

Abstract
Transition metal-catalyzed C–H functionalization and decarboxylative coupling are two of the most notable synthetic strategies developed in the last 30 years. Herein, we connect these two reaction pathways using bases and a simple Pd-based catalyst system to promote a para-selective C–H functionalization reaction from benzylic electrophiles. Experimental and computational mechanistic studies suggest a pathway involving an uncommon Pd-catalyzed dearomatization of the benzyl moiety followed by a base-enabled rearomatization through a formal 1,5-hydrogen migration. This reaction complements “C–H activation” strategies that convert inert C–H bonds into C–metal bonds prior to C–C bond formation. Instead, this reaction exploits an inverted sequence and promotes C–C bond formation prior to deprotonation. These studies provide an opportunity to develop general para-selective C–H functionalization reactions from benzylic electrophiles and show how new reactive modalities may be accessed with careful control of reaction conditions.
Graphical Abstract
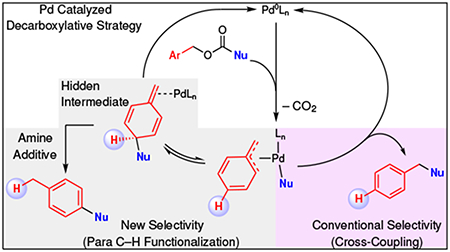
In recent decades, transition metal catalyzed cross-coupling reactions1 and unactivated C–H bond functionalization reactions2 have emerged as two of the most powerful strategies for constructing C–C bonds. These strategies have enabled the regioselective formation of reactive organometallic species derived from non-reactive groups, which has expanded possibilities for retrosynthetic analysis, and simplified synthetic routes. Additionally, these strategies have enabled the construction of hard-to-form bonds under mild conditions that tolerate a broad spectrum of functional groups. Both strategies have been successfully demonstrated with a variety of transition metal catalyst systems; however, Pd-based systems remain among the most common for both cross-coupling and C–H functionalization reactions, largely due to the low-cost relative to other precious metals and well-behaved 2 e− chemistry. Typically, decarboxylative coupling reactions occur under mild, neutral, and reductive conditions using a cycle starting with Pd(0), while C–H functionalization reactions occur with complimentary oxidative conditions using a cycle starting with Pd(II). Despite the countless number of catalyst systems designed for these two strategies, simple and creative ways to connect these two reaction paradigms would provide a significant advancement for synthetic chemistry. Herein, we report an unprecedented switch of a classical cross-coupling reaction into a para-selective C–H functionalization reaction by a simple additive.
The functionalization of C–H bonds represents an important and powerful strategy for converting simple arenes and hydrocarbons to functionalized molecules, thus providing an efficient atom- and step-economic strategy for increasing structural complexity in simple building blocks, and to modify bioactive molecules at a late stage of synthesis.3–5 However, the practical value of such C–H functionalization reactions depends on chemoselective activation of a single C–H bond over others. For aromatic systems, ortho-6–8 and even meta-selective9,10 C–H bond functionalization reactions normally exploit directing groups and/or specially designed ligands and catalysts. In contrast, reactions to functionalize C–H bonds at the para position of aromatic rings are frequently restricted to (Fig. 1): (a) electron-rich substrates that bear nucleophilic character and that typically provide mixtures of ortho- and para-substituted products;11–22 (b) template-assisted reactions of benzyl and phenol derivatives that require extra steps for the installation and removal of the large template, thus restricting broad application;23–26 (c) catalyst-controlled reactions that remain limited to reactions of electron-deficient pyridines and arenes,27–30 and (d) reactions proceeding through a specialized persistent sulfur-based radical that can engage in subsequent transformations.31 Considering the aforementioned challenges, alternate strategies to selectively functionalize the para C–H bond of aromatic rings, preferably through distinct mechanisms, are still highly desirable and rarely realized.
Fig. 1 |. C–H functionalization vs. decarboxylative cross-coupling.
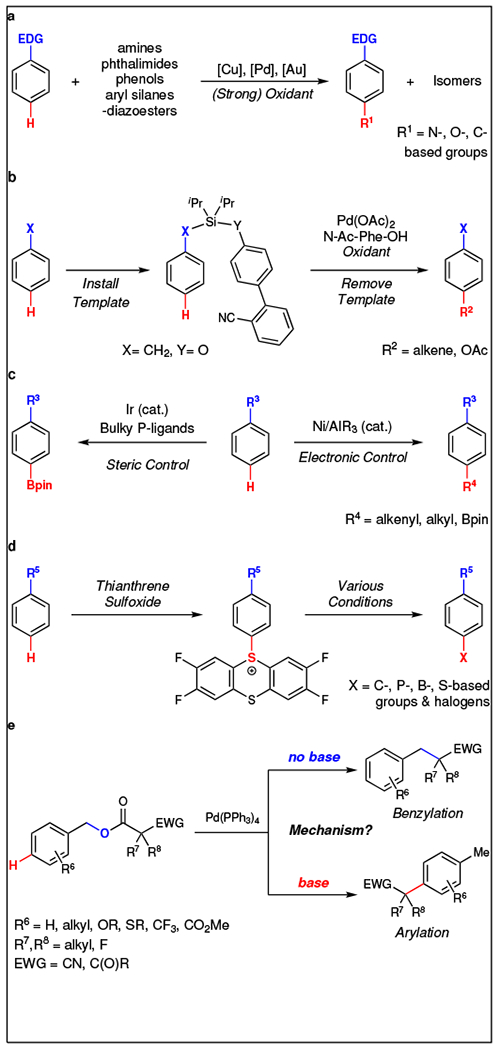
Current strategies for para-selective C–H functionalization of arenes involve: a, Intrinsic substrate-derived selectivity. b, Template-assisted selectivity. c, Catalyst-controlled selectivity. d, Reagent-controlled selectivity. e, This work: base additives override conventional decarboxylative coupling (benzylation) and enable para-selective C–H functionalization of arenes (arylation).
