine the safety and efficacy of gene therapy for haemophilia. Gene therapy for haemophilia is still in clinical investigation and there is a need for well‐designed clinical trials to assess the long‐term feasibility, success and risks of gene therapy for people with haemophilia.
Plain language summary
Gene therapy for haemophilia
Review question
We reviewed the evidence about the safety and effectiveness of gene therapy for treating people with haemophilia A or haemophilia B.
Background
Haemophilia is a bleeding disorder caused by defective genes that produce abnormal blood clotting proteins. Gene therapy modifies or replaces these defective genes with normal ones. We looked for trials that used this approach of modifying or replacing defective genes for producing normal blood clotting proteins for the treatment of haemophilia. This is an update of a published Cochrane Review.
Search date
The evidence is current to: 17 April 2020.
Key results
We found no trials to provide reliable evidence about the risks or benefits of gene therapy for haemophilia. There is a need for trials that assess the long‐term feasibility, success and risks of gene therapy for people with haemophilia.
Background
Description of the condition
Haemophilia is an X‐linked bleeding disorder characterized by spontaneous or provoked, often uncontrolled, bleeding into joints, muscles and other soft tissues, causing significant pain, swelling and permanent damage if left untreated. Haemophilia is caused by a genetic deficiency of specific proteins in the blood called clotting factors. Haemophilia A is caused by an inherited deficiency of factor VIII and haemophilia B is caused by a deficiency of factor IX. The incidence of haemophilia A is around 1 per 5000 to 10,000 male births, and about 1 per 60,000 male births for haemophilia B. Approximately 80% of affected children are born deficient in factor VIII (haemophilia A) and 20% are deficient in factor IX (haemophilia B) (Bi 1995; Wong 2011). Haemophilia is inherited in an X‐linked recessive manner. Hence, haemophilia occurs almost exclusively in males, but can also occur in some carrier females due to variances in inactivation of X chromosomes, or females with homozygosity or Turner syndrome.
The goal in managing patients with haemophilia is to control both the frequency and severity of bleeding episodes and ultimately to prevent permanent joint damage and death in severe cases. Replacement of the missing clotting factor from exogenous sources is the mainstay of therapy in haemophilia. Before World War II, treatment of haemophilia was limited to the transfusion of whole blood or fresh plasma. The first clotting factor concentrate was discovered in the 1960s in the form of cryoprecipitate. In the late 1960s, plasma‐derived clotting factors were isolated which created a paradigm shift in the management of haemophilia (Brinkhous 1968; Webster 1965). By 1980, the commercially available freeze‐dried factor VIII and IX concentrates increased the average lifespan of people with haemophilia from a mere 27 years in the 1940s to 60 years (Evatt 2006; Wong 2011). Early factor VIII and IX concentrates were derived from pooled human plasma from up to 20,000 donors.
Although hepatitis B and C were known risks of pooled human serum derived factor concentrates, they were considered acceptable due to a drastic improvement in the quality of life of people with haemophilia (Kasper 1972; Makris 1990; Mannucci 1977). In 1982 came the first reports of people with haemophilia succumbing to acquired immune deficiency syndrome (AIDS) and only later it was realized that plasma‐derived factor concentrates were responsible for transmission of the still unidentified infectious agent (Aronstam 1993; Gill 1983; Goedert 1989). Nearly 90% of Americans with severe haemophilia were reported to be infected with HIV in the 1980s via contaminated pooled serum products (NHF 2013). HIV was isolated in early 1984 and by early 1985 heating of factor concentrates became standard practice to kill the virus before transfusion (Wong 2011). Subsequently, several safeguards were employed to prevent donor derived factor transfusion induced infections, such as: donor screening; chromatographic purification; and viral inactivation (Wong 2011). Fortunately, since 1986, there have been no reported cases of HIV transmission through factor concentrates at least in the USA (NHF 2013). The human factor IX gene was cloned in 1982, followed by the production of human rFIX in Chinese hamster ovary cells (Anson 1984; Choo 1982). Factor VIII was cloned and sequenced in 1984 (Gitschier 1984; Toole 1984) and the first recombinant factor VIII (rFVIII) product was approved for clinical use in 1992 (Wong 2011).
The initial treatment modality was episodic or 'on demand' replacement of the missing factor as needed, after the onset of bleeding. However, several studies have shown that prophylactic administration of clotting factor concentrates to prevent bleeding episodes preemptively is more beneficial than episodic administration (Iorio 2011; Manco‐Johnson 2007). The currently used adjuvant therapies include the use of antifibrinolytic agents (in both haemophilia A and B) and desmopressin (in mild and moderate haemophilia A only). The antifibrinolytic agents such as aminocaproic acid and tranexamic acid exert their effect by inhibiting the proteolytic activity of plasmin and, therefore, inhibiting fibrinolysis (Wong 2011). Desmopressin works mainly by releasing von Willebrand factor from its storage sites and stabilizing the already present factor VIII in the plasma (Castaman 2008; Leissinger 2001).
Description of the intervention
It was first shown in 1989 that human factor IX could be synthesized and secreted into the circulation of laboratory animals after transplantation of genetically modified human fibroblasts using a human factor IX cDNA containing retrovirus (Nathwani 2004; Palmer 1989). Since then, several translational approaches have been developed for the clinical application of gene therapy in haemophilia, but most of these only resulted in transient benefits (Manno 2003; Manno 2006; Powell 2003; Roth 2001; VandenDriessche 1999; Xu 2003). Finally, in 2011, a new adeno‐associated virus (AAV)‐derived vector for factor IX gene transduction that could directly be given to a patient by a peripheral vein infusion was developed (Nathwani 2006; Nathwani 2011; Tuddenham 2012; Nathwani 2014). Subsequently, various AAV and lentivirus (LV)‐based gene therapy approaches have been developed and are currently in clinical investigation.
