


Abstract
Phosphorothioate modification is commonly introduced into therapeutic oligonucleotides, typically as a racemic mixture in which either of the two non-bridging phosphate oxygens is replaced by sulfur, which frequently increases affinities with proteins. Here, we used isothermal titration calorimetry and X-ray crystallography to investigate the thermodynamic and structural properties of the interaction between the primary DNA-binding domain (CUTr1) of transcription factor SATB1 and dodecamer DNAs with racemic phosphorothioate modifications at the six sites known to contact CUTr1 directly. For both the modified and unmodified DNAs, the binding reactions were enthalpy-driven at a moderate salt concentration (50 mM NaCl), while being entropy-driven at higher salt concentrations with reduced affinities. The phosphorothioate modifications lowered this susceptibility to salt, resulting in a significantly enhanced affinity at a higher salt concentration (200 mM NaCl), although only some DNA molecular species remained interacting with CUTr1. This was explained by unequal populations of the two diastereomers in the crystal structure of the complex of CUTr1 and the phosphorothioate-modified DNA. The preferred diastereomer formed more hydrogen bonds with the oxygen atoms and/or more hydrophobic contacts with the sulfur atoms than the other, revealing the origins of the enhanced affinity.
INTRODUCTION
Phosphorothioate and phosphorodithioate modifications are commonly introduced into chemically synthesized nucleic acids for therapeutic usages (1–4). In these modifications, one or both of the two non-bridging oxygens of the backbone phosphate are replaced by sulfur. The major benefits of introducing these modifications are that they enhance stability against endogenous nucleases and that they improve cellular uptake (4–6). In addition, it was repeatedly observed that the modifications enhanced binding affinities with proteins, by two to three orders of magnitude (3,4,7–12), which itself was highly desirable.
The interactions with proteins are important not only for decoy and aptamer nucleic acids, which mainly target specific proteins, but also for antisense oligonucleotide (ASO) and small interfering RNA (siRNA), which target RNAs in the cell. Namely, ASO and siRNA interact with ribonuclease H (RNase H) and Argonaute proteins in the RNA-induced silencing complex, respectively, for functioning (13,14), and also with numerous membrane and cytoplasmic proteins for cellular uptake and intracellular trafficking (15,16). It was indeed demonstrated that interactions of ASO with several cellular proteins required 10 or more of the phosphorothioate modifications within the ASO molecules (17). Nevertheless, these interactions with cellular proteins are sequence-nonspecific and have the promiscuous properties (4,18). Consequently, treatments with phosphorothioate-containing DNAs at high doses cause significant side effects (19,20).
It is, therefore, necessary to optimize the modification, so as to enhance the affinity with target molecule and to improve the efficiency in the cellular uptake, which should lead to the reduced toxicity by lowering doses. Efforts have been made to optimize the numbers and positions of phosphorothioate and additional 2′ modifications with regard to the protein interactions (17,21,22). It is especially noted that the chirality control of phosphorothioates in ASO enhanced interaction with RNase H and facilitated cleavage of target RNA (23), whereas it was also reported that it did not improve overall therapeutic profile of ASO (24).
To achieve target-specific affinity enhancement by the modifications, we need to understand the mechanism of their effects at the atomic level. There are several structural studies focused on the specific interactions of nucleic acids containing phosphorothioate or phosphorodithioate with proteins. It was observed for the Antennapedia homeodomain–DNA complex that replacement of oxygen by sulfur in the DNA at the contact site increased mobility of a hydrogen-bonded lysine residue and thereby enhanced the affinity in an entropy-driven manner (10,12). In contrast, for phosphorodithioate-modified siRNAs interacting with the Argonaute protein and for a restriction endonuclease catalyzing naturally occurring phosphorothioate DNAs, hydrophobic contacts between the sulfur atoms of phosphorodithioate and protein side chains were suggested to be the origin of the enhanced affinity (25,26). Also for an RNA aptamer, hydrophobic contacts between phosphorodithioate and protein side chains were observed in the structure, although the large polarizability of sulfur was regarded as the origin of the enhanced affinity (11). Thus, we noticed essential differences between these proposed mechanisms.
The phosphorothioate or phosphorodithioate nucleic acids used in the above-mentioned studies are stereochemically unique, although in most cases the phosphorothioate nucleic acids are racemic mixtures with regard to the modified positions in the phosphate group, where either of the two non-bridging oxygen atoms of a phosphate is replaced by sulfur. Therefore, it is worth investigating how such a racemic phosphorothioate affects the affinity with proteins. If an affinity-enhancing interaction arises from one of the two diastereomers, that diastereomer should be more preferentially bound to the target protein. We can then expect unequal populations of the diastereomers in the complex. This is, in turn, especially useful in understanding the cause of the affinity enhancement.
Here, we used transcription factor SATB1 (special AT-rich sequence binding protein 1), which was originally identified as a factor that binds to an AT-rich sequence in the matrix attachment region (MAR) of DNA (27). SATB1 regulates transcription of >1000 genes simultaneously and, therefore, is called ‘genome organizer’ (28). It recruits complexes of histone deacetylase or histone acetyltransferase to target MAR sites and triggers chromatin remodeling in the vicinity (29,30). Although the target systems controlled by SATB1 range widely, it is of particular therapeutic interest that expression of SATB1 is associated with the malignance of a variety of cancer species (28,31). For example, among breast cancer cell lines, those expressing SATB1 have more metastatic phenotypes than those that do not express SATB1, in which RNA interference against the SATB1 gene significantly reduced the phenotypes (28,31). Moreover, a decoy DNA possessing a sequence that can be specifically bound by SATB1 can also diminish the metastatic phenotypes (32).
SATB1 contains three major DNA-binding motifs, i.e. two CUT repeats (CUTr1 and CUTr2 from the N-terminus) and a homeodomain. Among them, CUTr1 is responsible for recognition of the target TAATA sequence appearing in many of the MAR regions, whereas CUTr2 has little DNA-binding activity and the homeodomain has sequence-nonspecific DNA-binding activity (33). We have determined the structures of the CUTr1 domain and the homeodomain by nuclear magnetic resonance and that of the complex of CUTr1 and a dodecamer DNA containing a TAATA sequence by crystallography (33–35). By examining the complex structure, we identified all the phosphate groups directly contacted by CUTr1.
In this study, we introduced racemic phosphorothioate modifications at the six phosphate groups that directly contact with CUTr1. Analysis by isothermal titration calorimetry (ITC) revealed that the modifications enhanced the affinity especially at a higher salt concentration, although only some DNA molecular species appeared to interact with CUTr1. We also observed in the crystal structure of the complex of CUTr1 and the phosphorothioate DNA that the population ratio of the two diastereomers significantly deviated from 1:1 at some sites. The preferred diastereomer formed more hydrogen bonds and/or hydrophobic contacts with the protein via oxygen and sulfur, respectively, than the other, which should be the major reason for the enhanced affinity.