To complement these known transformations, we report an unexpected catalytic para-selective C–H alkylation reaction of arenes controlled by simple bases (Fig. 1e). In this reaction, the use of amine bases overrides the conventional decarboxylative benzylation pathway, and provided products from a C–H functionalization pathway.32 Extensive screening, and mechanistic studies (quantum mechanical computations, cross-over experiments, kinetic isotopic effect measurements, and isotopic labeling studies) suggest a mechanism involving an uncommon reversible Pd-catalyzed dearomatization event to access a previously “hidden” dearomatized intermediate. In this process, the base differentiates the decarboxylative benzylation and C–H functionalization pathways by enabling an irreversible 1,5-proton migration that rearomatizes the system to provide the final product. Despite scattered reports of analogous reactions of electron-rich substrates (e.g. furan, naphthalenes) controlled by ligands33,34 or the intrinsic character of the substrate,35–41 the present work exploits a simple additive to convert the reaction pathway from cross-coupling to C–H functionalization of a broad set of substrates. Additionally, the experimental and computational studies provide new insights to support a previously speculative mechanism, and demonstrate the specific role of the base in rendering the aforementioned selectivity. Finally, this mechanism provides an alternate pathway relative to classical Cα–C(sp2) bond-forming reductive elimination at a metal center.42
Results and Discussion
Brønsted basicity controls selectivity.
Several factors, including solvents, ligands, and basic additives, affected the known decarboxylative coupling process and enabled the unusual C–H functionalization reaction. In early probing experiments, use of various Pd-based pre-catalysts afforded the α-aryl-α,α-difluoroketone product 2 in polar-coordinating solvents, as well as when exploiting electron-rich P-based ligands [e.g. P(p-C6H4OMe)3]. Although these catalyst systems improved the ratio of arylation to benzylation, the yields of arylated product 2 remained less than 40%, and provided varying yields of protonated enolate 4. However, the use of basic additives improved both the selectivity and yield of the reaction (Fig. 2a). Nitriles, inorganic bases, and weak bases, such as pyridine (Pyr) and anilines, exclusively provided benzylated product 3 (entries 2–7). In contrast, more basic amines, including N,N-dimethylaminopyridine (DMAP), tripropylamine (nPr3N), triethylamine (Et3N), and N,N,N’,N’-tetramethylethylene diamine (TMEDA), favored arylated product 2 (entries 8–11). Further exploration of aliphatic amines demonstrated a correlation between rigidity/hindrance and selectivity. The conformationally constrained base, quinuclidine, provided low yield of arylated product 2 (entry 12), while the bulkier N,N-diisopropylethylamine (iPr2NEt) favored benzylated product 3 (entry 13). Thus, Et3N, with a compromise between basicity and steric hindrance, afforded arylated product 2 in the highest yield (entry 10). Additionally, the stoichiometry of Et3N also influenced the yields of arylated product 2, with decreased amounts of Et3N (< 0.25 equiv.) decreasing the selectivity of 2 (entries 10, and 14–16). However, more than 1 equivalent of Et3N did not further increase the yield (entries 10 and 17). This trend matched initial kinetic rates of the reaction, in which increased equivalents of Et3N (0.05 ➔ 0.20 equiv.) increased the rate of reaction; while, above this range, increases in the equivalents of Et3N did not further accelerate the reaction (Supplementary Fig. 1). After further optimization, the final conditions [2.5 mol% of Pd(PPh3)4/1.0 equiv of Et3N/1,4-dioxane/100 °C] provided the desired arylated product 2 in 75% isolated yield (entry 18).
Fig. 2 |. Brønsted basicity controls selectivity.a.
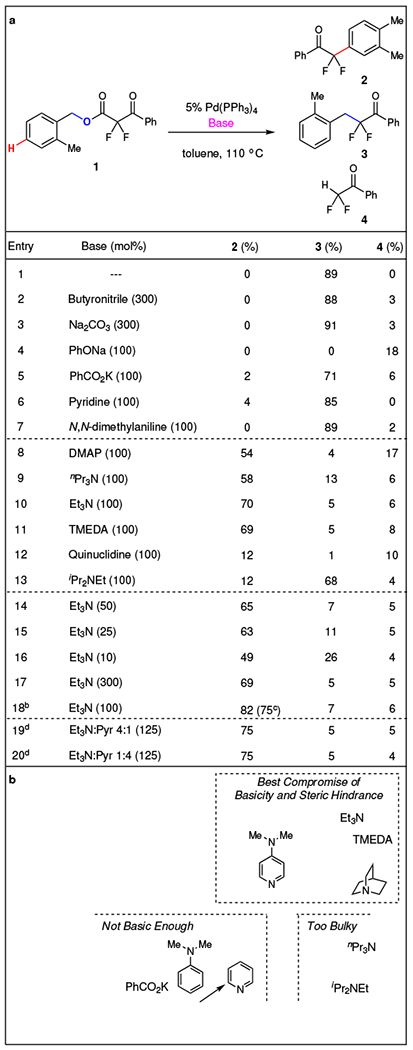
a, Though the addition of inorganic and non-basic amines favor generation of the benzylation product, the addition of basic amines inverts the selectivity and produces the arylated product. b, The selectivity for arylation (2) vs. benzylation (3) correlated with the basicity and the steric hindrance of the amine. Only unhindered basic amines (pKa > 8.0) favored the arylation product. a Reaction conditions: 1 (0.10 mmol), Pd(PPh3)4 (5.0 mol%), base, toluene (0.050 M), 110 °C, 24 h. 19F NMR yields (α,α,α-trifluorotoluene as standard). b Optimized conditions: 1 (0.50 mmol), Pd(PPh3)4 (2.5 mol%), Et3N (1.0 equiv.), 1,4-dioxane (0.050 M), 100 °C, 12 h. c Isolated yield. d 1 (0.10 mmol), Pd(PPh3)4 (2.5 mol%), Et3N:Pyr mixture (125 mol%), 1,4-dioxane (0.050 M), 100 °C.