Gene therapy is not without risk. The introduction of new genetic material via virus‐derived vectors may cause unpredictable outcomes due to the alteration of the host genetic material (insertional mutagenesis) (Hacein‐Bey‐Abina 2003; Miller 2005; Nakai 2005). There is a theoretical risk that the viral vector may regain its potential to produce new viral particles, or may cause harmful immune reactions (High 2011; Manno 2003; Mingozzi 2011). Also of concern is the development of neutralizing antibodies to factor VIII or IX after gene therapy and it may render the gene product inactive in such patients. Lastly, T‐cell mediated immune response by the immunocompetent host against the transgene, its product proteins, or the viral capsid can be potentially harmful for the recipients of the gene therapy (Nathwani 2011; Nathwani 2014). All these potential adverse effects, as well as the long‐term safety of gene therapy are yet to be evaluated in a large cohort for a sufficiently long period of time.
How the intervention might work
The currently available standard treatment for haemophilia is lifelong clotting factor replacement, a regimen that is both expensive and is not easily available in many countries (Iorio 2011; Ponder 2008). Worldwide, about 75% of haemophilia patients do not have access to adequate care (Ponder 2008; WFH 2013). Due to the short half‐life of the clotting factors in the blood, replacement is required usually every other day for haemophilia A and every two to three days for haemophilia B. Some of the recently approved factor formulations with an extended half life allow the convenience of once a week or once every other week dosing. These factor products not yet available in most countries. The frequency and mode of administration (intravenous (IV)) of these treatments poses a wide range of challenges to people affected with haemophilia and their families (need for IV catheters and related complications such as infections and thrombosis) (Santagostino 2010).
At USD 1 per unit of recombinant factor VIII, the annual cost of on‐demand factor replacement therapy for a single 50 kg adult patient of haemophilia is USD 150,000 in the United States of America (Manco‐Johnson 2007; Ponder 2011). This cost increases to about USD 300,000 for prophylactic therapy. This amounts to an individual lifetime cost of over USD 20 million in factor replacement costs alone (Ponder 2011). Such an exorbitantly expensive therapy is neither available frequently, nor affordable to almost 75% of the people with haemophilia in the world who live in low‐ and middle‐income countries (Ponder 2008; Ponder 2011; WFH 2013). These individuals continue to have significant morbidities and die young.
In comparison to the above treatment modalities, gene therapy holds substantial promise. The key advantage being that potentially a one time intervention can be permanently curative. This intervention will lead to complete avoidance of the need for IV infusions, reduced hospital visits, a decrease in the use of other interventions and their side effects and ultimately reduced costs (Nathwani 2011; Nathwani 2014). There is no commercially available gene therapy product for haemophilia currently. If the commercial cost of such a product was assigned to be USD 850,000 based on the cost of the only other gene therapy product (for a genetic cause of retinal dystrophy that leads to vision loss) that is currently available, it is estimated that gene therapy for the treatment of severe haemophilia would still be cost saving compared to prophylactic therapy (Machin 2018). Such a therapy would undoubtedly be life changing for people with haemophilia.
Why it is important to do this review
Gene therapy holds significant promise of a substantially better treatment modality. Apart from that, gene therapy offers the potential of a lifetime cure, a better quality of life and freedom from various related morbidities. Theoretically, once gene therapy is administered and achieves factor levels in the normal range, the affected individual will be asymptomatic with respect to haemophilia. There are still doubts with regards to long‐term sustenance of the effects, unintended consequences or adverse effects and the costs of therapy. We aim to conduct this review in the hopes of answering some of the above questions, specifically in terms of the benefits and safety of gene therapy in comparison to standard treatment in people affected with haemophilia. At this stage, when only phase 1 and 2 studies have been performed, this review is aimed at being a comprehensive resource about the available best evidence and status of ongoing and future research and trials in gene therapy for haemophilia. This is an update of a previously published Cochrane Review (Sharma 2016).
Objectives
To evaluate the safety and efficacy of gene therapy in the treatment of people affected with haemophilia A or haemophilia B.
Methods
Criteria for considering studies for this review
Types of studies
Randomised or quasi‐randomised clinical trials including controlled clinical trials.
Types of participants
Individuals with haemophilia A or B of all ages who do not have inhibitors to factor VIII or IX.
Types of interventions
Gene therapy (with or without standard treatment) compared with the current standard treatment (regular or long‐acting factor replacement given on an on‐demand schedule or prophylactically) or other 'curative' treatment such as liver transplantation.
Types of outcome measures
Primary outcomes
Bleeding episode(s) (bleeding frequency, number of bleeding episodes per year, or as reported in the trial)
Factor VIII or IX supplementation requirement (frequency as reported in the trials)
-
Serious adverse events (resulting in death or life threatening complications, inpatient hospitalisation, significant or permanent disability, or one that requires additional intervention to prevent permanent damage or disability)
Evidence of clotting factor antibody development
Evidence of organ toxicity
Evidence of tumour development
Secondary outcomes
-
Measures of haemostasis
Clotting factor plasma levels
Activated partial thromboplastin time (aPPT)
Non‐serious adverse events (any adverse event other than those mentioned above)
Joint damage (changes in: clinical joint function; orthopedic joint score; or radiologic joint score)
Quality of life (as measured by standardised instruments)
Search methods for identification of studies
We searched for all relevant published and unpublished trials without restrictions on language, year or publication status.
Electronic searches
We searched the Cystic Fibrosis & Genetic Disorders Group's Coagulopathies Trials Register using the terms: gene therapy AND haemophilia*.
The Coagulopathies Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of the Cochrane Library) and weekly searches of MEDLINE and the prospective handsearching of one journal ‐ Haemophilia. Unpublished work is identified by searching the abstract books of major conferences: the European Haematology Association conference; the American Society of Hematology conference; the British Society for Haematology Annual Scientific Meeting; the Congress of the World Federation of Hemophilia; the European Association for Haemophilia and Allied Disorders, the American Society of Gene and Cell Therapy and the International Society on Thrombosis and Haemostasis. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group's website.
Additionally, we directly searched PubMed and CENTRAL using a search strategy as described in appendices (Appendix 1; Appendix 2).
We also searched the following clinical trials registries: ClinicalTrials.gov; and the WHO ICTRP. For the full search strategies, please refer to the relevant appendix (Appendix 3). Date of the most recent search: 17 April 2020.
Date of the most recent search of the Cystic Fibrosis and Genetic Disorders Group's Coagulopathies Trials Register: 27 March 2020.
