

Abstract
Imaging neurons and neural circuits over large volumes at high speed and subcellular resolution is a difficult task. Incorporating a Bessel focus module into a two-photon fluorescence mesoscope, we achieved rapid volumetric imaging of neural activity over mesoscale with synaptic resolution. We applied it to calcium imaging of entire dendritic spans of neurons as well as neural ensembles within multiple cortical regions over two hemispheres of the awake mouse brain.
Neurons in the brain are highly connected. Within the neocortex, a single neuron may receive a multitude of synaptic inputs and send out projections traversing millimeters of tissue. Through these long-range connections, information flows through multiple cortical regions spanning two hemispheres. To understand computation by either a single neuron or an extended network, it is essential to monitor neural activity throughout cortical depth over large volumes, ideally at synaptic resolution. By combining a two-photon fluorescence mesoscope with 5-mm-diameter field of view (FOV)1 and a volumetric imaging module based on Bessel focus scanning2–5, here we demonstrated rapid synapse-resolving mesoscale volumetric imaging of neural activity in the adult mouse brain in vivo.
In Bessel focus scanning, an axially extended excitation focus is scanned laterally in two dimensions (2D) to provide a projected view of the volume defined by the scanning area and the axial extent of the focus. Such a focus can be generated by illuminating the microscope objective back focal plane with an annular pattern, with the resulting focal electric field amplitude approximating a Bessel function. Laterally confined yet axially extended, a Bessel-like focus provides synapse-resolving lateral resolution at a volumetric imaging rate of 2D scanning. We designed a compact Bessel optical module to fit between the excitation laser and the mesoscope, which generated a Bessel-like focus with its axial FWHM continuously adjustable from 37 to 116 µm (Supplementary Figs. 1, 2, Methods). Scanning the Bessel focus in 2D led to a projected view of the fluorescent structures within a volume defined by up to 20 mm2 area and the axial length of the focus. For in vivo experiments, we configured the module to generate a 0.4-NA Bessel focus with a 72-µm axial FWHM. Because focusing an annular illumination sharpens the lateral confinement of the light6, the 0.4-NA Bessel focus had lateral FWHMs of less than 0.6 µm or about 15% higher lateral resolution than the conventional 0.6-NA (“Gaussian”) focus across the entire imaging FOV (Supplementary Figs. 1,3). Compared to the 0.6-NA Gaussian focus, the Bessel focus elongated the axial extent of two-photon excitation by ~18 fold. Scanning this Bessel focus in 2D over the cortex of a Thy-1 GFP line M mouse in vivo, we imaged neurites within a 4.2 mm × 4.2 mm × 100 µm volume while maintaining the ability to resolve axonal boutons (i.e., varicosities along thinner and dimmer neurites7) and dendritic spines (i.e., protrusions from thicker and brighter neurites) over the entire FOV (Fig. 1). Such synapse-resolving capability of our volumetric mesoscope imaging was maintained even at an imaging depth of 472 µm below dura (Supplementary Fig. 4).
Figure 1. Mesoscope volumetric imaging with synaptic resolution.
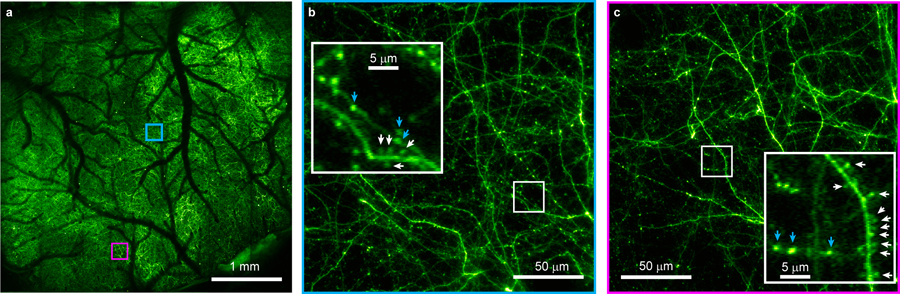
Scanning a Bessel focus in 2D imaged a 4.2 mm × 4.2 mm × 100 µm volume of L1 neurites in an awake Thy-1 GFP line M mouse cortex in vivo. (a) The entire image and (b,c) zoomed-in views (white arrows: spines; blue arrow: boutons).
Having validated the synapse-resolving capability of our microscope on the mesoscale, we next used it to monitor neural activity across entire dendritic spans of cortical neurons in head-fixed awake mice. Using the remote focusing module of the mesoscope to rapidly translate the Bessel focus along the axial direction, we monitored the calcium activity of the soma as well as dendrites of a GCaMP6s8-expressing infragranular neuron (depth of soma: 586 µm) in the mouse visual cortex at high speed and synaptic resolution (Fig. 2a–d, Supplementary Video 1). Imaging the 301 µm × 450 µm × 612 µm volume by six 2D scans of the Bessel focus (Fig. 2b), we achieved a volumetric imaging speed of 3.2 Hz (Supplementary Video 2). In comparison, scanning the same volume with remote focusing of the Gaussian focus gave rise to a volumetric rate of 0.06 Hz (Fig. 2a), a 50-fold reduction in imaging speed and too slow for calcium imaging. The dendritic spine images by the Bessel focus were sharper than those with the Gaussian focus (insets, Figs. 2a,b,d), reflecting the increased lateral resolution of the Bessel mesoscope. In total, we detected 145 dendritic spines from this neuron. Their calcium activity profiles were dominated by transients highly synchronized with those in its dendrites and soma (Figs. 2c,d). These global transients in spines and dendrites had faster temporal dynamics than those in soma. In some spines, we also detected transients independent of the global events (red arrows, Fig. 2d), likely evoked by local synaptic inputs.