MATERIALS AND METHODS
Sample preparation
DNA dodecamers with modification of phosphorothioate linkages (5′-GpsCpsTAATATATGC-3′/5′-GCATpsApsTpsApsTTAGC-3′; ‘ps’ stands for the position of phosphorothioate linkage) and those of the same sequences without the modification were chemically synthesized (Hokkaido System Science, Sapporo, Japan).
The CUTr1 domain of human SATB1 (Asn368–Leu452) containing additional four basic residues (Arg–Lys–Arg–Lys) at the C-terminus was produced by an Escherichia coli expression system and purified by chromatography as described previously (34,35). This fragment was used for our previous crystallography, because the four basic residues increase the net charge of CUTr1 from −1 to +3 at a neutral pH and enhance the affinity to DNAs (33,35). Hereafter, we refer to this fragment as ‘CUTr1’ unless otherwise stated.
ITC experiments
Interaction between SATB1-CUTr1 and DNAs was analyzed using a MicroCal VP-ITC calorimeter (Malvern, Worcestershire, UK) at 293 K as described previously (33). The buffer solution consisted of 50 mM sodium phosphate (pH 5.5) and 50–200 mM NaCl. Ten microliter quantities were repeatedly injected from a syringe containing ∼100 μM protein solution into the reaction cell of 1.427 mL volume containing 8–10 μM of dodecamer DNAs after degassing of the solutions. The resultant binding isotherms were analyzed using in-house programs as described previously (33).
Crystallography
Solutions of psDNA and SATB1-CUTr1 at a molar ratio 1.5:1.0 were subjected to crystallization by the sitting-drop vapor diffusion method at 293 K against 50 mM Tris–HCl (pH 8.5), 20% polyethylene glycol monomethyl ether 550 (Sigma-Aldrich), 20% ethylene glycol and 10 mM MgCl2. After one week, rod-like crystals with the longest axis of 0.2 mm were obtained.
Diffraction data up to 1.79 Å (dataset 1) and 2.16 Å (dataset 2) resolution were obtained under a nitrogen gas stream at 95 K on beamlines (NW12A and NE3A, respectively) at Photon Factory, High Energy Accelerator Research Organization (KEK; Tsukuba, Japan). The two diffraction datasets were processed with Mosfilm (36). The structure was determined by a molecular replacement method using MolRep (37) in the CCP4 program suite (38) with the structure of the complex of SATB1-CUTr1 and an unmodified DNA [Protein Data Bank (PDB) ID: 2O4A] (35) as the template model. The initial structure was built on dataset 2, while further refinement and model building were performed on dataset 1 using Refmac5 (39) and Coot (40), respectively.
For each phosphorothioate group, two alternative conformers corresponding to the diastereomers (Rp and Sp; Scheme 1) were set for the C3′, O3′, P, SP, OP, O5′ and C5′ atoms, since the conformation of the sugar–phosphate backbone may differ between the diastereomers. The relative occupancies of the conformers were determined as variables common to the above-mentioned atoms in the refinement process. The derived occupancy values were validated by the following index with regard to the constancy in B-factors for a rigid body, where values were derived after refinement with pre-fixed occupancies: ((BS(Rp) − BS(Sp))2 + (BO(Rp) − BO(Sp))2)1/2, where B(Rp) and B(Sp) stand for the B-factors in the two diastereomers, and the subscripts S and O indicate the sulfur and oxygen atoms, respectively.
Scheme 1.
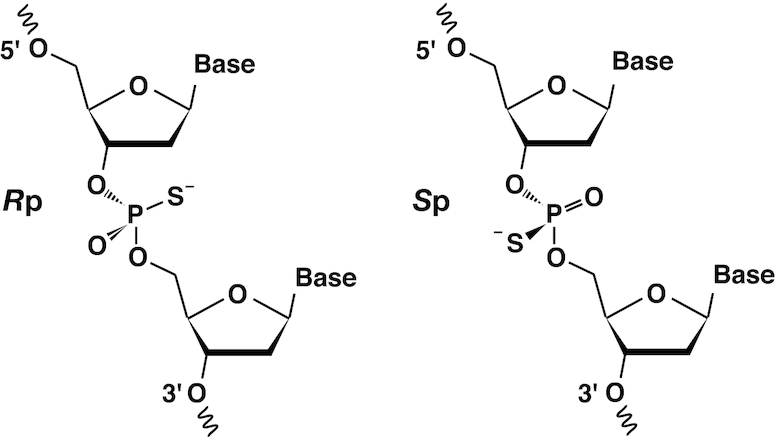
The Rp (left) and Sp (right) diastereomers of phosphorothioate.
The coordinate and structure factor files were deposited to the PDB under accession ID 6LFF. All drawings of 3D structures were rendered using PyMOL ver. 1.8 (Schrödinger, LLC, New York).
RESULTS AND DISCUSSION
ITC analyses on the interaction between SATB1-CUTr1 and DNAs
We analyzed the interaction between CUTr1 and DNAs with or without phosphorothioate modification (hereafter, psDNA and poDNA for the modified and unmodified DNAs, respectively) by ITC experiments (Figure 1, Table 1). The dodecamer DNAs used in this study contained a TAATA motif and possessed exactly the same sequence as that used in the previous crystallography for the CUTr1–DNA complex (35). For the modified DNA, we introduced the racemic phosphorothioate to the six sites that were known to be involved in direct contacts with CUTr1 as revealed in the crystal structure.
Figure 1.

ITC analyses on the interaction between SATB1-CUTr1 and DNA. Shown in the upper panels of the respective subfigures are calorimetric responses to titrations of CUTr1 to dodecamer DNAs containing a TAATA motif with phosphorothioate modifications at six protein-contacting sites (psDNA; A, B, C) or without the modifications (poDNA; D, E, F) (see Table 1). Salt concentrations are 50 mM (A, D), 100 mM (B, E) and 200 mM (C, F). In the corresponding lower panels, binding isotherms, i.e. relation between protein/DNA molar ratio and integrated heat per molar concentration of protein for the respective injections, are shown with fitting curves. Vertical broken lines show the apparent binding stoichiometry between protein and DNA.
Table 1.