Date of the most recent search of PubMed, Embase, CENTRAL and any other resources as listed below: 17 April 2020.
Searching other resources
We searched the reference lists and contact authors of included trials, to identify any additional, potentially relevant trials for inclusion.
Data collection and analysis
Two authors (VS and AS) undertook searches for eligible studies (see Figure 1). We were unable to identify any randomised controlled trials eligible for inclusion in this review. As a result, we were unable to perform the selection process and data analyses as planned. In the future, if we identify any trials and include them in the review, we will adhere to the methods described in detail below in the remainder of this section.
1.
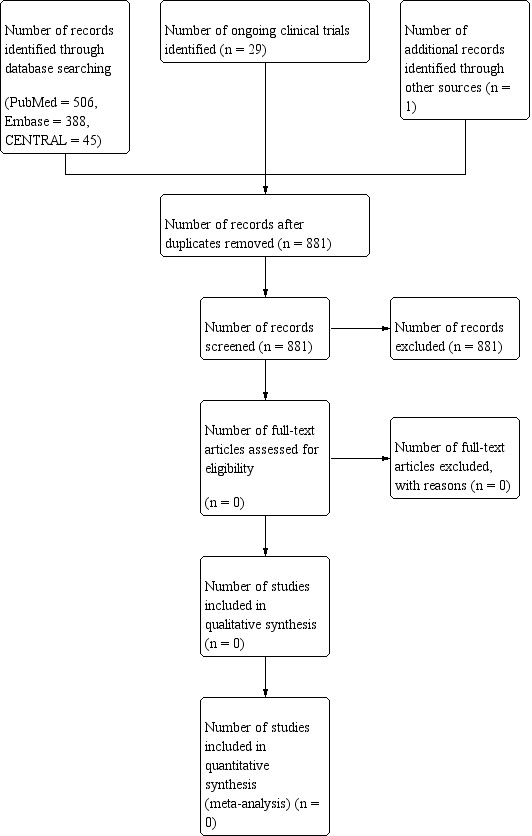
PRISMA flow diagram showing process of study selection.
Selection of studies
Search results from all sources will be put together in a reference manager software and duplicates will be excluded. Two review authors (AS and MEM) will then independently screen the search results using titles and abstracts to exclude irrelevant trials. We shall classify the remaining trials into included, excluded and those awaiting classification, using the predefined criteria for selecting trials. For those trials awaiting classification, we will evaluate the report(s) and then make the judgment regarding inclusion or exclusion. If we are not able to make a judgment from the report(s), we shall attempt to contact the trial authors and make the classification once we have obtained sufficient information. We will resolve any disagreements by a consensus meeting with the third author (VS). We shall clearly document the reasons for exclusion. We will also scrutinise each trial report to ensure that multiple publications from the same trial are included only once and all such reports will be linked to the original trial report in the reference list of included trials.
Data extraction and management
Two review authors (AS and MEM) will independently extract the data from the trials considered for inclusion using a structured data extraction form. We will resolve any disagreements by discussion and, in any cases where there are persisting disagreements, we will seek arbitration by another author (UR).
We will extract the following data.
General information: title; authors; source; country; year of publication; setting.
Trials characteristics: design; method of randomisation; allocation concealment; blinding of outcome assessors.
Patients: inclusion and exclusion criteria; underlying disease; sample size; baseline characteristics of patients; losses to follow up.
Interventions: type of vector; dose.
Additional treatment: immunosuppressive therapy.
Outcomes: as mentioned above (primary and secondary outcomes).
If the full text versions do not provide sufficient information, we will contact the corresponding author for further details.
Assessment of risk of bias in included studies
Two review authors (AS and MEM) will independently assess the risk of bias of the included trials on the basis of generation of the randomisation sequence, allocation concealment, blinding (of the participants, personnel and outcome assessors), attrition bias, and selective outcome reporting. We will assign judgments on the above domains of risk of bias into high, low and unclear, based on table 8.5d of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). We aim to resolve any disagreements by referring to the trial report and, if necessary, through arbitration with two other authors (VS and UR).
Measures of treatment effect
Different types of data will be analysed as below.
Rate data
When an outcome is measured in terms of rates we shall perform the analysis using rate ratios (RR) with their 95% confidence intervals (95% CI).
Continuous data
When outcome data are continuous, we shall extract means and standard deviations (SD). If the outcomes are reported using different continuous scales we shall use the standardized mean difference (SMD) and their associated 95% CIs to perform the analysis.
Dichotomous data
We shall extract the number of events and the total number of people reporting outcomes and analyse them using risk ratios (RR) and their associated 95% CI.
Unit of analysis issues
Trials with multiple treatment groups
In cases of trials with multiple treatment groups we shall ensure that a participant group is not represented more than once in the same analysis. We shall either combine groups to make pair wise comparisons if possible, or exclude groups by making a decision on which group is most clinically relevant and extract the data only for that group.
Cluster‐randomised trials
We do not expect to find cluster‐randomised trials for this intervention. However, if we do find such trials which report data adjusted for design effect, we shall use the information as such. If the effect estimates are not adjusted, we shall extract the intra cluster correlation coefficient (ICC), if provided in the trial report, or contact the authors for the same and adjust the effect estimate for the design effect.
Cross‐over trials
We do not expect to find cross‐over trials as successful gene therapy is irreversible.
Dealing with missing data
We will contact the primary investigators if information is missing or if there are unclear outcome data, summary data or individual data. We shall extract data for the number of people randomised and the number analysed for each outcome. If there is a difference, we shall calculate the percentage change and report it as loss to follow up.
For missing data for dichotomous outcomes, we will perform a per protocol analysis on the data.
For outcomes with continuous data that are missing SDs, we will either calculate these from other available data such as standard errors (SE), or will impute them, if possible. We shall impute SDs on the basis of SDs for the same outcome using the same scale, from within our review or from other similar studies, reviews or meta‐analyses (Higgins 2011b).
