


Abstract
During interphase centromeres often coalesce into a small number of chromocenters, which can be visualized as distinct, DAPI dense nuclear domains. Intact chromocenters play a major role in maintaining genome stability as they stabilize the transcriptionally silent state of repetitive DNA while ensuring centromere function. Despite its biological importance, relatively little is known about the molecular composition of the chromocenter or the processes that mediate chromocenter formation and maintenance. To provide a deeper molecular insight into the composition of the chromocenter and to demonstrate the usefulness of proximity-based biotinylation as a tool to investigate those questions, we performed super resolution microscopy and proximity-based biotinylation experiments of three distinct proteins associated with the chromocenter in Drosophila. Our work revealed an intricate internal architecture of the chromocenter suggesting a complex multilayered structure of this intranuclear domain.
INTRODUCTION
In many eukaryotes pericentromeric and centromeric chromatin of multiple chromosomes cluster in interphase to form a defined nuclear domain (1–3). Chromocenters have been initially described in the early 20th century by Heitz, who discovered them in mosses and later in somatic and larval polytene chromosomes from different Drosophila species (4). Though chromocenters have been described in different species, they differ substantially in their genomic composition. In Arabidopsis, similar to humans or mice, chromocenters are composed of the pericentric heterochromatin of individual chromosomes, which sometimes but not regularly associate (1,2,5). In Drosophila somatic pairing results in a coalescence of pericentromeric chromatin of multiple chromosomes to form a small number of chromocenters (6,7). Molecularly, chromocenters are composed of pericentromeric heterochromatin and CenpA containing centromeric chromatin, which are both rich in repetitive DNA and evolutionarily highly dynamic (8,9). They form and are held together by multiple components ranging from noncoding RNA (10) over DNA binding factors (11), protein–protein interactions (12) to histone posttranslational modifications (13). Interference with chromocenter formation results in an upregulation of transposable elements, mitotic defects and the formation of micronuclei (11,12). Despite their functional importance, the molecular description of the centromeric region is incomplete. This lack of information is mainly due to the fact that chromocenters are difficult to purify without disrupting their native conformation (14,15) and, like many other nuclear organelles, vary in size and shape during cell cycle and cellular differentiation (16). Although several chromocenter associated factors have been described and characterized, classical affinity-MS based approaches only identified stable interactors of centromeric chromatin or the inner kinetochore complexes but failed to describe the proteomic composition of this important nuclear compartment (17–20). This is very likely due to the fact that the chromocenter is held together by a multitude and rather weak interactions that do not withstand the harsh conditions used during purification. Recently, proximity based biotinylation has been shown to be able to reveal weak in vivo interactions thereby describing the composition of non-membranous compartments within a cell (21,22). Although an engineered ascorbate peroxidase from soybean (APEX2) has been targeted to satellite repeats using a CAS9-APEX2 fusion protein (23), it has not yet been systematically applied to a well characterized nuclear compartment such as the chromocenter.
The centromere as well as the pericentromeric repeats, which constitute the chromocenter, are highly divergent with regard to size, sequence and protein composition even in very closely related species (8,24,25). The rapid divergence of centromeric sequences is thought to be accompanied by an adaptive evolution of centromere binding proteins to counteract a meiotic drive, which would otherwise result in the potentially deleterious expansion of centromeric repeats (9,26). This ultimately results in substantial genomic and proteomic differences leading to a postzygotic incompatibility of hybrid animals (27,28). This genomic conflict hypothesis is supported by the fact that many of the gene products that cause hybrid incompatibility are either proteins or RNA molecules that bind to centromeric or pericentromeric heterochromatin (18,29–31), (32,33). One of the best characterised hybrid incompatibility factors is the Hybrid male rescue protein HMR (34). HMR interacts with the heterochromatin protein HP1a and is found in close vicinity to the centromere-specific H3 variant dCenpA in Drosophila melanogaster cell lines and imaginal disc cells (35). HMR binding is also observed at several euchromatic sites where it colocalizes with known boundary factors (36). Interestingly, the intracellular localization of HMR varies among different tissues. In interphase cells of larval brains it is primarily found at pericentromeric heterochromatin (30,37,38) whereas it also associates with telomeres on salivary gland polytene chromosomes (18,38). Mutations of Hmr results in an upregulation of transposable elements (18,38) and an increase in mitotic defects (18). Such a phenotype has also been observed upon knockdown of nucleoplasmin (NLP), which interacts with HMR and plays a role in centromere clustering (12,18). It is therefore possible that centromere clustering might not only be important for centromere function but also contributes to the formation of species.
To investigate the intricate structure of the Drosophila chromocenters, we performed confocal and super resolution microscopy using antibodies directed against HP1a, HMR and dCenpA and determined their proximal proteome using APEX2-based proximity biotinylation. Our results reveal the molecular map of the centromeric region and suggest that HMR is located at boundaries between HP1a containing heterochromatin and centromeric or transcriptionally active chromatin. Besides the proximity to heterochromatic and known centromeric factors, we also observe a close proximity of HMR to the condensin and cohesin complex and find a reduced CAPH2 binding to chromatin upon HMR overexpression. Furthermore, we observe that a part of the chromocenter is held together by dCenpC, which is found in proximity of HMR and dCenpA. These findings demonstrate a complex structure of the chromocenter and suggest an important role of HMR in the formation of this evolutionarily very dynamic domain. As a consequence, the differential regulation of HMR in different species of Drosophila might have resulted in the genetic instability of hybrid animals containing two different and separately evolved genomes.
MATERIALS AND METHODS
Cloning
APEX fusions were cloned into pMT vector (18), which was cut with XbaI and NotI. GST-APEX was cloned into pGEX-6P-1 vector (18), cut with EcoRI and NotI. RNAi-resistant dCenpC with two-point mutations (41H979E) was assembled from five fragments and cloned into pMT (18) vector digested with XbaI and NotI. Two RNAi-resistant fragments were designed with SeqMixer app of Tamas Schauer (https://tschauer.shinyapps.io/SeqMixer/) and synthesized by Eurofins Genomics. Mutants of RNAi-resistant dCenpC were also cloned into pMT vector digested with XbaI and NotI. Cloning was performed with In-Fusion cloning kit (Clontech). Details of ADD1-PA, HP5, XNP and CG8108 cloning into pMT vector are available upon request. The list of primers is available in the Supplementary Table S1.
Cell culture, transfection and generation of stable cell lines
Drosophila S2DGRC or L2–4 cells (18) were grown in Schneider medium supplemented with 10% fetal calf serum, penicillin and streptomycin at 26°C. To generate a stable cell line, 3–4 millions of cells were transfected with 2 μg of plasmid mixed with XtremeGENE HP (Roche) transfection reagent according to manufacturer's instructions. After transfection, cells were selected for 3 weeks with Hygromycin B (Invitrogen) at 100 μg/ml, and were selected on Hygromycin during further culture and experiments. Optional induction of cell lines with 250 μM CuSO4 was performed 12–24 h before experiments. To avoid effects of cells with extreme overexpression, dCenpAAP and HP1aAP cell lines were diluted and clones originating from several cells were selected as in (39). From dCenpAAP cell line clone 8 was used, from HP1aAP cell line—clone 29.
RNAi
Double-stranded (ds) RNAs were generated using MEGAscript RNA kit (Invitrogen) according to the manufacture's instructions. The list of primers is available in Supplementary Table S1. RNAi was performed as in (18). 1 million cells were seeded in a six-well plate and grown overnight; next day the medium was removed and 10 ug of dsRNA (5 ug each in case two different siRNAs were used) in 1 ml serum-free Schneider medium was added. Cells were gently rocked on a platform for 10 min at room temperature (RT) and left for additional 50 min at 26°C. Afterward 2 ml of medium was added. Cells were harvested on day 6. For rescue experiments cells were re-plated, medium was changed and plasmid transfection was performed on day 4 of RNAi and cells were harvested on day 7.
cDNA synthesis and RT-qPCR
RNA extraction was performed from 4 million cells using QIAGEN RNeasy kit according to manufacturer's protocol. cDNA was synthesized using SuperScript III First-Strand Synthesis System from Thermo Fischer Scientific according to manufacturer's instructions. cDNA was diluted 1:20 and used for RT-qPCR with PowerUp™ SYBR® Green Master Mix. Primers for RT-qPCR were designed with Primer3 (40) and listed in Supplementary Table S1.
APEX2 proximity biotinylation, nuclear extraction and immunoprecipitation
For biotinylation cells were grown in roller bottles (Greiner) to density of 5 × 106 cells/ml. For biotinylation in solution 5 × 108–109 cells were resuspended in 100–200 ml biotin-phenol/PBS or DMSO/PBS and incubated for 30 min at room temperature (RT). H2O2 was added to biotin-phenol treated cells to the concentration of 1 mM and cells were pelleted for 20 min at 250 g. The supernatant was aspirated, and cells were washed three times in quenching solution (10 mM sodium azide, 10 mM sodium ascorbate and 5 mM Trolox in PBS) or (untreated cells) in PBS. The last washing step was performed in 15 ml falcons. Nuclei isolation and extraction was performed as in (17) with modifications. All buffers were supplemented with freshly added protease inhibitors. Cells were swelled in three packed cell volumes (PCV) (e.g. 2.1 ml) of hypotonic buffer (10 mM Tris pH 7.6, 10 mM NaCl, 1.5 mM MgCl2, 0.1 mM EDTA) on ice for 30 min. Next, cells were centrifuged 10 min 250 g, and the pellet was resuspended in 3 PCV of hypotonic buffer supplemented with 0.2% NP-40. Subsequently, cells were incubated for 10 min at 4°C on a rotating wheel for the lysis of the plasma membrane. Nuclei were pelleted at 1000 g for 10 min and washed with 2 ml of quenching solution supplemented with protease inhibitors. After the wash nuclei were centrifuged for 10 min with 1500 g and the nuclear pellet was snap-frozen in liquid nitrogen. In the next step, nuclei were resuspended in 3 ml Tris-Ex100 buffer (10 mM Tris pH 7.6, 100 mM NaCl, 1.5 mM MgCl2, 0.5 mM EGTA and 10% v/v glycerol) supplemented with 1500 units Mnase, 1500 units Benzonase and 2 mM CaCl2. Chromatin was digested for 20 min at 26°C and the reaction was stopped by addition of EDTA and EGTA to 10 mM on ice. Nuclei were disrupted by Dounce homogenization using 10 strokes with a tight-fitting pestle. For chromatin extraction and solubilization, NaCl (to 600 mM) and detergents (Triton X-100 to 1%, sodiumdeoxycholate (SOD) to 0.5% and SDS to 0.1%) were added and chromatin was incubated for 1 h at 4°C. Nuclear extracts were cleared by centrifugation 20 min 10 000 g and dialyzed for 4 h at 4°C through 3.5 MWCO Millipore membranes against Tris-Ex100 with detergents and without glycerol, supplemented with 0.2 mM PMSF and 1 mM DTT. The obtained nuclear extract was snap-frozen. For streptavidin purification, nuclear extract with freshly added protease inhibitors was mixed with 500 μl of Pierce streptavidin magnetic beads, prewashed two times in Tris-Ex100 buffer with detergents. Immunoprecipitation was performed for 1.5 h at RT. Beads were washed two times with Tris-Ex100 buffer with detergents (with protease inhibitors), once with 2 M Urea 10 mM Tris, and again twice with Tris-Ex100 with detergents and protease inhibitors.