Figure 2. Extra-large volumetric functional imaging across the entire dendritic spans of neurons with synaptic resolution in awake mice.
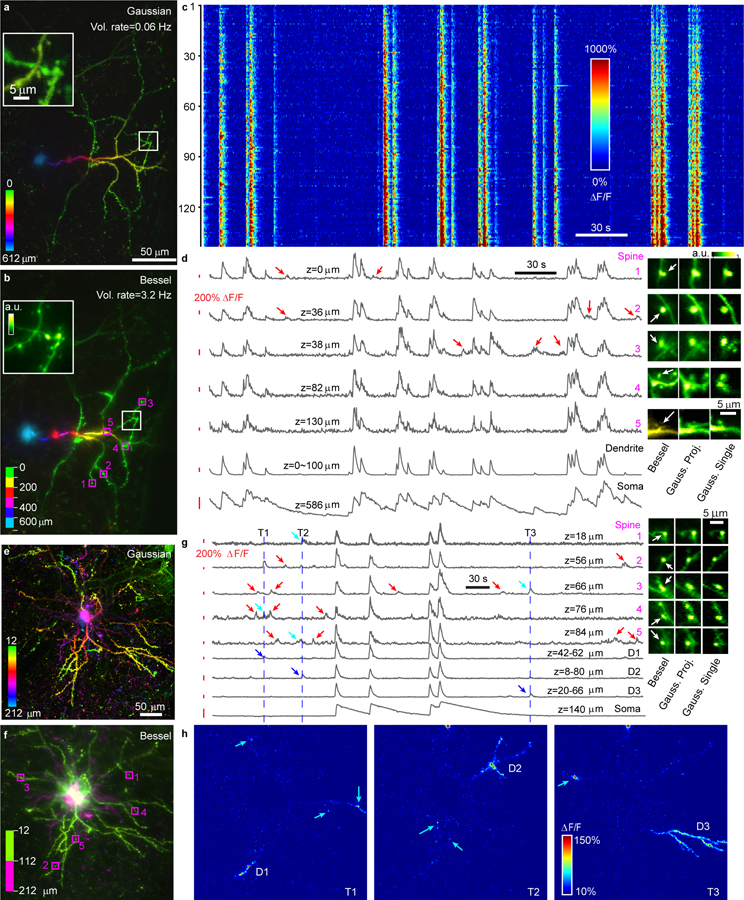
(a-d) Recording of an infragranular neuron in a 301 µm × 450 µm × 612 µm volume. (a) Maximum intensity projection of Gaussian image stack (307 images, 0.4-µm pixel, 2-µm z-step). (b) Composite of six Bessel images (0.4-µm pixel, 100-µm z-step). Insets in (a,b): zoomed-in views. (c) Calcium transient ΔF/F of 145 spines. (d) Calcium transients of example spines, dendrites, and soma. Red arrows: local synaptic transients. Insets: images of spines in (a), (b), and from a single Gaussian scan. (e-h) Recording of a L2/3 neuron in a 396 µm × 400 µm × 200 µm volume. (e) Maximum intensity projection of Gaussian image stack (101 images, 0.33-µm pixel, 2-µm z-step). (f) Composite of two Bessel images (0.33-µm pixel, 100-µm z step). (g) Calcium transients of example spines, dendrites, and soma. Red and blue arrows: local synaptic and dendritic transients, respectively. Blue lines: time points for local dendritic events. Insets: images of spines in (e), (f), and from a single Gaussian scan. (h) ΔF/F maps at three time points showing local dendritic and synaptic events (12 frame averages). Cyan arrows in (g,h): spines with activity coincident with dendritic branches D1, D2 or D3. Post-objective power: (a) 36–96 mW and (e) 24–40 mW, increased exponentially with depth; (b,f) 120 mW.
Two 2D scans of the Bessel focus imaged the dendrites of a L2/3 neuron within a volume of 396 µm × 400 µm × 200 µm (12 to 212 µm depth, Figs. 2e–h, Supplementary Videos 3, 4). Its dendritic spines exhibited both global responses synchronized with somatic firing and local isolated transients (red arrows, Fig. 2g). We also observed local transients spanning one or a few dendritic branches (blue arrows, Fig. 2g; ΔF/F map, Fig. 2h, Supplementary Fig. 5), indicative of dendritic spiking9,10. Interestingly, local synaptic transients on spines sometimes were synchronized with calcium events on distant dendritic branches and spines (cyan arrows, Figs. 2g,h), suggesting that they may share common inputs. As these two examples indicate, combining Bessel module with fast axial scanning allows mapping the complete synaptic and dendritic activity profile of neurons, thus serves as a powerful tool in investigating neuronal computation by individual neurons. The mesoscopic FOV of our system also enabled random-access volumetric imaging of neurites in distant volumes. We carried out simultaneous functional imaging of neurites in two 400 µm × 425 µm × 200 µm volumes 2-mm apart, and extracted calcium traces from combined 486 axonal boutons and dendritic spines (Supplementary Fig. 6, Supplementary Video 5).
