

Summary
Heavily transfused patients frequently develop human leukocyte antigen (HLA) allo-immunization resulting in platelet transfusion refractoriness and a high risk for life-threatening thrombocytopenia. Data suggest complement activation leading to the destruction of platelets bound by HLA allo-antibodies may play a pathophysiologic role in platelet refractoriness. Here we conducted a pilot trial to investigate the use of eculizumab, a monoclonal antibody that binds and inhibits C5 complement, to treat platelet transfusion refractoriness in allo-immunized patients with severe thrombocytopenia. A single eculizumab infusion was administered to 10 eligible patients, with four (40%) patients overcoming platelet refractories assessed measuring the corrected platelet count increment (CCI) 10–60 min and 18–24 h post transfusion. Responding patients had a reduction in the requirement for subsequent platelet transfusions and had higher post-transfusion platelet increments for 14 days following eculizumab administration. Remarkably, three of the four responders met CCI criteria for response despite receiving HLA-incompatible platelets. Our results suggest that eculizumab has the ability to overcome platelet transfusion refractoriness in patients with broad HLA allo-immunization. This study establishes proof of principle that complement inhibition can treat platelet transfusion refractoriness, laying the foundation for a large multicentre trial to assess the overall efficacy of this approach (ClinicalTrials.gov, identifier: NCT02298933).
Treatment of haematological and non-haematological diseases with chemotherapy and haematopoietic stem cell transplantation is not possible without allogeneic platelet transfusion support. Although human leukocyte antigen (HLA)-matched platelets can be lifesaving for allo-immunized patients with severe thrombocytopenia, HLA-matched platelet donors may be limited or unavailable for many patients. Consequently, a substantial proportion of allo-immunized patients develop platelet transfusion refractoriness (Doughty et al., 1994; Legler et al., 1997; Novotny, 1999; Stroncek & Rebulla, 2007), which is associated with major adverse outcomes including an increased risk of bleeding, longer hospital stays and higher inpatient hospital costs, and decreased survival (Meehan et al., 2000; Kerkhoffs et al., 2008).
When patients generate immunoglobulin (Ig)-G and IgM antibodies to HLA class I epitopes expressed on transfused allogeneic platelets, platelet destruction can occur (Pavenski et al., 2012). Recent data suggest complement activation leading to the destruction of platelets bound by HLA allo-antibodies may play an important pathophysiologic role in antibody-mediated platelet refractoriness (Kickler et al., 1990; Pavenski et al., 2012; Pavenski et al., 2013). Eculizumab is a monoclonal antibody that binds and inhibits C5 complement, blocking both the classic and alternative pathways of complement. Eculizumab is effective in blocking complement-mediated red blood cell (RBC) destruction associated with paroxysmal nocturnal haemoglobinuria and atypical haemolytic uremic syndrome (Hillmen et al., 2006; Legendre et al., 2013), and has been approved by the FDA for this use.
To test the hypothesis that complement inhibition could be used to overcome immune-mediated platelet transfusion refractoriness, we conducted a pilot trial utilizing eculizumab to treat HLA allo-immunized patients with severe thrombocytopenia and platelet transfusion refractoriness at risk of bleeding complications.
Patients and methods
Study design and participants
This single-arm pilot clinical trial was conducted between March 2015 and March 2017 in HLA allo-immunized patients who were receiving treatment at the National Institutes of Health (NIH) Clinical Center in Bethesda, MD, USA. Eligible adult patients aged 18 to 75 years had thrombocytopenia (due to congenital causes, bone marrow failure, haematologic malignancies, and treatment-related causes) requiring platelet transfusions for a platelet count of <10 × 109/l without bleeding or <30 × 109/l in the presence of active bleeding or with a history of life-threatening bleeding. Patients were required to have detectable anti-HLA A and/or B antibodies and immune-mediated platelet transfusion refractoriness, with refractoriness defined by at least two consecutive occurrences of less than adequate corrected platelet count increment (CCI): CCI < 7500 at 10–60 min (min) and CCI < 5000 at 18–24 h after each platelet transfusion (Novotny, 1999; Hod & Schwartz, 2008; Pavenski et al., 2012). Here, CCI, with a unit of (count/l) × m2/(count × 10−5), was calculated based on the standard formula taking into account the body surface area of the patient and number of platelets transfused, as the absolute platelet count increment (count/l) × body surface area (m2)/(10−5 × the number of platelets transfused) (Trial to Reduce Alloimmunization to Platelets Study Group, 1997; Davis et al., 1999; Novotny, 1999; Hod & Schwartz, 2008). We tested HLA antibodies and antigens, compliant with the regulations under the Clinical Laboratory Improvement Amendments, as described previously (Fasano et al., 2014; Goldspiel et al., 2014; Blau et al., 2015; Sissung et al., 2017). All the patients had evidence of HLA antibodies within one month of enrolment.