ITC analysis on DNA binding of SATB1-CUTr1
| DNA sequencesa | NaClb | K A c | ΔGc | ΔHc | ΔSc | Apparent stoichiometry |
|---|---|---|---|---|---|---|
| (mM) | (106 M−1) | (kJ mol−1) | (kJ mol−1) | (10−3 kJ mol−1 K−1) | (protein/DNA)c | |
| 1. GpsCpsTAATATATGC/GCATpsApsTpsApsTTAGC | 50 | 1.2 ± 0.2 | −34.1 ± 0.4 | −41.2 ± 2.8 | −24 ± 10 | 0.79 ± 0.02 |
| 100 | 0.51 ± 0.06 | −32.0 ± 0.3 | −38.9 ± 1.9 | −23 ± 7 | 0.68 ± 0.02 | |
| 200 | 0.62 ± 0.12 | −32.5 ± 0.5 | −18.7 ± 1.2 | 47 ± 6 | 0.50 ± 0.07 | |
| 2. GCTAATATATGC/GCATATATTAGC | 50 | 0.78 ± 0.02 | −33.1 ± 0.06 | −32.3 ± 0.6 | 2.7 ± 1.7 | 0.95 ± 0.14 |
| 100 | 0.54 ± 0.07 | −32.1 ± 0.3 | −18.9 ± 1.6 | 45 ± 6 | 1.07 ± 0.10 | |
| 200 | NAd | NAd | NAd | NAd | NAd |
aSequences of the two complementary strands are shown in the 5′ → 3′ direction. The recognition motif TAATA/TATTA is underlined. The positions for the phosphorothioate modification are indicated with ‘ps’.
bFor the concentrations of Na+, 50 mM from the sodium phosphate buffer should be added to the values.
cAverage values and standard deviations from triplicate experiments are shown.
dHeat release was too small to be analyzed.
Clear exothermic reactions were observed upon binding of CUTr1 to psDNA or poDNA under the moderate salt condition (50 mM NaCl) (Figure 1A and D). The binding constant (KA) is higher for psDNA (1.2 × 106 M–1) than for poDNA (0.78 × 106 M–1) (Table 1). For psDNA, the enthalpy change (ΔH = –41.2 kJ mol–1) exceeded the free energy change (ΔG = –34.1 kJ mol–1) deduced from KA, which showed that the binding is enthalpy-driven. Also, for poDNA, ΔH (–32.3 kJ mol–1) accounts for a large portion of ΔG (–33.1 kJ mol–1), indicating enthalpy-driven binding. Comparison of these thermodynamic parameters indicated that the enhancement of KA by the phosphorothioate modification was ascribed to ΔH under the moderate salt condition.
When the salt concentration was increased to 100 mM, heat release was reduced with both psDNA and poDNA (Figure 1B and E). For poDNA, ΔH decreased in terms of the absolute value, while the contribution of entropy (–TΔS = –13.2 kJ mol–1) increased to be comparable to ΔH (–18.9 kJ mol–1, Table 1). At 200 mM NaCl, heat release for poDNA became too small to analyze. In contrast, it was still analyzable with psDNA at 200 mM salt. The thermodynamic parameters for psDNA at 200 mM NaCl were very similar to those for poDNA at 100 mM salt (Table 1), showing that psDNA is less susceptible to salt than poDNA. It is thus evident that the phosphorothioate modification enhances the binding affinity of CUTr1 to DNA at 200 mM NaCl, which is close to the physiological salt concentration.
It should be noteworthy that the apparent stoichiometry for binding of CUTr1 to psDNA was less than that to poDNA and further decreased as the salt concentration was increased (Figure 1, Table 1). This implies that some portion of the racemic psDNA consisting of 64 (26) stereochemically unique diastereomers was unreactive with CUTr1 but was excluded from the system. We should mention that ITC is based on the detection of heat release upon binding and that the respective diastereomers may differ in it. However, those with small heat release would also contribute to increase the binding stoichiometry, if they retain the affinity. Therefore, it is likely that the affinities of some diastereomers indeed decreased considerably in comparison with others.
The mechanism of sequence-specific DNA binding of CUTr1 has been explained by a combination of electrostatic interactions and hydrophobic contacts, where the former includes hydrogen bonds and salt bridges (35). In principle, the hydrogen bonds contribute to the enthalpy-driven reactions, while the hydrophobic contacts and salt bridges contribute to the entropy-driven reactions (41). These are indeed affected by the salt concentration, i.e. the electrostatic interactions are weakened, while the hydrophobic contacts are strengthened by salt (42). Taking all these observations together, the salt-induced change of binding mode from enthalpy-driven to entropy-driven is likely to correspond to the situation that hydrogen bonds dominated over hydrophobic contacts under the low-salt condition and vice versa under the high-salt condition. We performed crystallography to investigate these interactions in the light of the 3D structure.
Crystal structure of the CUTr1–psDNA complex
Crystals of the CUTr1–psDNA complex were obtained under a condition similar to that for the CUTr1–poDNA complex in the previous study (35). Two diffraction datasets on the same crystal were collected at synchrotron beamlines (Table 2, Supplementary Table S1). The structure was determined by the molecular replacement method using the crystal structure of the CUTr1–poDNA complex as a model.
Table 2.
Data collection and refinement statistics in crystallography
| Crystallographic dataa | |
| Space group | P31 |
| Unit cell | |
| a/b/c (Å) | 45.32/45.32/97.89 |
| α/β/γ (°) | 90.0/90.0/120.0 |
| Wavelength (Å) | 1.000 |
| Resolution range (outer shell) (Å) | 48.94–1.79 (1.89–1.79) |
| Total reflections | 132 085 |
| Unique reflections | 19 724 |
| Completeness (outer shell) (%) | 93.2 (100.0) |
| Redundancy (outer shell) | 6.7 (6.1) |
| R merge (outer shell) (%) | 10.5 (35.3) |
| Average I/σ(I) (outer shell) | 11.6 (4.8) |
| Refinement | |
| R work/Rfree (%) | 23.0/27.1 |
| RMSD from ideal values | |
| Bond length (Å) | 0.004 |
| Bond angle (°) | 1.2 |
| Ramachandran plot (%) | |
| Favored | 94.1 |
| Allowed | 5.9 |
| Average B-factors (Å2) (number of atoms) | |
| Protein | |
| Chain A | 45.9 (676) |
| Chain B | 43.1 (670) |
| DNA | |
| Chain C | 32.1 (242) |
| Chain D | 31.8 (271) |
| Water | 44.5 (69) |
aParameters for the dataset used in the final refinement (dataset 1) are shown (see the ‘Materials and Methods’ section). Those for the dataset used in the initial molecular replacement (dataset 2) are shown in Supplementary Data, Supplementary Table S1.