With the extra-large FOV, the Bessel mesoscope also enabled activity measurements of very large neural ensembles over multiple cortical areas simultaneously. As an example, we studied spontaneous activity of cortical GABAergic neurons in a head-fixed quietly awake Gad2-IRES-cre mouse in the dark. Previous work studying tens of inhibitory neurons within a small cortical region reported that some neurons exhibited spontaneous activity highly correlated with arousal level4,11,12. Here, we monitored spontaneous activity of GABAergic neurons spanning multiple brain areas, including the primary visual cortex (V1), the anteromedial visual area (AVA), the posterior parietal association areas (PPAA), and somatosensory area (Fig. 3a, Supplementary Video 6), with the mouse’s pupil closely tracked and used as an indicator of arousal level. In cortex, because the GABAergic neurons are relatively sparsely distributed, within 100 µm thick tissue we detected minimal somatic overlaps in the projected images of neurons at different depths (Fig. 3b, Supplementary Fig. 7). Therefore, scanning and then axially translating the Bessel focus six times at 100 µm steps allowed recording the inhibitory neural activity within a volume of 3,020 µm × 1,500 µm × 600 µm (50 to 650 µm below the dura) at 1 Hz, a 50× gain in image rate (Fig. 3c, Supplementary Video 7). Because subpopulations of these cytosolic-GCaMP6s-expressing neurons exhibited synchronized activity, we observed highly-correlated background fluctuation across the FOV. Consequently, we found that correlation-based region-of-interest (ROI) segmentation algorithms13–16 failed to accurately detect active neurons. We modified CNMF-E16 to first segment ROIs based on local contrast of fluorescence intensity rather than local correlation and then model the neuropil background with a spatially smooth low-rank matrix (Methods, Supplementary Codes, Supplementary Video 8). The optimized CNMF-E method detected 9,247 active neurons within this volume and extracted their calcium transients without background neuropil activity (Fig. 3d). Across depths and cortical areas, we observed diverse spontaneous activity patterns positively or negatively correlated with pupil area (red and blue traces for highly positive and negative correlation, respectively, in Fig. 3e), as well as activity un-correlated with the arousal level (gray traces, Fig. 3e). We color-coded all 9,247 neurons based on their Pearson correlation coefficients (CCs) between their calcium transients and the pupil area (Fig. 3f). Within the entire neural ensemble, 16% of neurons exhibited highly correlated (CC>0.4) and 5% highly anti-correlated (CC<−0.4) spontaneous activity with arousal (Fig. 3g). We observed both cortical region and depth dependences in the CC distributions of these GABAergic neurons (Supplementary Fig. 8), with the average CCs increasing from posterior to anterior and cortical depth (Fig. 3h). Similar trends were observed when the same analyses were applied to 6,613 neurons segmented by hand (Supplementary Figs. 9,10). Finally, with the mesoscale FOV of the microscope, we measured neural activity from three volumes of 189 L2/3 callosal-projection neurons in visual cortices located in separate hemispheres at 3.1 volumes per second, 39× faster than 3D scanning of Gaussian focus (Methods, Supplementary Fig. 11, Supplementary Video 9).
Figure 3. Mesoscale volumetric functional imaging of GABAergic neuron ensembles within multiple cortical areas of an awake mouse.