Assessment of heterogeneity
We shall visually examine a forest plots for overlapping confidence intervals and use the Chi² test for homogeneity and the I² test for heterogeneity, to estimate the contribution of true inter trial variability. The Chi² statistic will be considered to represent significant heterogeneity at a level of 10% significance (P value < 0.1). We will use an I² statistic value of less than 40% to represent mild heterogeneity that can be ignored; 30% to 60% to represent moderate and 50% to 90% significant heterogeneity, the reasons for which we shall try to explore by subgroups. We shall consider an I² value of 75% or greater as substantial heterogeneity, in which case we shall not perform meta‐analysis.
Assessment of reporting biases
We shall assess for publication bias using funnel plots (provided that there are at least 10 included trials in an analysis). When funnel plots show asymmetry, we shall consider publication bias as a possible reason (Sterne 2011).
Data synthesis
We shall attempt to pool results only when there is no clinical heterogeneity and when there is no substantial statistical heterogeneity (I² < 75%). When heterogeneity is present we shall analyse it using the random‐effects model. If we have studies reporting effect estimates without summary data for each group, we shall attempt to pool the effect estimates using generic inverse variance method. We shall attempt to pool data of outcomes reported at the same point of follow up across trials. If this is not possible we shall pool trial data at one, two and five years.
If meta‐analysis is not possible, we shall perform a descriptive, qualitative critical appraisal of the outcome information from the included trials.
Subgroup analysis and investigation of heterogeneity
In case we observe moderate or substantial heterogeneity in any of the analyses, we plan to explore the possible causes by undertaking the following subgroup analyses:
age (previously treated paediatric patients, previously untreated paediatric patients and adult patients, paediatric patients will be defined as those less than 18 years of age);
type of haemophilia (haemophilia A, haemophilia B);
gene therapy technique (ex vivo or in vivo transduction);
vector used (different types of vectors used, e.g. AAV8 viral vector),
hepatitis C status (positive, negative).
We would like to compare, if possible, all outcomes following gene therapy in younger versus older individuals. This is because older individuals undergoing gene therapy may already have significant co‐morbidities from the ongoing disease process. Method and site of cellular transduction can also vary the adverse event profile and if there are adequate numbers of trials available then we will do a subgroup analysis comparing the different methods. If one particular approach seems to have more adverse effects than the other then we will conduct the meta‐analysis both with and without the given approach and report the result separately.
Sensitivity analysis
We will perform sensitivity analyses to assess the robustness of the review’s results by repeating the analysis after excluding studies with clearly a high risk of bias (e.g. inadequate allocation of concealment, randomisation, or blinding, or high attrition or missing data), provided there are sufficient numbers of studies to include.
Summary of findings and the quality of the evidence (GRADE)
In a post hoc change, we aim, in future updates when trials are included, to present summary of findings tables for each comparison included.
We aim to report on all seven outcomes as pre‐defined in 'Types of outcome measures'.
We will determine the quality of the evidence using the GRADE approach; and downgrade evidence in the presence of a high risk of bias in at least one trial, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results, high probability of publication bias. We will downgrade evidence by one level if we consider the limitation to be serious and by two levels if very serious.
Results
Description of studies
No trials were found that were eligible for inclusion in this review.
Results of the search
The search strategies used as described in the appendices below yielded a total of 881 unique records. Records were screened by their title, or if unclear, by the abstract, to identify if they matched the inclusion criteria. None of the records matched the inclusion criteria for this review and hence were not included.
Risk of bias in included studies
No trials were eligible for inclusion in the review.
Effects of interventions
No trials were eligible for inclusion in the review.
Discussion
Gene therapy involves making a change in the genetic material of certain target cells in the patient's body by means of a vector (usually modified viruses) or gene editing in such a way that the changed genetic material is functional and produces an adequate end product which corrects the underlying defect of the disease. In haemophilia, recombinant genetic material is introduced into the target cells or defective genes are modified to produce clotting factors deficient in the disease. The comprehensive search used in this review identified no eligible trials comparing gene therapy to currently recommended standard treatment, and hence at this point there is no robust evidence to support or refute the effectiveness and safety of gene therapy for people with haemophilia.
Although no randomised controlled trials were found that are eligible for inclusion in this review which compared gene therapy to factor replacement head‐to‐head, there are several ongoing phase 1 and 2 studies evaluating various factor IX or factor VIII gene therapy products. Several clinical trials have enrolled or are currently enrolling patients with haemophilia B (NCT01687608; NCT00979238; NCT02484092; NCT02396342; NCT03369444; NCT03569891), studying the safety or efficacy of various viral vector‐transgene constructs for the replacement of the defective factor IX gene. These studies have been using vectors derived from different adeno‐associated virus (AAV) serotypes for transfer of the gene product into the relevant human tissue. Depending upon the transgene delivered by the vector in these studies, successful treatment and the achievement of factor IX levels ranging from 2% to greater than 50% have been reported in the study participants (George 2016; George 2017; Miesbach 2018; Nathwani 2011; Nathwani 2014). An increase in factor IX level more than 50% is certainly enough to alleviate the symptoms of haemophilia, an increase of mere 2% also does improve the phenotype and reduces the requirement of factor replacement. These studies have reported a decrease in annual use of factor IX concentrate as well as a concomitant decrease in bleeding episodes. These studies also reported transient increases in markers of liver inflammation in a proportion of participants. This transient elevation of liver enzymes was observed early after vector infusion and resolved in all cases with a short course of oral corticosteroid therapy. No other significant toxicity has been observed to date following AAV‐mediated gene therapy in haemophilia. In addition to these trials, a phase 1 study (NCT02695160) that utilizes a novel gene editing technique using zinc finger nucleases has started recruiting participants. If successful, this technique will be a promising alternative to the approach of introducing a transgene to cure haemophilia, as discussed above.
