

Summary
Background:
Regulated protein synthesis is essential for megakaryocyte (MK) and platelet functions, including platelet production and activation. PDK1 (phospho-inositide-dependent kinase 1) regulates platelet functional responses and has been associated with circulating platelet counts. Whether PDK1 also directly regulates protein synthetic responses in MKs and platelets, and platelet production by MKs remains unknown.
Objective:
To determine if PDK1 regulates protein synthesis in MKs and platelets.
Methods:
Pharmacologic PDK1 inhibitors (BX-795) and mice where PDK1 was selectively ablated in MKs and platelets (PDK1−/−) were used. PDK1 signaling in MKs and platelets (human and murine) were assessed by immunoblots. Activation-dependent translation initiation and protein synthesis in MKs and platelets was assessed by probing for dissociation of eIF4E from 4EBP1, and using m7-GTP pulldowns and S35 methionine incorporation assays. Proplatelet formation by MKs, synthesis of Bcl-3 and MARCKs protein, and clot retraction were employed for functional assays.
Results:
Inhibiting or ablating PDK1 in MKs and platelets abolished the phosphorylation of 4EBP1 and eIF4E by preventing activation of the PI3K and MAPK pathways. Inhibiting PDK1 also prevented dissociation of eIF4E from 4EBP1, decreased binding of eIF4E to m7GTP (required for translation initiation), and significantly reduced de novo protein synthesis. Inhibiting PDK1 reduced proplatelet formation by human MKs and blocked MARCKs protein synthesis. In both human and murine platelets, PDK1 controlled Bcl-3 synthesis. Inhibition of PDK1 led to complete failure of clot retraction in vitro.
Conclusions:
PDK1 is a previously-unidentified translational regulator in MKs and platelets, controlling protein synthetic responses, proplatelet formation, and clot retraction.
Keywords: Platelets, Platelet Activation, Protein Translation, Signal Transduction, Thrombosis
Introduction
Megakaryocytes, which arise primarily in the bone marrow, are responsible for producing approximately 1×1011 platelets in adult humans daily under physiological conditions[1]. Each megakaryocyte produces between 1,000 and 3,000 platelets. Megakaryocytes accomplish this task by projecting proplatelets into bone marrow sinusoidal vessels, where shear releases platelets into the circulation. Translation of mRNAs, resulting in de novo protein synthesis, is a critical step in the process of proplatelet formation and platelet release[2–5]. Translation is generally divided into three steps: initiation, elongation, and termination. Of these three steps, initiation is often the rate-limiting step. Translation initiation is a tightly regulated series of events which begins when eukaryotic initiation factor 4F (eIF4F, a heterotrimeric protein complex) binds to the 5’ cap of mRNAs. The eIF4F protein complex is made up of three subunits: eIF4A, which processes ATPase and RNA helicase activities, the 5’ mRNA cap-binding protein eIF4E, and the scaffolding protein eIF4G, which connects the end of 5’terminus of the mRNA to other factors and mRNAs. In nucleated cells, the activity of eIF4E is regulated via its phosphorylation and binding to eIF4E-binding protein (4E-BP) repressor proteins. Previous work from our group has demonstrated that eIF4E is present and active in human platelets, and is regulated by outside-in signals delivered by integrins[6].
Nevertheless, the specific signaling events that control translational mechanisms and subsequent protein synthesis in MKs remain largely unknown. Phosphoinositide–dependent protein kinase 1 (PDK1) is a cytoplasmic, Ser/Thr protein kinase that phosphorylates and activates protein kinases from the AGC family, all which mediate cellular responses[7–9]. PDK1, which is also activated by PI3K, is known to play important roles in cell growth, metabolism, proliferation, and survival[9–12]. PDK1 is activated by binding to membrane-tethered PIP3. Activated PDK1 then phosphorylates Akt at Thr308, thereby activating its serine/threonine kinase activity. Many MK and platelet agonists signal through the PI3K/PDK1/AKT pathway[13, 14].
We have previously shown that in human platelets, PDK1 is active and, through selectively phosphorylating Akt at Thr308, regulates platelet aggregation, thromboxane generation, and clot retraction through outside-in signaling[15]. Moreover, in mice specifically lacking PDK1 in platelets and MKs, platelet counts are reduced by about 25%[7]. In this study, annexin V binding to platelets, which assesses for apoptosis, was not enhanced by PDK1 deficiency, suggesting that PDK1 regulates platelet production rather than platelet clearance. Nevertheless, how PDK1 regulates platelet production remains unknown.
In hepatocytes, activation of Akt induces protein synthesis through the mTORC1/4EBP1 pathway[16]. As PDK1 signaling in platelets results in Akt activation, this suggests to the possibility that PDK1 controls platelet production through regulation of protein synthetic events in MKs. However, whether PDK1 mediates protein synthesis in MKs and platelets has not been studied to date. In the current study, we use a combination of pharmacologic and genetic approaches to demonstrate that PDK1 plays a vital role in global protein synthesis in both MKs and platelets. We further show that these responses are conserved in human and murine MKs and results in impaired proplatelet formation. Moreover, we provide evidence that inhibiting or deleting PDK1 blocks translational control pathways, resulting in suppressed Bcl-3 synthesis and abolished clot retraction. Therefore, we conclude that PDK1 regulates protein synthesis in MKs and provide novel mechanistic findings of how PDK1 controls platelet production.
Materials and Methods
Reagents.
2MeSADP (Cat. no. 1624) was from Tocris (Minneapolis, MN, USA). Apyrase (type VII) was from Sigma (St Louis, MO, USA) and BX-795 (a specific PDK1 inhibitor we previously characterized in detail[15]) was from Selleckchem (Houston, TX, USA). Whatman protein nitrocellulose transfer membrane was from Fisher Scientific (Pittsburg, PA, USA). LI-COR Odyssey blocking buffer was from LI-COR Biosciences (Lincoln, NE, USA). Phospho-Akt (Thr308), phospho-ERK1/2 (Thr202/Tyr204), phospho-4EBP1 (Ser65), Phospho-eIF4E (Ser209) Total 4EBP1 and Total eIF4E antibodies were all from Cell Signaling Technology (Beverly, MA, USA). Bcl3 and β-actin antibodies were from Santa Cruz Biotechnologies (Santa Cruz, CA, USA).
Mice.
Platelets isolated from mice where PDK1 was specifically ablated in megakaryocytes and platelets were kindly provided by Dr. Oliver Borst from the University of Tübingen.
Human Platelet isolation.
Blood was collected from healthy volunteers into a one-sixth volume of acid/citrate/dextrose (2.5g sodium citrate, 2 g glucose, and 1.5 g citric acid in 100 ml deionized water). Platelet-rich plasma was obtained by centrifugation at 250 x g for 20 minutes at ambient temperature. Platelets were isolated from plasma by centrifugation at 980 x g for 10 minutes at ambient temperature and resuspended in Tyrode’s buffer pH 6.5 (138 mM NaCl, 2.7 mM KCl, 2 mM MgCl2, 0.42 mM NaH2PO4, 5 mM glucose, 10 mM PIPES (pH 6.5) containing 20 nM PGE1, 10 mM indomethacin, 500 mM EGTA and 0.2 U/ml apyrase,). Platelets were isolated from Tyrode’s buffer pH 6.5 by centrifugation at 980 x g for 10 minutes and resuspended in Tyrode’s buffer, pH 7.4 (138 mM NaCl, 2.7 mM KCl, 2 mM MgCl2, 0.42 mM NaH2PO4, 5 mM glucose, 10 mM HEPES and 0.2 U/ml apyrase, pH 7.4). The platelet count was adjusted to 2–2.5 × 108/ml. Approval was obtained from the institutional review board for these studies. Informed consent was provided prior to blood donation.
Primary MK cultures.