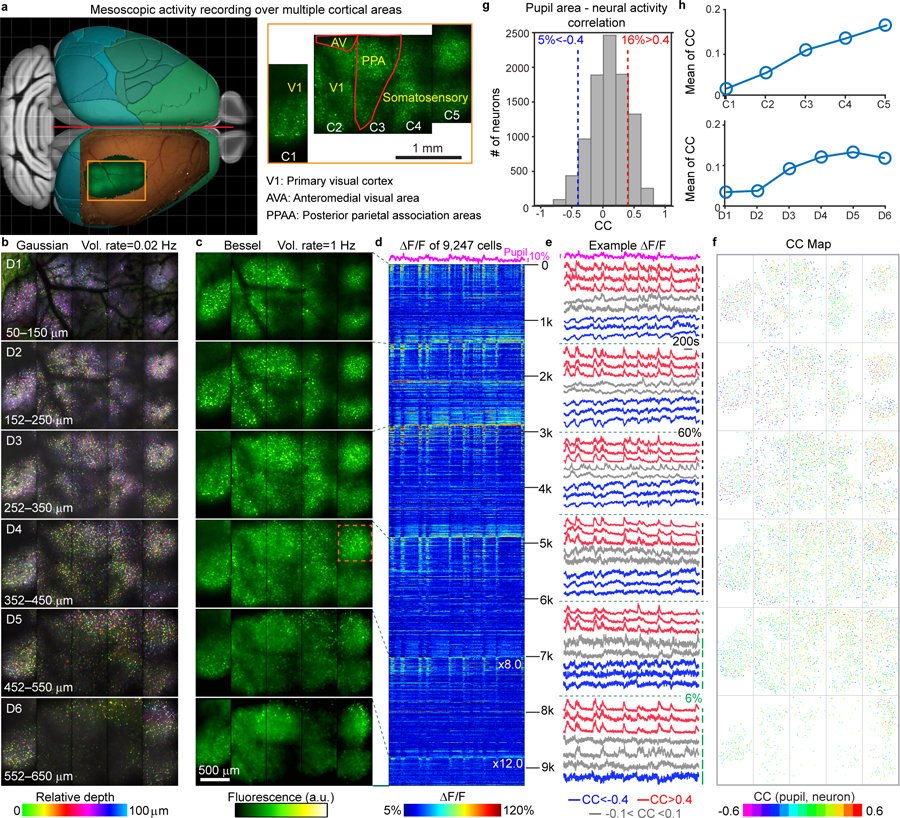
(a) Schematic drawing and two-photon fluorescence images showing FOV covering multiple cortical areas (C1-C5). (b) Maximum intensity projections of six (D1-D6) 100-µm-thick Gaussian image stacks covering a 3,020 µm × 1,500 µm × 600 µm volume (301 images, 2-µm pixel and z-step). Post-objective power: 24–96 mW increased exponentially with depth. (c) Six Bessel images of the same volume (2-µm pixel, 100-µm z-step size); Orange box: FOV in Supplementary Video 8. Post-objective power: 36, 51, 70, 99, 120, and 120 mW, respectively. (d) ΔF/F of 9,247 neurons in D1-D6, sorted by decreasing correlation with pupil area (top trace). In D5 and D6, ΔF/F traces were multiplied by 8× and 12×, respectively, to aid visibility. (e) Traces of the pupil area and example calcium transients. CC: Pearson’s correlation coefficient. Blue, red, and gray indicate varying correlation with pupil area. (f) Map of CC for individual neurons. (g) Histogram distribution of CC. (h) Mean of CCs of neurons in different cortical (top) areas and (bottom) depths. Data were taken from n = 1 mouse.
In summary, by combining Bessel-focus-based volumetric imaging with mesoscope and turning 2D frame rate into volume rate, we achieved fast activity imaging with synaptic resolution over large mesoscale volumes. A powerful tool to interrogate the input-output transformation by individual neurons, it simultaneously recorded synaptic, dendritic, and somatic calcium activity of neurons spanning 600 µm in depth in a head-fixed awake mouse. Such performance is difficult for other methods to achieve17, because of the simultaneous requirements of high spatial and temporal resolution large volumes of opaque tissue. Unlike light-sheet based methods which requires minimal scattering of the fluorescence photons, the nonlinear excitation and non-imaging detection of the two-photon mesoscope enabled us to resolve synapses at depth. The axial extension of the Bessel focus also renders the resulting images resistant to axial motion artifacts, without the need for complex motion compensation18.
On the circuit level, we took advantage of the mesoscale FOV and demonstrated simultaneous monitoring of the spontaneous activity of >9,000 GABAergic neurons within 600 µm cortical depth of four cortical areas, as well as callosal-projection neurons spanning two hemispheres. Compared to conventional 3D scanning approaches, in addition to an order-of-magnitude gain in imaging speed, Bessel focusing scanning with a mesoscope also generated information-rich datasets with much smaller data sizes, facilitating image processing and data analysis.
Bessel modules can also be combined with other mesoscope systems19,20, as well as three-photon fluorescence excitation21–23 to further increase imaging depth. Together, they will greatly benefit system neuroscientific investigations in live brains, where activity of neural circuits underlying animal behaviors can be studied at subcellular resolution on the mesoscale across brain areas and cortical depths.
ONLINE METHODS
Animals
All animal experiments were conducted according to the National Institutes of Health guidelines for animal research. Procedures and protocols on mice were approved by the Institutional Animal Care and Use Committee at Janelia Research Campus, Howard Hughes Medical Institute. Male or female mice (C57BL/6J background) aged two months and older were used in this study and included wild-type (Jackson Laboratories, Black 6, stock #:000664), Gad2-IRES-cre (Jackson Laboratories, Gad2tm2(cre)Zjh/J, stock #: 010802), and Thy1-GFP line M (Jackson Laboratories, Tg(Thy1-EGFP)MJrs/J, stock #: 007788) strains. Mice were housed in cages in groups of 1–5 before surgeries under reverse light cycle.