For biotinylation on plates, 800 ml of 5 × 109 million/ml cells were adhered on 40 15-cm plates for mammalian cells for 1 h. Cells were incubated in biotin-phenol/PBS (20 plates) or DMSO/PBS (20 plates) for 30 min, and H2O2 was added to biotin-phenol treated cells to the concentration of 1 mM for variable times as indicated. Solution was aspirated and quenching solution (or PBS for untreated cells) was added. Cells were scraped off, washed one more time in quenching solution and subjected to nuclear extraction as described above.
Mass spectrometry
Streptavidin beads from APEX pulldowns were washed 3 times with 500 μl of 50 mM Tris pH 7.5, 4 M urea and on-bead digestion was performed. Reduction was completed in 500 μl of 20 mM DTT in 50 mM Tris, 2 M urea pH 7.5 with Lys C 450 ng/sample at 27°C for 1 h. Subsequently, alkylation was performed with 50 mM iodoacetamide for 1 h 25°C, shaking 900 rpm, and was stopped by addition of DTT to 10 mM final concentration. The samples were incubated at 25°C shaking 900 rpm for two more hours. 300 μl water was added to reduce urea concentration to 1.5 M, and 1.5 μg trypsin and 2 mM final concentration CaCl2 were added for overnight incubation shaking 900 rpm. In the morning another 1.5 μg of trypsin was added, and the sample was incubated for another 4 h while shaking. After collection of the supernatant, the beads were washed two times with 100 μl of 20 mM Tris 50 mM NaCl 25% ACN in order to elute loosely-bound tryptic peptides from the beads. Washes were combined with supernatant, evaporated at <28°C, and desalted. The second elution of remaining peptides from the beads was performed with 300 μl 0.05% SDS, 0.1% formic acid (FA) at 80°C for 10 min. The second elution was evaporated and subjected to HILIC chromatography. After desalting and HILIC both elutions were combined, evaporated and resuspended in 45 μl 0.1% FA. Peptide mixtures were subjected to nanoRP-LC-MS/MS analysis on an Ultimate 3000 nano chromatography system coupled to a QExactive HF mass spectrometer (both Thermo Fisher Scientific) in 2–4 technical replicates (5 μl each). The samples were directly injected in 0.1% FA onto the separating column (120 × 0.075 mm, in house packed with ReprosilAQ-C18, Dr Maisch GmbH, 2.4 μm) at a flow rate of 0.3 μl/min. The peptides were separated by a linear gradient from 3% ACN to 40% ACN in 50 min. The outlet of the column served as electrospray ionization emitter to transfer the peptide ions directly into the mass spectrometer. The QExactive was operated in a TOP10 duty cycle, detecting intact peptide ion in positive ion mode in the initial survey scan at 60 000 resolution and selecting up to 10 precursors per cycle for individual fragmentation analysis. Therefore, precursor ions with charge state between 2+ and 5+ were isolated in a 2 Da window and subjected to higher-energy collisional fragmentation in the HCD-Trap. After MS/MS acquisition precursors were excluded from MS/MS analysis for 20 s to reduce data redundancy. Siloxane signals were used for internal calibration of mass spectra.
Protein MaxQuant search
The raw data files of APEX pulldowns were analysed with MaxQuant version 1.5.3.12 against dmel-all-translation-r6.08.fasta database from Flybase. All the parameters were set to default except choosing ‘Match between runs’ and LFQ and iBAQ quantitations. Technical replicates were assigned to one experiment (biological replicate). Experimental and control samples (treated with biotin-phenol and DMSO, respectively) were loaded into the same MaxQuant run. Samples from different cell lines and time points were run separately.
Data sources
ChIP-seq datasets were available from GEO with the following accession numbers: GSE86106 (HMR native and HP1a), GSE118291 (HMR induced), GSE54529 (Rad21 and CAPH2). GSE numbers of ChIP-seq profiles used for distance-to-peak measurements (Figure 4) are provided in Supplementary Table S6.
Figure 4.
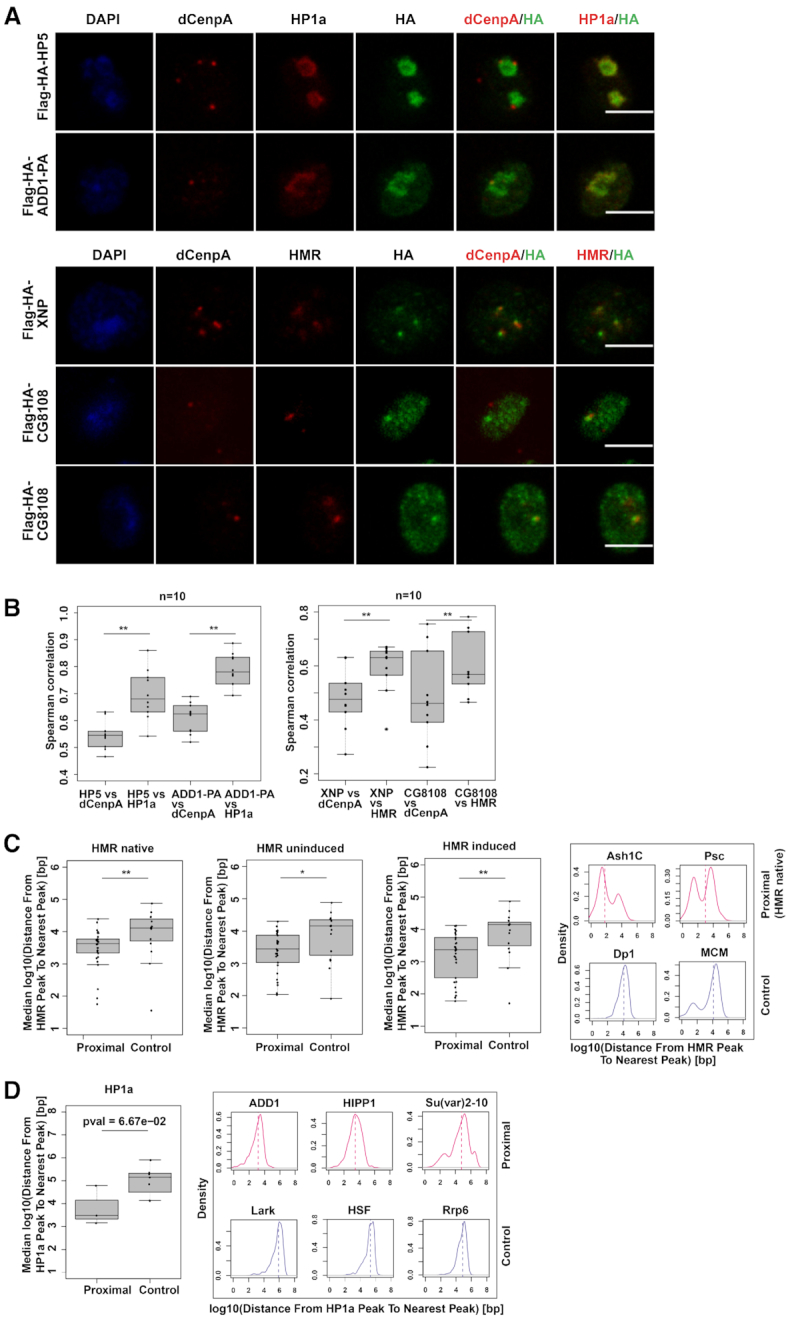
(A) dCenpA, HP1a/HMR and HA tag stainings (single stacks shown) of stable cell lines expressing Flag-HA- HP5, ADD1-PA, XNP and CG8108. Anti-HP1a mouse C1A9, anti-HMR rat 2C10, rabbit anti-dCenpA and anti-HA rat R001 antibodies were used. For XNP colocalization with HMR is observed in all cells, for CG8108 – in approx. 15–30% of cells with moderate protein overexpression (quantification done from single stacks). (B) Distributions of Spearman correlations of staining pairs. Wilcoxon signed-rank test was used for comparisons. For the second staining dCenpA and HA channels were recorded in parallel, HMR channel - separately. Images of 10 cells from 2–3 independent experiments were used. (C). (D) Distributions of median distances from the HMR/HP1a ChIP-seq peak to nearest protein ChIP-seq peak of HMR/HP1a proximal/anti-proximal (control) proteins. HMR native (36) and ectopically expressed HMR (uninduced, (68)), as well as HP1a (36) profiles are used for comparison. For HP1aAP enriched proteins found in two out of three time points are taken, for anti-enriched proteins – found in 2 out of 3 time points, with anti-enrichment cutoff of –0.3. Examples of distributions of the distances from the native HMR/HP1a peak to the nearest protein peak are given on the right. List of GSEs of ChIP-sequencing profiles used is available at Supplementary Table S6. Wilcoxon rank sum test is used for statistical analysis. *P-value < 0.05, **P-value < 0.01.
ChIP-Seq analysis
Raw reads were aligned to the reference genome (UCSC dm6) using bowtie2 (version 2.2.9) and quality filterered by samtools (version 1.3.1). ChIP-seq profiles were created by Homer (version 4.9) and normalized by the number of reads and the corresponding input. Peaks were called by Homer with parameters -style factor -size 200 -F 2 for HMR and CAPH2 and -F 4 for Rad21. Peak overlaps were plotted as Venn diagrams using the Vennerable R package (version 3.1.0.9). ChIP-seq profiles were aligned to the pool of native and induced HMR peaks and visualized as heatmaps. Heatmaps were ordered by HMR native enrichment and grouped by HP1a enrichment.
ChIP-seq profiles for distance comparison (see Supplementary Table S6 for GSE numbers) were processed using Homer as described above with optimized -F peak finding parameters (available upon request). Peak coordinates were imported to R and distances between peak centers were calculated using the distanceToNearest function (GenomicRanges package version 1.36.1). Distances for each factor were visualized either as density plots or their median was calculated for comparing proximal vs. control protein binding.
Novel CAPH2 and HMR ChIP-seq data (for HMR overexpression (HMRind)/dCenpC RNAi experiments) were processed using Homer as described above with -F 4 peak finding parameter. Peak finding was carried out on the pool of the reads from replicates. ChIP-seq tracks were visualized as average plots centered at the pool of HMR peaks or CAPH2 peaks in a 2 kb window. Statistical analysis was performed on the mean ChIP signal at the center of the peak for each replicate. Paired t-test was performed on the mean values. Scatterplots comparing RNAi/overexpression to control conditions show the ChIP-seq enrichment at the center of the peaks. The color is proportional to the density of the datapoints (heatscatter function, LSD package version 4.0-0). Figures were created by R base graphics.