The crystal structure contains two molecules of the protein and one molecule of the double-stranded DNA in an asymmetric unit (Figure 2A). The two protein molecules bind to the major groove of the psDNA molecule in the standard B form. One of them, protein molecule 1, binds to the TAATA motif similarly to that in the CUTr1–poDNA complex (35) as expected, while the other, molecule 2, is positioned in a reversed orientation (Figure 2B). Molecule 1 retains most of the contacts to bases and phosphates of the DNA (Figure 2C). For molecule 2, many of the contacts to phosphate are retained, although some contacts to bases, i.e. Leu404 to a T base and Glu407 to a T base, are disrupted due to the lack of the bases at the expected position (Figure 2D). Because the six phosphorothioate groups contact only with molecule 1, the affinity enhancement by the modification should be attributed to the interactions with this molecule. The ITC results showing the enhanced affinity and apparent stoichiometry of ∼1.0 rather than 2.0 (Table 1), therefore, indicate that only molecule 1 binds to psDNA in solution. On the other hand, molecule 2, which has fewer sequence-specific contacts, is likely to be coincidental due to the crystal packing and the semi-palindromic sequence of the DNA.
Figure 2.

Crystal structure of the complex of SATB1-CUTr1 and psDNA. (A) Overall structure of the 2:1 complex of protein and DNA in a stereo view. Protein molecules 1 and 2 are shown in cyan and yellow, respectively. Also, DNA strands 1 and 2 (the former and latter sequences in Table 1) are shown in red and blue, respectively, where the phosphorothioates are shown by orange spheres. (B) Structure comparison of the present 2:1 complex, colored as in (A), and the 1:1 complex of SATB1-CUTr1 and poDNA (35), where the protein is shown in magenta and DNA strands are shown in orange and green. (C, D) Summary of contacts between protein molecule 1 (C) or molecule 2 (D) and psDNA drawn from the major groove side of DNA. DNA bases are numbered for strand 1 and the pairing bases in strand 2 are indicated with corresponding numbers with primes. Yellow and orange circles represent DNA phosphates and phosphorothioates, respectively (phosphorus and bridging/non-bridging oxygen and/or sulfur), while those in cyan with ‘W’ represent water molecules. Bases contacted by the protein are highlighted in green. Cyan, red and yellow lines represent hydrogen bonds (distance of hydrogen donor and acceptor <3.5 Å), hydrophobic contacts (C–C or C–S distance <4.5 Å, unless an adjacent N or O atom is closer to the counterpart) and salt bridges (N+–O− or N+–S− distance <5.0 Å), respectively, between protein and DNA. Contacts mediated by hydrogen bonds with the same water molecule are also indicated. Yellow broken circles indicate that contacts are formed with 5′ oxygens, although these terminals have no phosphorus.
The populations of the two diastereomers of racemic phosphorothioate (Rp and Sp; Scheme 1; two ensembles of diastereomers, more correctly for the nucleic acid), i.e. occupancies, were obtained by optimization as variable parameters during the refinement process (Figure 3). To validate these values, we also examined the constancy in the B-factors at different population ratios, essentially based on the rigid-body assumption (see the ‘Materials and Methods’ section; Supplementary Figure S1). This is so because B-factors should be erroneously larger than those of atoms in the vicinity when the population was overestimated and vice versa. The results were largely consistent with the values of occupancy calculated in the structure refinement.
Figure 3.

Diastereomer-specific intermolecular contacts. In (A) and (B), parts of DNA strands including the six phosphorothioate-modified sites are shown, where omit maps contoured at 2.5σ of the Rp and Sp diastereomers are indicated (green and orange, respectively). A water molecule specifically observed for the Sp diastereomer at A8′(PS)T7′ is also indicated with the omit map. In (C)–(F), the two panels on the left show diastereomer-specific protein–DNA interactions in four selected sites, where dashed lines indicate hydrogen bonds with the non-bridging oxygen atoms (cyan), hydrophobic contacts with sulfur atoms (red) and salt bridges with sulfur atoms [yellow; most close pairs of (partially) charged atoms are connected], as defined in Figure 2. In the panels on the right, contacts to the equivalent phosphates in the CUTr1–poDNA complex (35) are shown. Population ratios of the diastereomers are also shown.
In the following descriptions of the interactions at the respective sites, we consider hydrogen bonds primarily with the oxygen atoms, without excluding the possibility of those with the sulfur atoms, because they may also act as hydrogen acceptors (43). For hydrophobic contacts, we consider those with sulfur but not with oxygen. Also for salt bridges, we consider those with sulfur, which has a negative charge (Scheme 1) (44). We should mention that the alternative conformations did not necessarily have to be set for the protein side chains even though they would interact with the phosphorothioates, since the electron densities fitted well to those of single conformers.
Among the six modified sites (Figure 3A and B), the largest difference in the population of diastereomers was observed at the phosphorothioate between G1 and C2 [G1(PS)C2; similar terms will be used hereafter], where the Rp diastereomer dominated over the Sp diastereomer at an approximate ratio of 4:1 (Figure 3C). A hydrogen bond is formed between the oxygen atom of the more populated Rp diastereomer and Asn425 Nδ2, which is also seen in the original CUTr1–poDNA complex (35) (Figure 3C, Supplementary Figure S2). In addition, the sulfur atom of the Rp diastereomer is found at a position to form hydrophobic contacts with side chain carbons and backbone carbons of Leu404 and Leu422. In contrast, the less populated Sp diastereomer lacks the hydrogen bond as above with oxygen, while the sulfur atom, in turn, is at the position in place of oxygen. Hydrophobic contacts by the sulfur atom are less extensive as well, while a salt bridge is observed between the sulfur atom and Arg400. Thus, the difference in the population is likely to be due to a hydrogen bond with the oxygen atom and/or hydrophobic contacts with the sulfur atom. Note that the conformation of Arg400 is altered from that in the complex with poDNA, causing disruption of hydrogen bonds. This is likely to be due to the extensive interactions with C2(PS)T3, as follows.
For C2(PS)T3, the Sp diastereomer dominated over the Rp diastereomer with an approximate ratio of 7:3 (Figure 3D). The more populated Sp diastereomer forms two hydrogen bonds with Thr401 N and Oγ1 with the oxygen atom (Figure 3D, Supplementary Figure S2). Also, a salt bridge between the sulfur and Arg400 is observed for the Sp diastereomer. In contrast, extensive hydrophobic contacts are formed with the sulfur atoms of both the diastereomers. The sulfur atom of the Rp diastereomer is located at a position capable of forming the above hydrogen bonds, whereas it is not likely to contribute to the population increment of the Rp diastereomer.
The Sp diastereomer was also more populated at T7′(PS)A6′ with a ratio of ∼2:1 (Figure 3E). Its oxygen atom forms two hydrogen bonds with Ser406 Oγ and Gln390 Nδ2 as in the complex with poDNA. It is noteworthy that the positions of the phosphorus atoms of the two diastereomers slightly deviate from each other (0.4 Å), so as to move the oxygen atom closer to the hydrogen donors of the hydrogen bonds as above (Supplementary Figure S2). Similarly to the case of C2(PS)T3, the sulfur atom of the Rp diastereomer is located at a position capable of forming hydrogen bonds, without contributing to the population increment.