Treatment schedule
Patients received a single intravenous infusion of eculizumab (1200 mg over 30–40 min) followed by the first platelet transfusion within 48 h of eculizumab administration. All patients received meningococcal vaccination and/or antibiotics for 14 days before eculizumab was administered as prophylaxis for Neisseria meningitidis.
Outcome and statistical analysis
To monitor for a response following eculizumab administration, platelet counts were obtained immediately pre-transfusion, 10–60 min and 18–24 h after the first two platelet transfusions. The primary endpoint was the response to the eculizumab therapy. Patients were defined as responding to therapy if one of the first two platelet transfusions following eculizumab resulted in a 10–60 min CCI > 7500 together with a 18–24 h CCI > 5000 post transfusion. Patients were taken off study 14 days following eculizumab treatment. Responding patients with recurrent platelet refractoriness were eligible to re-enrol on study but were not counted in the primary endpoint evaluation.
We used a Simon’s minimax two-stage design to test the null hypothesis that the proportion of patients achieving response to therapy in the study was 20% or lower versus the alternative hypothesis that it was 60% or higher, with a type I error of 0·05% and 83% power. We established that a sample size of 10 patients evaluable for the primary endpoint was needed.
Total complement (CH50), as the most reliable and consistent measure for complement inhibition (Costabile, 2010), was measured before and immediately after eculizumab administration. The numbers of platelet transfusions given two weeks before and after eculizumab administration were obtained for each patient. For each transfusion, the 1-h post-transfusion platelet increments were calculated as the difference between the 10–60 min post-transfusion platelet count and the pre-transfusion platelet count; and the days to next transfusion were calculated as the interval between two consecutive transfusions.
Data are summarized as frequency (percentage), mean ± standard deviation or median (range) as appropriate. The differences in CH50 before and after eculizumab administration were assessed by the Wilcoxon signed-rank test because the data are skewed. The difference in the numbers of platelet transfusion before and after eculizumab was compared between the patients responding to eculizumab and the non-responders using Welch’s two-sample t-test. For comparing the platelet transfusion response between the patients responding to eculizumab and non-responders, linear mixed models were used to account for the within-subject correlation among the repeated transfusions for the same subject. Analysis was performed using r statistical software version 3.6.0 (R Foundation for Statistical Computing, Vienna, Austria).
Results