CD34+ hematopoietic stem cells (HSCs) were isolated from human umbilical vein cord blood. For culturing CD34+ hematopoietic stem cell-derived MKs, human CD34+ cells from human umbilical cord blood were isolated as described previously. The CD34+ cells were placed in X-Vivo 20 media that contained 40 ng/mL recombinant human stem cell factor (SCF; Invitrogen, Carlsbad, CA), 50 ng/mL recombinant thrombopoietin (TPO; Invitrogen), and 10 ng/mL recombinant human interleukin-3 (IL-3; Invitrogen). Every 2–3 days, the cells were re-suspended in media with fresh growth factors, with the exception that IL-3 was removed at day 5. CD34+ derived MKs were incubated on culture day 11–13 with DMSO or BX-795. MKs were isolated and used for proplatelet formation and protein synthesis assays.
Protein synthesis assays.
Gradient-isolated, mature CD34+ megakaryocytes (culture day 13) were suspended for 1 hour (37°C) in DMEM that lacked methionine and cysteine (MP Biomedical). After 1 hour, EasyTag™ EXPRESS35S protein labelling mix (0.06 mCI total) (Perkin Elmer) was subsequently added to CD34+ megakaryocytes and after a brief incubation period (15 minutes), the megakaryocytes were activated by adhering on a fibrinogen-coated plate (10mg/ml) for 2 hours in the presence or absence of BX-795. After 2 hours the cells, were carefully removed and centrifuged at 20,000 x g for 5 minutes. The cell pellets and supernatants were then collected in clean Eppendorf tubes. The CD34+ MKs were washed three times in complete media and then lysed in radio immunoprecipitation assay buffer (RIPA) (1X PBS with 1% NP-40, 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulfate). Lysates were cleared and then Trichloroacetic acid (TCA) precipitated using 20% TCA on ice for 30 minutes. The precipitated proteins were loaded onto a Whatman grade GF/C glass microfiber filter (VWR) and washed five times with 10% TCA and five times with 95% ethanol. Filter papers were then read using a liquid scintillation counter.
Western blotting.
Platelets were stimulated with agonists in the presence of inhibitors (or the vehicle control) for the appropriate time under stirring conditions at 37˚C and the reaction was stopped by the addition of 0.6 N HClO4. The resulting acid precipitate was collected and kept on ice. The samples were centrifuged at 13,000 x g for 4 minutes followed by re-suspending in 0.5 ml of deionized water. The protein was again pelleted by centrifugation at 13,000 x g for 4 minutes. The protein pellets were solubilized in sample buffer containing 0.1 M Tris, 2% SDS, 1% (v/v) glycerol, 0.1% bromophenol blue, and 100 mM DTT, and then boiled for 10 minutes. Proteins were resolved by SDS polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Whatman Protran). Membranes were blocked with Odyssey blocking buffer for 1 hour at ambient temperature, incubated overnight at 4oC with the desired primary antibody, and then washed 4 times with TBS-T. Membranes were incubated with the appropriate secondary infrared dye-labeled antibody for 60 minutes at room temperature and washed 4 times with 10% TBS-T. Membranes were examined with a Li-Cor Odyssey infrared imaging system.
Proplatelet formation.
Plates were coated with fibrinogen (10mg/ml) and megakaryocytes (culture day 13) were incubated overnight at 37°C. The cells were fixed with equal volume of 4% PFA for 10 minutes and washed 3 times with sterile PBS solution. The cells were treated with phalloidin dye for 30 minutes at room temperature and washed 3 times with sterile PBS. The plates were analyzed using confocal microscopy and images were processed using ImageJ software.
Co-immunoprecipitation assay.
CD34+ hematopoietic stem cells (HSCs) were isolated from human umbilical vein cord blood. MKs were cultured from CD34+ HSCs ex-vivo as described above. Plates were coated with fibrinogen (10mg/ml) and megakaryocytes (culture day 13) were incubated overnight at 37°C for 2 hours. Cells were lysed using equal volumes of chilled 2X NP-40 lysis buffer (50 mM N-2-hydroxyethylpiperazine-N′−2-ethanesulfonic acid, 100 mM sodium chloride, 2% NP-40, 2 mM ethyleneglycoltetraacetic acid, and 100 mL of 2X Halt Protease and Phosphatase Cocktail solution; Pierce, Rockford, IL). Cells were gently rocked for 30 minutes at 4°C and then centrifuged at 10,000g for 10 minutes at 4°C to remove any non-lysed cells. Twenty microliters of agarose-conjugated, normal rabbit IgG (a control IgG) or rabbit 4EBP1 polyclonal IgG was added to the samples and incubated overnight with gentle rocking at 4°C. The samples were then incubated with agarose G beads (IgG control or 4EBP1) for 2 hours at 4°C. Samples were centrifuged at 5,000g for 30 seconds at 4°C to pellet down the agarose beads. The agarose beads were then washed 3 times using 1X NP-40 lysis buffer and washed once using phosphate-buffered saline. Proteins were solubilized in 4X sample buffer, separated by SDS-PAGE, and transferred to a nitrocellulose membrane.
m7-GTP Pull down assay.
Cells (megakaryocytes or platelets, depending on the experiment) were lysed in 10 mM Tris (pH 8), 150 mM NaCl, 10% glycerol, and 1% NP-40. Cell lysates were incubated with 7-methyl-GTP agarose beads (Jene bioscience) for 2 hours, in accordance with the manufacturer’s recommendations. Samples were then washed gently three times with PBS to remove any unbound beads followed by centrifugation (1,500 x g for 5 minutes). Next, 2x laemmli buffer was added to the beads. Cell lysates were subjected to 15% SDS–PAGE, and resolved proteins were transferred onto nitrocellulose membranes for immunoblotting.
Clot retraction.
Human platelets were isolated as described above and clot retraction was measured. Briefly, 500 μL platelets (5 × 108 /ml) were added to a glass cuvette and mixed with 1 mM CaCl2, 100 μg/ml of batroxabin (convert fibrinogen to fibrin to induce clot retraction), and 0.1 mg/ml fibrinogen. A total of 100nM of 2MeSADP was then added to initiate clot retraction. Platelets were allowed to retract at room temperature and photographed at the indicated time points.
Statistics.
Each experiment was independently repeated at least 3 times. Results were expressed as means ± SD. Data were analyzed using Prism software. Significant differences were determined using Student’s t-test. Differences were considered significant at a two-tailed p<0.05.
Results
PDK1 regulates stimulation-dependent protein synthesis in human megakaryocytes
Previous studies from our lab and others have shown that PDK1 regulates Akt activity, a downstream effector of the PI3K pathway[15]. Interestingly, studies in other cells have shown that Akt regulates translation control pathways via regulating mTORC1[17–19]. Additionally, we previously demonstrated that PDK1 activates the MAPK pathway in platelets[20, 21]. In nucleated cells, activation of the MAPK pathway results in phosphorylation of eIF4E and subsequent translational initiation[22]. However, whether activation of the PI3K and MAPK pathways in megakaryocytes and their progeny - anucleate platelets – controls translational events is unknown.
We hypothesized that PDK1 regulates protein synthesis in megakaryocytes and platelets through PI3K and MAPK pathways. In order to evaluate this hypothesis, we examined the effect of PDK1 inhibition on translation in megakaryocytes using an S35 methionine incorporation assay (which labels newly synthesized proteins). Previous studies have shown that megakaryocyte adhesion to fibrinogen induces activation of translation as well as proplatelet formation, in a mechanism requiring integrin receptor αΙΙb[23]. We therefore used fibrinogen adhesion as an activating signal for megakaryocytes. As shown in Figure 1A, treatment of fibrinogen-adherent, CD34+ derived, mature megakaryocytes with the PDK1 inhibitor BX-795 significantly decreased S35 methionine incorporation into newly synthesized proteins. These results indicate that PDK1 regulates signal-dependent protein synthesis in human megakaryocytes.
Figure 1: PDK1 regulates protein synthesis in CD34+ derived, cultured, human megakaryocytes.