Both the steric bulk and the strength of the base affected the selectivity of the reaction (Fig. 2b). The use of weak bases with pKa’s ranging from 4.0 – 8.0 (e.g. Pyr or N,N-dimethylaniline) and bulky trialkyl amine bases (e.g. iPr2NEt) favored the benzylation product.43 Arylation products only formed using trialkyl amine bases with pKa 8.0 – 10.5. Additional experiments confirm that base strength governed the product distribution. Specifically, reactions conducted with mixtures of Pyr and Et3N (4:1 and 1:4) all afforded the arylation product, thus confirming that the Brønsted basicity dictated the reaction outcome (Fig. 2a, entries 19–20).
Substrate scope.
A variety of substrates bearing distinct electronic properties and substitution patterns on the benzylic moieties underwent the decarboxylative arylation to incorporate a α,α-difluoroketone group at the para position of arenes relative to the original benzyl position (Table 1). Generally, ortho-substituted electron-rich (5a–b) and -deficient (5c–d) substrates provided the arylated products (6a–d) in good yields and selectivities (> 10:1). Particularly in the case of substrate 5d, no benzylated product was observed by 19F NMR. The reaction of substrate 5e bearing a bulky phenyl group on the ortho-position of the aromatic ring gave product 6e in modest yield. This reaction generated 35% of 9H-fluorene as the major side product, which might derive from an intramolecular cyclization reaction. Moreover, substrates bearing an ortho coordinating group (5f–g) and two ortho groups (5h) tolerated the present transformation and produced the arylated products (6f–h) in good yields, though requiring higher catalyst loading and increased reaction temperatures. Even a non-substituted benzylic substrate (5i) was transformed to the arylated product (6i) in good yield and selectivity. This example demonstrated that the selectivity arose from Et3N rather than a substituent effect. The regioselectivity of the arylation products was confirmed by extensive 2D NMR characterization of adducts 2, 6b, 6d and 6f (Supplementary Fig. 2 and Supplementary Tables 1–4).
Table 1 |.
Substrate scope of benzyl esters.
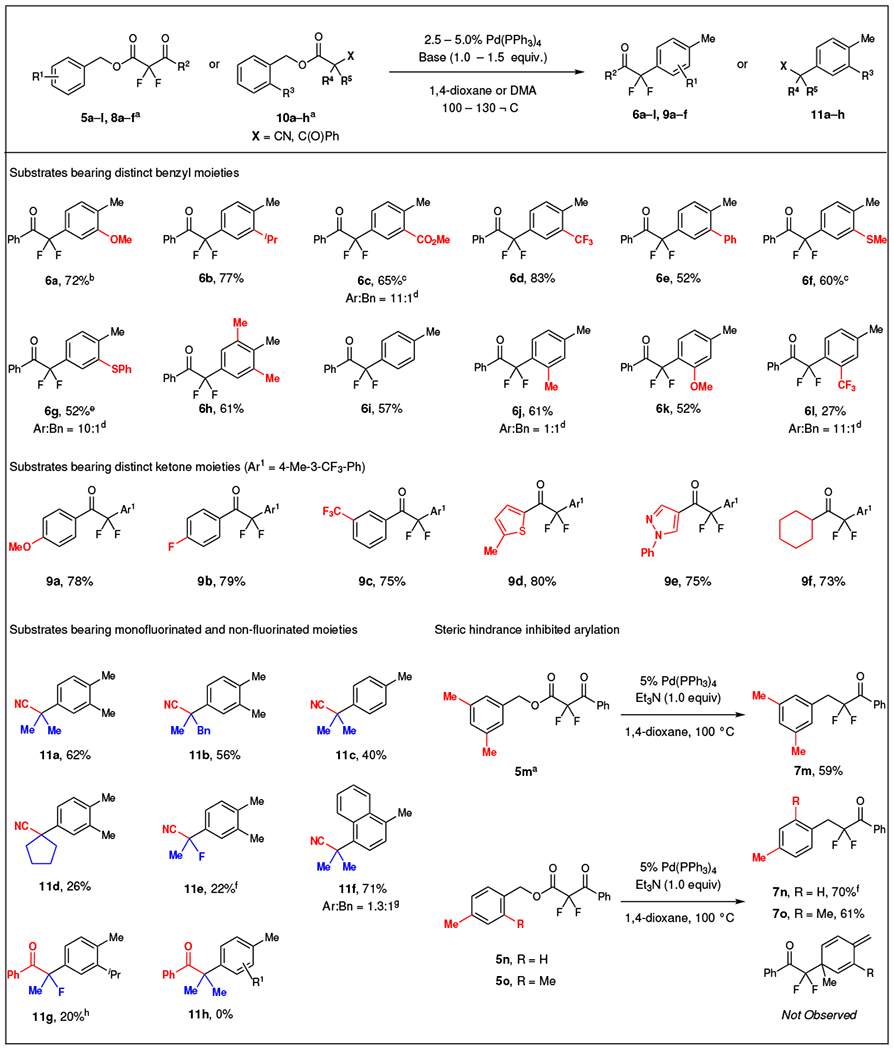 |
Reaction conditions: 5a–o or 8a–f (0.50 mmol), Pd(PPh3)4 (2.5 – 5.0 mol%), Et3N (1.0 equiv.), 1,4-dioxane (0.050 M), 100 °C, 12 – 24 h. For 10a–e,h (0.50 mmol), Pd(PPh3)4 (5.0 mol%), 2-tbutyl tetramethylguanidine (tBu-TMG, 1.5 equiv.), N,N-dimethylacetamide (0.25 M), 110 °C, 15 h; For 10f (0.50 mmol), Pd(PPh3)4 (5.0 mol%), (4-CF3Ph)3P (10 mol%), tetramethylguanidine (TMG, 2.0 equiv.), toluene (0.25 M), 110 °C, 15 h. Isolated yields.