We enrolled and treated 10 HLA allo-immunized patients (median age 39·5 years, range 20–71 years; all were positive for HLA class I antibodies with a median panel reactive antibody [PRA] of 77·5%, range 11–98%) who had severe thrombocytopenia and platelet refractoriness with a single eculizumab infusion. Patients had an underlying diagnosis of severe aplastic anaemia (n = 4), refractory/relapsed acute myeloid leukaemia (n = 3; one patient had just finished conditioning for a haplo-identical transplant at time of enrolment, two post-multiple chemotherapy treatment), relapsed acute lymphoblastic leukaemia (n = 2; post-matched unrelated and matched related transplants, respectively) and high-risk myelodysplastic syndrome (n = 1) (Table I). No patient had splenomegaly or had undergone a splenectomy. Most patients did not have other common non-immune-mediated causes for platelet refractoriness, such as fever, sepsis or antibiotic therapy, within two weeks of receiving eculizumab treatment (Table SI).
Table I.
Patient characteristics and treatment response.⋆
| Patient no. | Sex | Age | Race | Diagnosis | BSA (m2) | HLA Class I (% PRA) | Pretreatment platelet count (×103/μl) | Transfusion #1 post eculizumab treatment | Transfusion #2 post eculizumab treatment | Response | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Platelet Product† | Platelets transfused (×1011) | CCI (10–60 min) | CCI (18–24 h) | Platelet Product† | Platelets transfused (×1011) | CCI (10–60 min) | CCI (18–24 h) | |||||||||
| 1 | F | 24 | Black | SAA | 1·82 | Positive (20%) | 8 | Compatible | 2·8 | 17 207⋆ | 7942⋆ | Incompatible | 3·3 | 28 127⋆ | 13 236⋆ | Yes |
| 1R‡ | - | - | - | - | 1·87 | Positive (20%) | 1 | Compatible | 3·3 | 18 133⋆ | 11 900⋆ | Compatible | 4·4 | 15 725⋆ | 7650⋆ | Yes |
| 2 | M | 71 | Asian | MDS | 1·88 | Positive (29%) | 6 | Incompatible | 4·4 | 8545 | 4273 | Incompatible | 3·3 | 18 230 | −5127 | No |
| 3 | F | 46 | White | AML | 1·66 | Positive (56%) | 3 | Compatible | 2·2 | 6791 | 3773 | Compatible | 3·3 | 5030 | 2515 | No |
| 4 | F | 10 | Asian | SAA | 1·41 | Positive (73%) | 4 | Incompatible | 2·8 | 8716 | 2051 | Incompatible | 2·8 | 513 | NE | No |
| 5 | F | 32 | White | ALL | 1·95 | Positive (82%) | 3 | Compatible | 2·8 | 16 309⋆ | 7091⋆ | Incompatible | 2·2 | 10 636 | 2659 | Yes |
| 6 | F | 39 | Black | AML | 1·87 | Positive (11%) | 6 | Compatible | 2·8 | 4766 | NE | Compatible | 2·8 | 4085 | NE | No |
| 7 | F | 57 | Black | SAA | 1·97 | Positive (98%) | 3 | Incompatible | 2·2 | 3582 | 0 | Incompatible | 3·3 | 18 506⋆ | 13 133⋆ | Yes |
| 8 | F | 36 | White | ALL | 1·73 | Positive (82%) | 8 | Incompatible | 2·8 | 16 985⋆ | 11 953⋆ | Incompatible | 3·3 | 12 582⋆ | 5767⋆ | Yes |
| 9 | M | 68 | Asian | AML | 1·77 | Positive (95%) | 6 | Incompatible | 2·8 | 644 | 644 | Incompatible | 2·8 | 0 | NE | No |
| 10 | M | 20 | Asian | SAA | 2·06 | Positive (98%) | 1 | Incompatible | 3·9 | 1070 | 0 | Incompatible | 2·8 | 3745 | 0 | No |
BSA, Body surface area; PRA, panel reactive antibody; CCI, corrected platelet count increment after transfusion with a unit of (count/μl) × m2/(count × 10−11); F, female; M, male; SAA, severe aplastic anaemia; MDS, myelodysplastic syndrome; AML, acute myeloid leukaemia; ALL, acute lymphoblastic leukaemia.
NE: not evaluable for the 18–24 h CCI when patients received another transfusion <24 h of the prior platelet transfusion.
To highlight the patients responded to treatment are bolded.
Met the combined response criteria of CCI > 7500 at 10–60 min together with a CCI > 5000 18–24 h post transfusion.
HLA incompatible: administered platelet product expressed an HLA allele for which an HLA antibody had been detected in the patient’s serum. HLA-compatible platelets were defined as those that matched the HLA A and HLA B antigens of the patient or when they were at least matched for the cognate HLA antigens, to which the patient carried HLA antibodies.