Statistical analysis of proteomics data
Proteins detected only under biotinylation conditions in at least two biological replicates and neither detected in control nor in APEXNLS samples were pre-selected as enriched proteins prior subsequent analysis. Log2-transformed LFQ values were imputed with the impute.MinProb function (imputeLCMD R package v2.0) with parameter q = 0.05, tune.sigma = 0.1 and normalized by median normalization. Moderated t-statistics were computed by fitting a linear model with empirical Bayes moderation using the limma R package (version 3.34.9). The data were visualized as volcano plots using R base graphics and Venn diagrams using the Vennerable R package (version 3.1.0.9). Code is available upon request. GO-term analysis was done using Gene Ontology Consortium (http://geneontology.org) tool, and only the terms with the lowest hierarchy were considered. The protein-protein interaction network was visualized using Cytoscape. For network analysis those proteins were considered, which are known to be localized to the nucleus and were specifically enriched in HMRAP or dCenpAAP but not in APEX2NLS. Protein-protein interaction data were taken from the STRING database (41) using experiments, databases, gene fusion, co-expression and co-occurrence as interaction sources and minimum required interaction score of 0.7. Additional interactions were taken from the database Flybase and indicated as dotted lines.
ChIP-sequencing was performed as described in (36). Libraries were prepared from 1 ng DNA with Microplex Diagenode kit without size selection.
Immunofluorescent staining
Cells were adhered on coverslips for 30 min, washed with PBS and fixed in 0.3% Triton X-100/3.7% formaldehyde/PBS for 12 min at RT (or in 3.7% formaldehyde/PBS for 10 min at RT for staining after RNAi). After wash with PBS cells were permeabilized in 0.25% Triton/PBS on ice for 6 min. Cells were rinsed two times and washed again twice with PBS, following blocking in Image-iT FX signal enhancer (Invitrogen) for 45 min at RT. Primary antibodies, diluted in 5% normal goat serum (NGS) (Dianova), were incubated with coverslips 1 h at RT (or at 4°C overnight for high resolution microscopy). Coverslips were washed with 0.1% Triton X-100/PBS and PBS and incubated with secondary antibody in 5% NGS 1 h at RT. Following a wash in 0.1% Triton X-100/PBS and two washes in PBS, cells were stained in DAPI/PBS (200 ng/ml or 50 ng/ml for high resolution microscopy) and washed again in PBS. Cells were mounted in VECTASHIELD (Vector Labs) or ProLong™ Diamond Antifade (Thermo Fisher Scientific), for widefield and confocal or super resolution microscopy, respectively.
For biotinylation followed by immunofluorescence on coverslips, 106 cells were adhered on coverslips for 30 min, incubated with biotin-phenol/PBS or DMSO/PBS for 30 min, followed by (optional) addition of 1 mM H2O2 for denoted time. Cells were next washed in quenching solution and subjected to immunofluorescent staining as described above. Biotinylation in solution was performed with 106 cells in 200 μl biotin-phenol/PBS or DMSO/PBS and 1 mM H2O2, added for denoted time. Cells were pelleted during biotinylation 250 g, resuspended in 200 μl quenching solution, adhered on coverslips for 15 min and processed for IF as described above.
Image acquisition was performed on a Zeiss Axiovert 200 epifluorescence microscope with a CCD Camera (AxioCam MR, Zeiss). Confocal microscopy was performed with a Leica TCS SP5 (with 63× objective with 1.3 NA) or at the Core Facility Bioimaging of the Biomedical Center with an inverted Leica SP8X STED 3D microscope, equipped with a 405 nm Laser and a pulsed white light Laser (470 - 670 nm). Gated-STED Images were acquired with a 100×/1.4 NA oil immersion objective, pixel size was set to 24–25 nm. The following spectral settings were used: DAPI (excitation 405 nm; emission 415–470 nm), Alexa Fluor 594 (590 nm; 600–625) and Abberior STAR 635P (635; 645–720). Recording was done line sequentially to avoid bleed-through and potential channel misalignment caused by drift. Signals were recorded with hybrid photo detectors (HyDs) in counting mode. STED and confocal images (except images in Figures 5 and 6) were deconvolved using Huygens 17.10 p2. All images were processed using ImageJ.
Figure 5.

(A) Peak overlaps of HMR, Rad21 and CAPH2 (see GSE accession numbers in Materials and Methods). (B) Heatmaps of HMR (36), overexpressed HMR (HMRind), HP1a (36), Rad21, and CAPH2 ChIP-seqs (100). All the peaks are centered at HMR binding sites and clustering is performed as in (36). (C) Examples of CAPH2 ChIP-seq tracks in CuSO4-treated untransfected L2–4 cells and CuSO4-treated (induced) L2–4 stable cell line overexpressing HMR+LHR (replicate 1). (D) Composite ChIP-seq plot of CAPH2 signal centered at CAPH2 peaks in CuSO4-treated untransfected L2–4 cells and CuSO4-treated (induced) L2–4 stable cell line overexpressing HMR + LHR.
Figure 6.
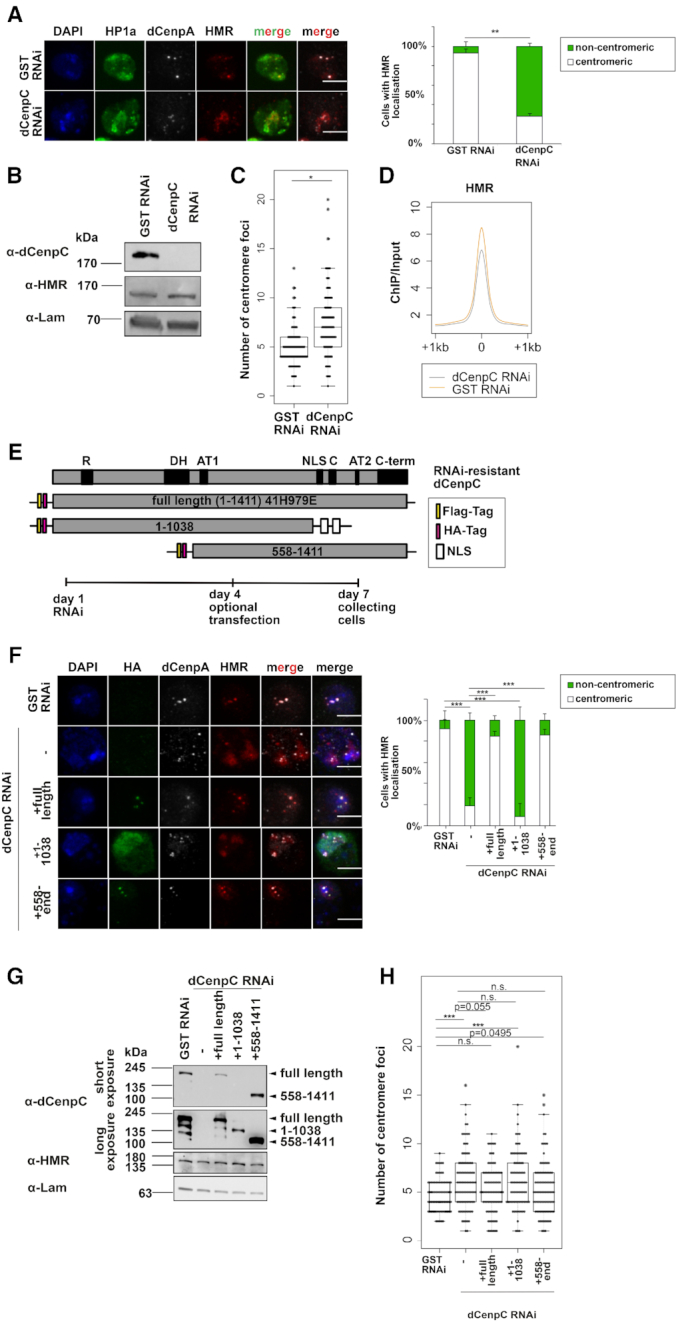
(A) Left panel: Immunofluorescent stainings (maximum intensity projections shown) against HP1a, HMR and dCenpA upon GST and dCenpC knockdowns. Scale bars represent 5 μm. Antibodies used for staining were anti-HMR 2C10, anti-HP1a C1A9 and rabbit anti-dCenpA. Right panel: Quantification of centromeric and heterochromatic localization of HMR. Unpaired T-test on fractions of cells with centromeric HMR localization was used for statistical analysis. Error bars represent standard deviation. (B) Western blots of GST RNAi and dCenpC RNAi whole cell extracts indicating dCenpC knockdown and unchanged HMR levels. Lamin was used as a loading control. (C) Distributions of centromere foci number upon GST and dCenpC RNAi. P-values were calculated by fitting a generalized mixed effect model on condition as fixed effect and independent experiments as random effect. (D) Composite plot of HMR ChIP-sequencing signal centered at HMR peaks upon GST and dCenpC RNAi. (E) Domains’ scheme of dCenpC and RNAi-resistant constructs used in rescue experiments, as well as the scheme of rescue experiment. R - arginine-rich region, DH—a highly conserved region in the dCenpC homologs; AT1/2—predicted AT hooks; C—the dCenpC motif; C-term—the metazoan-like C-terminal region; NLS—nuclear localization signal (101). (F) Immunofluorescent stainings (maximum intensity projections shown) against HA tag, HMR and dCenpA upon GST and dCenpC RNAi. Scale bars represent 5 μm. Antibodies used for staining were anti-HA mouse 12CA5, anti-HP1a C1A9 and rabbit anti-dCenpA. Right panel: Quantification of centromeric and non-centromeric localization of HMR. Linear model on fractions of cells with centromeric HMR localization was used for statistical analysis. Error bars represent standard deviation. (G) Western blot indicating dCenpC RNAi and expression of RNAi-resistant dCenpC proteins upon transient transfection, as well as HMR levels in all conditions. Lamin was used as a loading control. (H) Distributions of centromere foci number upon GST RNAi, dCenpC RNAi and dCenpC RNAi + transient transfection of RNAi-resistant dCenpC proteins. P-values were calculated as in (C). N.S., non-significant; *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001.
For analysis of HMR distribution in RNAi and rescue experiments, maximum intensity projections were used. Localization was considered heterochromatic/non-centromeric if <20% of cell centromeres overlapped with HMR or very low signal of dCenpA staining was observed. For centromere declustering calculation the number of foci was quantified from maximum intensity projections of stainings with monoclonal rat anti-dCenpA 7A2 antibody. Different exposures were used for GST and dCenpC RNAi images, since dCenpA signal is reduced upon dCenpC RNAi. In cohesins’ and condensins’ RNAi experiments different exposures might be taken for dCenpA and HP1a. For RNAi experiments 50 cells, for rescue experiments ∼40–50 cells from two independent replicates were counted. For rescue experiments only transfected cells (with positive nuclear HA tag signal) were considered for calculation.
Western blotting of whole cell extracts
Fifteen million cells from each cell line (or 2 million cells of each RNAi condition) were collected, washed two times in PBS and resuspended on ice in 80 μl (or 20 ul for cells after RNAi) RIPA buffer with freshly added protease inhibitors and 30 units benzonase. Lysates were left on ice for 30 min, and afterwards 20 μl (or 5 ul for lysates after RNAi) of 5× Laemmli buffer was added. Lysates were boiled 10 min 96°C before loading on the gel (10 μl).