The use of AAV vectors in gene therapy for haemophilia A has lagged behind due to challenges associated with the large size and inefficient expression of the human factor VIII coding sequence. However, a single phase 1/2 clinical trial studying the efficacy and safety of escalating doses of an AAV5 gene therapy product for haemophilia A recently concluded enrolling participants (NCT02576795, Rangarajan 2017). In their results, the investigators reported achieving a median factor VIII activity of 77% with a range of 19‐164% after a single infusion of vector at 1 year after infusion (Rangarajan 2017). While this is the first report of a successful gene therapy product for haemophilia A in humans, it is certainly not the only clinical trial. There are now at least six independent agents under clinical investigation in phase 1/2 clinical trials including approaches using AAV‐derived vectors and lentiviral vectors (NCT03734588; NCT03001830; NCT03061201; NCT03370172; NCT03818763; NCT03217032; NCT03588299; NCT03003533)
Despite this initial success, long‐term safety of these gene transfer methods is yet to be ascertained. As a new paradigm in the treatment of haemophilia, gene therapy will need to undergo long‐term follow‐up studies to demonstrate sustained efficacy and affordability. Long‐term safety of the gene therapy vectors in terms of insertional mutagenesis or host immune response and tolerance, will have to be demonstrated. Availability of newer factor concentrates, with much longer half‐lives, requiring less frequent infusions as compared to the existing factor concentrates, and other non‐factor products are changing the standard of patient care already (Carcao 2014; Konkle 2015; Young 2015; Powell 2013; Santagostino 2016; Mahlangu 2018). New drugs have been developed which have shown promise in the treatment of cystic fibrosis and Duchenne and Becker muscular dystrophy by preventing premature termination of gene translation due to nonsense mutations (which produce premature stop codons) (NCT00803205; NCT00847379). Similar drugs may be used for treating haemophilia caused by nonsense mutations in the future (Hamed 2006; NCT00947193). These new therapies, in conjunction with other advances in the field of haemophilia, may obviate the need for gene therapy. Conversely, gene therapy may prove to be a safe, affordable, convenient and sustainable treatment for patients with haemophilia. Future studies will hopefully form the basis for an objective assessment of the benefits versus the risks of gene therapy over the current standard of care in haemophilia.
Authors' conclusions
Implications for practice.
No randomised controlled trials of gene therapy for haemophilia were found for inclusion in this review. A few first‐in‐human studies of gene therapy in haemophilia have been conducted with limited short‐term success; however, long‐term safety and efficacy data are lacking. It is yet to be known whether one‐time infusion of these viral vectors is successful in 'curing' this disease or several infusions over a period of time are needed to control the symptoms. Further, the safety and economic burden of this technique vis‐a‐vis current standard of care will only be known once the previous questions are answered. We are currently unable to draw any conclusions regarding the effects of gene therapy for haemophilia.
Implications for research.
This systematic review has identified the need for further well‐designed and long‐term clinical trials to assess the feasibility, benefits and risks of gene therapy for haemophilia. The primary outcome for these trials would be reduction in symptoms or factor infusion requirement for the haemophilia patients. Also, because of the potential side effects of using viruses as vectors in gene therapy, long‐term follow up studies of existing vectors and further progress in new non‐viral vector development is necessary. Hence, this review identifies areas of research involving safety, efficacy, long‐term follow up and economic burden of gene therapy compared to current standard of care.
What's new
| Date | Event | Description |
|---|---|---|
| 17 April 2020 | New search has been performed | For this update, we re‐ran the literature searches, but no eligible trials were identified for inclusion in the review. There have been some new references added to the 'Additional references' section which have been referenced in the 'Background' and 'Discussion' sections of the review. |
| 7 April 2020 | New citation required but conclusions have not changed | Minor changes have been made throughout several sections of the review and the methods have been updated regarding 'Summary of findings' table(s). |
History
Protocol first published: Issue 11, 2013 Review first published: Issue 11, 2014
| Date | Event | Description |
|---|---|---|
| 16 December 2016 | New citation required but conclusions have not changed | Minor changes have been made throughout this update. |
| 16 December 2016 | New search has been performed | A new search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register did not identify any potentially relevant trials for inclusion in this update. |
Acknowledgements
We would like to thank the South Asian Cochrane Network & Centre for providing logistical and technical support. We are also grateful to the Cochrane Cystic Fibrosis and Genetic Disorders Group for their support throughout the development of this review. Lastly, we would also like to acknowledge Jessica Neely, MD and Sasank Kalipatnapu, MBBS for their contributions to the original review (Sharma 2014).
Appendices
Appendix 1. PubMed search strategy
#1 randomized controlled trial [pt] #2 controlled clinical trial [pt] #3 randomized [tiab] #4 placebo [tiab] #5 drug therapy [sh] #6 randomly [tiab] #7 trial [tiab] #8 groups [tiab] #9 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 #10 animals [mh] NOT humans [mh] #11 #9 NOT #10 #12 (gene or genetic) and (therapy or therapies) and (hemophilia* or haemophilia*) #13 #11 AND #12
Note: Lines #1‐#11 are the Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE: sensitivity‐ and precision‐maximizing version (2008 revision); PubMed format.
PubMed searched via NCBI PubMed website www.ncbi.nlm.nih.gov/pubmed/
Appendix 2. CENTRAL search strategy
(gene or genetic) and (therapy or therapies) and (hemophilia* or haemophilia*)
Note: CENTRAL searched via the Cochrane Library (//crso.cochrane.org/)
Appendix 3. Search of trial registries
| Registry | Search terms |
| ClinicalTrials.gov | Search terms: (hemophilia OR haemophilia) AND gene therapy |
| WHO ICTRP | hemophilia AND gene therapy |
Differences between protocol and review
The 'Summary of findings and the quality of the evidence (GRADE)' section of the review has been added at the 2020 update.
Contributions of authors
| Roles and responsibilities | |
| Task | Who will undertake this task? |
| Protocol stage: draft the protocol | AS, MEM |
| Review stage: select which trials to include (2 + 1) | AS, MEM, VS |
| Review stage: extract data from trials (2 + 1) | AS, MEM, UR |
| Review stage: enter data into RevMan | AS |
| Review stage: carry out the analysis | AS, MEM |
| Review stage: interpret the analysis | AS, MEM, UR |
| Review stage: draft the final review | AS, UR |
| Update stage: update the review | AS, MEM, VS, UR |
Sources of support
Internal sources
No sources of support supplied
External sources
Cochrane Cystic Fibrosis & Genetic Disorders Group, UK
-
National Institute for Health Research, UK
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
-
St. Jude and ALSAC, USA
This work was supported by St. Jude Children’s Research Hospital and American Lebanese Syrian Associated Charities (ALSAC).