Stereotaxic surgery for in vivo imaging
All surgeries were performed using a stereotaxic apparatus (Model 1900, David Kopf Instruments, CA, USA) and aseptic technique, as described previously1,4. Briefly, mice were anaesthetized with isoflurane (1–2% by volume in O2) and given the analgesic buprenorphine (SC, 0.3 mg per kg of body weight). A craniotomy was made over one dorsal hemisphere of mice or across the two hemispheres with dura left intact. For mice that required virus injection, a glass pipette (Drummond Scientific Company) beveled at 45° with a 15–20-μm opening was back-filled with mineral oil. A fitted plunger controlled by a hydraulic manipulator (Narishige, MO10) was inserted into the pipette and used to load and slowly inject 10–20 nl viral solution into the brain at ~200–400 μm below pia. The following injection coordinates (in mm) were chosen to label brain regions in one or both hemispheres: site 1 (Bregma: −4.6, midline: 2.1), site 2 (Bregma: −4.6, midline: 2.8), site 3 (Bregma: −4.1, midline: 2.1), site 4 (Bregma: −4.1, midline: 2.8), site 5 (Bregma: −3.5, midline: 2.1), site 6 (Bregma: −3.5, midline: 2.8), site 7 (Bregma: −3.0, midline: 2.1), site 8 (Bregma: −3.0, midline: 2.8).
For sparse labeling of neurons in one hemisphere, AAV2/1.syn.FLEX.GCaMP6s (1×1013 GC/ml) was mixed with AAV2/1.syn.Cre (1×1013 GC/ml diluted 10,000 times) at 1:1 for injection into wildtype mice. For the labeling of GABAergic neurons, AAV2/1.syn.FLEX.GCaMP6s (1×1013 GC/ml) was used for injection into Gad2-IRES-cre mice. For the labeling of callosal projection neurons in both hemispheres, AAV2/1.syn.FLEX.GCaMP6s (1×1013 GC/ml) was mixed with retroAAV.CAG.FlipO (2×1012 GC/ml) at 1:1 for injection into left hemisphere at the aforementioned coordinates, and AAV2/1.CAG.FRT.GCaMP6s (3.9×1012 GC/ml) was mixed with retroAAV.syn.Cre (1×1013 GC/ml) at 1:1 for injection into right hemisphere at the above coordinates in the wild-type mouse. At the completion of viral injections or craniotomy without viral injections, a 450-µm-thick custom-made glass window that fit the shape of the craniotomy was embedded and sealed in place with dental acrylic. A titanium head-post was then attached to the skull with cyanoacrylate glue and dental acrylic. In vivo imaging was carried out after at least two weeks of recovery with single or paired housing and habituation for head fixation. All imaging experiments were carried out on head-fixed awake mice.
Two-photon fluorescence mesoscope with a Bessel module
The two-photon fluorescence mesoscope (Supplementary Fig. 1a) was described in detail previously1. In its conventional operation mode (“Gaussian mode”), after beam expansion, the excitation laser first entered the remote focusing (RF) unit and was focused by an objective onto a mirror mounted on a voice coil. A polarized beam splitter and a quarter-wave plate directed the back-reflected light towards the scanning optics. Pupil relay PR1 imaged the back focal plane of the RF objective onto a resonant scanner that scanned the focus along x axis at 24 kHz line rate. After pupil relay PR2, the excitation light was scanned by a virtually conjugated galvo pair (VCGP), consisting of two x galvo mirrors and one y galvo mirror. The two x galvo mirrors scanned synchronously to keep the beam stationary at the y gavlo mirror without needing a pupil relay made of refractive optics. Directing the excitation laser to different locations in the FOV, the VCGP unit enabled the transition time between two imaging areas to within 3.4 ms. Finally, the y galvo was optically conjugated by pupil relay PR3 to the back focal plane of the imaging objective of an illumination NA of 0.6 and collection NA of 1.0.
Using the optical prescriptions of the mesoscope, we designed an axicon-based compact Bessel module following the principles detailed previously5. The module was composed of an axicon, three lenses (L1, L2, and L3), and an annular mask, and fit between the excitation laser and the scanning optics of a mesoscope (Supplementary Fig. 1a, Methods). The module was mounted on a 12” × 12” breadboard and attached to the mesoscope using two angle brackets (see Supplementary Information for a complete part list and solid models in Autodesk Inventor®, Supplementary Fig. 1). The module contained two mirrors mounted on a motorized stage. When inserted into the path of the excitation laser, mirror M1 directed the light into the Bessel module, and after it propagated through the module, mirror M2 directed the excitation light back into the mesoscope. Together, they enabled us to conveniently and reproducibly switch between the Bessel and Gaussian focus scanning modalities without requiring realignment of the optical path. In the Bessel module, after the excitation laser was directed to propagate through the axicon (i.e., a conical lens), it formed an annular illumination at the focal plane of lens L1 (FL: 125 mm), where an annular mask was placed to spatially filter the annular illumination to achieve a desired axial intensity profile. The lens pair L2 and L3 then relayed the filtered annular illumination onto the back focal plane of the RF objective and eventually the back focal plane of the imaging objective via PR1, PR2 and PR3 (Supplementary Fig. 1a). In order not to change the optical layout of the mesoscope, lens L3 had to be at least 330 mm away from the back focal plane of the RF objective. To make the module compact, we chose L2 and L3 to have relatively short focal lengths (FL: L2, 75 mm; L3, 250 mm). To maintain optical conjugation between the mask and the RF objective back focal plane, instead of the usual 4f configuration for L2 and L3, we used the following distances: annular mask – L2: 75 - (330–250) / (330/75)2 mm= 70.8 mm; L2 – L3: 325 mm; L3 – RF back focal plane: 330 mm. The radius and thickness of the annulus determined the lateral resolution and axial extent of the focus, respectively4. To adjust the axial length of the Bessel beam focus, we either displaced L2 along the direction of beam propagation5 or varied the beam size on the axicon24 (Supplementary Fig. 2). We measured the excitation point spread functions (PSFs) across the mesoscope FOV for both the Gaussian and Bessel configurations (Supplementary Fig. 3) with 0.2-µm-diameter fluorescent beads.