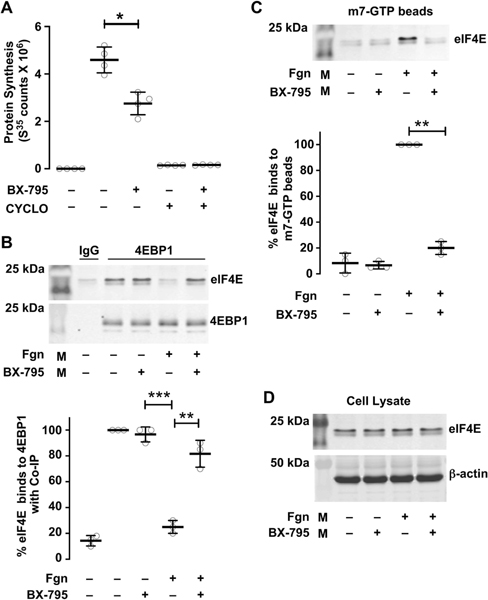
A. CD34+ megakaryocytes were left alone or treated with BX-795 (1μM) from culture day 11 – 13. Cells were collected on Day 13 and adhered to fibrinogen in media that contains S35 methionine for 2 hours. Cyclohexamide (Cyclo) was used as a positive control. Protein synthesis was measured using scintillation counts. B. On culture day 13, CD34+ megakaryocytes were adhered to a fibrinogen coated surface for 60 min in the presence or absence of BX-795 (1μM). Cell lysates were immunoprecipitated with 4EBP1 and samples were probed for eIF4E using an anti-eIF4E mouse mAb. C. CD34+ megakaryocytes were left alone or adhered to fibrinogen (60 min) in the presence or absence of BX-795. Cells were lysed and incubated with m7 GTP- agarose beads. The beads were eluted to collect the eIF4E protein. Samples were analyzed for eIF4E binding using western blot (**P < 0.05) (***P < 0.01).
Next, we determined how PDK1 controls critical steps of the translation initiation process. Previous studies have shown that activated Akt phosphorylates 4EBP1 through mTORC1, thereby disrupting the eIF4E/4EBP1 complex. This dissociation frees eIF4E, allowing eIF4E to form the translation initiation complex. As shown in our previous studies, PDK1 inhibition significantly reduces Akt activity[15]. Therefore, we evaluated the effects of PDK1 inhibition on eIF4E/4EBP1 complex dissociation in megakaryocytes. As shown in Figure 1B, in unstimulated human megakaryocytes both eIF4E and 4EBP1 are complexed together. However, in fibrinogen-stimulated megakaryocytes, eIF4E is not complexed with 4EBP1. This dissociation is reversed, and eIF4E and 4EBP1 complex together, when PDK1 is inhibited with BX-795 in fibrinogen stimulated megakaryocytes (Figure 1B). These data indicate that PDK1 –prevents 4EBP1 dissociation from eIF4E. As dissociated eIF4E is necessary for translational initiation, this suggests that PDK1 serves as a mechanism to regulate translational control.
The majority of eukaryotic mRNAs have a 7-methyl-guanosine 5’ cap structure (m7GpppX, where X is any nucleotide) at their 5’ end. This cap structure mediates mRNA splicing, stability, and translational efficiency. Once eIF4E has dissociated from its complex with 4EBP1, eIF4E facilitates localizing ribosomes to the mRNA cap structure - a necessary and rate limiting step of translation initiation. Our group has previously demonstrated that in human platelets, eIF4E is capable of interacting with a methylated mRNA cap homolog (m7GTP) [6]. Based on our findings (Figures 1A–B), we hypothesized that eIF4E interactions with m7GTP (which are necessary for translation initiation) would also be regulated by PDK1. Indeed, pharmacologically inhibiting PDK1 with BX-795 in human megakaryocytes adherent to fibrinogen significantly and completely decreased binding of eIF4E to m7GTP (Figure 1C). Total eIF4E protein levels were unaffected by PDK1 inhibition (Figure 1D), in contrast, demonstrating that PDK1 selectively controls eIF4E binding to mRNA caps.
PDK1 signaling regulates translation control pathways in human megakaryocytes
Fibrinogen mediated adhesion and signaling is an important component of megakaryocyte differentiation, development, and proplatelet formation[24–26]. Fibrinogen signaling also activates translation control pathways, including the PI3K and MAPK pathways[27, 28]. As noted above, in other cells PDK1 regulates two pathways that converge on translation: first, Akt, which activates mTORC1 mediated translation[15, 29, 30] and secondly, ERK1/2, which activates MAPK mediated translation[31]. Therefore, we hypothesized that PDK1 would regulate translation control pathways and thereby protein synthesis in human megakaryocytes. As shown in Figure 2, pharmacologically inhibiting PDK1 in megakaryocytes adherent to fibrinogen near-completely abolished both PI3K and MAPK pathway activation (as measured by phosphorylation of Akt and ERK1/2, respectively). This was accompanied by inhibition of phosphorylation of 4EBP1 and eIF4E, which are translational regulators downstream of Akt and ERK1/2. Phosphorylation of 4EBP1 and eIF4E is necessary for their activity.
Figure 2: Inhibition of PDK1 effects translation control pathways.
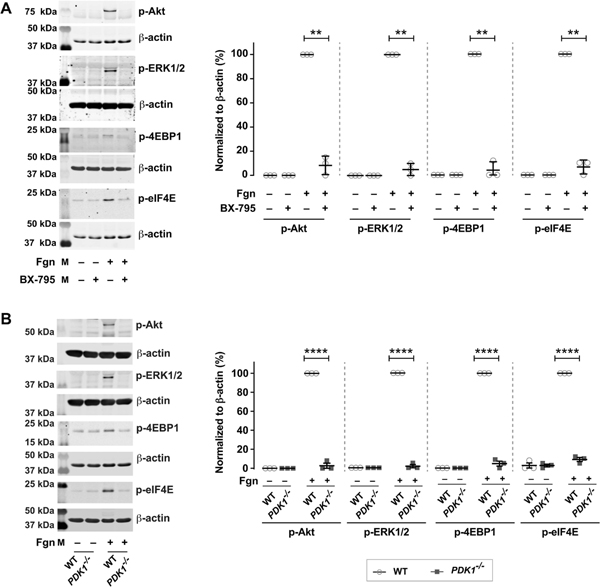
A. Day 13 CD34+ megakaryocytes were incubated with or without BX-795 (1μM) under adherent or non-adherent conditions on fibrinogen. CD34+ megakaryocyte proteins were separated by SDS-PAGE and probed for phosphorylation of p-Akt (T308), extracellular signal-regulated kinase 1/2 (p-ERK1/2 (T202/Y204), p-4EBP1 (T37) and p-eIF4E (S209). The immunoblots are representative of three independent experiments. (**P < 0.05). B. Day 5 bone marrow derived mouse megakaryocytes from wild type or platelet specific PDK1 knockout mice were analyzed for p-Akt (T308), extracellular signal-regulated kinase 1/2 (p-ERK1/2 (T202/Y204), p-4EBP1 (T37) and p-eIF4E (S209) under adherent or non-adherent conditions on fibrinogen. The western blots shown are representative of three independent experiments. (****P < 0.0001).
To complement our pharmacologic studies, we also evaluated the role of PDK1 in translation control pathways by using bone marrow derived megakaryocytes from platelet-specific PDK1-deficient mice (KO). We used both fibrinogen adherent and non–adherent conditions to understand the role of PDK1 in megakaryocyte function. Consistent with pharmacologic inhibitor studies using BX-795 (Figure 2A), phosphorylation of Akt, ERK1/2, 4EBP1, and eIF4E was blocked in fibrinogen adherent megakaryocytes from PDK1 KO mice, compared to wild type (WT) mice (Figure 2B). These data indicate that PDK1 activates translation control pathways in human, fibrinogen-stimulated, megakaryocytes.