Declarations of interest
Akshay Sharma: none known. Manu Easow Mathew: none known. Vasumathi Sriganesh: is the Founder and Hon CEO of Quality Medical (QMed) Knowledge Foundation, a not‐for‐profit Trust in Mumbai, India. Through the Foundation she and her colleagues offer online courses, training and support for literature search, referencing and writing activities to health sciences students and professionals. Ulrike M Reiss: none known.
New search for studies and content updated (no change to conclusions)
References
Additional references
Anson 1984
- Anson DS, Choo KH, Rees DJ, Giannelli F, Gould K, Huddleston JA, et al. The gene structure of human anti-haemophilic factor IX. EMBO Journal 1984;3:1053-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Aronstam 1993
- Aronstam A, Congard B, Evans DI, Gazengel CF, Herberg U, Hill FG, et al. HIV infection in haemophilia--a European cohort. Archives of Disease in Childhood 1993;68(4):521-4. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bi 1995
- Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH Jr. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nature Genetics 1995;10(1):119-21. [DOI] [PubMed] [Google Scholar]
Brinkhous 1968
- Brinkhous KM, Shanbrom E, Roberts HR, Webster WP, Fekete L, Wagner RH. A new high-potency glycine-precipitated antihemophilic factor (AHF) concentrate. Treatment of classical hemophilia and hemophilia with inhibitors. JAMA 1968;205(9):613-7. [PubMed] [Google Scholar]
Carcao 2014
- Carcao M. Changing paradigm of prophylaxis with longer acting factor concentrates. Haemophilia 2014;20 Suppl 4:99-105. [PMID: ] [DOI] [PubMed] [Google Scholar]
Castaman 2008
- Castaman G. Desmopressin for the treatment of haemophilia. Haemophilia 2008;14 Suppl 1:15-20. [DOI] [PubMed] [Google Scholar]
Choo 1982
- Choo KH, Gould KG, Rees DJ, Brownlee GG. Molecular cloning of the gene for human anti-haemophilic factor IX. Nature 1982;299(5879):178-80. [DOI] [PubMed] [Google Scholar]
Evatt 2006
- Evatt BL. The tragic history of AIDS in the hemophilia population, 1982-1984. Journal of Thrombosis and Haemostasis 2006;4(11):2295-301. [DOI] [PubMed] [Google Scholar]
George 2016
- George LA, Sullivan S, Teitel J, Cuker A, Luk A, Wright F, et al. Preliminary results of a phase 1/2 trial of SPK-9001, a hyperactive FIX variant delivered by a novel capsid, demonstrate consistent factor IX activity levels at the lowest dose cohort. Haemophilia 2016;22(Suppl 4):151-2. [DOI: 10.1111/hae.13068] [DOI] [Google Scholar]
George 2017
- George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, Cuker A, et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. New England Journal of Medicine 2017;377(23):2215-27. [DOI: 10.1056/NEJMoa1708538] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gill 1983
- Gill JC, Menitove JE, Wheeler D, Aster RH, Montgomery RR. Generalized lymphadenopathy and T cell abnormalities in hemophilia A. Journal of Pediatrics 1983;103(1):18-22. [PMID: ] [DOI] [PubMed] [Google Scholar]
Gitschier 1984
- Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, Eaton DH, et al. Characterization of the human factor VIII gene. Nature 1984;312:326-30. [DOI] [PubMed] [Google Scholar]
Goedert 1989
- Goedert JJ, Kessler CM, Aledort LM, Biggar RJ, Andes WA, White GC 2nd, et al. A prospective study of human immunodeficiency virus type 1 infection and the development of AIDS in subjects with hemophilia. New England Journal of Medicine 1989;321(17):1141-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hacein‐Bey‐Abina 2003
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003;302(5644):415-9. [DOI] [PubMed] [Google Scholar]
Hamed 2006
- Hamed SA. Drug evaluation: PTC-124--a potential treatment of cystic fibrosis and Duchenne muscular dystrophy. IDrugs 2006;9(11):783-9. [PMID: ] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC, on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Higgins 2011b
- Higgins JPT, Deeks JJ, Altman DG. Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
High 2011
- High KA. Gene therapy for haemophilia: a long and winding road. Journal of Thrombosis and Haemostasis 2011;9 Suppl 1:2-11. [DOI] [PubMed] [Google Scholar]
Iorio 2011
- Iorio A, Marchesini E, Marcucci M, Stobart K, Chan AK. Clotting factor concentrates given to prevent bleeding and bleeding-related complications in people with hemophilia A or B. Cochrane Database of Systematic Reviews 2011, Issue 9. [DOI: 10.1002/14651858.CD003429.pub4] [PMID: ] [DOI] [PubMed] [Google Scholar]
Kasper 1972
- Kasper CK, Kipnis SA. Hepatitis and clotting-factor concentrates. JAMA 1972;221(5):510. [PMID: ] [PubMed] [Google Scholar]
Konkle 2015
- Konkle BA, Stasyshyn O, Chowdary P, Bevan DH, Mant T, Shima M, et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood 2015;126(9):1078-85. [DOI: 10.1182/blood-2015-03-630897] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leissinger 2001
- Leissinger C, Becton D, Cornell C Jr, Cox Gill J. High-dose DDAVP intranasal spray (Stimate) for the prevention and treatment of bleeding in patients with mild haemophilia A, mild or moderate type 1 von Willebrand disease and symptomatic carriers of haemophilia A. Haemophilia 2001;7(3):258-66. [DOI] [PubMed] [Google Scholar]
Machin 2018
- Machin N, Ragni MV, Smith KJ. Gene therapy in hemophilia A: a cost-effectiveness analysis. Blood Advances 2018;2(14):1792-98. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mahlangu 2018
- Mahlangu J, Oldenburg J, Paz-Priel I, Negrier C, Niggli M, Mancuso ME, et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. New England Journal of Medicine 2018;379(9):811-22. [DOI: 10.1056/NEJMoa1803550] [PMID: ] [DOI] [PubMed] [Google Scholar]
Makris 1990
- Makris M, Preston FE, Triger DR, Underwood JC, Choo QL, Kuo G, et al. Hepatitis C antibody and chronic liver disease in haemophilia. Lancet 1990;335(8698):1117-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Manco‐Johnson 2007
- Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. New England Journal of Medicine 2007;357(6):535-44. [DOI] [PubMed] [Google Scholar]
Manno 2003
Manno 2006
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nature Medicine 2006;12(3):342-7. [DOI] [PubMed] [Google Scholar]
Mannucci 1977
- Mannucci PM, Ruggeri ZM, Pareti FI, Capitanio A. 1-Deamino-8-d-arginine vasopressin: a new pharmacological approach to the management of haemophilia and von Willebrands' diseases. Lancet 1977;1(8017):869-72. [PMID: ] [DOI] [PubMed] [Google Scholar]
Miesbach 2018
- Miesbach W, Meijer K, Coppens M, Kampmann P, Klamroth R, Schutgens R, et al. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood 2018;131(9):1022-31. [DOI: 10.1182/blood-2017-09-804419] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Miller 2005
- Miller DG, Trobridge GD, Petek LM, Jacobs MA, Kaul R, Russell DW. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. Journal of Virology 2005;79(17):11434-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mingozzi 2011
- Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nature Reviews. Genetics 2011;12(5):341-55. [DOI] [PubMed] [Google Scholar]
Nakai 2005
- Nakai H, Wu X, Fuess S, Storm TA, Munroe D, Montini E, et al. Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. Journal of Virology 2005;79(6):3606-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nathwani 2004
- Nathwani AC, Davidoff AM, Tuddenham EG. Prospects for gene therapy of haemophilia. Haemophilia 2004;10(4):309-18. [DOI] [PubMed] [Google Scholar]
Nathwani 2006
- Nathwani AC, Gray JT, Ng CY, Zhou J, Spence Y, Waddington SN, et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 2006;107(7):2653-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nathwani 2011
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. New England Journal of Medicine 2011;365(25):2357-65. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nathwani 2014
- Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. New England Journal of Medicine 2014;371(21):1994-2004. [DOI: 10.1056/NEJMoa1407309] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00803205
- NCT00803205. Study of ataluren (PTC124™) in cystic fibrosis. //clinicaltrials.gov/ct2/show/study/NCT00803205 (accessed 1 September 2014). [NCT00803205]
NCT00847379
- NCT00847379. Phase 2b extension study of ataluren (PTC124) in Duchenne/Becker muscular dystrophy (DMD/BMD). //clinicaltrials.gov/ct2/show/NCT00847379 (accessed 1 September 2014). [NCT00847379]
NCT00947193
- NCT00947193. Study of ataluren (PTC124®) in hemophilia A and B. //clinicaltrials.gov/ct2/show/NCT00947193 (accessed 1 September 2014). [NCT00947193]
NCT00979238
- NCT00979238. An open label dose-escalation study of a self complementary adeno-associated viral vector (scAAV 2/8-LP1-hFIXco) for gene transfer in hemophilia B. //clinicaltrials.gov/show/NCT00979238 (accessed 26 April 2014). [NCT00979238]
NCT01687608
- NCT01687608. A phase 1/2 open-label, single ascending dose trial of a self-complementing optimized adeno-associated virus serotype 8 factor IX gene therapy (AskBio009) in adults with hemophilia B. //clinicaltrials.gov/show/NCT01687608 (accessed 26 April 2014). [NCT01687608]
NCT02396342
- NCT02396342. Trial of AAV5-hFIX in Severe or Moderately Severe Hemophilia B. //clinicaltrials.gov/ct2/show/NCT02396342 (accessed 05 November 2016). [NCT02396342]
NCT02484092
- NCT02484092. A Gene Therapy Study for Hemophilia B. //clinicaltrials.gov/ct2/show/NCT02484092 (accessed 05 November 2016). [NCT02484092]
NCT02576795
- NCT02576795. Gene Therapy Study in Severe Haemophilia A Patients. //clinicaltrials.gov/ct2/show/NCT02576795 (accessed 05 November 2016). [NCT02576795]
NCT02695160
- NCT02695160. Ascending Dose Study of Genome Editing by the Zinc Finger Protein (ZFP) Therapeutic SB-FIX in Severe Hemophilia B. //clinicaltrials.gov/ct2/show/NCT02695160 (accessed 05 November 2016). [NCT02695160]
NCT03001830
- NCT03001830. Gene Therapy for Haemophilia A. //clinicaltrials.gov/ct2/show/NCT03001830 (accessed 25 January 2019). [NCT03001830]
NCT03003533
- NCT03003533. A Gene Transfer Study for Hemophilia A. //clinicaltrials.gov/ct2/show/NCT03003533 (accessed 25 January 2019). [NCT03003533]
NCT03061201
- NCT03061201. Dose-Ranging Study of Recombinant AAV2/6 Human Factor 8 Gene Therapy SB-525 in Subjects With Severe Hemophilia A. //clinicaltrials.gov/ct2/show/NCT03061201 (accessed 25 January 2019). [NCT03061201]
NCT03217032
- NCT03217032. Gene Modified Stem Cells for Hemophelia A and B. //clinicaltrials.gov/ct2/show/NCT03217032 (accessed 25 January 2019). [NCT03217032]
NCT03369444
- NCT03369444. A Factor IX Gene Therapy Study (FIX-GT). //clinicaltrials.gov/ct2/show/NCT03369444 (accessed 25 January 2019). [NCT03369444]
NCT03370172
- NCT03370172. Safety and Dose Escalation Study of an Adeno-Associated Viral Vector for Gene Transfer in Hemophilia A Subjects. //clinicaltrials.gov/ct2/show/NCT03370172 (accessed 25 January 2019). [NCT03370172]
NCT03569891
- NCT03569891. HOPE-B: Trial of AMT-061 in Severe or Moderately Severe Hemophilia B Patients. //clinicaltrials.gov/ct2/show/NCT03569891 (accessed 25 January 2019). [NCT03569891]
NCT03588299
- NCT03588299. Factor VIII Gene Therapy Study in Patients With Hemophilia A. //clinicaltrials.gov/ct2/show/NCT03588299 (accessed 25 January 2019). [NCT03588299]
NCT03734588
- NCT03734588. Dose-finding Study of SPK-8016 Gene Therapy in Patients With Hemophilia A to Support Evaluation in Individuals With FVIII Inhibitors. //clinicaltrials.gov/ct2/show/NCT03734588 (accessed 25 January 2019). [NCT03734588]
NCT03818763
- NCT03818763. Gene Therapy Trial for Platelet Derived Factor VIII Production in Hemophilia A. //clinicaltrials.gov/ct2/show/NCT03818763 (accessed 25 January 2019). [NCT03818763]
NHF 2013
- National Hemophilia Foundation. Fast Facts. http://www.hemophilia.org/NHFWeb/MainPgs/MainNHF.aspx?menuid=259&contentid=476 (accessed 26 April 2014).