1R: patient 1 with recurrent platelet transfusion refractoriness was re-enrolled two months after her first response.
After eculizumab treatment, CH50 decreased significantly to <10 000 U/l in all patients (compared to pre-treatment, median 55 000 U/l, range 39 000–126 000; P = 0·006, Fig 1A). The corrected platelet count increment (CCI), was calculated for the first two transfusions (the first transfusion occurred on days 0–1 and the second transfusion occurred days 0–6) after eculizumab was administered to evaluate the response to this agent. Four of 10 (40%) transfusion-refractory patients had a response to therapy, with refractoriness resolving following five eculizumab administrations (Table I and Fig 1B). Patient 1 received a second eculizumab treatment for recurrent transfusion refractory thrombocytopenia two months after her first response, and again had resolution of refractoriness based on CCI criteria (Table I). Importantly, we observed that in three out of four patients where refractoriness was overcome, the administered platelet product given immediately after eculizumab expressed an HLA allele for which an HLA antibody had been detected in the patient’s serum (Table II). No drug-related adverse events were observed.
Fig 1.
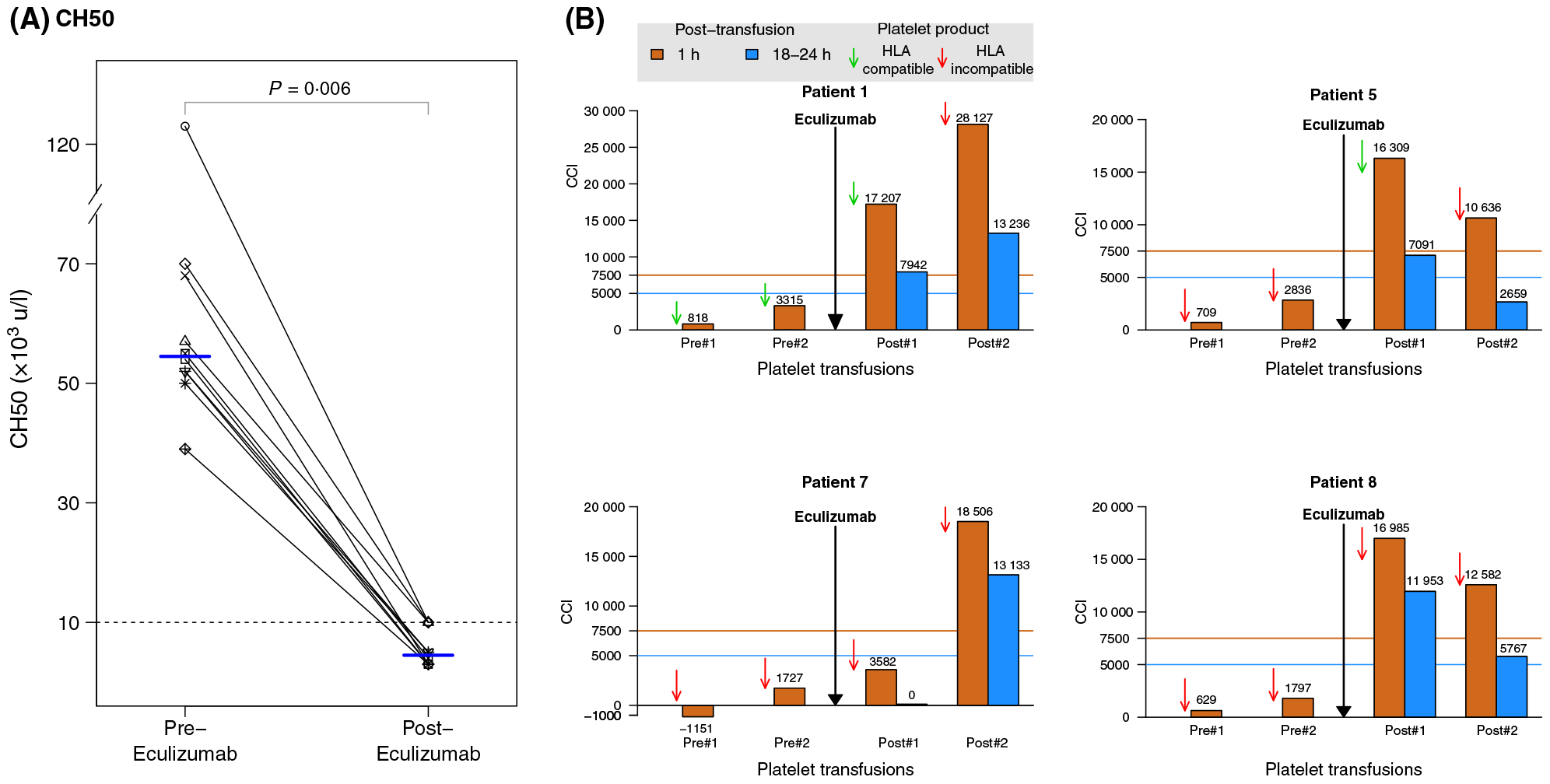
(A) Total complement (CH50) levels pre and post eculizumab treatment. Median (horizontal lines) are shown at the two time points. (B) Response to platelet transfusion refractoriness post eculizumab in the four responding patients. One-hour (10–60-min, orange bars) and 18–24 h (blue bars) post platelet transfusion CCIs are shown in patients before and after receiving eculizumab. Responses were defined by 1-h CCI > 7500 together with the 18–24-h CCI > 5000 following a platelet transfusion. Green and red arrows indicate HLA-compatible and HLA-incompatible products, respectively.
Table II.
Responding patients’ HLA antibodies and the HLA type of the transfused platelet product.
| HLA A and B antibodies detected in patients | HLA alleles of the incompatible platelet product given immediately after eculizumab | |
|---|---|---|
| Patient 1- treatment 1 | A: 02, 34 | HLA A: 23, 24 |
| B: 42, 67, 55, 8, 54, 81, 41*, 07*, 82* | HLA B: 38, 41 | |
| Patient 5 | A: 02, 69, 68, 24, 23, 01, 11 | HLA A: 01, 2901 |
| B: 51, 35, 53, 71, 49, 75, 8, 78, 50, 77, 56, 52, 57, 72, 62, 63, 58, 46, 59 | HLA B: 8, 1302 | |
| Patient 7 | A: 02, 68, 69, 80, 24, 11, 01, 66, 34, 25, 26, 3, 32, 36, 43, 33, 29, 74, 31 | HLA A: 23, 32 |
| B: 07, 49, 42, 67, 81, 55, 56, 82, 57, 60, 50, 13, 61, 27, 58, 44, 45, 41, 48, 52, 73, 47, 51, 54, 62, 76, 63, 8, 72, 59, 71, 77, 53 | HLA B: 35, 39 | |
| Patient 8 | A: 23, 32, 24, 25, 01, 31*, 30*, 33*, 74* | HLA A: 02, 03 HLA B: 07, 35 |
| B: 57, 44, 49, 63, 76, 58, 45, 51, 77, 52, 13, 59, 53, 27, 38, 62, 47, 41, 37, 50, 61, 72, 60, 75, 82, 56, 55, 42, 67, 8, 07*, 81*, 65*, 64*, 48*, 39*, 73*, 18*, 46* | HLA A: 03, 68 HLA B: 14†, 35 |
Numbers shown in bold represent HLA A and B antibodies detected in the patient and HLA A and B alleles expressed in the transfused HLA-incompatible platelet product.
HLA antibodies detected historically in the serum of thrombocytopenic patients.
B64 and 65 are sero-equivalents of B14.