110 °C.
120 °C.
Ratio of isolated product determined by 19F NMR.
1,4-dioxane (0.10 M), 120 °C.
19F NMR yields (α,α,α-trifluorotoluene as standard).
Ratio of products determined by GC-FID.
For 10g (0.10 mmol), Pd(PPh3)4 (5.0 mol%), TMG (3.0 equiv.), Cs2CO3 (3.0 equiv.), o-xylene (0.10 M), 130 °C, 12 h.
Steric hindrance on the benzyl moiety influenced the yields and selectivity of the reaction (Table 1). Specifically, meta-substituted substrates provided lower yields of the arylated products relative to their ortho-substituted counterparts (6j vs 2, 6k vs 6a, and 6l vs 6d). This trend likely arose from the steric hindrance induced by the meta-substituent that disfavored the attack of α,α-difluoroketone enolates at the para position, thus reducing the yields and selectivity of arylated products. Accordingly, substitution of both meta positions, only provided benzylation product 7m. Further, substitution of the para position exclusively afforded the benzylation product (7n–o), with no evidence of dearomatized species.
The decarboxylative arylation reaction also selectively converted substrates bearing a variety of aryl, heteroaryl, and alkyl α,α-difluoroketones into the products of C–H functionalization (Table 1). Substrates bearing electron-rich (8a), -neutral (8b), and -deficient (8c) aryl α,α-difluoroketone moieties provided the arylated products (9a–c) in high yields and selectivities (>20:1). Even, heteroaryl-containing α,α-difluoroketone substrates (5d–e) worked well under the standard reaction conditions. The tolerance to S- and N-containing heterocycles suggests that the current reaction can apply toward accessing fluorinated analogues of biologically active molecules. Additionally, the reaction of an aliphatic α,α-difluoroketone substrate (8f) afforded a good yield of the arylated product (9f) without further optimization. In general, products derived from C–C bond formation at the meta and ortho positions were not detected.
The reaction demonstrated more broad utility, and was not restricted to α-aryl-α,α-difluoroketones (Table 1). Reaction of α,α-dialkyl and α-fluoro-α-alkyl nitrile-derived substrates were successful (11a–f). However, these substrates required reoptimization of the reaction conditions to achieve appropriate reactivities. Most notably, these reactions required use of a stronger base, N-tert-butyl-N’,N’,N”,N”-tetramethyl guanidine (tBu-TMG), to deliver arylated products in good yields and excellent selectivities (>25:1, except for bulky naphthyl derivative 11f). Further, the decarboxylative arylation delivered an α-fluoro-α-methyl ketone (11g), though this reaction was complicated by protonation of the in situ generated enolate and homocoupling of the benzyl moieties. Nonetheless, isolation and characterization of product 11g confirms that the reaction is not limited simply to difluorinated ketones. Finally, α,α-dialkyl ketone substrate 10h afforded benzylated product without observation of arylated product 11h. Combined, these examples (11a–g) suggest that this strategy can be further developed to deliver a broad subset of products, though identification of appropriate bases might prove crucial to further expand the scope.
Mechanistic investigations.
The general mechanism of the C–H functionalization process to furnish α-aryl-α,α-difluoroketone or α-benzyl-α,α-difluoroketone products is depicted in Fig. 3 (also see Supplementary Figure 3). An initial oxidative addition of the Pd(0) catalyst to the substrate forms the Pd-Benzyl-Carboxylate ion pair that reversibly dissociates. Upon decarboxylation, the Pd-Benzyl-Carboxylate formed Pd-Enolate complexes, of which the O-Bound and C-Bound states exist in equilibrium. The C-Bound Pd-Enolate irreversibly reductively eliminates to directly form the Benzylation Product. In contrast, the O-Bound Pd-Enolate undergoes reversible C–C bond formation at the para position to generate the Dearomatized Intermediate. Aromatization would provide the Arylation Product. At the beginning of this study, the Et3N-controlled switch of selectivity was not understood; Et3N could either bind to Pd and facilitate the arylation process over the benzylation process (Pd Ligand Sphere Inset, Fig. 3), or Et3N could facilitate the rearomatization of the Dearomatized Intermediate. To investigate these processes in deeper detail, we conducted cross-over, isotopic labeling, kinetic isotope effect (KIE), and computational studies.
Fig. 3 |. General reaction mechanism.