Palmer 1989
- Palmer TD, Thompson AR, Miller AD. Production of human factor IX in animals by genetically modified skin fibroblasts: potential therapy for hemophilia B. Blood 1989;73(2):438-45. [PubMed] [Google Scholar]
Ponder 2008
- Ponder KP, Srivastava A. Walk a mile in the moccasins of people with haemophilia. Haemophilia 2008;14(3):618-20. [DOI] [PubMed] [Google Scholar]
Ponder 2011
- Ponder KP. Merry christmas for patients with hemophilia B. New England Journal of Medicine 2011;365(25):2424-5. [DOI] [PubMed] [Google Scholar]
Powell 2003
- Powell JS, Ragni MV, White GC 2nd, Lusher JM, Hillman-Wiseman C, Moon TE, et al. Phase 1 trial of FVIII gene transfer for severe hemophilia A using a retroviral construct administered by peripheral intravenous infusion. Blood 2003;102(6):2038-45. [DOI] [PubMed] [Google Scholar]
Powell 2013
- Powell JS, Pasi KJ, Ragni MV, Ozelo MC, Valentino LA, Mahlangu JN, et al. Phase 3 study of recombinant factor IX Fc fusion protein in hemophilia B. New England Journal of Medicine 2013;369(24):2313-23. [DOI: 10.1056/NEJMoa1305074] [PMID: ] [DOI] [PubMed] [Google Scholar]
Rangarajan 2017
- Rangarajan S, Walsh L, Lester W, Perry D, Madan B, Laffan M, et al. AAV5-factor VIII gene transfer in severe hemophilia A. New England Journal of Medicine 2017;377(26):2519-30. [DOI: 10.1056/NEJMoa1708483] [PMID: ] [DOI] [PubMed] [Google Scholar]
Roth 2001
- Roth DA, Tawa NE Jr, O'Brien JM, Treco DA, Selden RF, Factor VIII Transkaryotic Therapy Study Group. Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. New England Journal of Medicine 2001;344(23):1735-42. [DOI] [PubMed] [Google Scholar]
Santagostino 2010
- Santagostino E, Mancuso ME. Venous access in haemophilic children: choice and management. Haemophilia 2010;16 Suppl 1:20-4. [DOI: 10.1111/j.1365-2516.2009.02156.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
Santagostino 2016
- Santagostino E, Martinowitz U, Lissitchkov T, Pan-Petesch B, Hanabusa H, Oldenburg J, et al. Long-acting recombinant coagulation factor IX albumin fusion protein (rIX-FP) in hemophilia B: results of a phase 3 trial. Blood 2016;127(14):1761-9. [DOI: 10.1182/blood-2015-09-669234] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D, on behalf of the Cochrane Bias Methods Group. Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook forSystematic Reviews of Interventions Version 5.1 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Toole 1984
- Toole JJ, Knopf JL, Wozney JM, Sultzman LA, Buecker JL, Pittman DD, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312(5992):342-7. [DOI] [PubMed] [Google Scholar]
Tuddenham 2012
- Tuddenham E. Gene therapy for haemophilia B. Haemophilia 2012;18 Suppl 4:13-7. [DOI] [PubMed] [Google Scholar]
VandenDriessche 1999
- VandenDriessche T, Vanslembrouck V, Goovaerts I, Zwinnen H, Vanderhaeghen ML, Collen D, et al. Long-term expression of human coagulation factor VIII and correction of hemophilia A after in vivo retroviral gene transfer in factor VIII-deficient mice. Proceedings of the National Academy of Sciences of the United States of America 1999;96(18):10379-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Webster 1965
- Webster WP, Roberts HR, Thelin GM, Wagner RH, Brinkhous KM. Clinical use of a new glycine-precipitated antihemophilic fraction. American Journal of the Medical Sciences 1965;250(6):643-51. [DOI] [PubMed] [Google Scholar]
WFH 2013
- World Federation of Hemophilia. Annual Report 2013. http://www1.wfh.org/publications/files/pdf-1578.pdf (accessed 02 December 2016).
Wong 2011
- Wong T, Recht M. Current options and new developments in the treatment of haemophilia. Drugs 2011;71(3):305-20. [DOI] [PubMed] [Google Scholar]
Xu 2003
- Xu L, Gao C, Sands MS, Cai SR, Nichols TC, Bellinger DA, et al. Neonatal or hepatocyte growth factor-potentiated adult gene therapy with a retroviral vector results in therapeutic levels of canine factor IX for hemophilia B. Blood 2003;101(10):3924-32. [DOI] [PubMed] [Google Scholar]
Young 2015
- Young G, Mahlangu J, Kulkarni R, Nolan B, Liesner R, Pasi J et al. Recombinant factor VIII Fc fusion protein for the prevention and treatment of bleeding in children with severe hemophilia A. Journal of Thrombosis and Haemostasis 2015;13(6):967-77. [DOI: 10.1111/jth.12911] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Sharma 2014
- Sharma A, Easow Mathew M, Sriganesh V, Neely JA, Kalipatnapu S. Gene therapy for haemophilia. Cochrane Database of Systematic Reviews 2014, Issue 11. [DOI: 10.1002/14651858.CD010822.pub2] [DOI] [PubMed] [Google Scholar]
Sharma 2016
- Sharma A, Mathew ME, Sriganesh V, Reiss UM. Gene therapy for haemophilia. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD010822.pub3] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]