The positional deviation of the phosphorus atom was most prominent at A8′(PS)T7′ (0.7 Å), where the oxygen atoms of both the diastereomers became closer to each other so as to form hydrogen bonds with the backbone amide of Gln390 (Figure 3B and F, Figure 4, Supplementary Figure S2). These hydrogen bonds appear to be strengthened by a hydrogen-bonding cascade within an α-helix (Figure 4A), being classified into resonance-assisted hydrogen bonds (45). Namely, the negative charge originally located on the phosphorothioate sulfur is delocalized to distant carbonyl oxygens through resonance that involves rearrangements of covalent bonds (Figure 4B). This presumable electron transfer fully meets the dipole moment of the helix with the negative charge at the C-terminus (46), which should be stabilized by an electrostatic interaction with Arg400. Although this scheme is not specific to psDNA, it causes a large positional deviation of the phosphorus, which strongly suggests that hydrogen bonds are formed in a favorable manner with oxygen, but not with sulfur. In addition, the more populated Rp diastereomer (58%) forms extensive hydrophobic contacts with carbons of the protein and DNA base via the sulfur atom. These are likely to contribute to the population increment of this diastereomer. The position equivalent to this sulfur is occupied by a water molecule in the case of the Sp diastereomer or in the case of the original poDNA, forming hydrogen bonds with the protein backbone amide and the phosphate oxygen at the same time, i.e. water-mediated intermolecular hydrogen bonds. The hydrophobic effects as above would be enhanced by the neutralization of the charge on sulfur (Figure 4B), since the negative charge strengthens the hydrogen bonds with water (see also discussions in the next section) (45).
Figure 4.
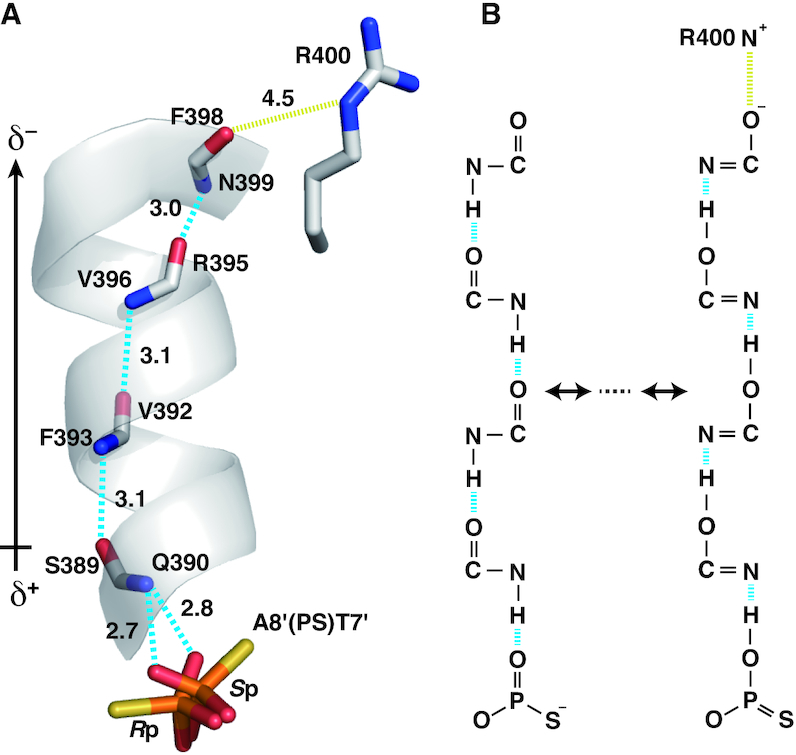
Resonance-assisted hydrogen bonds at A8′(PS)T7′. (A) Hydrogen-bonding cascade (cyan lines) involving an α-helix of the protein and a presumed salt bridge (yellow lines). Distances for these interactions are shown in Å. The probable dipole moment for the α-helix is indicated on the left. (B) Possible resonance in the cascade provoking electron delocalization, where the negative charge at the C-terminus would be stabilized by a salt bridge.
At the two other sites, i.e. A6′(PS)T5′ and T9′(PS)A8′, the populations deviated slightly (55% for the Rp diastereomer, at both the sites; Figure 3). The more populated Rp diastereomer at A6′(PS)T5′ forms hydrophobic contacts and a salt bridge with Arg410, whereas the less populated Sp diastereomer at T9′(PS)A8′ forms a salt bridge to Arg448 (Supplementary Figure S2).
The observations above clearly indicate that (i) hydrogen bonds are preferentially formed on the oxygen atoms of phosphorothioate and contribute primarily to the population increment of the relevant diastereomer and (ii) hydrophobic contacts formed on the sulfur atoms of phosphorothioate also contribute to the population increment. The combinations of these factors should account for the affinity enhancement of psDNA, for which further discussions based on the thermodynamic aspects are presented in the next section.
The differences in the population ratios in the crystal structure indicate that a selection similar to the ITC experiment (Table 1) occurred during the crystal formation. Namely, accumulation of the difference in the population ratios at the six sites as above (for each site, a portion of 1/(1 + |FRp − FSp|) remains active, where FRp and FSp are fractions of the Rp and Sp diastereomers with an assumption that as many DNA molecules as possible went into the crystal) accounts for 24% of the active population, which is less than the apparent stoichiometry in ITC (50% at 200 mM NaCl). Because the crystal condition that includes precipitant is different from those used for the ITC experiments, it is not appropriate to directly compare the values themselves. We may say, however, that selection of particular diastereomers was indeed observed in the crystal structure, which is likely to be due to the difference in the affinity and explains the decrease in the apparent stoichiometry at least qualitatively.
Chemical and thermodynamic implications of the affinity enhancement
The Pauling electronegativity of sulfur (2.58) is less than that of oxygen (3.44) and rather similar to that of carbon (2.55) (47). Despite this property, the negative charge is located on the sulfur atom in the phosphorothioate (Scheme 1), which was ascribed to the larger size and polarizability of sulfur than oxygen (44). When a hydrogen bond with the sulfur atom as the acceptor is considered, it must be strengthened by the negative charge on the acceptor, being classified into a charge-assisted hydrogen bond (45). Nevertheless, the bond energy for the hydrogen bonds with sulfur was estimated to be significantly smaller than that with oxygen (43). As a consequence, we observed in the crystal structure that hydrogen bonds were formed preferentially with the oxygen atoms. This should also be directly related to the hydrophobic contacts made by sulfur in that the tendency not to form hydrogen bonds with water leads to the hydrophobicity. Therefore, if the sulfur becomes neutral, hydrophobic effects should be enhanced as described earlier.