Mean platelet trajectories were calculated using 221 available platelet counts for two weeks following eculizumab (Fig 2A). In responding patients, resolution of platelet refractoriness resulted in a clinically meaningful reduction in the requirement for platelet transfusions: the mean number of transfusions given two weeks before and two weeks after the eculizumab infusion in responding patients decreased from 9·3 to 5·3 transfusions, respectively (Δ = −3·5 ± 1·4); For non-responders (CCI-based criteria), no difference was observed (Δ = 4·0 ± 4·1). The changes in the number of platelet transfusions were significantly different between the responding patients and non-responders (P = 0·004, Fig 2B).
Fig 2.
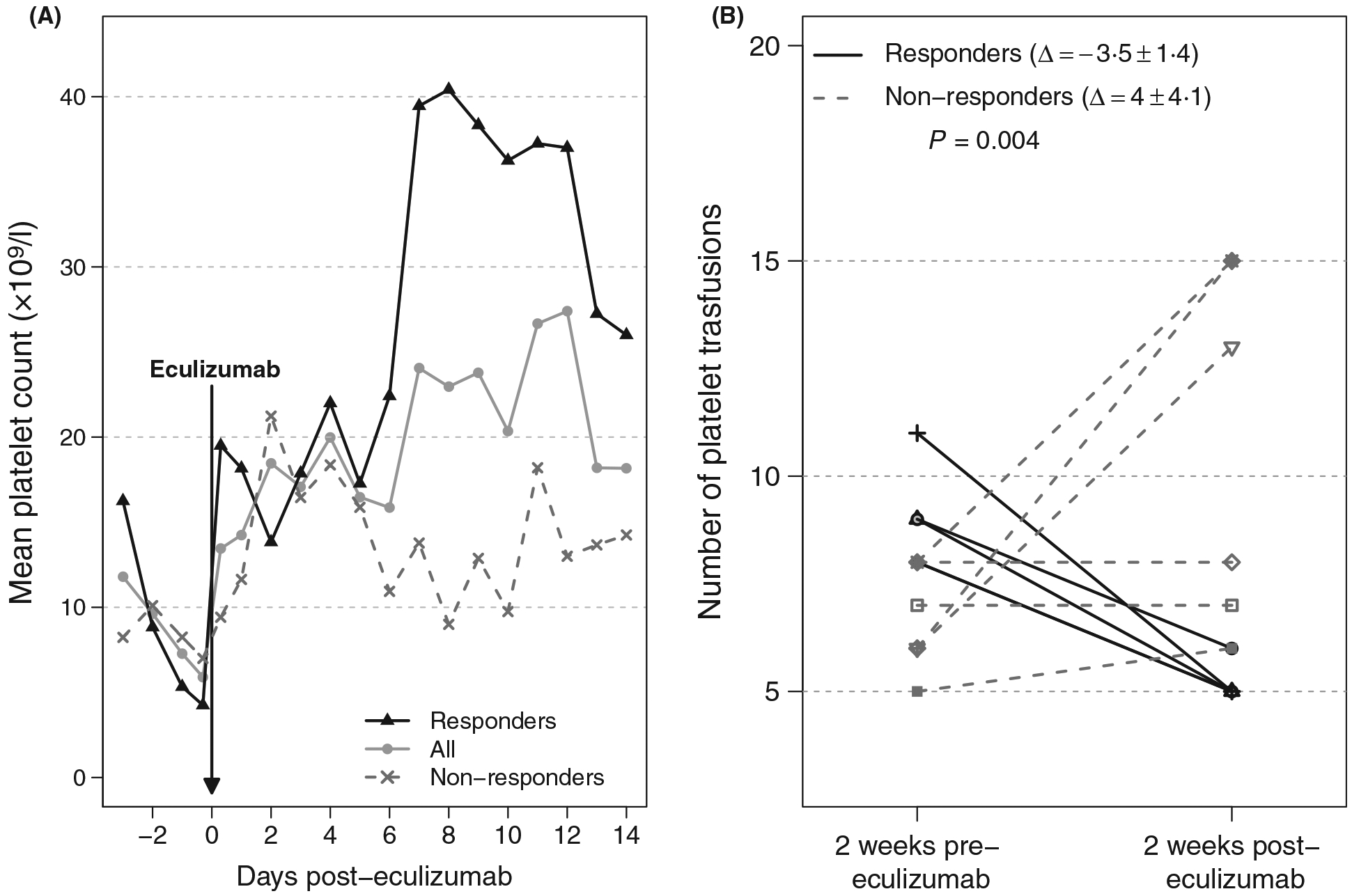
(A) Mean platelet count trajectories calculated using 221 available platelet counts measured for two weeks following eculizumab. (B) Number of platelet transfusions given two weeks pre and two weeks post eculizumab treatment.