Assembly and alignment of the Bessel module
Detailed documentation on assembling and aligning the mesoscope with conventional Gaussian focus scanning can be found at https://wiki.janelia.org/wiki/display/mesoscopy/Documentation. Below we provide a detailed assembly and alignment procedure for the Bessel module (laser safety guidelines should be followed at all time):
After installing the breadboard containing the Bessel module onto the mesoscope, set up all mechanical mounts and cage system (Supplementary Fig. 1b) and install all optical components except the axicon, mask, or lenses. Adjust the mirrors so that the excitation light passes through the center of the cage system. Install the axicon, mask, and lenses L1-L3.
Adjust the xy position of the axicon, so that the excitation light hits its center. The transmitted light should be circularly symmetric when viewed immediately before L1 with a near-infrared detector card (VRC4, Thorlabs).
Adjust the xyz position of the mask to maximize its transmission power throughput.
Place the near-infrared detector card below the focal plane of the mesoscope objective to visualize the transmitted annular illumination. Adjust mirror M2 till the annular illumination is circularly symmetric and centers at the same lateral position as the transmitted Gaussian beam.
Translate lens L2 along its optical axis (made easy by the cage system) to control the length of the Bessel focus (upwards for shorter, and downwards for longer foci).
Automatic detection of ROIs and extraction of calcium transients from mesoscopic volumetric activity data
All in vivo images were registered in 2D with either a piecewise25 or full-frame iterative cross-correlation-based rigid motion correction algorithm26. Because neuron shapes here are consistently quasi-ellipsoidal and neurons are not densely packed, we can easily use a template-matching method to detect cell locations and create ROI masks for individual neurons. However, the neurons’ activity cannot be simply extracted by averaging pixels within each ROI, because the background fluctuations are highly correlated with the neural signal (with some correlation coefficients higher than 0.7) and individual neural signals are highly correlated as well. We developed a simple solution to separate individual neurons’ signals from the background, based on the empirical observation that the background is spatially smoother than the shape of a single neuron; see Ref. 16 for a related approach.
We begin by estimating the background values of the pixels outside of all neurons’ ROIs with a low-rank matrix factorization model, and then fill the background values with neurons’ ROIs by interpolating the pixel values on ROI boundaries. The low-rank approximation ensures that the background sources have large spatial ranges and the interpolating strategy preserves the spatial smoothness of these sources. Once the background is estimated, extracting a single neuron’s shape and activity can be simply done by running a rank-1 matrix factorization on the background-subtracted data within each ROI.
We summarize the details below:
Compute the mean image of the video over time and filter the image with a mean-centered truncated 2D Gaussian template (gaussian width pixels and the truncated size is pixels).
In the resulting image following step 1, find all pixels that are local maxima within an area of pixels. All pixels with values above a manually selected threshold will be selected as neuron centers.
Create ROI masks for each neuron by creating a circular region (diameter=10 pixels, 20 μm) surrounding the identified neuron centers.
Mask all ROIs out from the raw video data (d is the number of pixels and T is the number of frames) and fit the model (note that has no neural signals after we mask all ROIs out) by running a truncated-k (k=3 here) singular value decomposition on the masked video data . Both and have missing values for pixels inside the ROIs.
Run MATLAB built-in function regionfill to fill the missing values in the estimated and , and then reconstruct the background signal , as shown in the “background” panel in Supplementary Video 8..
Let (“Raw-BG” panel in Supplementary Video 8). For every detected neuron in step 2, fit a model by requiring and , where is constructed from by only selecting pixels in the -th neuron’s ROI. After we process one neuron, we update by subtracting . We also fit the estimated with a 2D Gaussian function and delete the neurons with R-squared values (variance explained by the model / total variance) smaller than a user-specified value (~0.55); these deleted neurons typically have non-neural shapes or can sometimes correspond to merges of multiple cell shapes.
Compute for all neurons. Calcium dynamics for the -th neuron without neuropil contamination is first reconstructed as ∑ plus the sum of within the -th neuron’s ROI. We then determine and from the reconstructed traces, from which “Denoised” and “Demixed” panels in Supplementary Video 8 are calculated.