PDK1 regulates proplatelet formation in megakaryocytes by controlling MARCKs protein synthesis
Circulating platelet counts in PDK1 null mice are about 25% reduced[7]. Recent study has shown that PDK1 plays a critical role in platelet production by regulating cytoskeletal dynamics[23]. Our lab and others have shown that, regulated protein synthesis is critical for proplatelet formation by mature megakaryocytes[2, 32]. Thus, we next wondered whether inhibiting PDK1 in megakaryocytes impacted proplatelet formation by regulating expression of certain proteins that are important for proplatelet formation. As shown in Figure 3A, pharmacologic inhibition of PDK1 with BX-795 modestly, yet significantly, reduced proplatelet formation by human megakaryocytes. BX-795 did not impact megakaryocyte viability compared to DMSO vehicle control (82.70 ± 1.94% vs. 85.20 ± 1.89% cell viability, respectively; p=0.37) (Supplementary figure 1).
Figure 3: PDK1 inhibition regulates proplatelet formation and MARCKs expression.
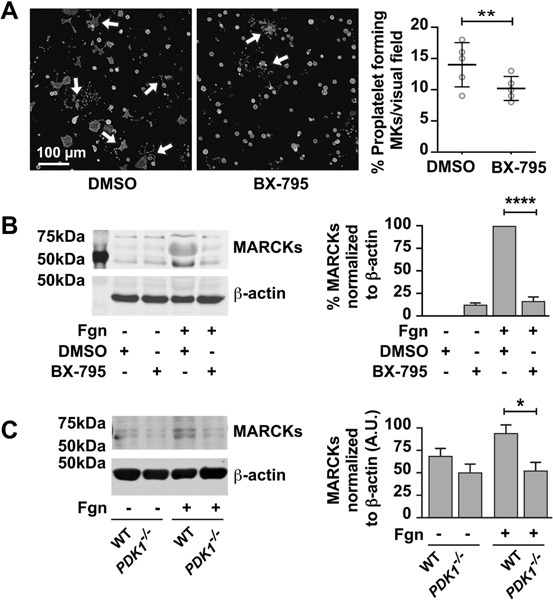
A. CD34+ megakaryocytes are treated with BX795 (1μM) from day 11 – 13. Cells are collected on Day 13 and adheared to fibrinogen coated plates and incubated over night at 370C. The cells are then treated with phalloidin dye and observed for proplatelet formation using confocal microscopy. The number of proplatelet forming cells were counted and graphs are plotted. B. CD34+ megakaryocytes are treated with BX795 (1μM) from day 11 – 13. Cells are collected on Day 13 and adheared to fibrinogen for 2 hours. Proteins were separated by using SDS-PAGE and probed from MARCKs protein. The immunoblot is representative of three independent experiments (****P < 0.05). C. Day 5 bone marrow derived mouse megakaryocytes from wild type or platelet specific PDK1 knockout mice were analyzed for expression of MARCKs proteins under adherent or non-adherent conditions on fibrinogen using SDS-PAGE. The western blots shown are representative of three independent experiments. (*P < 0.05).
MARCKs is translationally upregulated during proplatelet formation, and deletion of MARCKs results in decreased proplatelet formation[2]. Therefore, we evaluated the expression of MARCKs in mature megakaryocytes adherent to fibrinogen that are treated with PDK1 inhibitor or from PDK1 knockout mice. MARCKs protein expression in human and mouse megakaryocytes significantly increased upon adhesion to fibrinogen while inhibition or deletion of PDK1 blocked fibrinogen-triggered upregulation of MARCKs protein (Figure 3B and C). Thus, these findings demonstrate that PDK1 regulates MARCKs protein expression and proplatelet formation by human and mouse megakaryocytes.
Inhibition or genetic deletion of PDK1 abolishes activation of PI3K and MAPK mediated translation control pathways in platelets
Previous pharmacologic and genetic studies targeting PDK1 in platelets have shown that PDK1 plays an important role in platelet functional responses, including aggregation and clot retraction[7, 15, 21, 33]. Clot retraction is dependent upon the mTOR-driven synthesis of B-cell lymphoma 3 (Bcl-3) protein[5]. However, direct pathways linking PDK1 and Bcl-3 protein synthesis in platelets thus far have not been elucidated.
Our data in human megakaryocytes demonstrates that PDK1 controls eIF4E dissociation from 4EBP1 and its subsequent activation, binding of eIF4E to m7GTP caps, protein synthetic responses, and proplatelet formation (Figures 1–3). We therefore sought to determine whether PDK1 similarly regulates translation control pathways in human platelets.
We previously demonstrated that PDK1 signals through the ADP receptor in human platelets[21]. Pretreating isolated, human platelets with BX-795 abolished both PI3K (as measured by phosphorylation of Akt) and MAPK (as measured by phosphorylation of ERK1/2) signaling pathways induced by 2MeSADP (Figure 4A). Prior studies from our lab and others have shown that the PI3K and MAPK pathways trigger protein synthesis through mTORC1 mediated translation[34–38]. We therefore evaluated the effects of both mTORC1 and mTORC2 inhibition on eIF4E activation using Torin1, a specific mTORC1 and mTORC2 inhibitor. Torin1 inhibited phosphorylation of eIF4E in platelets stimulated with either thrombin or 2MesADP (Supplemental Figure 1). Thus, we next evaluated if inhibiting PDK1 in platelets blocked activation-induced phosphorylation of 4EBP1 and eIF4E, two proteins that are important downstream translation pathway targets of Akt (which signals to mTORC1) and ERK1/2 (which signals to MAPK). As shown in Figure 4A, PDK1 inhibition rapidly and completely prevented phosphorylation of 4EBP1 and eIF4E in activated platelets.
Figure 4: PDK1 regulates translation control pathways in human and murine platelets.
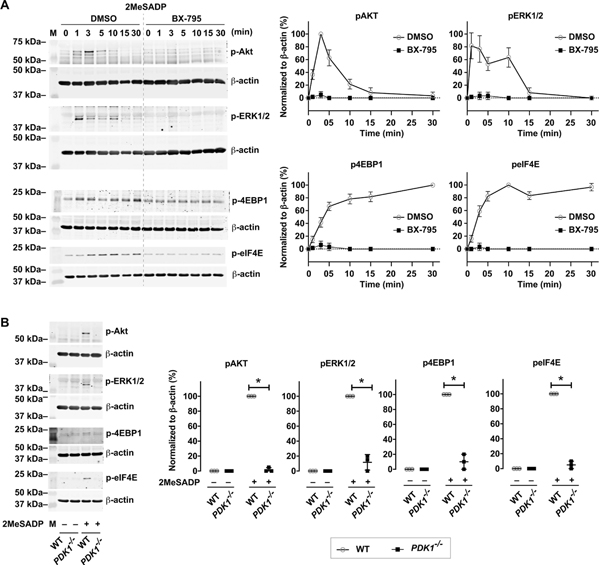
A. Washed human platelets were untreated or pretreated with the PDK1 inhibitor BX-795 (1μM) for 5 min, followed by stimulation with 2-methylthio-ADP (2MeSADP) (50 nM) under stirring for different time points. Western blots were then probed for phosphorylated Akt (T308), ERK1/2 (T202/Y204), 4EBP1(T37), and eIF4E (S209). The western blots shown are representative of three independent experiments. B. Washed wild type (WT) or PDK1 null murine platelets (PDK1−/−) were stimulated with 2-methylthio-ADP (2MeSADP) (50 nM) under stirring for 5 min. Western blots were then probed for phosphorylated Akt (T308), ERK1/2 (T202/Y204), 4EBP1(T37), and eIF4E (S209). The western blots shown are representative of three independent experiments. Graphs represent mean ± standard error of the mean from at least three different experiments (*P < 0.05).