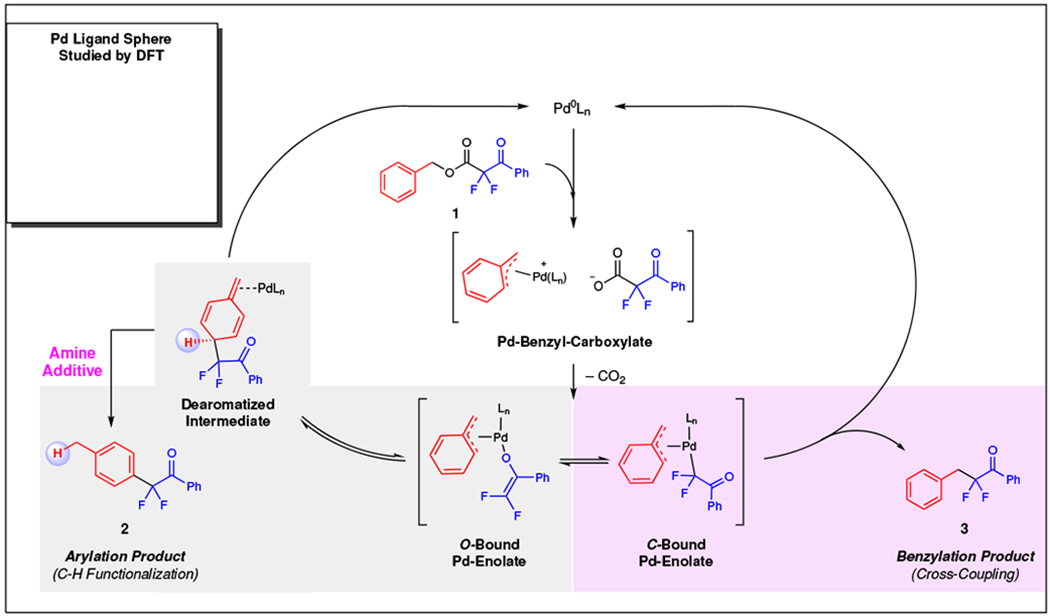
The arylation (2) and benzylation (3) products derive from a common Pd-Benzyl-Enolate intermediate for which the O-Bound and C-Bound Enolates exist in equilibrium. While the C-bound Pd-enolate generates the expected benzylation product through a traditional cross-coupling mechanism, O-bound Pd-enolate rearranges to form the Dearomatized Intermediate that bears an acidic methine proton, and an appropriate base facilitates rearomatization.
Dissociation of the Pd-benzyl – Carboxylate Ion Pair.
Support for the reversible dissociation of the Pd-Benzyl-Carboxylate ion pair derived from cross-over experiments (Fig. 4a and Supplementary Table 5). Subjection of equimolar amounts of substrates 1 and 12 to standard reaction conditions provided all four possible products in equivalent yields. This result implicated a fast scrambling of the Pd-Benzyl-Carboxylate ion pair, as has been previously observed in decarboxylative allylation reactions.44,45
Fig. 4 |. Mechanistic experiments.
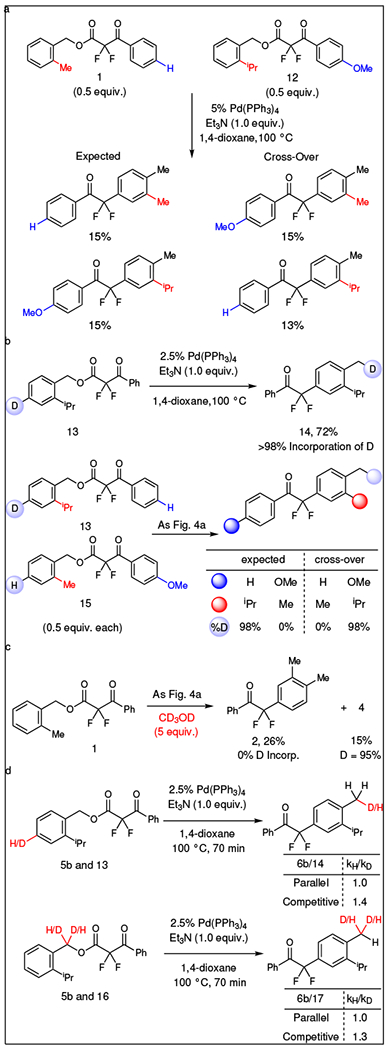
a, The detection of cross-over products supports dissociation of the Pd-Benzyl-Carboxylate ion pair. b, The para hydrogen atom migrated to the benzylic position at a late stage of the reaction, as indicated using a 2H-labeled substrate. Using a double-labeled substrate the 2H does not migrate away from the iPr-containing benzyl ring, suggesting that C–C bond formation occurs prior to H-migration, and that the H-migration occurs within the solvent sphere of the substrate. c, Upon addition of MeOH-d4, the lack of deuteration excludes reaction pathways involving discrete Pd–H and/or Pd–Ar intermediates that might be formed by direct C–Hpara palladation. d, The experimental KIE values at the para and benzyl positions indicate that the rate determining step occurs prior to the C–Hpara bond cleavage and C–Hbenzyl bond formation.
Nature of C–H functionalization step.
In this reaction, the hydrogen atom initially at the para position of the substrate migrated to the benzylic position of the final arylation product. This migration was unequivocally observed using para-deuterated substrate 13, which provided product 14 with full transfer of deuterium to the benzylic position (Fig. 4b). This proton transfer occurred in an intramolecular fashion after the C–C bond forming event, as evidenced by a second cross-over experiment using deuterated substrate 13 and unlabeled substrate 15. In this experiment, the yields and distribution of cross-over products closely matched those observed previously (Fig. 4a); however, the deuterium label did not cross away from the iPr-bearing benzyl moiety (Fig. 4b). Therefore, the hydrogen atom migration occurred inside the solvent-cage and after formation of the C–C bond. Computational studies suggest that this hydrogen transfer-rearomatization sequence is enabled by the base (vide infra),40,46,47,48 as opposed to hydride transfer mediated by Pd.34 Attempts to detect and/or isolate the Dearomatized Intermediate and study its reactivity were unsuccessful, likely because conversion to the Arylation Product occurs at a sufficiently fast rate to minimize observation under reaction conditions.