For the most part, the extracted ΔF/F does not include surrounding neuropil activity, due to the background subtraction described above. However, signal contamination by out-of-focus excitation of fluorescence structures near the cortical surface constituted a time-invariant additional background signal, which was absorbed into the baselines (F) of detected ROIs and remained throughout the processing pipeline. As this out-of-focus background fluorescence increased exponentially with imaging depth, neurons in 450 – 650 µm depth had artefactually small ΔF/F values (Fig. 3c,d), which nevertheless did not affect the correlation computation or the conclusions related to the data shown in Fig. 3.
To validate the automatic neuropil subtraction approach employed here, we compared these calcium transients with those obtained with the more established method of neuropil subtraction (“surrounding-pixel neuropil subtraction”), where fluorescence signal from pixels surrounding each ROI was used to calculate neuropil contribution4,8,27. We found that the calcium transients calculated for the same neuron show high correlation coefficient across all depths (Supplementary Fig. 12 and Supplementary Video 10), indicating that our CNMF-E pipeline produced very similar traces to those from the standard method of neuropil subtraction. A closer look at the 2% of neurons with correlation coefficients less than 0.6 indicates that they fell within three categories: (1) neurons whose neuropil mask contained nearby cell bodies, causing errors to the surrounding-pixel subtraction method; (2) neurons close to image borders with fewer surround pixels for background estimation; and (3) neurons with very noisy activity traces. For both (1) and (2), the CNMF-E pipeline should provide more accurate estimation, as it does not rely exclusively on the local pixels. For case (3), these neurons do not generally provide useful information, thus can be ignored in final analysis.
Additional data processing
For manually segmented ROIs representing neuronal cell bodies (Supplementary Fig. 11, Supp. Fig. 7), neuropil background contamination was removed from the signal using surround-pixel neuropil subtraction. Because this approach is only accurate when the pixels surrounding the ROIs are not over other cell bodies, we excluded neurons with contiguous neighboring cells from our analysis.
The ΔF/F traces in Fig. 2g were smoothed by a gaussian filter with size = 3 and s.d. = 2. A Gaussian filter with size = 5 and s.d. = 3 was used to smooth the ΔF/F traces extracted from both automatically (Fig. 3) and manually (Supplementary Fig. 11, Supp. Fig. 7) segmented ROIs, as well as the trace of pupil area. Correlation coefficients were calculated using the smoothed traces.
See “Life Sciences Reporting Summary” for additional information.
Upper bound for labeling density for volumetric imaging using Bessel focus scanning
To unambiguously detect and identify an active structure (bouton, spine, or soma) in a Bessel imaging time series, during the response duration T of the activity indicator, there should only be a single active structure within the volume A*L, where A is the area of the structure in the lateral plane and L is the axial length of the Bessel focus. Given ρ as volume density of labeled structures, and an activity rate of P (# of activity events per second), the above requirement translates into A*L*ρ*P*T≤ 1. This leads to an upper bound on the labeling density:
Clearly, this upper bound is inversely proportional to area of the structure, the length of the Bessel focus, the response time of the sensor, and the frequency of active events. In other words, smaller structures, shorter Bessel beam, faster sensors, and lower firing probability allow higher densities of neuronal structures to be studied with the Bessel focus scanning method. (Here we assume that the ΔF due to a single active structure can be detected above the baseline fluorescence F from multiple labeled structures at different depths, for which activity sensors with high contrast and low basal fluorescence are advantageous.)
We can estimate the upper bound for the following hypothetical condition. Given cell body area A of 100 µm2, a Bessel focus that probes 100 µm depth range, T being 2 sec for GCaMP6s, and P being 2 action potential per second, the maximum ρ is 25,000 cell bodies per mm3. (In comparison, the density of the GCaMP6s+ GABAergic neurons in Fig. 3 is 3,402 neurons per mm3.)”
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank N. Sofroniew for demos on cranial window preparation, D. Flickinger for troubleshooting, optical alignments, and helpful discussions, B. Mohar and A. Kerlin for help with system alignments and ScanImage operations, V. Custard for administrative support, J. Zheng for assistance with ScanImage, M. Pachitariu for sharing the mesoscope and helpful discussions. R.L., Y.L., K.S., and N.J. were supported by Howard Hughes Medical Institute. G.M. and N.J. were supported by National Institutes of Health (NINDS U01NS103489). P.Z. and L.P. were supported by National Institute of Health (R01-EB22913, U01NS103489), NSF (NeuroNex award DBI-1707398), and the Gatsby Foundation.
Footnotes
COMPETING FINANCIAL INTERESTS STATEMENT: The mesoscope (K.S.) and the Bessel focus scanning (R.L. and N.J.) intellectual property has been licensed to Thorlabs, Inc by Howard Hughes Medical Institute.
DATA AVAILABILITY STATEMENT: The data that support the findings of this study are available from the corresponding author upon reasonable request.
CODE AVAILABILITY STATEMENT: The custom CNMF-E processing codes in MATLAB® are included in Supplementary Information.