Antibodies
For immunofluorescence and western blotting, rat anti-HMR 2C10 antibody (Helmholtz Zentrum München, (18)) was used in dilution 1:25 (or 1:5 for high resolution microscopy); rat anti-dCenpA 7A2 (Helmholtz Zentrum München, (12)) 1:100 (or 1:50 for high resolution microscopy); rabbit anti-dCenpA (Immunofluorescence grade, Actif Motif) 1:500 (or 1:250 for high resolution microscopy); mouse anti-HP1a C1A9 (42) 1:100; mouse anti-HA 12CA5 (Helmholtz Zentrum München) 1:1000; rat anti-HA R001 (Helmholtz Zentrum München) 1:100; anti-APEX2 20H10 (raised in this study, Helmholtz Zentrum München) 1:50; Streptavidin-Alexa555 (Thermo Fisher Scientific) 1:400; rabbit anti-histone H3 (Abcam) 1:3000, mouse anti-FLAG (Sigma M2, 1 mg/ml) 1:100. Rabbit anti-dCenpC antibody was kindly provided by Christian Lehner and used in dilution 1:1000 for high resolution microscopy and 1:5000 for western blotting. Rabbit anti-CAPH2 antibody was kindly provided by Giovanni Bosco and 6 ul of the antibody were used per one ChIP sample.
Antibody generation
Wistar rats were immunized subcutaneously (s.c.) and intraperitonially (i.p.) with 50 μg of GST-APEX2 fusion protein dissolved in 500μl PBS, 5 nmol CpG2006 (TIB MOLBIOL) an equal volume of incomplete Freund's adjuvant. Six weeks after immunization a 50 μg boost injection was applied i.p. and s.c. 3 days before fusion. Fusion of the splenic B cells and the myeloma cell line P3 × 63Ag8.653 was performed using polyethylene glycol 1500 according to standard protocols (43). Hybridoma supernatants were tested by solid-phase enzyme-linked immunoassay (ELISA) using the recombinant GST-fusion protein and verified by Western blotting of whole cell extracts from APEX2 fusions-expressing cell lines (Figure 2B). Hybridoma cell line from specifically reacting supernatants were cloned twice by limiting dilution. Experiments in this study were performed with clone 20H10 (rat IgG2a/κ).
Figure 2.

(A) APEX2 fusion constructs used for proximity based labeling of the chromocenter. (B) Schematic display of a proximity based biotinylation experiment. (C) Biotinylation experiment performed for 1 minute with 5 mM biotin-phenol using the HMRAP cell line (top four panels) in the absence of biotin phenol and hydrogen peroxide, the presence of either biotin phenol or hydrogen peroxide or both reagents. Biotinylation experiment performed for 1 min with 5 mM biotin-phenol using the dCenpAAP cell line (middle panel). The bottom panel shows a treatment of non APEX2 expressing L2–4 cells treated with biotin phenol and hydrogen peroxide. For selected cells (indicated by an asterisk) the inlet shows an approx. 2.3-fold zoom of the nucleus. (D) Proximity based biotinylation of the HP1aAP and the APEX2NLS cell lines using 0.5 mM biotin-phenol. Stainings were performed with anti-APEX2 20H10 antibody and anti-Streptavidin Alexa555. Scale bars represent 5 μm.
RESULTS
HMR and dCenpA form an interdigitated centromeric network
To confirm the previously detected centromeric localization of HMR in Drosophila cells with an unrelated antibody, we performed immunofluorescent staining using a FLAG antibody in a cell line where HMR is endogenously tagged with the FLAG epitope at the C-terminus using CRISPR/Cas9 (36). Consistent with our previous results (18) most of the FLAG signals co-localize with the signal we obtained when using an anti-dCenpA antibody, which confirms the close proximity of HMR and centromeric chromatin during interphase (Figure 1A). Notably, the quantitation of 122 interphase centromere clusters indicated that 57.4% (70) of all counted centromere foci perfectly overlap with HMR foci and another 13.9% (17) overlap partially. However, we also detected 28.7% (35) HMR clusters that did not overlap with dCenpA. We observed a trend that nuclei with fewer dCenpA positive spots showed a stronger degree of overlap, whereas we found more non overlapping HMR dots in nuclei with many centromeric spots. This suggests that the localization of HMR to the centromere is either cell cycle regulated or specific for a subset of centromeres (Figure 1B) and that the formation of these nuclear domains is highly regulated. This variability might also explain the differences observed when staining HMR in different tissues (18,30). Interestingly, when we investigated the HMR bound interphase centromeres and their surrounding chromatin using super resolution STED microscopy we found the chromocenter to be composed of a structural meshwork of interdigitated dCenpA, dCenpC and HMR proteins (Figure 1C and D, Supplementary Movie S1) where dCenpA showed a much higher correlation with dCenpC than with HMR (Figure 1E). This domain is formed by large lobes of HP1a constituting the pericentromeric heterochromatin surrounding the chromocenter (Supplemental Figure S1A). Strikingly, the HP1a containing lobes are frequently bordered by regions of strong HMR binding, which is consistent with our ChIP-Seq data where HMR binding is frequently detected at boundaries between HP1a containing heterochromatin and actively transcribed genes (36). As we have previously shown that HMR and LHR can block enhancer action in transient transfection (18), it is tempting to speculate that HMR constitutes a physical boundary between heterochromatin and actively transcribed centromeric chromatin (44). To further unravel the details of this architectural meshwork at the chromocenter, we aimed at determining the protein composition of these nuclear domains.
Figure 1.

(A) Staining of S2 FLAG-HMR line (36) with mouse anti-FLAG (Sigma), anti-HMR 2C10 and rabbit anti-dCenpA antibodies. Maximum intensity projections are shown. Scale bars represent 3 μm. (B) Quantification of centromeres overlapping with HMR. Experiments were performed in a dCenpA-GFP expressing cell line (61). In total 25 cells and 122 centromeres identified using an anti-GFP antibody were counted. (C) Confocal (top panel) and stimulated emission depletion (STED) microscopy (middle panel) images of dCenpA and HMR. The bottom panel shows a zoom in of the centromeric region. Single stacks are shown. Scale bars represent 3 μm, for zooms – 0.5 μm. (D) Plot profiles of the highest fluorescence intensities of STED images of the chromocenter from S2 cells using rabbit/rat 7A2 anti-dCenpA, rabbit anti-dCenpC and rat 2C10 anti-HMR antibodies. Line profile plots were built in ImageJ and normalized to one of the maximum peaks. (E) Distribution of pair-wise Spearman correlations for quantifying the relationship between dCenpA, dCenpC and HMR. Images of 10 cells from two independent experiments were used. Wilcoxon rank sum test is used for comparison (N.S., non-significant; ***P-value < 0.001).
HMR, dCenpA and HP1a APEX2 fusion proteins localize similarly to the endogenous proteins
Several groups including us have performed large scale affinity purification experiments coupled to mass spectrometry (AP-MS) using antibodies specific against dCenpA (17,19), HMR (18) or HP1a (45,46) to identify putative interactors of these proteins. However, in contrast to other well characterized nuclear protein complexes such as the nucleosome remodelers or the histone acetyltransferases (47,48), these three proteins have been refractory to classical biochemical purification. In fact, when comparing the AP-MS data from different laboratories the overlap in proteins identified is marginal (17–19,45,46,49). We reasoned that this is due to the fragility of the interactions that do not withstand the harsh condition of cell lysis and subsequent purification. We therefore generated stable cell lines expressing HMR, dCenpA and HP1a fused to APEX2 that enables biotin labelling of factors surrounding the fusion protein (Figure 2A). Upon treatment of APEX2 expressing cells with biotin phenol and hydrogen peroxide, a localized burst of diffusible biotin–phenoxyl radicals is generated. These radicals then react with nearby (<20 nm) electron rich amino acid side chains leading to the biotinylation of neighboring proteins that can be subsequently purified and identified using shot gun mass spectrometry (23,50–55). We confirmed the expression of ectopic proteins by Western blotting using antibodies against HMR, dCenpA, HP1a (Supplemental Figure S2A) and APEX2 (Supplemental Figure S2B). The APEX2 protein was fused to a double nuclear localization signal to determine the non-targeted nuclear proteome. Since the fusion proteins are expressed under a copper inducible promoter, we were able to tune their expression to match the endogenous levels (Supplemental Figures S2A–D and S1B). Hence, we performed the biotinylation reaction under non-inducing conditions for HMRAP and dCenpAAP expression but induced the expression of HP1aAP (Supplemental Figure S2B–D). For the APEX2NLS cell line we induced the expression of APEX2NLS to match HP1aAP expression levels (Supplemental Figure S2D). At the expression level used, HMRAP and dCenpAAP localize to centromeres, marked by the staining of centromere-specific histone variant dCenpA (Supplemental Figures S2C and S1B), HP1aAP occupies a domain in the nucleus, which coincides with endogenous HP1a staining and APEX2NLS localizes to the nucleus (Supplemental Figure S2D), showing a proper nuclear localization of the fusion proteins under the conditions used.
HMR, dCenpA and HP1a APEX2 fusion proteins biotinylate defined nuclear domains
To evaluate whether the biotinylation is spatially restricted to the surrounding of the expressed fusion proteins, we stained the cell lines for APEX2 and biotin after performing an in situ biotinylation reaction (Figure 2B). Consistent with the limited diffusion of the phenoxyl radicals generated by APEX2, we observed a strong and highly localized biotinylation signal colocalizing with HMRAP and dCenpAAP in cells treated for 25 minutes with hydrogen peroxide at a high concentration of biotin-phenol (5 mM) (Figure 2C and Supplemental Figure S3). Since HP1a has previously been shown to be very dynamic (56–58) and the APEX2-fusion protein is expressed at higher levels, we tested whether varying the concentration of biotin-phenol and the duration of hydrogen peroxide treatment would result in a spatially more confined biotinylation of the neighboring proteome. This was achieved when treating HP1aAP cells for only 1 or 5 min with 0.5 mM biotin–phenol (Supplemental Figures S3 and S4). These experiments clearly revealed the need of a thorough titration of the labelling time and reagent concentration dependent on the APEX2 fusion proteins used.