To complement our pharmacologic studies, we also evaluated the role of PDK1 in protein synthetic events by using platelet-specific PDK1-deficient mice (KO). We have previously demonstrated that PDK1 is completely absent in these KO mice[21]. Consistent with pharmacologic inhibitor studies using BX-795 (Figure 4A), phosphorylation of Akt, ERK1/2, 4EBP1, and eIF4E was blocked in 2MeSADP-stimulated platelets from PDK1 KO mice, compared to wild type (WT) mice where PDK1 was present and active (Figure 4B). These data indicate that PDK1 is necessary for 2MeSADP-induced activation of the mTORC1 and MAPK pathways in human platelets. As we identified in megakaryocytes (Figure 1C–D), in activated human platelets inhibiting PDK1 near-completely blocked binding of eIF4E to m7GTP, without altering total eIF4E levels (Figure 5A–B).
Figure 5: PDK1 regulates eIF4E activation and binding to an mRNA cap homologue in activated platelets.
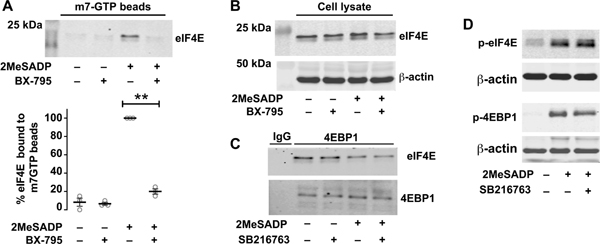
A, B Washed human platelets were untreated or pretreated with the PDK1 inhibitor BX-795 (1μM) for 5 min, followed by stimulation with 2-methylthio-ADP (2MeSADP) (50nM) under stirring condition for 15 min. The platelets were lysed and part of the lysate was separated and used as loading control before adding m7 GTP-Sepharose beads. The platelet lysate was incubated with m7 GTP-Sepharose beads. The beads were eluted to collect the eIF4E protein. Samples were analyzed for eIF4E binding using western blot analysis. This experiment is a representation of three experiments (**P < 0.05). C. Washed human platelets were left alone or pre-treated with the GSK3β inhibitor SB216763, followed by stimulation with 2MeSADP. Cell lysates were immunoprecipitated with 4EBP1 and samples were probed for total eIF4E using an anti-eIF4E mouse mAb. D. Washed human platelets were left alone or pre-treated with the GSK3β inhibitor SB216763 for 5 min, followed by stimulation with 2MeSADP (50 nM) for 5 min. Western blots were then probed for phosphorylated 4EBP1(T37) and eIF4E (S209). The western blots shown are representative of three independent experiments (* P< 0.05).
Previous studies have shown that PDK1 induced platelet activation is regulated by GSK3β.[7] However, to the best of our knowledge the role of GSK3β in translation control pathway has not been studied. We therefore used SB216763, a GSK3β specific inhibitor, to study its role in eIF4E and 4EBP1 signaling.[21] As shown in Figures 5C and D, GSK3β inhibition did not substantially alter either eIF4E and 4EBP1 phosphorylation or dissociation. These results suggest that GSK3β role in activation of platelet translation control pathways induced by 2MeSADP is minimal.
eIF4E and 4EBP1 can be activated by αIIbβ3-mediated outside-in signaling in platelets stimulated with fibrinogen. Consistent with this, inhibition of αIIbβ3-mediated outside-in signaling with GR-144053 (a well characterized αIIbβ3 antagonist) near-completely abolished eIF4E phosphorylation by fibrinogen (Supplemental Figure 2). In contrast, and as expected, GR-144053 did not significantly reduce 2MeSADP induced eIF4E phosphorylation.
PDK1 regulates Bcl3 protein synthesis and clot-retraction in activated platelets
Bcl-3, a member of the Ikβα family of factors, is a canonical example of a protein under signal-dependent translational control in human platelets[5]. In unstimulated platelets, Bcl-3 protein is expressed at lower levels constitutively. However, upon activation and engagement of integrin αIIbβ3, Bcl-3 is rapidly translated, yielding new protein. Translation of Bcl-3 in human platelets is under the control of the mTOR signaling pathway.
Given our findings that PDK1 regulates translational initiation and global protein synthesis through the mTOR pathway (Figures 1–5), we next sought to determine whether PDK1 controlled de novo synthesis of Bcl-3 protein in platelets. As shown in Figure 6A, activation of human platelets with 2MeSADP significantly increased the expression of Bcl-3 protein. Moreover, inhibiting PDK1 in platelets fully abolished Bcl-3 synthesis (Figure 6A). Similar results were obtained with platelets from PDK1-null mice (Figure 6B).
Figure 6: PDK1 regulates Bcl-3 protein synthesis and clot-retraction in activated platelets.
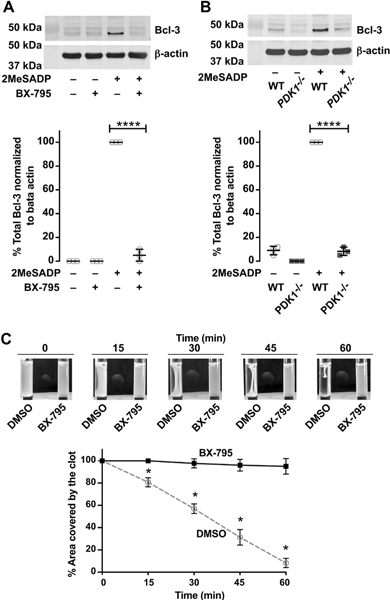
A. Washed human platelets were untreated or pretreated with the PDK1 inhibitor BX-795 (1μM) for 5 min, followed by stimulation with 2-methylthio-ADP (2MeSADP) (50nM) under stirring. Western blots were then probed for total Bcl-3 protein. Beta actin was used as loading control. The western blots shown are representative of three independent experiments. Graphs represent mean ± standard error of the mean from at least three different experiments (****P < 0.05). B. Washed wild type (WT) or PDK1 knockout (PDK1−/−) murine platelets were stimulated with 2-methylthio-ADP (2MeSADP) (50nM) under stirring for 5 min. Western blots were then probed total Bcl-3. The western blots shown are representative of three independent experiments. Graphs represent mean ± standard error of the mean from at least three different experiments (****P < 0.05). C. Washed human platelets were incubated with BX-795 for 5 min prior to initiating clot retraction as described in methods. Photographs were taken at the times indicated. Data are representative of three independent experiments.
Previous studies from our lab have shown that activated platelets synthesize B-cell lymphoma-3 (Bcl3) protein, which then functions critically to mediate clot retraction[4]. We therefore determined whether PDK1 inhibition functionally impacted clot-retraction in vitro. In these experiments, we used batroxobin (an enzyme derived from snake venom also known as retiplase) which has thrombin-like activity converting fibrinogen to fibrin, thereby inducing clot retraction. Batroxobin does not induce platelet activation and thereby allows us to specifically examine the effects of 2MeSADP activated platelets on clot retraction in the presence or absence of PDK1 inhibition.
We treated 2MeSADP-activated platelets with BX-795 and measured clot retraction induced by fibrinogen and batroxobin. Batroxobin by itself did not trigger clot retraction. As shown in Figure 6C, PDK1 inhibition abolish clot retraction similar to rapamycin-treated platelets. These results indicate that PDK1 induced Bcl3 protein synthesis in platelets stimulated with 2MeSADP is essential for clot retraction.
Discussion
Platelets are specialized anucleate cells produced primarily from bone marrow megakaryocytes[39, 40]. Protein synthesis plays a critical role in platelet production and function[2, 32, 40, 41]. However, the molecular mechanisms and the translational control pathways that regulate these processes remain incompletely understood. We and others have also shown that mTORC1 is functionally active in megakaryocytes and platelets. mTORC1 activation results in phosphorylation of 4EBP1 and the initiation of translation in platelets[4, 42]. The activation of mTORC1 is regulated, in part, by the Akt pathway[43] [15]. Akt, in turn, is controlled by PDK1[15]. Interestingly, prior studies have demonstrated that ablation of PDK1 in platelets or embryonic stem cells leads to defects in signal transduction pathways and cellular activation responses[7, 21], many of which regulate RNA translation[15, 20, 21, 44].