The ITC experiments showed that interaction of psDNA with protein was more enthalpy-driven than that of poDNA when compared under the same condition (50 or 100 mM NaCl, Table 1). This is somewhat puzzling because the introduced sulfur atoms are involved in hydrophobic contacts and salt bridges, which contribute to the entropic term (41). We propose here a mechanism based on the crystal structure in which the hydrogen bonds are formed with oxygen but not with sulfur (Figure 5). In poDNAs, the non-bridging oxygens are negatively charged by −0.5 on average and attract cations. Therefore, the cations stabilize the free state of DNA, and thereby destabilize hydrogen bonds formed on these oxygens. In contrast, non-bridging oxygens in psDNAs are neutral, because the negative charge is located on sulfur. The cations are attracted to the sulfur atom and, therefore, the hydrogen bonds formed with these oxygens should be stable even under a high-salt condition. Note that differences in ΔH are larger at the higher salt concentration (∼9 and ∼20 kJ mol–1 under the 50 and 100 mM NaCl conditions, respectively; Table 1).
Figure 5.
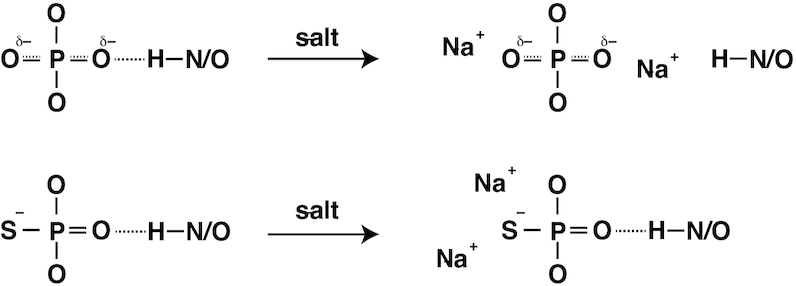
A mechanism for salt-resistant hydrogen bonds for phosphorothioate.
The above scheme implies the lack of release of cations upon interaction in psDNA, which explains the disadvantage in the entropic term. Under the 200 mM NaCl condition, however, interaction of psDNA with the protein became entropy-driven (Table 1). Considering that the hydrophobic contacts are enhanced by salt (42), they should be the major force of the interaction. Indeed, we observed in the crystal structure that many of the hydrophobic contacts are formed with the DNA base in a sequence-specific manner (Figure 2C) and that some are formed with the sulfur atoms of the phosphorothioates, which appeared to contribute to selection of the particular diastereomer (Figure 3).
Relating to the switch in the driving force depending on the salt concentration, we should consider a possibility that the geometry in the protein–DNA binding may also be altered. However, the sequence-specific binding should not be largely affected under the higher salt condition, because the base recognition involves hydrophobic contacts as stated earlier. Therefore, the presumable alterations will be limited, which are likely to be comparable to the observed positional shifts for the phosphorus atoms (<1.0 Å, Supplementary Figure S2), occurring to maximize the favorable contacts.
The hydrophobicity of ASOs has been considered important in the interactions with proteins during cellular uptake and trafficking (15,16). It was indeed shown that 10 or more of phosphorothioate modifications were necessary for interaction of ASOs with Hsp90, and that hydrophobic mid-domain of Hsp90 was responsible for this interaction (17). These are consistent with the present observation of the extensive hydrophobic contacts with sulfur atoms.
Relation to the previous structural observations
The present ITC and crystallographic analyses have revealed two distinct mechanisms by which the phosphorothioate modification enhances affinity between oligonucleotides and proteins. One is enthalpy-driven, depending on hydrogen bonds between the remaining oxygen atom and amino acids containing N–H or O–H moieties. The other is entropy-driven, depending on hydrophobic contacts between the sulfur atom and mostly aliphatic amino acids of proteins. The former is dominant under the lower salt condition, while the latter is dominant under the higher salt condition. Note that only the latter scheme is applicable to the phosphorodithioate modification.
Iwahara et al. observed an entropy-driven enhancement of the affinity between phosphorodithioate-modified double-stranded DNA and Antennapedia homeodomain (10). They ascribed the origin of the effect to an increase in mobility of the Lys57 side chain amide that formed a hydrogen bond with the phosphate in the unmodified DNA. However, a re-examination of their crystal structures (PDB entries 4XIC and 4XID) indicates that the sulfur atoms of the phosphorodithioate are close to Met54 Cϵ forming hydrophobic contacts, while the hydrogen bond/electrostatic attraction with the Lys57 amide is fully disrupted at least in one of the two molecules in the asymmetric unit. Therefore, the entropy-driven affinity enhancement can be ascribed also to the hydrophobic contacts. Indeed, their analyses on the two phosphorothioate-modified DNAs showed that the affinity was more enhanced for the Rp diastereomer with the hydrophobic contacts via the sulfur atom as above (PDB entry 5JLW) (12).
For an endonuclease that cleaves naturally occurring phosphorothioate DNA, the sulfur atom of the phosphorothioate of the substrate was recognized via a number of hydrophobic contacts with non-polar atoms of the protein, while the oxygen atom was contacted via a hydrogen bond with an Arg residue (PDB entry 5ZMO) (26). Therefore, this is fully consistent with our observation that sulfur and oxygen are favored in the hydrophobic contacts and hydrogen bonds, respectively.
The RNA aptamer against thrombin acquired a 1000-fold affinity enhancement by phosphorodithioate modification, which was ascribed to the higher polarizability of sulfur compared to oxygen (11,48). In their crystal structure of the complex, the conformation around the phosphorodithioate changed significantly so as to make both the sulfur atoms involved in the extensive hydrophobic contacts. However, considering this extent of the amplification, which is much larger than that by phosphorothioate, the particular geometry involving interaction between sulfur and the edge of the aromatic ring, involving the polarization of sulfur, is likely to be the key factor in this case. This was confirmed by a rigorous quantum mechanical calculation (49).
Stereocontrolled synthesis as a strategy to develop effective therapeutic oligonucleotides
Use of chiral reagents enabled stereocontrolled synthesis of phosphorothioate in nucleic acids (50,51). By such a method, a series of stereochemically unique ASOs were synthesized and tested for their activities (23). Surprisingly, a pattern of 3′-SpSpRp-5′ in the antisense strand enhanced the activity of the RNase H-dependent RNA cleavage and improved drug efficacy in mice. They ascribed the effect to the affinity enhancement with RNase H, and identified several contacts in a structural model based on the crystal structure of the complex of RNase H and RNA/DNA duplex (52). Here, we revisited the model in the light of the present findings (Supplementary Figure S3). In sites 1 and 3 (the first and third positions of 3′-SpSpRp-5′, respectively), we noticed more hydrogen bonds formed with the oxygen atoms in this preferred diastereomer compared with the alternative diastereomer. Also in site 2, extensive hydrophobic contacts are formed with the sulfur atom in the preferred diastereomer. Therefore, the enhanced affinity of the 3′-SpSpRp-5′-containing ASO with RNase H can be well explained in the present scheme. In addition, we noticed that the edge of the indole ring of Trp225 is close to the sulfur in site 1 (Supplementary Figure S3A), which may also contribute to the affinity enhancement through the polarization scheme (11,48,49). Nonetheless, it was reported recently that the effect of the stereocontrolled synthesis on the RNase H activity is not sufficient for the total therapeutic profile, which presumably reflects the complex mechanism of drug activity involving the cellular uptake, etc.