Proteomic analysis of HMR, dCenpA and HP1a containing domains
To unravel the chromocenter composition, we compared the biotinylated proteomes from the different APEX2-fusion protein expressing cell lines upon biotin–phenol and H2O2 versus DMSO treatment only. Using label-free quantitation and a statistical analysis of the proximity-based proteomes from at least four biological replicates upon 25-min biotinylation, we identified 325, 314, 259 and 273 proteins that were specifically biotinylated in dCenpAAP, HMRAP, HP1aAP and APEX2NLS expressing cells respectively (Supplementary Table S2). A comparative analysis of the proximity-proteomes revealed a strong overlap between the HP1aAP and the APEX2NLS proximity-proteome (72%), which was not the case for dCenpAAP and APEX2NLS (50%), or HMRAP and APEX2NLS (37%) (Figure 3A, B). The latter finding may reflect the lower expression level of dCenpA and HMR, the lower mobility and the more specific localization of dCenpAAP and HMRAP compared to HP1aAP (Figure 2C). Proteins that are found in the proximity proteome of at least three out of four factors were often highly abundant nuclear factors constituting the splicing machinery or structural proteins such as NLP, D1 or Lamin. These factors are likely to be in close proximity to most nuclear proteins but may nevertheless fulfill a function connected to the three proteins we investigated. To identify proteins that localize close to dCenpAAP, HMRAP and HP1aAP and are not distributed throughout the entire nucleus, we selected nuclear proteins that were preferentially biotinylated in the dCenpAAP, HMRAP or the HP1aAP cell line but not or only to a much lesser degree in the APEX2NLS line (Supplementary Table S3, Figure 3A). This resulted in a reduction of proteins to 115, 203 and 31 for dCenpA, HMR and HP1a respectively. For these factors, we compared the enriched GO terms using the Gene Ontology Consortium tool (http://www.geneontology.org) (Supplementary Table S4). For HP1aAP we found the GO terms such as chromatin organization and RNA and/or DNA metabolic processes across all three time points of biotinylation. For HMRAP, we found the GO terms cell cycle checkpoint, mitotic sister chromatid segregation and both negative and positive regulation of transcription by RNA polymerase II, which were consistent with the observed phenotypes of HMR mutations in Drosophila melanogaster (18,30,38,59,60). For dCenpAAP, we found the GO terms mitotic sister chromatid segregation and terms related to transcription by RNA-polymerase II, which were consistent with the known role of dCenpA in centromere function and the observation of centromeric transcription (33,44,61–63). In the HMRAP proximity proteome we identified several insulator factors (Su(hw), HIPP1, pita and Ibf2) two of which (Su(hw) and an isoform of Ibf2, Ibf1) are also detected in proximity of dCenpA. We also identify a different set of insulator proteins in the HP1a proximity proteome (pita, HIPP1 and mod(mdg4)). We therefore suggest that HMR and HP1a reside on different edges of a putative insulator complex, which is also consistent with the genome wide mapping of HMR and HP1a binding sites which frequently border each other (36). We therefore think that our findings support the hypothesis that HMR localizes in between dCenpA and HP1a containing chromatin.
Figure 3.

(A) Volcano plots of proteins enriched in the bait pulldown vs APEX2NLS. Selected protein groups were colored. (B) Venn diagrams of proteins found enriched in the individual purifications (biotinylated versus control). (C) Network diagram of nuclear proteins specifically enriched in dCenpAAP but not in APEX2NLS. Solid lines were provided by the STRING databases using the high stringency settings (0.7). Dotted lines were manually added based on information from Flybase. The size of the node reflects the enrichment value. (D) Network diagram of nuclear proteins specifically enriched in HMRAP but not in APEX2NLS.
As 25-min hydrogen peroxide treatment resulted in a broader and less confined distribution of biotinylation with the HP1aAP construct, we also measured the HP1aAP proximity proteome upon 1.5 and 5 min of biotinylation. When comparing the proteins under those different conditions we obtained the strongest enrichment of the previously published HP1a interactors already after 1.5 minutes of biotinylation (45,46). However, these interactors were also the ones with the highest abundancy suggesting that although 1.5 min biotinylation is most specific it has a significantly lower sensitivity (Supplementary Figure S5). As the HP1aAP specific nuclear proximity proteome showed a large overlap with the proteome in proximity to APEX2NLS, only very few chromatin-associated factors were significantly closer to HP1aAP than to APEX2NLS (Supplementary Table S3), which probably reflects the high mobility and concentration of HP1aAP and APEX2NLS in the nucleus. For dCenpAAP and HMRAP, in contrast, we detected a substantial number of nuclear proteins being closer to dCenpAAP and HMRAP than to APEX2NLS.The network graphs shown in Figure 3C and D visualize the similarities and differences between the dCenpAAP and HMRAP proximity proteomes, which displays individual proteins as nodes and previously published interactions as edges. Consistent with their close spatial localization within the nucleus, the proximity-based proteome of dCenpAAP and HMRAP shows a substantial overlap with proteins associated with this specific nuclear domain (93, Supplemental Table S5). Besides known centromeric proteins such as dCenpC or prod (17) we identify a large fraction of nucleolar factors as well as components of the nuclear pore in proximity to HMR and dCenpA. Interestingly, we also find proteins associated with active transcription, such as components of the mediator complex or histone acetyltransferases and proteins involved in DNA replication residing next to HMR and dCenpA. Since HMR’s localization is not centromere restricted (Figure 1), it is not surprising we detected a larger network of HMR neighboring genes when compared to dCenpA.
Proximity based proteomics and AP-MS datasets have limited overlap
A comparison of the proximity-based proteome with published AP-MS datasets revealed a rather limited overlap. In the case of HMR we identified 11 proteins by AP-MS as well as by proximity based biotinylation. Among those are the two speciation factors HMR and LHR, the centromere associated factor dCenpC, five Zn-finger containing proteins (CG33213, Su(var)3–7, Su(Hw), CG144388 and CG8108), the putative lysine specific demethylase NO66, the nucleolar polynucleotide kinase and an uncharacterized protein (CG16972). Notably, eight of these proteins have been tested for localization and five of these proteins have been shown to localize to centromeric chromatin (HMR, LHR, dCenpC, CG144388 and Su(var)3–7 (17,18,64–66). In case of dCenpA we identified nine proteins that were strongly enriched in the proximity proteome and in dCenpA containing chromatin. Six of them had been tested for localization and 5 were shown to localize to the chromocenter in interphase (dCenpA, dCenpC, Med30, sub and prod). We also observed a kinase that regulates heterochromatin fusion in Oocytes (SRPK), two putative RNA binding factors (YT521-B and caz) as well as a putative member of the nuclear membrane (CG4972). We assume that the reason for poor overlap of the different datasets using the same protein as a bait might be a much higher efficiency of biotinylation at domains where the bait is highly concentrated compared to regions of lower concentration (Supplementary Figure S6). This hypothesis is supported by the fact that we capture more than half of the 23 known proteins that have been experimentally shown to localize to interphase Drosophila melanogaster centromeres by immunofluorescence (12,17,65–67) (Supplementary Table S6) using two different baits localizing to the centromeric network (dCenpAAP and HMRAP). In case of HP1a the overlap between the proximity proteome and an HP1a pulldown from Alekseyenko et al. was limited to two factors HIPP1 and ADD1, both of which have been shown to functionally interact with HP1a (45,68,69). The overlap with the data from Swenson et al. in contrast revealed another 10 factors (46). This discrepancy in protein identification can have multiple reasons. An obvious one is the fact that many of the proteins identified in proximity based biotinylation but not in the AP-MS experiments cannot be purified from an extract due to instable interaction. Alternatively, they are interactions that also occur with APEX as it is the case for NLP and HP1a, which both interact with HMR in AP-MS experiments (18,35). Similarly, the proteins identified in AP-MS but not in the proximity proteome could be non-specific binders to beads, proteins that are too far away from the APEX fusion protein (>20 nm) or otherwise not susceptible to biotinylation due to the lack of surface exposed electron rich side chains.
Immunohistochemistry and genome wide ChIP-Seq data validate proximity based biotinylation assays
To validate our proximity based biotinylation we generated 4 transgenic cell lines expressing tagged versions of two factors in proximity to HP1a (HP5 and ADD1) and two proteins we found colocalizing with HMR (XNP and CG8108) (Figure 4A and B). In agreement with their stronger biotinylation by HP1aAP and HMRAP but not by dCenpAAP, the localization of these factors correlated more with the corresponding proteins than with dCenpA upon immunofluorescent staining (Figure 4B). As an alternative way to validate the proximity labeling, we compared the ChiP-seq profile of HMR with 29 available high-quality ChIP-seq profiles of proteins found in proximity to HMRAP and 15 ChIP-seq profiles of control proteins that showed a higher degree of biotinylation by APEXNLS. For the two groups of ChIP-seq profiles we calculated the median distance of HMR peaks to the nearest peak in the profiles. The genomic distance between HMR and the proteins we detected in proximity is on average 2.5–4 and 10–12.5 kb for the control proteins (Figure 4C). For many proteins found in proximity to HMR we detect a clear bimodal distribution with a substantial fraction of the HMR peaks being in close proximity to the protein of interest and a second fraction mapping more distantly (right panel of Figure 4C). This distribution indicates that only a subfraction of these proteins is actually residing close to HMR whereas another fraction resides in other nuclear domains. Most of the control proteins do not show such a distribution with a few exceptions such as the MCM proteins, which are probably too abundant to be significantly enriched in the HMRAP proximity proteome. We also did a similar comparison for HP1a, taking into account proteins enriched or depleted below a –0.3-enrichment cutoff (log2) in at least two out of three timepoints. We observed a trend (P < 0.067) that HP1a proximal proteins are on median 2.8 kb away from the nearest HP1a peak, while proteins enriched in the APEXNLS dataset are located about 126 kb from the nearest HP1a peak (Figure 5D).
Cohesin and condensin factors reside in proximity to HMR
Larval brain cells in HMR or LHR mutants show an increased number of anaphase defects (30), which has also been observed in fly strains carrying a mutation in genes encoding for subunits of the cohesin complex. Defects in chromosome condensation are also detected in hybrids between D. melanogaster and D. simulans (30,59). Therefore, we were particularly intrigued by the proximity of HMR to subunits of the condensin and the cohesin complexes (Figure 3D) and investigated the overlap of ChIP-Seq data between HMR (36), the cohesin subunit Rad21 and the condensin subunit CAPH2 (70) in greater detail. Consistent with the proximity labelling result, approximately half (51%) of the HMR binding sites colocalize with binding sites of the cohesin complex and 20% with the CAPH2 subunit of the condensin complex (Figure 5A). HMR binding sites can be separated in two classes one that flanks HP1a binding regions (class I) and another one that overlaps with binding sites for the insulator binding protein Su(Hw) (class II) (36). When we aligned ChIP-seq profiles with respect to the HMR binding sites, we detect strong CAPH2 binding at class I HMR sites (Figure 5B) and an inverse correlation of CAPH2 binding intensity at class II binding sites. Rad21-binding does not discriminate between the two types of HMR binding sites. We conclude from these findings that HMR frequently resides at loci that are also bound by members of the condensin and cohesin complex, which have been shown to play an important role in bookmarking the genome (71,72). However, this binding appears to be strongly context dependent and maybe even mutually exclusive between HMR and CAPH2 at class II HMR binding sites.