Responses to 73 and 88 platelet transfusions two weeks before and two weeks following eculizumab, respectively, are shown in Table III. Compared to non-responders, the responding patients had a greater mean increment in 1-h post-transfusion platelet counts (26·0 ± 17·3 vs. 9·4 ± 9·5 × 109/l, P = 0·038) and a trend towards having a longer time to the next transfusion (2·5 days ± 1·3 vs.1·4 ± 1·2 P = 0·10) after eculizumab treatment.
Table III.
Platelet transfusion responses within the two weeks before and after eculizumab administration.
| All patients (n = 10) | Responders (n = 4) | Non-responders (n = 6) | P⋆ | |
|---|---|---|---|---|
| Before eculizumab (N = 73 transfusions) | ||||
| Pretransfusion PLT count (×109/l) | 8·2 ± 5·2 | 7·8 ± 5·4 | 8·6 ± 50 | 0·72 |
| I h post-transfusion PLT count (×109/l) | 191 ± 15·3 | 20·9 ± 16·2 | 17·3 ± 14·3 | 0·25 |
| Platelet increment (×109/l) | 11 ± 14·5 | 13·2 ± 15·1 | 8·7 ± 13·7 | 0·22 |
| Days to next transfusion | 1·4 ± 1·3 | 1·4 ± 1·3 | 1·5 ± 1·3 | 0·85 |
| After eculizumab (N = 88 transfusions) | ||||
| Pretransfusion PLT count (×109/l) | 9·5 ± 7·8 | 11·5 ± 9·9 | 8·8 ± 6·8 | 0·49 |
| I h post-transfusion PLT count (×109/l) | 22·9 ± 15·8 | 36·3 ± 16·6 | 18·1 ± 12·5 | 0·033 |
| Platelet increment (×109/l) | 13·8 ± 140 | 26·0 ± 17·3 | 9·4 ± 9·5 | 0·038 |
| Days to next transfusion | 1·7 ± 1·3 | 2·5 ± 1·3 | 1·4 ± 1·2 | 0·10 |
Data are given as mean ± SD. PLT, platelet.
To highlight it is statistically significant are bolded.
P value for comparing responders and non-responders (CCI criteria) using the linear mixed models to account for within-subject correlation.
Discussion
The full elucidation of the pathophysiology mediating platelet refractoriness in the context of HLA allo-immunization remains elusive. When patients generate IgG and IgM against HLA A and B class I epitopes expressed on allogeneic platelets, platelet destruction is thought to occur by one of the known antibody-dependent mechanisms, namely eradication via the reticuloendothelial system (RES), antibody-dependent cellular cytotoxicity (ADCC), and complement activation. Some data suggest complement activation may play a more important role in the pathophysiology of platelet HLA antibody-mediated refractoriness than was previously appreciated (Kickler et al., 1990; Pavenski et al., 2012; Pavenski et al., 2013). Fc regions of IgG HLA antibodies are capable of binding the first component (C1q) of the classical (antibody-dependent) complement pathway resulting in complement deposition (Thomas et al., 2015). The ability of anti-HLA antibodies to activate complement leading to kidney graft destruction has previously been established (Loupy et al., 2013). The inability to overcome platelet refractoriness by multiple sequential infusions of platelets, high dose intravenous immunoglobulin (IVIG), and splenectomy have established that full saturation of the RES and FcRIIIA (resulting in full inhibition of natural killer cell-mediated ADCC) are alone insufficient to prevent antibody-mediated platelet destruction, highlighting the important role complement plays in this process (Pavenski et al., 2012).
To the best of our knowledge, this is the first systematic study to evaluate complement inhibition as a method to overcome platelet refractoriness in thrombocytopenic patients who are HLA-allo-immunized. As expected, all patients receiving eculizumab had substantial complement inhibition as measured by CH50, with four (40%) having a response to therapy and overcoming platelet refractoriness. Moreover, three of the four responders who were HLA-allo-immunized overcame refractoriness despite receiving an HLA-mismatched platelet product. This finding is of important clinical relevance for patients with uncommon HLA types who have multiple different HLA antibodies or when the donor apheresis pool is limited, as it may take several days for blood banks to acquire HLA-matched platelet products or recruit HLA-matched platelet donors. Such delays put thrombocytopenic patients with HLA allo-immunization at high risk for severe and life-threatening bleeding. Additionally, for the 14 days following eculizumab treatment, responding patients had a clinically meaningful reduction in the requirement for subsequent platelet transfusions by nearly 50% and an increase in 1-h post-transfusion platelet increments.