Further evidence of the late-stage irreversible C–H bond cleavage / hydrogen migration sequence derived from kinetic isotopic effect (KIE) experiments and reactions with a deuterated additive (Fig. 4c). To probe the nature of the C–H bond cleavage, the reaction was run in the presence of CD3OD, a tool widely applied to identify metal hydride intermediates and reversible C–H abstractions.49,50 Unlike many C–H functionalization reactions, the present reaction formed no deuterated product, which discounted mechanisms involving reversible C–H bond activation and/or Pd–H and Pd–Ar intermediates (Fig. 4c). Additionally, KIE’s were explored at both the benzylic and para positions to investigate the sequence of C–C bond formation, Cpara–H bond cleavage, and Cbenzyl–H bond formation (Supplementary Figs. 4–6). In parallel experiments, a KIE value of 1.0 was measured at both the benzylic and para positions, which suggests that the rate-determining step does not involve C–H bond cleavage (Fig. 4d).49 In competitive experiments, 2° KIE values of 1.3–1.4 at the same positions support a reaction for which the rate determining step does not involve cleavage of the C–H bond, but is still influenced by the C–H vs. C–D bond strengths. Combined, both experiments suggest that the rate-determining step occurs prior to the C–Hpara bond cleavage and C–Hbenzyl bond formation (Fig. 3).36,51
Computations.
An extensive computational investigation clarified the mechanism and the role of the Et3N in switching the selectivity. These results were validated by crossover experiments, labelling studies, and reactivity trends (Fig. 4). All conformational and ligand isomers of species leading from the Pd-Enolates were computed (Fig. 5a). The results revealed that, uniformly, amine-Pd coordination is disfavored over the phosphine-Pd coordination complexes (>2.5 kcal/mol, Supplementary Fig. 7). Thus, the amine does not regulate selectivity by changing the hapticity of the benzyl or enolate ligands, but rather by acting as a base to trap a hidden intermediate. More specifically, the Arylation-TS is kinetically favored by 4.1 kcal/mol over the Benzylation-TS (23.6 kcal/mol vs 27.7 kcal/mol, respectively), and upon dearomatization, the critical Dearomatized Intermediate along the arylation pathway bears an acidic methine proton adjacent to two allyl and one fluoroalkyl groups. The pKa of this proton suggests the need for stronger amine bases than Pyr (Fig. 2). In the presence of trialkyl amine base, rapid rearomatization favors the Arylation Product (26.5 kcal/mol barrier for Me3N). This distinct dearomatization/H-migration mechanism for reductive elimination complements the recently reported reductive elimination from a [LnPd(CF2COR)(Ar)] complex, which also generates a C(α)–C(sp2) bond through a standard cross-coupling paradigm.41 In the absence of an appropriate base, the slower rearomatization (32.1 kcal/mol for Pyr) favors formation of the Benzylation Product (27.7 kcal/mol). These results match experimental observations in which the use of Et3N provides the arylation products, but the use of Pyr or no base provides the benzylation products.
Fig. 5 |. Energetics of key steps.
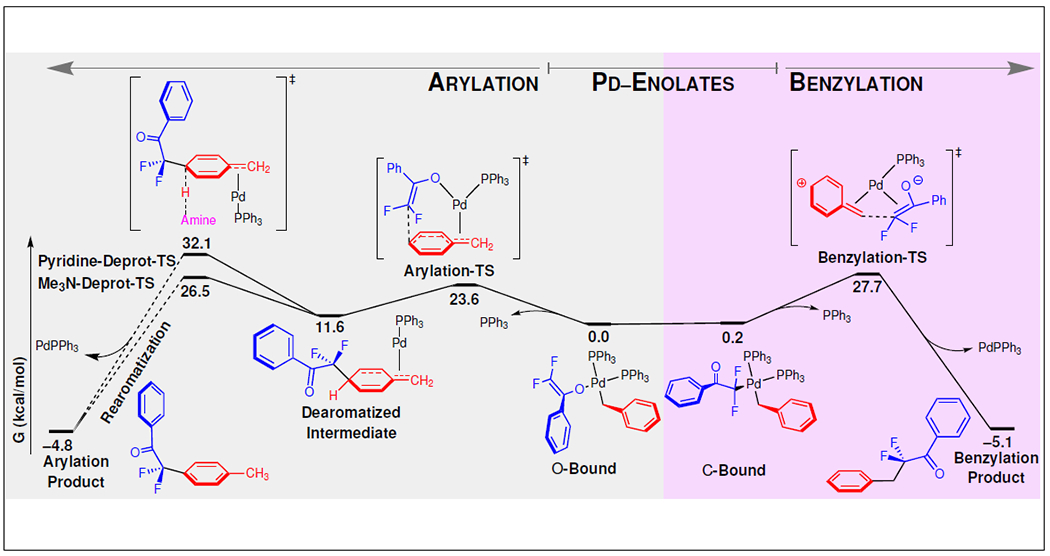
The computed reaction coordinate diagram for the formation of arylation and benzylation products from Pd-enolate complexes suggests a process controlled by Curtin-Hammett kinetics, in which the O-Bound and C-Bound enolates and the previously “hidden” Dearomatized Intermediate exist in equilibrium, and the product distribution is regulated by the energies for C–C reductive elimination vs. rearomatization. Specifically, the selectivity for arylation vs. benzylation is controlled by the ability of the amine additive to lower the energy for rearomatization (Pyridine-Deprot-TS and Me3N-Deprot-TS) relative to C–C reductive elimination (Benzylation-TS).
Overall, the selectivity of this process is ultimately controlled by the relative kinetics between Benzylation-TS and Deprot-TS, as the trialkyl amine base governs the reaction by providing a low-energy pathway for rearomatization, which cannot otherwise occur in the absence of an appropriate base. Further, this mechanism also explains why sterically bulky bases were ineffective in rendering arylation products, as these bases are too hindered to deprotonate the methine position. This controlling feature not only applies to α,α-difluoroketone-based substrates for which the fluorine atoms lower the pKa of the methine proton in Deprot-TS and enable use of a weaker base (NEt3), but also to nitrile-derived substrates (Table 1) bearing less-acidic protons on the analogous Dearomatized Intermediate that require stronger bases (e.g. TMG and tBu-TMG) to facilitate proton transfer and rearomatization. To date, reactions of α,α-dialkyl ketone-derived substrates generated benzylation products, which was supported by computational studies (Supplementary Fig. 8) that predicted that the benzylation (Benzylation-TS ΔG‡ = 33.7 kcal/mol) is favored over arylation using tBu-TMG (tBu-TMG-Deprot-TS ΔG‡ = 47.2 kcal/mol) and trialkyl amine (Me3N-Deprot-TS ΔG‡ = 43.1 kcal/mol). Computational studies also suggest that the excellent para selectivity derives from inability of the benzylic cation to distribute charge to the meta position, as well as the unfavorable orientations of the benzyl and enolate moieties that would be required to form C–C bonds at the ortho position (Supplementary Fig. 9).