CAPH2 condensin subunit binding to chromatin is reduced upon increased levels of HMR/LHR complex
Hybrids from D.mel mothers and D.sim fathers express increased levels of HMR and LHR (18), which results in a block in G1 or G2 phase (59) and frequently an aberrant chromatin morphology (73) in larval brain cells. Such a phenotype has also been reported upon reducing condensin or increasing cohesin levels (74,75) In D.mel tissue culture cells the overexpression of HMR and LHR also results in an increased number of mitotic defects that are somewhat reminiscent of the defects seen in diploid hybrid cells (18,30). To simulate the hybrid situation, we therefore induced HMR and LHR expression in S2 cells to investigate whether an increased HMR expression has an effect on CAPH2 binding at HMR binding sites. Similar to what we detect in flies (18,76) we observe increased HMR binding in cells expressing higher levels of HMR and LHR. This increased HMR binding was particularly obvious in class II binding sites that overlapped with mapped binding sites of CAPH2, essentially eliminating the inverse correlation between HMR and CAPH2 we observe under native conditions (Figure 5B compare HMR with HMRind, (76)). We therefore tested whether HMR overexpression has an effect on CAPH2 binding by performing a CAPH2 ChIP-seq experiment upon HMR and LHR overexpression. Strikingly, we observed a global reduction of CAPH2 binding at a large number of binding sites (Figure 5C, D and Supplementary Figure S7A, B), suggesting that high levels of HMR/LHR complex destabilize chromatin bound condensin II.
dCenpC is necessary for HMR localization to the centromere
The formation of defined clusters in proximity to the centromere in tissue culture cells, early embryos and in larval neuroblasts is a striking feature of HMR (18,30,37). This clustered localisation is difficult to reconcile with the binding of HMR at multiple sites along the chromosomal arms even when HMR is overexpressed (18). Based on the detection of dCenpC in proximity to HMR and dCenpA we therefore hypothesized that dCenpC might be important to cluster HMR binding sites at or around the centromere. To test this hypothesis, we depleted dCenpC from L2–4 cells using an RNAi approach and measured HMR localization as well as the degree of centromere clustering (Figure 6A–C). As previously reported (66,77), the removal of dCenpC leads to a reduction of dCenpA staining in the nucleus (Figure 6A). The remaining dCenpA positive dots decluster and fail to colocalize with HMR suggesting that dCenpC is required for HMRs localization to the chromocenter. This was not due to a reduction of HMR levels (Figure 6B) or to a loss of chromatin binding as HMR can be detected localizing to HP1a rich heterochromatin (Figure 6A). Furthermore, we performed the ChIP-sequencing of HMR upon dCenpC RNAi and did not observe prominent change of HMR-binding sites (Figure 6D, Supplementary Figure S7C), suggesting that HMR sites decluster and diffuse into heterochromatin upon dCenpC knockdown. This is not seen upon depletion of either condensin or cohesin subunits (Supplementary Figure S8).
To demonstrate that the dCenpC loss is directly responsible for HMR mislocalization and centromere de-clustering, we generated RNAi resistant dCenpC expression constructs and used them to rescue HMR localization. The expression of the full-length construct reverted the HMR mislocalization phenotype and partially rescued the centromere declustering phenotype (Figure 6E–H). This was also achieved by expressing the C-terminal fragment, while the N-terminal half failed to rescue the phenotypes (Figure 6E–H). As dCenpC was previously reported to be a direct interactor of HMR (18), we suggest that interaction with HMR occurs at the C-terminus or the middle part of dCenpC.
DISCUSSION
The eukaryotic nucleus lacks internal membranes and many proteins transverse through it within seconds by simple diffusion (78). Nevertheless, it contains a number of distinct nuclear subcompartments and chromatin domains that represent cell type specific active or silenced states of the genome (79). The molecular description of these nuclear compartments so far escaped detailed analysis since classical interaction partner screenings often disrupt weak interactions and genetic screens often identify very indirect interactors. By applying proximity based biotinylation of three proteins associated with the chromocenter in Drosophila, we tried to fill this gap by an in vivo labelling method that requires a close physical proximity without the necessity of a stable enough interaction to withstand multiple purification steps. Our data reveal a potentially multilayered structure of the chromocenter, a well characterized nuclear subcompartment, and suggest that this method can be applied to similar nuclear bodies that have not been extensively characterized. Moreover, based on our results we hypothesize that the product of the speciation gene Hmr forms a boundary between centromeric dCenpA containing centromeric chromatin and pericentric heterochromatin and plays an important role in chromocenter biology.
In comparison to classical proteomic approaches where a specific antibody is used to purify and identify specific interactors of a given protein (AP-MS), which result in the identification of a small number of stable interactors (49), we find a much larger number of proteins in proximity to the three factors we investigated. Moreover, these classical AP-MS experiments show a rather limited overlap with the proximity-based proteomes. This may be due to the weak or transient nature of the interaction on one hand or a lack of electron rich amino acids in proximity to the APEX2 enzyme. Another reason for the lack of overlap even when comparing classical AP-MS experiments performed in different laboratories (45,46) could be the fact that these proteins do not form stable and defined protein complexes in vivo. In light of the recent findings that HP1 can undergo liquid-liquid phase separation (80,81) this hypothesis has been strengthened making proximity-based proteomics the method of choice to describe the composition of such highly flexible and dynamic nuclear domains. Unfortunately, the proximity-based proteome of HP1a is very similar to the one detected when expressing only APEX2 in the nucleus with only very few proteins being much closer to HP1a than to APEX2. This is very likely due to the relatively high concentration and fast diffusion of HP1 in the nucleus (56,58,82), by which it can sample the entire nuclear space within the labeling period. This is supported by our finding that we achieve the highest specificity of the HP1a proximity proteome using short labeling times, which on the other hand suffer from lower sensitivity. Therefore, we conclude that proteins such as HMR and dCenpA that form more distinct nuclear domains are better suited to characterize the proteomic composition of such domains by proximity-based biotinylation than the highly dynamic ones like HP1a. Our results also show that, depending on the APEX2 fusion protein used, expression levels, labelling time and the concentration of biotin-phenol have to be carefully titrated in order to come to meaningful conclusions.
Consistent with HMR’s clustering in proximity to centromeric chromatin (18,30,37,38), we find a substantial but not complete overlap between the proximity proteomes of HMR and dCenpA. In addition to the majority of centromere annotated proteins of D.mel (83) we find a substantial fraction of nucleolar proteins in proximity to dCenpA and HMR. This could be due to the proximity of the rDNA locus to pericentromeric heterochromatin in Drosophila. However, it has also been shown that centromeres localize to the nucleolar periphery during interphase (12,84,85) where they are anchored by two prominent nucleolar proteins NLP and Nph. NLP is detectable in proximity to HMR and dCenpA. However, due to it's high nuclear abundance, it is also labelled by APEX2NLS making it difficult to unambiguously identify it as being in closer proximity to HMR or dCenpA than to APEX2. This finding is nevertheless supported by the recent findings of Anselm and colleagues, which observe a similar cytological staining pattern of HMR and NLP/Nph oligomers and an HMR dependency of NLP’s binding to the centromere (35).
Besides these nucleolar proteins we also observe a number of components of the nuclear pore and factors involved in RNA transcription in proximity to both factors. The nuclear pore as well as the transcriptional machinery have been shown to play a functional role in centromere assembly and maintenance (33,44,86) as well as heterochromatin organisation (87), hence the proximity of these factors to dCenpA and HMR is not unexpected. In case of the nuclear pore, it is worth mentioning that although HMR and dCenpA both are in proximity to the nuclear pore, HMRAP preferentially biotinylates components of the nuclear basket whereas dCenpAAP shows a preference for the ring complex of the nuclear pore. This difference in proximity to specific areas of a large macromolecular assembly such as the nuclear pore suggests that this method could in fact be used to describe the higher order geometry of nuclear domains.
Besides the proteins that are in proximity to HMR and dCenpA, we also identified several factors, which were solely in proximity to HMR. Among those are members of the PcG family, components of the condensin and the cohesin complex and well-known boundary factors. All three of these complexes have been shown to regulate the 3D organization of interphase chromatin (88–90) (91,92), suggesting that HMR may also play a role in modulating higher order chromatin structure. Such a role of HMR in organizing chromatin within the nucleus is also supported by the striking discrepancy between the number of HMR binding sites observed in ChIP-Seq experiments and the few cytologically visible clusters (35,36,76). The fact that HMR is frequently observed in proximity to HP1 binding sites (36), the recent finding of Lee et al. (93) that euchromatic HP1a sites cluster together in the nucleus and the apparent localization of HMR between HP1 and dCenpA further supports this hypothesis. The proximity of HMR to members of the condensin and cohesin complexes and the higher overlap of their binding sites also suggest a role of HMR in interphase chromatin organization (94). Based on our data we assume that HMR uses dCenpC as a docking site to cluster HP1a rich heterochromatin around the interphase chromocenter until HMR is lost from chromatin at the onset of mitosis (18,30) when the kinetochore assembles on centromeric chromatin. The lack of HMR (or its lack of centromere association) results in a moderate declustering of the chromocenter, which in turn would lead to an increased number of chromosome breaks at boundaries between eu- and heterochromatin (where HMR binds) and a subsequent chromosome entanglement followed by a defect in anaphase (30). Such increased DNA damage has been shown to enhance the activity of transposable elements (95–97) and could therefore explain the increased transposon activity (18,38) and the similar phenotype of HMR and piwi mutations (98) despite the apparent lack of HMR binding to the regulated transposons or members of the RNAi machinery. As a disruption of transposon silencing in the drosophila female germline results in an increased genome instability in the progeny (95), it may also be the cause for the observed increase in female sterility in HMR mutants (60).
Can HMR’s proximity proteome also help explaining the hybrid phenotype? In fact, hybrid lethality is caused by a gain of HMR’s function rather than a loss of it. The apparent inverse relationship between HMR binding and the binding of the condensin II subunit CapH2 as well as the decrease in CapH2 binding to chromatin upon HMR overexpression suggest it can. Though it is unclear how higher levels of HMR reduce CapH2 binding this effect may very well explain the phenotype of fuzzy and irregularly condensed chromosomes (30,59), which is reminiscent of a defect in condensin II (94). It will be interesting in the future to analyze why higher levels of HMR results in a decrease in condensin II binding considering that HMR is bound in proximity to several condensin subunits.