For aptamer and decoy nucleic acids, which target proteins, it is still worth pursuing the affinity enhancement in the target-specific manner. If we specify the preferable diastereomers, we can generate the affinity-enhanced oligonucleotide by stereocontrolled synthesis. The extent of the enhancement should be at most twice for the respective contact sites, which increases multiplicatively over the sites. We can thereby reduce the treatment dose, to minimize the side effects. Therefore, analysis of the structure of the complex with racemic oligonucleotide to observe the unequal populations of diastereomers, as in this study, should be one of the effective strategies to improve therapeutic oligonucleotides.
DATA AVAILABILITY
The coordinate and structure factor files were deposited to the Protein Data Bank (PDB) under accession ID 6LFF.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr K. Furukawa of AIST for the support in the ITC measurement. The X-ray diffraction data were obtained at KEK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Japan Society for the Promotion of Science [24570144 to KY and MM]. Funding for open access charge: AIST internal funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Eckstein F. Nucleoside phosphorothioates. Annu. Rev. Biochem. 1985; 54:367–402. [DOI] [PubMed] [Google Scholar]
- 2. Bielinska A., Shivdasani R.A., Zhang L.Q., Nabel G.J.. Regulation of gene expression with double-stranded phosphorothioate oligonucleotides. Science. 1990; 250:997–1000. [DOI] [PubMed] [Google Scholar]
- 3. Marshall W.S., Caruthers M.H.. Phosphorodithioate DNA as a potential therapeutic drug. Science. 1993; 259:1564–1570. [DOI] [PubMed] [Google Scholar]
- 4. Stein C.A. Exploiting the potential of antisense: beyond phosphorothioate oligodeoxynucleotides. Chem. Biol. 1996; 3:319–323. [DOI] [PubMed] [Google Scholar]
- 5. Marcus-Sekura C.J., Woerner A.M., Shinozuka K., Zon G., Quinnan G.V. Jr.. Comparative inhibition of chloramphenicol acetyltransferase gene expression by antisense oligonucleotide analogues having alkyl phosphotriester, methylphosphonate and phosphorothioate linkages. Nucleic Acids Res. 1987; 15:5749–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014; 24:374–387. [DOI] [PubMed] [Google Scholar]
- 7. Marshall W.S., Beaton G., Stein C.A., Matsukura M., Caruthers M.H.. Inhibition of human immunodeficiency virus activity by phosphorodithioate oligodeoxycytidine. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:6265–6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sharma H.W., Perez J.R., Higgins-Sochaski K., Hsiao R., Narayanan R.. Transcription factor decoy approach to decipher the role of NF-kappa B in oncogenesis. Anticancer Res. 1996; 16:61–69. [PubMed] [Google Scholar]
- 9. Yang X., Fennewald S., Luxon B.A., Aronson J., Herzog N.K., Gorenstein D.G.. Aptamers containing thymidine 3′-O-phosphorodithioates: synthesis and binding to nuclear factor-kappaB. Bioorg. Med. Chem. Lett. 1999; 9:3357–3362. [DOI] [PubMed] [Google Scholar]
- 10. Zandarashvili L., Nguyen D., Anderson K.M., White M.A., Gorenstein D.G., Iwahara J.. Entropic enhancement of protein–DNA affinity by oxygen-to-sulfur substitution in DNA phosphate. Biophys. J. 2015; 109:1026–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abeydeera N.D., Egli M., Cox N., Mercier K., Conde J.N., Pallan P.S., Mizurini D.M., Sierant M., Hibti F.-E., Hassell T. et al.. Evoking picomolar binding in RNA by a single phosphorodithioate linkage. Nucleic Acids Res. 2016; 44:8052–8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nguyen D., Zandarashvili L., White M.A., Iwahara J.. Stereospecific effects of oxygen-to-sulfur substitution in DNA phosphate on ion pair dynamics and protein–DNA affinity. ChemBioChem. 2016; 17:1636–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crooke S.T. Molecular mechanisms of action of antisense drugs. Biochim. Biophys. Acta. 1999; 1489:31–44. [DOI] [PubMed] [Google Scholar]
- 14. Peters L., Meister G.. Argonaute proteins: mediators of RNA silencing. Mol. Cell. 2007; 26:611–623. [DOI] [PubMed] [Google Scholar]
- 15. Juliano R.L., Ming X., Carver K., Laing B.. Cellular uptake and intracellular trafficking of oligonucleotides: implications for oligonucleotide pharmacology. Nucleic Acid Ther. 2014; 24:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H.. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:230–237. [DOI] [PubMed] [Google Scholar]
- 17. Liang X.H., Shen W., Sun H., Kinberger G.A., Prakash T.P., Nichols J.G., Crooke S.T.. Hsp90 protein interacts with phosphorothioate oligonucleotides containing hydrophobic 2′-modifications and enhances antisense activity. Nucleic Acids Res. 2016; 44:3892–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khaled Z., Benimetskaya L., Zeltser R., Khan T., Sharma H.W., Narayanan R., Stein C.A.. Multiple mechanisms may contribute to the cellular anti-adhesive effects of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 1996; 24:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Srinivasan S.K., Iversen P.. Review of in vivo pharmacokinetics and toxicology of phosphorothioate oligonucleotides. J. Clin. Lab. Anal. 1995; 9:129–137. [DOI] [PubMed] [Google Scholar]
- 20. Agrawal S. Antisense oligonucleotides: towards clinical trials. Trends Biotechnol. 1996; 14:376–387. [DOI] [PubMed] [Google Scholar]
- 21. Brown D.A., Kang S.H., Gryaznov S.M., DeDionisio L., Heidenreich O., Sullivan S., Xu X., Nerenberg M.I.. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J. Biol. Chem. 1994; 269:26801–26805. [PubMed] [Google Scholar]
- 22. Mou T.C., Gray D.M.. The high binding affinity of phosphorothioate-modified oligomers for Ff gene 5 protein is moderated by the addition of C-5 propyne or 2′-O-methyl modifications. Nucleic Acids Res. 2002; 30:749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iwamoto N., Butler D.C.D., Svrzikapa N., Mohapatra S., Zlatev I., Sah D.W.Y., Meena, Standley S.M., Lu G., Apponi L.H. et al.. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:845–851. [DOI] [PubMed] [Google Scholar]