Conclusion
In summary, amine additives transformed a benchmark decarboxylative-coupling benzylation reaction into a unique non-chelation-controlled para-selective C–H functionalization reaction. The two reaction paradigms typically require orthogonal substrates and conditions, but can now be accessed using complementary reaction conditions, namely by simply adding an inexpensive base to a readily available Pd-based catalyst system. Mechanistically, the reaction involved dearomative C–C bond formation to generate a “hidden” dearomatized intermediate. This process simultaneously acidified a typically inert proton, and provided a low-energy pathway for base-mediated rearomatization to para-substituted toluene derivatives from substrates that would typically provide benzyl-substituted products. This dearomatized intermediate might exist in other catalytic processes,32 and exploitation of this strategy should enable the construction of new and unique products as yet inaccessible by current means.
Supplementary Material
Acknowledgements
We thank the donors of the Herman Frasch Foundation for Chemical Research (701-HF12), and the National Science Foundation (CHE-1455163) and National Institute of General Medical Sciences (R35 GM124661) for supporting this work. NMR Instrumentation was provided by NIH (S10OD016360, S10RR024664, P20GM103418) and NSF Grants (9977422, 0320648). PHYC acknowledges financial support from the Bert and Emelyn Christensen Professorship and the Vicki & Patrick F. Stone family. PHYC, TF, MAG and ACB acknowledge the National Science Foundation (NSF, CHE-1352663) and the computing infrastructure in part provided by the NSF Phase-2 CCI, Center for Sustainable Materials Chemistry (NSF CHE-1102637). TF acknowledges Summer Fellowship Award from the department of Chemistry at OSU.
Footnotes
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information files.
Competing Interests
The authors declare no competing interests.
References
- 1.Johansson Seechurn CCC, Kitching MO, Colacot TJ & Snieckus V Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel prize. Angew. Chem. Int. Ed 51, 5062–5085 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Labinger JA & Bercaw JE Understanding and exploiting C–H bond activation. Nature 417, 507 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Hartwig JF Evolution of C–H bond functionalization from methane to methodology. J. Am. Chem. Soc 138, 2–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gensch T, Hopkinson MN, Glorius F & Wencel-Delord J Mild metal-catalyzed C-H activation: examples and concepts. Chem. Soc. Rev 45, 2900–2936 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi J, Yamaguchi AD & Itami K C–H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed 51, 8960–9009 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Engle KM, Mei T-S, Wasa M & Yu J-Q Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc. Chem. Res 45, 788–802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neufeldt SR & Sanford MS Controlling site selectivity in palladium-catalyzed C–H bond functionalization. Acc. Chem. Res 45, 936–946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang F & Spring DR Arene C-H functionalisation using a removable/modifiable or a traceless directing group strategy. Chem. Soc. Rev 43, 6906–6919 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Yang J Transition metal catalyzed meta-C-H functionalization of aromatic compounds. Org. Biomol. Chem 13, 1930–1941 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Dey A, Agasti S & Maiti D Palladium catalysed meta-C-H functionalization reactions. Org. Biomol. Chem 14, 5440–5453 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Ciana C-L, Phipps RJ, Brandt JR, Meyer F-M & Gaunt MJ A highly para-selective copper(II)-catalyzed direct arylation of aniline and phenol derivatives. Angew. Chem. Int. Ed 50, 458–462 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Leow D & Yu J-Q Pd(II)-catalyzed para-selective C–H arylation of monosubstituted arenes. J. Am. Chem. Soc 133, 13864–13867 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ball LT, Lloyd-Jones GC & Russell CA Gold-catalyzed direct arylation. Science 337, 1644–1648 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Wu Z et al. Palladium-catalyzed para-selective arylation of phenols with aryl iodides in water. Chem. Commun 49, 7653–7655 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Yu Z et al. Highly site-selective direct C–H bond functionalization of phenols with α-aryl-α-diazoacetates and diazooxindoles via gold catalysis. J. Am. Chem. Soc 136, 6904–6907 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Berzina B, Sokolovs I & Suna E Copper-catalyzed para-selective C–H amination of electron-rich arenes. ACS Catalysis 5, 7008–7014 (2015). [Google Scholar]
- 17.Marchetti L, Kantak A, Davis R & DeBoef B Regioselective gold-catalyzed oxidative C–N bond formation. Org. Lett 17, 358–361 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu H, Shang M, Dai H-X & Yu J-Q Ligand-controlled para-selective C–H arylation of monosubstituted arenes. Org. Lett 17, 3830–3833 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Yang Z, Qiu F-C, Gao J, Li Z-W & Guan B-T Palladium-catalyzed oxidative arylation of tertiary benzamides: para-selectivity of monosubstituted arenes. Org. Lett 17, 4316–4319 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Sokolovs I & Suna E Para-selective Cu-catalyzed C–H aryloxylation of electron-rich arenes and heteroarenes. J. Org. Chem 81, 371–379 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Ma B et al. Highly para-selective C–H alkylation of benzene derivatives with 2,2,2-trifluoroethyl α-aryl-α-diazoesters. Angew. Chem. Int. Ed 56, 2749–2753 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Luan Y-X et al. Amide-ligand-controlled highly para-selective arylation of monosubstituted simple arenes with arylboronic acids. J. Am. Chem. Soc 139, 1786–1789 