In summary, our results describe the interphase chromocenter as a heterogeneous multilayered nuclear subcompartment in an unprecedented detail. The proteins found at or in proximity of the chromocenter support some of the previous functional studies and allow the development of new hypothesis regarding novel functions of the chromocenter such as the postzygotic separation of species.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (99) partner repository with the dataset identifier PXD012551. Novel CAPH2 and HMR ChIP-seq data were deposited to GEO with accession number GSE137194.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Christian Lehner and Sarah Elgin for the Rabbit anti-dCenpC and the mouse anti-HP1a antibody respectively, as well as Giovanni Bosco for rabbit anti-CAPH2 antibody. We furthermore would like to thank Nitin Phadnis and Patrick Heun for critical comments on the manuscript as well as Anne-Claude Gingras for advise on proteomic analysis, Ignasi Forne for mass-spectrometry analysis, Tobias Straub for initial bioinformatic analysis, Irene Vetter for cloning HP1aAP construct, Angelika Zabel for ChIP-sequencing libraries’ preparation, Raffaella Villa for advice on ChIP-sequencing and the entire Imhof group/Becker department for helpful discussions. We would also like to thank Stefan Krebs and Helmut Blum from LAFUGA facility for sequencing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft (DFG) [CIPSMand AI23/9-1 to A.I.]; Graduate School of Quantitative Biosciences Munich (QBM) (to N.K. and A.L.). Funding for open access charges: DFG [CRC 1064/Z03].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jones K.W. Chromosomal and nuclear location of mouse satellite DNA in individual cells. Nature. 1970; 225:912–915. [DOI] [PubMed] [Google Scholar]
- 2. Fransz P., De Jong J.H., Lysak M., Castiglione M.R., Schubert I.. Interphase chromosomes in Arabidopsis are organized as well defined chromocenters from which euchromatin loops emanate. Proc. Natl Acad. Sci. U.S.A. 2002; 99:14584–14589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jones K.W., Robertson F.W.. Localisation of reiterated nucleotide sequences in Drosophila and mouse by in situ hybridisation of complementary RNA. Chromosoma. 1970; 31:331–345. [DOI] [PubMed] [Google Scholar]
- 4. Heitz E. Über totale und partielle somatische Heteropyknose, sowie strukturelle Geschlechtschromosomen bei Drosophila funebris. Cell Tissue Res. 1933; 19:720–742. [Google Scholar]
- 5. Politz J.C.R., Scalzo D., Groudine M.. Something silent this way forms: the functional organization of the repressive nuclear compartment. Annu. Rev. Cell Dev. Biol. 2013; 29:241–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jagannathan M., Cummings R., Yamashita Y.M.. The modular mechanism of chromocenter formation in Drosophila. eLife. 2019; 8:e43938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jagannathan M., Cummings R., Yamashita Y.M.. A conserved function for pericentromeric satellite DNA. eLife. 2018; 7:1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jagannathan M., Warsinger-Pepe N., Watase G.J., Yamashita Y.M.. Comparative analysis of satellite DNA in the Drosophila melanogaster species complex. G3 (Bethesda). 2017; 7:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Malik H.S., Henikoff S.. Major evolutionary transitions in centromere complexity. Cell. 2009; 138:1067–1082. [DOI] [PubMed] [Google Scholar]
- 10. Casanova M., Pasternak M., Marjou El, F., Le Baccon P., Probst A.V., Almouzni G.. Heterochromatin reorganization during early mouse development requires a single-stranded noncoding transcript. CellReports. 2013; 4:1156–1167. [DOI] [PubMed] [Google Scholar]
- 11. Jagannathan M., Cummings R., Yamashita Y.M.. A conserved function for pericentromeric satellite DNA. eLife. 2018; 7:e34122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Padeken J., Mendiburo M.J., Chlamydas S., Schwarz H.-J., Kremmer E., Heun P.. The nucleoplasmin homolog nlp mediates centromere clustering and anchoring to the nucleolus. Mol. Cell. 2013; 50:236–249. [DOI] [PubMed] [Google Scholar]
- 13. Peters A.H., O’Carroll D., Scherthan H., Mechtler K., Sauer S., Schöfer C., Weipoltshammer K., Pagani M., Lachner M., Kohlmaier A. et al.. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001; 107:323–337. [DOI] [PubMed] [Google Scholar]
- 14. Prusov A.N., Zatsepina O.V.. Isolation of the chromocenter fraction from mouse liver nuclei. Biochemistry Mosc. 2002; 67:423–431. [DOI] [PubMed] [Google Scholar]
- 15. Ostromyshenskii D.I., Chernyaeva E.N., Kuznetsova I.S., Podgornaya O.I.. Mouse chromocenters DNA content: sequencing and in silico analysis. BMC Genomics. 2018; 19:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wachsmuth M., Knoch T.A., Rippe K.. Dynamic properties of independent chromatin domains measured by correlation spectroscopy in living cells. Epigenet. Chromatin. 2016; 9:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barth T.K., Schade G.O.M., Schmidt A., Vetter I., Wirth M., Heun P., Imhof A., Thomae A.W.. Identification of Drosophila centromere associated proteins by quantitative affinity purification-mass spectrometry. Data Brief. 2015; 4:544–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomae A.W., Schade G.O.M., Padeken J., Borath M., Vetter I., Kremmer E., Heun P., Imhof A.. A pair of centromeric proteins mediates reproductive isolation in drosophila species. Dev. Cell. 2013; 27:412–424. [DOI] [PubMed] [Google Scholar]
- 19. Boltengagen M., Huang A., Boltengagen A., Trixl L., Lindner H., Kremser L., Offterdinger M., Lusser A.. A novel role for the histone acetyltransferase Hat1 in the CENP-A/CID assembly pathway in Drosophila melanogaster. Nucleic Acids Res. 2015; 44:2145–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Samejima I., Spanos C., de Lima Alves F., Hori T., Perpelescu M., Zou J., Rappsilber J., Fukagawa T., Earnshaw W.C.. Whole-proteome genetic analysis of dependencies in assembly of a vertebrate kinetochore. J. Cell Biol. 2015; 211:1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Youn J.-Y., Dunham W.H., Hong S.J., Knight J.D.R., Bashkurov M., Chen G.I., Bagci H., Rathod B., MacLeod G., Eng S.W.M. et al.. High-Density proximity mapping reveals the subcellular organization of mRNA-Associated granules and bodies. Mol. Cell. 2018; 69:517–532. [DOI] [PubMed] [Google Scholar]
- 22. Markmiller S., Soltanieh S., Server K.L., Mak R., Jin W., Fang M.Y., Luo E.-C., Krach F., Yang D., Sen A. et al.. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell. 2018; 172:590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao X.D., Tu L.-C., Mir A., Rodriguez T., Ding Y., Leszyk J., Dekker J., Shaffer S.A., Zhu L.J., Wolfe S.A. et al.. C-BERST: defining subnuclear proteomic landscapes at genomic elements with dCas9-APEX2. Nat. Methods. 2018; 15:433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Talbert P.B., Kasinathan S., Henikoff S.. Simple and complex centromeric satellites in drosophila sibling species. Genetics. 2018; 208:977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clark A.G., Eisen M.B., Smith D.R., Bergman C.M., Oliver B., Markow T.A., Kaufman T.C., Kellis M., Gelbart W., Iyer V.N. et al.. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007; 450:203–218. [DOI] [PubMed] [Google Scholar]
- 26. Henikoff S., Malik H.S.. Centromeres: selfish drivers. Nature. 2002; 417:227–227. [DOI] [PubMed] [Google Scholar]
- 27. Sawamura K. Chromatin evolution and molecular drive in speciation. Int. J. Evol.Biol. 2012; 2012:301894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crespi B., Nosil P.. Conflictual speciation: species formation via genomic conflict. Trends Ecol. Evol. 2013; 28:48–57. [DOI] [PubMed] [Google Scholar]
- 29. Ross B.D., Rosin L., Thomae A.W., Hiatt M.A., Vermaak D., la Cruz de, A.F.A., Imhof A., Mellone B.G., Malik H.S.. Stepwise evolution of essential centromere function in a drosophila neogene. Science. 2013; 340:1211–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blum J.A., Bonaccorsi S., Marzullo M., Palumbo V., Yamashita Y.M., Barbash D.A., Gatti M.. The hybrid incompatibility genes lhr and hmr are required for sister chromatid detachment during anaphase but not for centromere function. Genetics. 2017; 207:1457–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bayes J.J., Malik H.S.. Altered heterochromatin binding by a hybrid sterility protein in drosophila sibling species. Science. 2009; 326:1538–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ferree P.M., Prasad S.. How can satellite DNA divergence cause reproductive isolation? Let us count the chromosomal ways. Genet. Res. Int. 2012; 2012:430136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rošić S., Köhler F., Erhardt S.. Repetitive centromeric satellite RNA is essential for kinetochore formation and cell division. J. Cell Biol. 2014; 207:335–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barbash D.A., Siino D.F., Tarone A.M., Roote J.. A rapidly evolving MYB-related protein causes species isolation in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:5302–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anselm E., Thomae A.W., Jeyaprakash A.A., Heun P.. Oligomerization of Drosophila Nucleoplasmin-Like Protein is required for its centromere localization. Nucleic Acids Res. 2018; 46:11274–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gerland T.A., Sun B., Smialowski P., Lukacs A., Thomae A.W., Imhof A.. The Drosophila speciation factor HMR localizes to genomic insulator sites. PLoS One. 2017; 12:e0171798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maheshwari S., Barbash D.A.. Cis-by-trans regulatory divergence causes the asymmetric lethal effects of an ancestral hybrid incompatibility gene. PLoS Genet. 2012; 8:e1002597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Satyaki P.R.V., Cuykendall T.N., Wei K.H.-C., Brideau N.J., Kwak H., Aruna S., Ferree P.M., Ji S., Barbash D.A.. The hmr and lhr hybrid incompatibility genes suppress a broad range of heterochromatic repeats. PLos Genet. 2014; 10:e1004240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Böttcher R., Hollmann M., Merk K., Nitschko V., Obermaier C., Philippou-Massier J., Wieland I., Gaul U., Förstemann K.. Efficient chromosomal gene modification with CRISPR/cas9 and PCR-based homologous recombination donors in cultured Drosophila cells. Nucleic Acids Res. 2014; 42:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rozen S., Skaletsky H.. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000; 132:365–386. [DOI] [PubMed] [Google Scholar]
- 41. Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P. et al.. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017; 45:D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. James T.C., Elgin S.C.. Identification of a nonhistone chromosomal protein associated with heterochromatin in Drosophila melanogaster and its gene. Mol. Cell Biol. 1986; 6:3862–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Köhler G., Milstein C.. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975; 256:495–497. [DOI] [PubMed] [Google Scholar]
- 44. Bobkov G.O.M., Gilbert N., Heun P.. Centromere transcription allows CENP-A to transit from chromatin association to stable incorporation. J. Cell Biol. 2018; 217:1957–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alekseyenko A.A., Gorchakov A.A., Zee B.M., Fuchs S.M., Kharchenko P.V., Kuroda M.I.. Heterochromatin-associated interactions of Drosophila HP1a with dADD1, HIPP1, and repetitive RNAs. Genes Dev. 2014; 28:1445–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Swenson J.M., Colmenares S.U., Strom A.R., Costes S.V., Karpen G.H.. The composition and organization of Drosophila heterochromatin are heterogeneous and dynamic. eLife. 2016; 5:1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eberharter A., Becker P.B.. ATP-dependent nucleosome remodelling: factors and functions. J. Cell Sci. 2004; 117:3707–3711. [DOI] [PubMed] [Google Scholar]
- 48. Lee K.K., Workman J.L.. Histone acetyltransferase complexes: one size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007; 8:284–295. [DOI] [PubMed] [Google Scholar]