References
- 1.Sofroniew NJ, Flickinger D, King J & Svoboda K A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging. eLife 5, e14472–e14472, doi: 10.7554/eLife.14472 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Botcherby EJ, Juškaitis R & Wilson T Scanning two photon fluorescence microscopy with extended depth of field. Opt Commun 268, 253–260, doi: 10.1016/j.optcom.2006.07.026 (2006). [DOI] [Google Scholar]
- 3.Theriault G, Cottet M, Castonguay A, McCarthy N & De Koninck Y Extended two-photon microscopy in live samples with Bessel beams: steadier focus, faster volume scans, and simpler stereoscopic imaging. Front Cell Neurosci 8, 139, doi: 10.3389/fncel.2014.00139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu R et al. Video-rate volumetric functional imaging of the brain at synaptic resolution. Nature Neuroscience 20, 620–628, doi: 10.1038/nn.4516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu R, Tanimoto M, Koyama M & Ji N 50 Hz volumetric functional imaging with continuously adjustable depth of focus. Biomed Opt Express 9, 1964–1976, doi: 10.1364/BOE.9.001964 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welford WT Use of annular apertures to increase focal depth. Journal of the Optical Society of America 50, 749–752, doi: 10.1364/JOSA.50.000749 (1960). [DOI] [Google Scholar]
- 7.De Paola V et al. Cell type-specific structural plasticity of axonal branches and boutons in the adult neocortex. Neuron 49, 861–875, doi: 10.1016/j.neuron.2006.02.017 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Chen T-W et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300, doi: 10.1038/nature12354 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Häusser M, Spruston N & Stuart GJ Diversity and Dynamics of Dendritic Signaling. Science 290, 739, doi: 10.1126/science.290.5492.739 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Manita S, Miyakawa H, Kitamura K & Murayama M Dendritic Spikes in Sensory Perception. Front Cell Neurosci 11, 29, doi: 10.3389/fncel.2017.00029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reimer J et al. Pupil fluctuations track fast switching of cortical states during quiet wakefulness. Neuron 84, 355–362, doi: 10.1016/j.neuron.2014.09.033 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pakan JM et al. Behavioral-state modulation of inhibition is context-dependent and cell type specific in mouse visual cortex. Elife 5, doi: 10.7554/eLife.14985 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pachitariu M et al. Suite2p: beyond 10,000 neurons with standard two-photon microscopy. bioRxiv, doi: 10.1101/061507 (2016). [DOI] [Google Scholar]
- 14.Liu H, Wu Z, Li X, Cai D & Huang TS Constrained Nonnegative Matrix Factorization for Image Representation. IEEE Trans Pattern Anal Mach Intell 34, 1299–1311, doi: 10.1109/TPAMI.2011.217 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Pnevmatikakis, Eftychios A et al. Simultaneous Denoising, Deconvolution, and Demixing of Calcium Imaging Data. Neuron 89, 285–299, doi: 10.1016/j.neuron.2015.11.037 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou P et al. Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. eLife 7, e28728, doi: 10.7554/eLife.28728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji N, Freeman J & Smith SL Technologies for imaging neural activity in large volumes. Nat Neurosci 19, 1154–1164, doi: 10.1038/nn.4358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szalay G et al. Fast 3D Imaging of Spine, Dendritic, and Neuronal Assemblies in Behaving Animals. Neuron 92, 723–738, doi: 10.1016/j.neuron.2016.10.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stirman JN, Smith IT, Kudenov MW & Smith SL Wide field-of-view, multi-region, two-photon imaging of neuronal activity in the mammalian brain. Nature Biotechnology 34, 857–862, doi: 10.1038/nbt.3594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai PS et al. Ultra-large field-of-view two-photon microscopy. Optics Express 23, 13833–13833, doi: 10.1364/OE.23.013833 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horton NG et al. In vivo three-photon microscopy of subcortical structures within an intact mouse brain. Nature Photonics 7, 205–205, doi: 10.1038/nphoton.2012.336 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ouzounov DG et al. In vivo three-photon imaging of activity of GCaMP6-labeled neurons deep in intact mouse brain. Nat Methods 14, 388–390, doi: 10.1038/nmeth.4183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez C, Liang Y, Lu R & Ji N Three-photon fluorescence microscopy with an axially elongated Bessel focus. Opt Lett 43, 1914–1917, doi: 10.1364/OL.43.001914 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thériault G, De Koninck Y & McCarthy N Extended depth of field microscopy for rapid volumetric two-photon imaging. Optics Express 21, 10095–10104, doi: 10.1364/OE.21.010095 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Pnevmatikakis EA & Giovannucci A NoRMCorre: An online algorithm for piecewise rigid motion correction of calcium imaging data. J Neurosci Methods 291, 83–94, doi: 10.1016/j.jneumeth.2017.07.031 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Guizar-Sicairos M, Thurman ST & Fienup JR Efficient subpixel image registration algorithms. Optics Letters 33, 156–158, doi: 10.1364/OL.33.000156 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Kerlin AM, Andermann ML, Berezovskii VK & Reid RC Broadly tuned response properties of diverse inhibitory neuron subtypes in mouse visual cortex. Neuron 67, 858–871, doi: 10.1016/j.neuron.2010.08.002 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.