Our results should be viewed within the context of the limitations of this pilot study. First, a limitation of utilizing eculizumab to treat platelet refractoriness is the high cost of the drug. Therefore, such therapy, would be intended to be reserved for platelet-refractory patients with either active bleeding or for those defined to be at an extremely high risk for bleeding. Secondly, the cause of platelet refractoriness is often multifactorial (Stroncek & Rebulla, 2007). It is important to consider that confounding non-immune factors such as fever, sepsis and bleeding may all contribute to platelet refractoriness in HLA-allo-immunized patients which could preclude a response to targeted complement inhibition with eculizumab (Table SI). Of note, patient 2 achieved a 10–60 min CCI > 7500 with both post-treatment transfusions but had a poor 18–24 h CCI. The patient, therefore, did not meet the strict response criteria defined in the protocol (Table I). It is possible that other clinical factors associated with this patient may have contributed to him not meeting the response criteria at the 24- h timepoint. After eculizumab treatment, the patient developed persistent neutropenic fever, requiring treatment with meropenem and vancomycin. His persistent fever may have led to ‘excess platelet consumption’ causing the CCI to be low at the 24-h timepoint. Although we could not consider him to have had a response per protocol, there was some evidence suggesting a clinical benefit as he also met the 10–60 min CCI > 7 500 and 18–24 h CCI > 5 000 after the third and fourth transfusion of platelets given after eculizumab treatment (Table SII). Since only 10 patients were treated in our trial, the study was not powered to estimate the response rate precisely and is unable to identify unique patient characteristics leading to refractoriness and factors predicting response to eculizumab (Slichter et al., 2005; Solves et al., 2018). Nevertheless, the resolution of platelet refractoriness in a subset of responding patients treated with eculizumab provides proof of concept and data supporting complement as playing a pathophysiologic role in platelet refractoriness. Additional biological correlative studies characterizing complement activation, particularly on platelets, could potentially help distinguish patients who are most likely to benefit from complement blockade.
Taken altogether, our study provides the first data showing complement inhibition with eculizumab may have a therapeutic role in thrombocytopenic patients who are HLA-allo-immunized and refractory to platelet transfusions, potentially buying time to acquire better or fully HLA-matched platelet products or until platelet recovery occurs following chemotherapy or haematopoietic stem cell transplantation. Based on the promising results of this pilot trial, we believe a large multicentre study is warranted to establish the overall efficacy of eculizumab as a therapeutic for HLA antibody-mediated platelet transfusion refractoriness.
Supplementary Material
Table SI. Patient information on non-immune factors reported to be related to platelet transfusion refractoriness.⋆
Table SII. Response to platelet transfusions in patient #2 before and after eculizumab treatment.
Acknowledgements
The authors would like to thank all the patients who participated in this trial. We acknowledge Alexion for providing the study drug and comments on a draft of this manuscript. This research was supported by the Intramural Research Program of the National Heart, Lung and Blood Institute (NHLBI), the Clinical Center, National Institutes of Health in Bethesda, Maryland, and the United States Public Health Service Commissioned Corps.
Footnotes
Conflict of interest disclosure
The authors declare no competing financial interests.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- Blau JE, Abegg MR, Flegel WA, Zhao X, Harlan DM & Rother KI (2015) Long-term immunosuppression after solitary islet transplantation is associated with preserved C-peptide secretion for more than a decade. American Journal of Transplantation, 15, 2995–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costabile M (2010) Measuring the 50% haemolytic complement (CH50) activity of serum. Journal of Visualized Experiments: JoVE, 29, e1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KB, Slichter SJ & Corash L (1999) Corrected count increment and percent platelet recovery as measures of posttransfusion platelet response: problems and a solution. Transfusion, 39, 586–592. [DOI] [PubMed] [Google Scholar]
- Doughty HA, Murphy MF, Metcalfe P, Rohatiner AZ, Lister TA & Waters AH (1994) Relative importance of immune and non-immune causes of platelet refractoriness. Vox Sanguinis, 66, 200–205. [DOI] [PubMed] [Google Scholar]