Here, we demonstrate for the first time that PDK1 regulates mRNA translation control pathways in megakaryocytes and platelets. We found that either pharmacologic inhibition or genetic ablation of PDK1 in megakaryocytes and platelets blocked the phosphorylation of 4EBP1, as well as its dissociation from eIF4E. This dissociation occurs when 4EBP1 is phosphorylated by functionally active Akt through the mTORC1 complex. Dissociation of eIF4E and 4EBP1 complex, which also requires activation of the upstream PI3K and MAPK kinase pathways, is essential for formation of the translation initiation complex on mRNAs [42, 45–47]. We recently reported that that PDK1 regulates both PI3K and MAPK pathways in platelets[21]. Consistent with this, we found that inhibition or ablation of PDK1 completely blocked phosphorylation of Akt and ERK1/2 and, in parallel, prevented binding of the m7 cap to eIF4E (necessary for translation initiation) and significantly reduced de novo protein synthesis in platelets. Signal dependent mRNA translation in platelets regulates platelet functional responses such as clot retraction[4]. In the current study, we found that PDK1 inhibition or genetic ablation was accompanied by inhibition of the synthesis of Bcl-3 and concordant defects in clot retraction.
Through the use of both pharmacologic inhibitors of PDK1 and murine models where PDK1 is genetically ablated, we and others have shown that PDK1 regulates platelet aggregation, secretion, and thromboxane generation[20, 21]. However, in addition to platelet functional defects, PDK1 knockout mice have decreased platelet counts[7], although the mechanistic link between PDK1 and platelet counts was previously unknown. Here, we show that PDK1 inhibition also reduced de novo protein synthesis in megakaryocytes and, in parallel, reduced proplatelet formation by megakaryocytes. Recent studies using proteomics-based approaches reported that the synthesis of MARCKs protein is upregulated in late-stage, murine megakaryocytes during proplatelet formation[2]. Consistent with these reports, we also found that inhibition of PDK1 reduced the expression of MARCKs protein in mature, human megakaryocytes adherent to fibrinogen (which triggers proplatelet production). Our findings suggest that the signal-dependent expression of MARCKs protein in is regulated, at least in part, by PDK1.
A recent study published while our manuscript was under review demonstrated that PDK1 mediates platelet production by regulating cytoskeletal dynamics. In mice, PDK1 deletion caused defects in actin polymerization with disrupted F-actin assembly, reduced podosome numbers, and impaired spreading[23]. Previous studies in other cells have shown that MARCKs plays an important role in cytoskeletal development and regeneration as well as actin crosslinking[48–50].
PDK1 is positioned at the crossroads of the PI3K/MAPK pathways and activated by growth factors. As such, PDK1 is being investigated as a potential target for cancer therapy, with clinical trials using PDK1 inhibitors actively recruiting patients[10, 51–55]. Thrombocytopenia is a known side effect for many cancer therapies[56], although it is not known yet whether drugs targeting PDK1 may have similar effects on platelet counts. Understanding the potential off-target effects of PDK1 inhibition on platelet production by megakaryocytes may provide useful insights relevant to clinical settings, as the results of these trials are reported.
Our studies provide build on and extend these published studies by providing new evidence that PDK1 is a regulator of translational control pathways in platelets and megakaryocytes. While our data support a role for PDK1 in signal-dependent translation control pathways, we recognize that blocking or ablating PDK1 did not completely abolish protein synthesis. This is not unexpected, as other translational control pathways, such as mTOR, are known to be present and active in platelets and megakaryocytes.
By using complementary systems in both human and murine platelets, our results also demonstrate the conservation of these pathways across species. Perhaps somewhat surprisingly, our findings that PDK1 in platelets and megakaryocytes controls 4EBP1 phosphorylation and mRNA translation appear to contrast with results in embryonic stem cells[44], where PDK1 deletion has minimum effects on 4EBP1 phosphorylation and mTOR activity. These differences suggest that the role of PDK1 in regulating translational events may vary between cell types. As platelets are one of only two anucleate cells (the other being red blood cells), it is also possible that platelets retain this regulatory pathway as one mechanism of regulating its portfolio of proteins in response to activating signals.
Conclusions
In conclusion, the present study unravels PDK1 as a crucial regulator of signal dependent mRNA translation both in megakaryocytes and platelets. We also provide new evidence that PDK1 serves a previously-unknown role for regulating platelet production by megakaryocytes.
Supplementary Material
Figure 7:
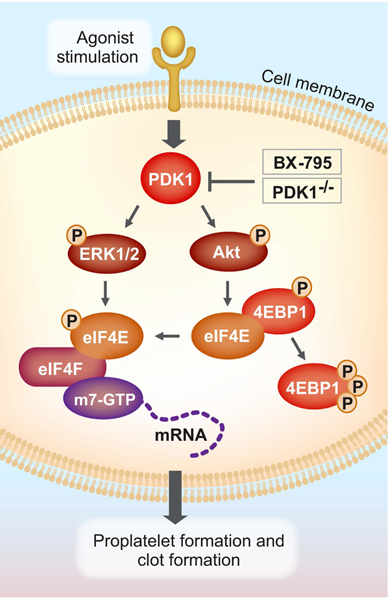
Model representation of PDK1 regulation of translation in megakaryocytes and platelets.
Essentials:
PDK1 regulates translation control pathways in megakaryocytes and platelets.
PDK1 regulates activation of translation control pathways in megakaryocytes.
PDK1 is crucial for 2MeSADP-induced platelet protein synthesis and clot retraction.
Acknowledgements
This work was supported by the NHLBI, NIA, VA merit (HL142804, AG059877, HL145237 to M. T. Rondina and A. S. Weyrich, and AG048022, I01CX001696 to M. T. Rondina). This work is also supported by AHA (18POST34030020 to B. K. Manne). This material is the result of work supported with resources and the use of facilities at the George E. Wahlen VA Medical Center, Salt Lake City, UT, USA. The contents do not represent the views of the US Department of Veterans Affairs or the United States Government.
Footnotes
Addendum
Bhanu Kanth Manne designed and performed experiments, analyzed and interpreted data, and wrote the manuscript. Seema Bhatlaker performed experiments, and analyzed data. Elizabeth Middleton performed experiments. Andrew S. Weyrich provided reagents and reviewed the manuscript. Oliver Borst provided reagents, performed experiments, and reviewed the manuscript. Matthew T. Rondina designed experiments, and analyzed and interpreted data.
Disclosure of Conflict of Interests
The authors state that they have no relevant conflict of interest.