- 24. Ostergaard M.E., De Hoyos C.L., Wan W.B., Shen W., Low A., Berdeja A., Vasquez G., Murray S., Migawa M.T., Liang X.H. et al.. Understanding the effect of controlling phosphorothioate chirality in the DNA gap on the potency and safety of gapmer antisense oligonucleotides. Nucleic Acids Res. 2020; 48:1691–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pallan P.S., Yang X.B., Sierant M., Abeydeera N.D., Hassell T., Martinez C., Janicka M., Nawrot B., Egli M.. Crystal structure, stability and Ago2 affinity of phosphorodithioate-modified RNAs. RSC Adv. 2014; 4:64901–64904. [Google Scholar]
- 26. Liu G., Fu W., Zhang Z., He Y., Yu H., Wang Y., Wang X., Zhao Y.-L., Deng Z., Wu G. et al.. Structural basis for the recognition of sulfur in phosphorothioated DNA. Nat. Commun. 2018; 9:4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dickinson L.A., Joh T., Kohwi Y., Kohwi-Shigematsu T.. A tissue-specific MAR/SAR DNA-binding protein with unusual binding-site recognition. Cell. 1992; 70:631–645. [DOI] [PubMed] [Google Scholar]
- 28. Han H.-J., Russo J., Kohwi Y., Kohwi-Shigematsu T.. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature. 2008; 452:187–193. [DOI] [PubMed] [Google Scholar]
- 29. Yasui D., Miyano M., Cai S.T., Varga-Weisz P., Kohwi-Shigematsu T.. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature. 2002; 419:641–645. [DOI] [PubMed] [Google Scholar]
- 30. Pavan K.P., Purbey P.K., Sinha C.K., Notani D., Limaye A., Jayani R.S., Galande S.. Phosphorylation of SATB1, a global gene regulator, acts as a molecular switch regulating its transcriptional activity in vivo. Mol. Cell. 2006; 22:231–243. [DOI] [PubMed] [Google Scholar]
- 31. Kohwi-Shigematsu T., Poterlowicz K., Ordinario E., Han H.-J., Botchkarev V.A., Kohwi Y.. Genome organizing function of SATB1 in tumor progression. Semin. Cancer Biol. 2013; 23:72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamayoshi A., Yasuhara M., Galande S., Kobori A., Murakami A.. Decoy-DNA against special AT-rich sequence binding protein 1 inhibits the growth and invasive ability of human breast cancer. Oligonucleotides. 2011; 21:115–121. [DOI] [PubMed] [Google Scholar]
- 33. Yamasaki K., Yamasaki T.. The combination of sequence-specific and nonspecific DNA-binding modes of transcription factor SATB1. Biochem. J. 2016; 473:3321–3339. [DOI] [PubMed] [Google Scholar]
- 34. Yamaguchi H., Tateno M., Yamasaki K.. Solution structure and DNA-binding mode of the matrix attachment region-binding domain of the transcription factor SATB1 that regulates the T-cell maturation. J. Biol. Chem. 2006; 281:5319–5327. [DOI] [PubMed] [Google Scholar]
- 35. Yamasaki K., Akiba T., Yamasaki T., Harata K.. Structural basis for recognition of the matrix attachment region of DNA by transcription factor SATB1. Nucleic Acids Res. 2007; 35:5073–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Battye T.G., Kontogiannis L., Johnson O., Powell H.R., Leslie A.G.. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D: Biol. Crystallogr. 2011; 67:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vagin A., Teplyakov A.. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997; 30:1022–1025. [Google Scholar]
- 38. Potterton E., Briggs P., Turkenburg M., Dodson E.. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D: Biol. Crystallogr. 2003; 59:1131–1137. [DOI] [PubMed] [Google Scholar]
- 39. Murshudov G.N., Vagin A.A., Dodson E.J.. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D: Biol. Crystallogr. 1997; 53:240–255. [DOI] [PubMed] [Google Scholar]
- 40. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D: Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kauzmann W. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 1959; 14:1–63. [DOI] [PubMed] [Google Scholar]
- 42. Melander W., Horvath C.. Salt effects on hydrophobic interactions in precipitation and chromatography of proteins: interpretation of lyotropic series. Arch. Biochem. Biophys. 1977; 183:200–215. [DOI] [PubMed] [Google Scholar]
- 43. Bhattacharyya S., Bhattacherjee A., Shirhatti P.R., Wategaonkar S.. O–H···S hydrogen bonds conform to the acid–base formalism. J. Phys. Chem. A. 2013; 117:8238–8250. [DOI] [PubMed] [Google Scholar]
- 44. Frey P.A., Sammons R.D.. Bond order and charge localization in nucleoside phosphorothioates. Science. 1985; 228:541–545. [DOI] [PubMed] [Google Scholar]
- 45. Gilli G., Gilli P.. Towards an unified hydrogen-bond theory. J. Mol. Struct. 2000; 552:1–15. [Google Scholar]
- 46. Hol W.G., van Duijnen P.T., Berendsen H.J.. The alpha-helix dipole and the properties of proteins. Nature. 1978; 273:443–446. [DOI] [PubMed] [Google Scholar]
- 47. Haynes W.M., Lide D.R., Bruno T.J.. CRC Handbook of Chemistry and Physics. 2017; 97th ednBoca Raton, FL: CRC Press. [Google Scholar]
- 48. Yang X.B., Abeydeera N.D., Liu F.W., Egli M.. Origins of the enhanced affinity of RNA–protein interactions triggered by RNA phosphorodithioate backbone modification. Chem. Commun. 2017; 53:10508–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Egli M., Lybrand T.P.. Enhanced dispersion and polarization interactions achieved through dithiophosphate group incorporation yield a dramatic binding affinity increase for an RNA aptamer–thrombin complex. J. Am. Chem. Soc. 2019; 141:4445–4452. [DOI] [PubMed] [Google Scholar]
- 50. Oka N., Wada T., Saigo K.. An oxazaphospholidine approach for the stereocontrolled synthesis of oligonucleoside phosphorothioates. J. Am. Chem. Soc. 2003; 125:8307–8317. [DOI] [PubMed] [Google Scholar]
- 51. Knouse K.W., deGruyter J.N., Schmidt M.A., Zheng B., Vantourout J.C., Kingston C., Mercer S.E., McDonald I.M., Olson R.E., Zhu Y. et al.. Unlocking P(V): reagents for chiral phosphorothioate synthesis. Science. 2018; 361:1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nowotny M., Gaidamakov S.A., Ghirlando R., Cerritelli S.M., Crouch R.J., Yang W.. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol. Cell. 2007; 28:264–276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinate and structure factor files were deposited to the Protein Data Bank (PDB) under accession ID 6LFF.