- Fasano RM, Mamcarz E, Adams S, Donohue Jerussi T, Sugimoto K, Tian X, Flegel WA & Childs RW (2014) Persistence of recipient human leucocyte antigen (HLA) antibodies and production of donor HLA antibodies following reduced intensity allogeneic haematopoietic stem cell transplantation. British Journal of Haematology, 166, 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldspiel BR, Flegel WA, DiPatrizio G, Sissung T, Adams SD, Penzak SR, Biesecker LG, Fleisher TA, Patel JJ, Herion D, Figg WD, Lertora JJ & McKeeby JW (2014) Integrating pharmacogenetic information and clinical decision support into the electronic health record. Journal of the American Medical Informatics Association, 21, 522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, Roth A, Szer J, Elebute MO, Nakamura R, Browne P, Risitano AM, Hill A, Schrezenmeier H, Fu CL, Maciejewski J, Rollins SA, Mojcik CF, Rother RP & Luzzatto L (2006) The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. New England Journal of Medicine, 355, 1233–1243. [DOI] [PubMed] [Google Scholar]
- Hod E & Schwartz J (2008) Platelet transfusion refractoriness. British Journal of Haematology, 142, 348–360. [DOI] [PubMed] [Google Scholar]
- Kerkhoffs JL, Eikenboom JC, van de Watering LM, van Wordragen-Vlaswinkel RJ, Wijer-mans PW & Brand A (2008) The clinical impact of platelet refractoriness: correlation with bleeding and survival. Transfusion, 48, 1959–1965. [DOI] [PubMed] [Google Scholar]
- Kickler T, Braine HG, Piantadosi S, Ness PM, Herman JH & Rothko K (1990) A randomized, placebo-controlled trial of intravenous gammaglobulin in alloimmunized thrombocytopenic patients. Blood, 75, 313–316. [PubMed] [Google Scholar]
- Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, Eitner F, Feldkamp T, Fouque D, Furman RR, Gaber O, Herthelius M, Hourmant M, Karpman D, Lebranchu Y, Mariat C, Menne J, Moulin B, Nurnberger J, Ogawa M, Remuzzi G, Richard T, Sberro-Soussan R, Severino B, Sheerin NS, Trivelli A, Zimmerhackl LB, Goodship T & Loirat C (2013) Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. New England Journal of Medicine, 368, 2169–2181. [DOI] [PubMed] [Google Scholar]
- Legler TJ, Fischer I, Dittmann J, Simson G, Lynen R, Humpe A, Riggert J, Schleyer E, Kern W, Hiddemann W & Kohler M (1997) Frequency and causes of refractoriness in multiply transfused patients. Annals of Hematology, 74, 185–189. [DOI] [PubMed] [Google Scholar]
- Loupy A, Lefaucheur C, Vernerey D, Prugger C, Duong van Huyen JP, Mooney N, Suberbielle C, Fremeaux-Bacchi V, Mejean A, Desgrandchamps F, Anglicheau D, Nochy D, Charron D, Empana JP, Delahousse M, Legendre C, Glotz D, Hill GS, Zeevi A & Jouven X (2013) Complement-binding anti-HLA antibodies and kidney-allograft survival. New England Journal of Medicine, 369, 1215–1226. [DOI] [PubMed] [Google Scholar]
- Meehan KR, Matias CO, Rathore SS, Sandler SG, Kallich J, LaBrecque J, Erder H & Schulman KA (2000) Platelet transfusions: utilization and associated costs in a tertiary care hospital. American Journal of Hematology, 64, 251–256. [DOI] [PubMed] [Google Scholar]
- Novotny VM (1999) Prevention and management of platelet transfusion refractoriness. Vox Sanguinis, 76, 1–13. [DOI] [PubMed] [Google Scholar]
- Pavenski K, Freedman J & Semple JW (2012) HLA alloimmunization against platelet transfusions: pathophysiology, significance, prevention and management. Tissue Antigens, 79, 237–245. [DOI] [PubMed] [Google Scholar]
- Pavenski K, Rebulla P, Duquesnoy R, Saw CL, Slichter SJ, Tanael S & Shehata N (2013) Efficacy of HLA-matched platelet transfusions for patients with hypoproliferative thrombocytopenia: a systematic review. Transfusion, 53, 2230–2242. [DOI] [PubMed] [Google Scholar]
- Sissung TM, McKeeby JW, Patel J, Lertora JJ, Kumar P, Flegel WA, Adams SD, Eckes EJ, Mickey F, Plona TM, Mellot SD, Baugher RN, Wu X, Soppet DR, Barcus ME, Datta V, Pike KM, DiPatrizio G, Figg WD & Goldspiel BR (2017) Pharmacogenomics implementation at the National Institutes of Health Clinical Center. Journal of Clinical Pharmacology, 57, S67–S77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slichter SJ, Davis K, Enright H, Braine H, Gernsheimer T, Kao KJ, Kickler T, Lee E, McFarland J, McCullough J, Rodey G, Schiffer CA & Woodson R (2005) Factors affecting posttransfusion platelet increments, platelet refractoriness, and platelet transfusion intervals in thrombocytopenic patients. Blood, 105, 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solves P, Sanz J, Freiria C, Santiago M, Villalba A, Gomez I, Montesinos P, Montoro J, Pinana JL, Lorenzo JI, Puig N, Sanz GF, Sanz MA & Carpio N (2018) Factors influencing platelet transfusion refractoriness in patients undergoing allogeneic hematopoietic stem cell transplantation. Annals of Hematology, 97, 161–167. [DOI] [PubMed] [Google Scholar]
- Stroncek DF & Rebulla P (2007) Platelet transfusions. The Lancet, 370, 427–438. [DOI] [PubMed] [Google Scholar]
- Thomas KA, Valenzuela NM & Reed EF (2015) The perfect storm: HLA antibodies, complement, FcgammaRs, and endothelium in transplant rejection. Trends in Molecular Medicine, 21, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trial to Reduce Alloimmunization to Platelets Study Group (1997) Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. New England Journal of Medicine, 337, 1861–1869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table SI. Patient information on non-immune factors reported to be related to platelet transfusion refractoriness.⋆
Table SII. Response to platelet transfusions in patient #2 before and after eculizumab treatment.