- 49. Guruharsha K.G., Rual J.-F., Zhai B., Mintseris J., Vaidya P., Vaidya N., Beekman C., Wong C., Rhee D.Y., Cenaj O. et al.. A protein complex network of drosophila melanogaster. Cell. 2011; 147:690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lam S.S., Martell J.D., Kamer K.J., Deerinck T.J., Ellisman M.H., Mootha V.K., Ting A.Y.. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods. 2015; 12:51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hung V., Zou P., Rhee H.-W., Udeshi N.D., Cracan V., Svinkina T., Carr S.A., Mootha V.K., Ting A.Y.. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell. 2014; 55:332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rhee H.-W., Zou P., Udeshi N.D., Martell J.D., Mootha V.K., Carr S.A., Ting A.Y.. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013; 339:1328–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zuzow N., Ghosh A., Leonard M., Liao J., Yang B., Bennett E.J.. Mapping the mammalian ribosome quality control complex interactome using proximity labeling approaches. Mol. Biol. Cell. 2018; 29:1258–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mick D.U., Rodrigues R.B., Leib R.D., Adams C.M., Chien A.S., Gygi S.P., Nachury M.V.. Proteomics of primary cilia by proximity labeling. Dev. Cell. 2015; 35:497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gupta R., Somyajit K., Narita T., Maskey E., Stanlie A., Kremer M., Typas D., Lammers M., Mailand N., Nussenzweig A. et al.. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell. 2018; 173:972–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cheutin T., McNairn A.J., Jenuwein T., Gilbert D.M., Singh P.B., Misteli T.. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science. 2003; 299:721–725. [DOI] [PubMed] [Google Scholar]
- 57. Festenstein R., Pagakis S.N., Hiragami K., Lyon D., Verreault A., Sekkali B., Kioussis D.. Modulation of heterochromatin protein 1 dynamics in primary Mammalian cells. Science. 2003; 299:719–721. [DOI] [PubMed] [Google Scholar]
- 58. Müller K.P., Erdel F., Caudron-Herger M., Marth C., Fodor B.D., Richter M., Scaranaro M., Beaudouin J., Wachsmuth M., Rippe K.. Multiscale analysis of dynamics and interactions of heterochromatin protein 1 by fluorescence fluctuation microscopy. Biophys. J. 2009; 97:2876–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bolkan B.J., Booker R., Goldberg M.L., Barbash D.A.. Developmental and cell cycle progression defects in Drosophila hybrid males. Genetics. 2007; 177:2233–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Aruna S., Flores H.A., Barbash D.A.. Reduced fertility of Drosophila melanogaster hybrid male rescue (Hmr) mutant females is partially complemented by Hmr orthologs from sibling species. Genetics. 2009; 181:1437–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Heun P., Erhardt S., Blower M.D., Weiss S., Skora A.D., Karpen G.H.. Mislocalization of the Drosophila centromere-specific histone CID promotes formation of functional ectopic kinetochores. Dev. Cell. 2006; 10:303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Probst A.V., Okamoto I., Casanova M., Marjou El, F. Le Baccon, Almouzni G.. A strand-specific burst in transcription of pericentric satellites is required for chromocenter formation and early mouse development. Dev. Cell. 2010; 19:625–638. [DOI] [PubMed] [Google Scholar]
- 63. Chan F.L., Wong L.H.. Transcription in the maintenance of centromere chromatin identity. Nucleic Acids Res. 2012; 40:11178–11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cléard F., Delattre M., Spierer P.. SU(VAR)3-7, a Drosophila heterochromatin-associated protein and companion of HP1 in the genomic silencing of position-effect variegation. EMBO J. 1997; 16:5280–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jankovics F., Bence M., Sinka R., Faragó A., Bodai L., Pettkó-Szandtner A., Ibrahim K., Takács Z., Szarka-Kovács A.B., Erdélyi M.. Drosophila small ovarygene is required for transposon silencing and heterochromatin organization, and ensures germline stem cell maintenance and differentiation. Development. 2018; 145:dev170639. [DOI] [PubMed] [Google Scholar]
- 66. Erhardt S., Mellone B.G., Betts C.M., Zhang W., Karpen G.H., Straight A.F.. Genome-wide analysis reveals a cell cycle-dependent mechanism controlling centromere propagation. J. Cell Biol. 2008; 183:805–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen C.-C., Bowers S., Lipinszki Z., Palladino J., Trusiak S., Bettini E., Rosin L., Przewloka M.R., Glover D.M., O’Neill R.J. et al.. Establishment of centromeric chromatin by the CENP-A assembly factor CAL1 requires FACT- mediated transcription. Dev. Cell. 2015; 34:73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Glenn S.E., Geyer P.K.. Investigation of the developmental requirements of drosophila HP1 and insulator protein partner, HIPP1. G3 (Bethesda). 2019; 9:345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chavez J., Murillo-Maldonado J.M., Bahena V., Cruz A.K., Castañeda-Sortibrán A., Rodriguez-Arnaiz R., Zurita M., Valadez-Graham V.. dAdd1 and dXNP prevent genome instability by maintaining HP1a localization at Drosophila telomeres. Chromosoma. 2017; 126:697–712. [DOI] [PubMed] [Google Scholar]
- 70. Van Bortle K., Nichols M.H., Li L., Ong C.-T., Takenaka N., Qin Z.S., Corces V.G.. Insulator function and topological domain border strength scale with architectural protein occupancy. Genome Biol. 2014; 15:R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fudenberg G., Imakaev M., Lu C., Goloborodko A., Abdennur N., Mirny L.A.. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016; 15:2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Doyle B., Fudenberg G., Imakaev M., Mirny L.A.. Chromatin loops as allosteric modulators of enhancer-promoter interactions. PLoS Comput. Biol. 2014; 10:e1003867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Orr H.A., Madden L.D., Coyne J.A., Goodwin R., Hawley R.S.. The developmental genetics of hybrid inviability: a mitotic defect in Drosophila hybrids. Genetics. 1997; 145:1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Savvidou E. Drosophila CAP-D2 is required for condensin complex stability and resolution of sister chromatids. J. Cell Sci. 2005; 118:2529–2543. [DOI] [PubMed] [Google Scholar]
- 75. Oliveira R.A., Kotadia S., Tavares A., Mirkovic M., Bowlin K., Eichinger C.S., Nasmyth K., Sullivan W.. Centromere-independent accumulation of cohesin at ectopic heterochromatin sites induces chromosome stretching during anaphase. PLoS Biol. 2014; 12:e1001962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cooper J.C., Lukacs A., Reich S., Schauer T., Imhof A., Phadnis N.. Altered localization of hybrid incompatibility proteins in drosophila. Mol. Biol. Evol. 2019; 36:1783–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Orr B., Sunkel C.E.. Drosophila CENP-C is essential for centromere identity. Chromosoma. 2011; 120:83–96. [DOI] [PubMed] [Google Scholar]
- 78. Phair R.D., Scaffidi P., Elbi C., Vecerová J., Dey A., Ozato K., Brown D.T., Hager G., Bustin M., Misteli T.. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell Biol. 2004; 24:6393–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. et al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Strom A.R., Emelyanov A.V., Mir M., Fyodorov D.V., Darzacq X., Karpen G.H.. Phase separation drives heterochromatin domain formation. Nature. 2017; 547:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Larson A.G., Elnatan D., Keenen M.M., Trnka M.J., Johnston J.B., Burlingame A.L., Agard D.A., Redding S., Narlikar G.J.. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature. 2017; 547:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cheutin T., Gorski S.A., May K.M., Singh P.B., Misteli T.. In vivo dynamics of Swi6 in yeast: evidence for a stochastic model of heterochromatin. Mol. Cell Biol. 2004; 24:3157–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Barth T.K., Schade G.O.M., Schmidt A., Vetter I., Wirth M., Heun P., Thomae A.W., Imhof A.. Identification of novel Drosophila centromere-associated proteins. Proteomics. 2014; 14:2167–2178. [DOI] [PubMed] [Google Scholar]
- 84. Mellone B., Erhardt S., Karpen G.H.. The ABCs of centromeres. Nat. Cell Biol. 2006; 8:427–429. [DOI] [PubMed] [Google Scholar]
- 85. Foltz D.R., Jansen L.E.T., Black B.E., Bailey A.O., Yates J.R., Cleveland D.W.. The human CENP-A centromeric nucleosome-associated complex. Nat. Cell Biol. 2006; 8:458–469. [DOI] [PubMed] [Google Scholar]
- 86. Talbert P.B., Henikoff S.. Transcribing centromeres: Noncoding RNAs and kinetochore assembly. Trends Genet. 2018; 34:587–599. [DOI] [PubMed] [Google Scholar]
- 87. Gozalo A., Duke A., Lan Y., Pascual-Garcia P., Talamas J.A., Nguyen S.C., Shah P.P., Jain R., Joyce E.F., Capelson M.. Core components of the nuclear pore bind distinct states of chromatin and contribute to polycomb repression. Mol. Cell. 2019; 77:67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gerasimova T.I., Byrd K., Corces V.G.. A chromatin insulator determines the nuclear localization of DNA. Mol. Cell. 2000; 6:1025–1035. [DOI] [PubMed] [Google Scholar]
- 89. Rowley M.J., Lyu X., Rana V., Ando-Kuri M., Karns R., Bosco G., Corces V.G.. Condensin II counteracts cohesin and RNA Polymerase II in the establishment of 3D chromatin organization. CellReports. 2019; 26:2890–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bushey A.M., Dorman E.R., Corces V.G.. Chromatin insulators: regulatory mechanisms and epigenetic inheritance. Mol. Cell. 2008; 32:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gerasimova T.I., Corces V.G.. Polycomb and trithorax group proteins mediate the function of a chromatin insulator. Cell. 1998; 92:511–521. [DOI] [PubMed] [Google Scholar]
- 92. Ciabrelli F., Cavalli G.. Chromatin-driven behavior of topologically associating domains. J. Mol. Biol. 2015; 427:608–625. [DOI] [PubMed] [Google Scholar]
- 93. Lee Y.C.G., Ogiyama Y., Martins N.M.C., Beliveau B.J., Acevedo D., Wu C.T., Cavalli G., Karpen G.H.. Pericentromeric heterochromatin is hierarchically organized and spatially contacts H3K9me2/3 islands located in euchromatic genome. 2019; bioRxiv doi:21 January 2019, preprint: not peer reviewed 10.1101/525873. [DOI] [PMC free article] [PubMed]
- 94. Rosin L.F., Nguyen S.C., Joyce E.F.. Condensin II drives large-scale folding and spatial partitioning of interphase chromosomes in Drosophila nuclei. PLoS Genet. 2018; 14:e1007393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Durdevic Z., Pillai R.S., Ephrussi A.. Transposon silencing in the Drosophilafemale germline is essential for genome stability in progeny embryos. Life Sci. Alliance. 2018; 1:e201800179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang Z., Schwacke R., Kunze R.. DNA Damage-Induced transcription of transposable elements and long non-coding rnas in arabidopsis is rare and ATM-Dependent. Mol. Plant. 2016; 9:1142–1155. [DOI] [PubMed] [Google Scholar]
- 97. Klattenhoff C., Bratu D.P., McGinnis-Schultz N., Koppetsch B.S., Cook H.A., Theurkauf W.E.. Drosophila rasiRNA pathway mutations disrupt embryonic axis specification through activation of an ATR/Chk2 DNA damage response. Dev. Cell. 2007; 12:45–55. [DOI] [PubMed] [Google Scholar]
- 98. Kelleher E.S., Edelman N.B., Barbash D.A.. Drosophila interspecific hybrids phenocopy piRNA-pathway mutants. PLoS Biol. 2012; 10:e1001428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Vizcaíno J.A., Deutsch E.W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J.A., Sun Z., Farrah T., Bandeira N. et al.. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014; 32:223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Van Bortle K., Ramos E., Takenaka N., Yang J., Wahi J.E., Corces V.G.. Drosophila CTCF tandemly aligns with other insulator proteins at the borders of H3K27me3 domains. Genome Res. 2012; 22:2176–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Heeger S., Leismann O., Schittenhelm R., Schraidt O., Heidmann S., Lehner C.F.. Genetic interactions of separase regulatory subunits reveal the diverged Drosophila Cenp-C homolog. Genes Dev. 2005; 19:2041–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (99) partner repository with the dataset identifier PXD012551. Novel CAPH2 and HMR ChIP-seq data were deposited to GEO with accession number GSE137194.