Reference
- 1.Machlus KR, Italiano JE Jr, The incredible journey: From megakaryocyte development to platelet formation. J Cell Biol. 2013; 201: 785–96. 10.1083/jcb.201304054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Machlus KR, Wu SK, Stumpo DJ, Soussou TS, Paul DS, Campbell RA, Kalwa H, Michel T, Bergmeier W, Weyrich AS, Blackshear PJ, Hartwig JH, Italiano JE Jr, Synthesis and dephosphorylation of MARCKS in the late stages of megakaryocyte maturation drive proplatelet formation. Blood. 2016; 127: 1468–80. 10.1182/blood-2015-08-663146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Machlus KR, Thon JN, Italiano JE Jr, Interpreting the developmental dance of the megakaryocyte: a review of the cellular and molecular processes mediating platelet formation. Br J Haematol. 2014; 165: 227–36. 10.1111/bjh.12758. [DOI] [PubMed] [Google Scholar]
- 4.Weyrich AS, Dixon DA, Pabla R, Elstad MR, McIntyre TM, Prescott SM, Zimmerman GA. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci U S A. 1998; 95: 5556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weyrich AS, Denis MM, Schwertz H, Tolley ND, Foulks J, Spencer E, Kraiss LW, Albertine KH, McIntyre TM, Zimmerman GA. mTOR-dependent synthesis of Bcl-3 controls the retraction of fibrin clots by activated human platelets. Blood. 2007; 109: 1975–83. 10.1182/blood-2006-08-042192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindemann S, Tolley ND, Eyre JR, Kraiss LW, Mahoney TM, Weyrich AS. Integrins regulate the intracellular distribution of eukaryotic initiation factor 4E in platelets. A checkpoint for translational control. J Biol Chem. 2001; 276: 33947–51. 10.1074/jbc.M104281200. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Zhang Y, Wang Y, Li D, Zhang L, Wang K, Luo X, Yang Z, Wu Y, Liu J. PDK1 regulates platelet activation and arterial thrombosis. Blood. 2013; 121: 3718–26. 10.1182/blood-2012-10-461897. [DOI] [PubMed] [Google Scholar]
- 8.Wang W, Sun X, Hu T, Wang L, Dong S, Gu J, Chu Y, Wang X, Li Y, Ru Y, Cheng T, Yuan W. PDK1 regulates definitive HSCs via the FOXO pathway during murine fetal liver hematopoiesis. Stem Cell Res. 2018; 30: 192–200. 10.1016/j.scr.2018.05.020. [DOI] [PubMed] [Google Scholar]
- 9.Baracho GV, Cato MH, Zhu Z, Jaren OR, Hobeika E, Reth M, Rickert RC. PDK1 regulates B cell differentiation and homeostasis. Proc Natl Acad Sci U S A. 2014; 111: 9573–8. 10.1073/pnas.1314562111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo D, Xu X, Li J, Chen C, Chen W, Wang F, Xie Y, Li F. The PDK1/cJun pathway activated by TGFbeta induces EMT and promotes proliferation and invasion in human glioblastoma. Int J Oncol. 2018; 53: 2067–80. 10.3892/ijo.2018.4525. [DOI] [PubMed] [Google Scholar]
- 11.Hu T, Li C, Wang L, Zhang Y, Peng L, Cheng H, Chu Y, Wang W, Ema H, Gao Y, Ju Z, Yang Z, Wang X, Cheng T, Yuan W. PDK1 plays a vital role on hematopoietic stem cell function. Sci Rep. 2017; 7: 4943 10.1038/s41598-017-05213-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge Q, Wang H, Xu X, Xu L, Zhai L, Tao R. PDK1 promotes apoptosis of chondrocytes via modulating MAPK pathway in osteoarthritis. Tissue Cell. 2017; 49: 719–25. 10.1016/j.tice.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Nakao T, Geddis AE, Fox NE, Kaushansky K. PI3K/Akt/FOXO3a pathway contributes to thrombopoietin-induced proliferation of primary megakaryocytes in vitro and in vivo via modulation of p27(Kip1). Cell Cycle. 2008; 7: 257–66. 10.4161/cc.7.2.5148. [DOI] [PubMed] [Google Scholar]
- 14.Chanprasert S, Geddis AE, Barroga C, Fox NE, Kaushansky K. Thrombopoietin (TPO) induces c-myc expression through a PI3K- and MAPK-dependent pathway that is not mediated by Akt, PKCzeta or mTOR in TPO-dependent cell lines and primary megakaryocytes. Cell Signal. 2006; 18: 1212–8. 10.1016/j.cellsig.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 15.Dangelmaier C, Manne BK, Liverani E, Jin J, Bray P, Kunapuli SP. PDK1 selectively phosphorylates Thr(308) on Akt and contributes to human platelet functional responses. Thromb Haemost. 2014; 111: 508–17. 10.1160/TH13-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang C, Cigliano A, Jiang L, Li X, Fan B, Pilo MG, Liu Y, Gui B, Sini M, Smith JW, Dombrowski F, Calvisi DF, Evert M, Chen X. 4EBP1/eIF4E and p70S6K/RPS6 axes play critical and distinct roles in hepatocarcinogenesis driven by AKT and N-Ras proto-oncogenes in mice. Hepatology. 2015; 61: 200–13. 10.1002/hep.27396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruggero D, Sonenberg N. The Akt of translational control. Oncogene. 2005; 24: 7426–34. 10.1038/sj.onc.1209098. [DOI] [PubMed] [Google Scholar]
- 18.Nandagopal N, Roux PP. Regulation of global and specific mRNA translation by the mTOR signaling pathway. Translation (Austin). 2015; 3: e983402. 10.4161/21690731.2014.983402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Showkat M, Beigh MA, Andrabi KI. mTOR Signaling in Protein Translation Regulation: Implications in Cancer Genesis and Therapeutic Interventions. Mol Biol Int. 2014; 2014: 686984. 10.1155/2014/686984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel P, Golla K, Naik UP. PDK1 governs thromboxane generation and thrombosis in platelets by regulating activation of Raf1 in the MAPK pathway: comment. J Thromb Haemost. 2018; 16: 1901–4. 10.1111/jth.14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manne BK, Munzer P, Badolia R, Walker-Allgaier B, Campbell RA, Middleton E, Weyrich AS, Kunapuli SP, Borst O, Rondina MT. PDK1 governs thromboxane generation and thrombosis in platelets by regulating activation of Raf1 in the MAPK pathway. J Thromb Haemost. 2018; 16: 1211–25. 10.1111/jth.14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shveygert M, Kaiser C, Bradrick SS, Gromeier M. Regulation of eukaryotic initiation factor 4E (eIF4E) phosphorylation by mitogen-activated protein kinase occurs through modulation of Mnk1-eIF4G interaction. Mol Cell Biol. 2010; 30: 5160–7. 10.1128/MCB.00448-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geue S, Aurbach K, Manke MC, Manukjan G, Munzer P, Stegner D, Brahler C, Walker-Allgaier B, Marklin M, Borst CE, Quintanilla-Fend L, Rath D, Geisler T, Salih HR, Seizer P, Lang F, Nieswandt B, Gawaz M, Schulze H, Pleines I, Borst O. Pivotal role of PDK1 in megakaryocyte cytoskeletal dynamics and polarization during platelet biogenesis. Blood. 2019; 134: 1847–58. 10.1182/blood.2019000185. [DOI] [PubMed] [Google Scholar]
- 24.Larson MK, Watson SP. Regulation of proplatelet formation and platelet release by integrin alpha IIb beta3. Blood. 2006; 108: 1509–14. 10.1182/blood-2005-11-011957. [DOI] [PubMed] [Google Scholar]
- 25.Eto K, Nishikii H, Ogaeri T, Suetsugu S, Kamiya A, Kobayashi T, Yamazaki D, Oda A, Takenawa T, Nakauchi H. The WAVE2/Abi1 complex differentially regulates megakaryocyte development and spreading: implications for platelet biogenesis and spreading machinery. Blood. 2007; 110: 3637–47. 10.1182/blood-2007-04-085860. [DOI] [PubMed] [Google Scholar]