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Bag S et al. Remote para-C–H functionalization of arenes by a D-shaped biphenyl template-based assembly. J. Am. Chem. Soc 137, 11888–11891 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Patra T et al. Palladium-catalyzed directed para C–H functionalization of phenols. Angew. Chem. Int. Ed 55, 7751–7755 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Maji A et al. Experimental and computational exploration of para-selective silylation with a hydrogen-bonded template. Angew. Chem. Int. Ed 56, 14903–14907 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Li M et al. Remote para-C–H acetoxylation of electron-deficient arenes. Org. Lett 21, 540–544 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Nakao Y, Yamada Y, Kashihara N & Hiyama T Selective C-4 alkylation of pyridine by nickel/lewis acid catalysis. J. Am. Chem. Soc 132, 13666–13668 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Tsai C-C et al. Bimetallic nickel aluminun mediated para-selective alkenylation of pyridine: direct observation of η2,η1-pyridine Ni(0)–Al(III) intermediates prior to C–H bond activation. J. Am. Chem. Soc 132, 11887–11889 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Saito Y, Segawa Y & Itami K Para-C–H borylation of benzene derivatives by a bulky iridium catalyst. J. Am. Chem. Soc 137, 5193–5198 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Okumura S et al. Para-selective alkylation of benzamides and aromatic ketones by cooperative nickel/aluminum catalysis. J. Am. Chem. Soc 138, 14699–14704 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Berger F et al. Site-selective and versatile aromatic C–H functionalization by thianthrenation. Nature 567, 223–228 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Yang M-H, Hunt JR, Sharifi N & Altman RA Palladium catalysis enables benzylation of α,α-difluoroketone enolates. Angew. Chem. Int. Ed 55, 9080–9083 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Recio IIIA, Heinzman JD & Tunge JA Decarboxylative benzylation and arylation of nitriles. Chem. Commun 48, 142–144 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Mendis SN & Tunge JA Decarboxylative dearomatization and mono-α-arylation of ketones. Chem. Commun 52, 7695–7698 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Jaeger CW & Kornblum N New type of substitution at a saturated carbon atom. J. Am. Chem. Soc 94, 2545–2547 (1972). [Google Scholar]
- 36.Bao M, Nakamura H & Yamamoto Y Facile allylative dearomatization catalyzed by palladium. J. Am. Chem. Soc 123, 759–760 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Lu S, Xu Z, Bao M & Yamamoto Y Carbocycle synthesis through facile and efficient palladium-catalyzed allylative de-aromatization of naphthalene and phenanthrene allyl chlorides. Angew. Chem. Int. Ed 47, 4366–4369 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Peng B, Zhang S, Yu X, Feng X & Bao M Nucleophilic dearomatization of chloromethyl naphthalene derivatives via η3-benzylpalladium intermediates: a new strategy for catalytic dearomatization. Org. Lett 13, 5402–5405 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Ueno S, Komiya S, Tanaka T & Kuwano R Intramolecular SN′-type aromatic substitution of benzylic carbonates at their para-position. Org. Lett 14, 338–341 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Zhang S, Wang Y, Feng X & Bao M Palladium-catalyzed amination of chloromethylnaphthalene and chloromethylanthracene derivatives with various amines. J. Am. Chem. Soc 134, 5492–5495 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Zhang S, Yu X, Feng X, Yamamoto Y & Bao M Palladium-catalyzed regioselective allylation of five-membered heteroarenes with allyltributylstannane. Chem. Commun 51, 3842–3845 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Arlow SI & Hartwig JF Synthesis, characterization, and reactivity of palladium fluoroenolate complexes. J. Am. Chem. Soc 139, 16088–16091 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bordwell FG Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res 21, 456–463 (1988). [Google Scholar]
- 44.Trost BM, Xu J & Schmidt T Palladium-catalyzed decarboxylative asymmetric allylic alkylation of enol carbonates. J. Am. Chem. Soc 131, 18343–18357 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weaver JD, Recio A, Grenning AJ & Tunge JA Transition metal-catalyzed decarboxylative allylation and benzylation reactions. Chem. Rev 111, 1846–1913 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie H, Zhang H & Lin Z DFT studies on the palladium-catalyzed dearomatization reaction between chloromethylnaphthalene and the cyclic amine morpholine. Organometallics 32, 2336–2343 (2013). [Google Scholar]
- 47.Boga C, Del Vecchio E, Forlani L & Tozzi S Evidence of reversibility in azo-coupling reactions between 1,3,5-tris(N,N-dialkylamino)benzenes and arenediazonium salts. J. Org. Chem 72, 8741–8747 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Forlani L, Boga C, Del Vecchio E, Ngobo A-LTD & Tozzi S Reactions of Wheland complexes: base catalysis in re-aromatization reaction of σ complexes obtained from 1,3,5-tris(N,N-dialkylamino)benzene and arenediazonium salts. J. Phys. Org. Chem 20, 201–205 (2007). [Google Scholar]
- 49.Simmons EM & Hartwig JF On the interpretation of deuterium kinetic isotope effects in C–H bond functionalizations by transition-metal complexes. Angew. Chem. Int. Ed 51, 3066–3072 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Ma S, Villa G, Thuy-Boun PS, Homs A & Yu J-Q Palladium-catalyzed ortho-selective C–H deuteration of arenes: evidence for superior reactivity of weakly coordinated palladacycles. Angew. Chem. Int. Ed 53, 734–737 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Ariafard A & Lin Z DFT studies on the mechanism of allylative dearomatization catalyzed by palladium. J. Am. Chem. Soc 128, 13010–13016 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.