- 26.Balduini A, Badalucco S, Pugliano MT, Baev D, De Silvestri A, Cattaneo M, Rosti V, Barosi G. In vitro megakaryocyte differentiation and proplatelet formation in Ph-negative classical myeloproliferative neoplasms: distinct patterns in the different clinical phenotypes. PLoS One. 2011; 6: e21015. 10.1371/journal.pone.0021015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luff SA, Papoutsakis ET. Megakaryocytic Maturation in Response to Shear Flow Is Mediated by the Activator Protein 1 (AP-1) Transcription Factor via Mitogen-activated Protein Kinase (MAPK) Mechanotransduction. J Biol Chem. 2016; 291: 7831–43. 10.1074/jbc.M115.707174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guerriero R, Parolini I, Testa U, Samoggia P, Petrucci E, Sargiacomo M, Chelucci C, Gabbianelli M, Peschle C. Inhibition of TPO-induced MEK or mTOR activity induces opposite effects on the ploidy of human differentiating megakaryocytes. J Cell Sci. 2006; 119: 744–52. 10.1242/jcs.02784. [DOI] [PubMed] [Google Scholar]
- 29.Dan HC, Ebbs A, Pasparakis M, Van Dyke T, Basseres DS, Baldwin AS. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IkappaB kinase alpha (IKKalpha). J Biol Chem. 2014; 289: 25227–40. 10.1074/jbc.M114.554881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012; 485: 109–13. 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carriere A, Ray H, Blenis J, Roux PP. The RSK factors of activating the Ras/MAPK signaling cascade. Front Biosci. 2008; 13: 4258–75. [DOI] [PubMed] [Google Scholar]
- 32.Machlus KR, Johnson KE, Kulenthirarajan R, Forward JA, Tippy MD, Soussou TS, El-Husayni SH, Wu SK, Wang S, Watnick RS, Italiano JE Jr, Battinelli EM CCL5 derived from platelets increases megakaryocyte proplatelet formation. Blood. 2016; 127: 921–6. 10.1182/blood-2015-05-644583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munzer P, Walker-Allgaier B, Geue S, Geuss E, Hron G, Rath D, Eissler D, Winter S, Schaeffeler E, Meinert M, Schaller M, Greinacher A, Schwab M, Geisler T, Kleinschnitz C, Lang F, Gawaz M, Borst O. PDK1 Determines Collagen-Dependent Platelet Ca2+ Signaling and Is Critical to Development of Ischemic Stroke In Vivo. Arterioscler Thromb Vasc Biol. 2016; 36: 1507–16. 10.1161/ATVBAHA.115.307105. [DOI] [PubMed] [Google Scholar]
- 34.Kalous J, Tetkova A, Kubelka M, Susor A. Importance of ERK1/2 in Regulation of Protein Translation during Oocyte Meiosis. Int J Mol Sci. 2018; 19 10.3390/ijms19030698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geddis AE, Fox NE, Kaushansky K. Phosphatidylinositol 3-kinase is necessary but not sufficient for thrombopoietin-induced proliferation in engineered Mpl-bearing cell lines as well as in primary megakaryocytic progenitors. J Biol Chem. 2001; 276: 34473–9. 10.1074/jbc.M105178200. [DOI] [PubMed] [Google Scholar]
- 36.Miyazaki R, Ogata H, Kobayashi Y. Requirement of thrombopoietin-induced activation of ERK for megakaryocyte differentiation and of p38 for erythroid differentiation. Ann Hematol. 2001; 80: 284–91. [DOI] [PubMed] [Google Scholar]
- 37.Mazharian A, Watson SP, Severin S. Critical role for ERK1/2 in bone marrow and fetal liver-derived primary megakaryocyte differentiation, motility, and proplatelet formation. Exp Hematol. 2009; 37: 1238–49 e5. 10.1016/j.exphem.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pelzl L, Tolios A, Schmidt EM, Alesutan I, Walker B, Munzer P, Borst O, Gawaz M, Lang F. Translational regulation of the serum- and glucocorticoid-inducible kinase-1 (SGK1) in platelets. Biochem Biophys Res Commun. 2012; 425: 1–5. 10.1016/j.bbrc.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 39.Schulze H, Shivdasani RA. Mechanisms of thrombopoiesis. J Thromb Haemost. 2005; 3: 1717–24. 10.1111/j.1538-7836.2005.01426.x. [DOI] [PubMed] [Google Scholar]
- 40.Deutsch VR, Tomer A. Megakaryocyte development and platelet production. Br J Haematol. 2006; 134: 453–66. 10.1111/j.1365-2141.2006.06215.x. [DOI] [PubMed] [Google Scholar]
- 41.Italiano JE Jr., Shivdasani RA Megakaryocytes and beyond: the birth of platelets. J Thromb Haemost. 2003; 1: 1174–82. [DOI] [PubMed] [Google Scholar]
- 42.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 1999; 13: 1422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and −2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002; 99: 13571–6. 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tominaga Y, Tamguney T, Kolesnichenko M, Bilanges B, Stokoe D. Translational deregulation in PDK-1−/− embryonic stem cells. Mol Cell Biol. 2005; 25: 8465–75. 10.1128/MCB.25.19.8465-8475.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yanagiya A, Suyama E, Adachi H, Svitkin YV, Aza-Blanc P, Imataka H, Mikami S, Martineau Y, Ronai ZA, Sonenberg N. Translational homeostasis via the mRNA cap-binding protein, eIF4E. Mol Cell. 2012; 46: 847–58. 10.1016/j.molcel.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Josse L, Xie J, Proud CG, Smales CM. mTORC1 signalling and eIF4E/4E-BP1 translation initiation factor stoichiometry influence recombinant protein productivity from GS-CHOK1 cells. Biochem J. 2016; 473: 4651–64. 10.1042/BCJ20160845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sekiyama N, Arthanari H, Papadopoulos E, Rodriguez-Mias RA, Wagner G, Leger-Abraham M. Molecular mechanism of the dual activity of 4EGI-1: Dissociating eIF4G from eIF4E but stabilizing the binding of unphosphorylated 4E-BP1. Proc Natl Acad Sci U S A. 2015; 112: E4036–45. 10.1073/pnas.1512118112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Myat MM, Anderson S, Allen LA, Aderem A. MARCKS regulates membrane ruffling and cell spreading. Curr Biol. 1997; 7: 611–4. 10.1016/s0960-9822(06)00262-4. [DOI] [PubMed] [Google Scholar]
- 49.El Amri M, Fitzgerald U, Schlosser G. MARCKS and MARCKS-like proteins in development and regeneration. J Biomed Sci. 2018; 25: 43 10.1186/s12929-018-0445-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature. 1992; 356: 618–22. 10.1038/356618a0. [DOI] [PubMed] [Google Scholar]
- 51.Li D, Mullinax JE, Aiken T, Xin H, Wiegand G, Anderson A, Thorgeirsson S, Avital I, Rudloff U. Loss of PDPK1 abrogates resistance to gemcitabine in label-retaining pancreatic cancer cells. BMC Cancer. 2018; 18: 772 10.1186/s12885-018-4690-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu N, He C, Zhu B, Jiang J, Chen Y, Ma T. 3-Phosphoinositide Dependent Protein Kinase-1 (PDK-1) Promotes Migration and Invasion in Gastric Cancer Cells Through Activating the NF-kappaB Pathway. Oncol Res. 2017; 25: 1153–9. 10.3727/096504017X14845839228545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Blasio L, Gagliardi PA, Puliafito A, Primo L. Serine/Threonine Kinase 3-Phosphoinositide-Dependent Protein Kinase-1 (PDK1) as a Key Regulator of Cell Migration and Cancer Dissemination. Cancers (Basel). 2017; 9 10.3390/cancers9030025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wada M, Horinaka M, Yasuda S, Masuzawa M, Sakai T, Katoh N. PDK1 is a potential therapeutic target against angiosarcoma cells. J Dermatol Sci. 2015; 78: 44–50. 10.1016/j.jdermsci.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 55.Chinen Y, Kuroda J, Shimura Y, Nagoshi H, Kiyota M, Yamamoto-Sugitani M, Mizutani S, Sakamoto N, Ri M, Kawata E, Kobayashi T, Matsumoto Y, Horiike S, Iida S, Taniwaki M. Phosphoinositide protein kinase PDPK1 is a crucial cell signaling mediator in multiple myeloma. Cancer Res. 2014; 74: 7418–29. 10.1158/0008-5472.CAN-14-1420. [DOI] [PubMed] [Google Scholar]
- 56.Liebman HA. Thrombocytopenia in cancer patients. Thromb Res. 2014; 133 Suppl 2: S63–9. 10.1016/S0049-3848(14)50011-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.