

Abstract
There is an urgent need for new biomarkers that address the shortcomings of current screening methods which fail to detect a large proportion of cases with hepatocellular carcinoma (HCC) at early stage. To develop a robust, multiple‐biomarker panel based on multiple reaction monitoring–mass spectrometry with high performance in detecting early‐stage HCC within at‐risk populations. In the discovery set, 150 samples were analyzed to identify candidate biomarkers. The resulting list of candidates was tested in the training set (713 samples) to establish a multimarker panel, which was evaluated in the validation set (305 samples). We identified 385 serum HCC biomarker candidates in the discovery set and developed a multimarker panel consisting of 28 peptides that best differentiated HCC from controls. The area under the receiver operating characteristic curve of multimarker panel was significantly higher than alpha‐fetoprotein (AFP) in the training (0.976 vs. 0.804; P < 0.001) and validation (0.898 vs. 0.778; P < 0.001) sets. In the validation set, this multimarker panel, compared with AFP, showed significantly greater sensitivity (81.1% vs. 26.8%; P < 0.001) and lower specificity (84.8% vs. 98.8%; P < 0.001) in detecting HCC cases. Combining AFP with the multimarker panel did not significantly improve the area under the receiver operating characteristic curve compared with the panel alone in the training (0.981 vs. 0.976; P = 0.37) and validation set (0.906 vs. 0.898; P = 0.75). Conclusion: The multiple reaction monitoring–mass spectrometry multimarker panel consisting of 28 peptides discriminates HCC cases from at‐risk controls with high performance and may have potential for clinical application in HCC surveillance.
Abbreviations
- AFP
alpha‐fetoprotein
- ALT
alanine aminotransferase
- AUROC
area under the receiver operating characteristic curve
- BCLC
Barcelona Clinic Liver Cancer
- CHB
chronic hepatitis B
- CHC
chronic hepatitis C
- CI
confidence interval
- CT
computed tomography
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- LC
liver cirrhosis
- MBL
mannan‐binding lectin
- MRM‐MS
multiple reaction monitoring–mass spectrometry
- US
ultrasonography
Primary liver cancer is the second‐most common cause of cancer‐related mortality globally.( 1 ) Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and the fastest‐rising cause of cancer‐related deaths in the western hemisphere over the past 2 decades.( 2 , 3 ) Hepatitis B virus (HBV) and hepatitis C virus (HCV) account for the vast majority of HCC‐related mortalities.( 4 )
The prognosis for patients with HCC is extremely poor, with a 5‐year survival rate less than 20%. Patient outcomes depend largely on the tumor stage at detection, as curative treatments are only available for early‐stage patients.( 5 , 6 ) However, even for patients with early‐stage HCC, the opportunity for curative local ablation (such as radiofrequency ablation), the most cost‐effective treatment for HCC,( 7 ) is often limited to very‐early stage disease (a single lesion <2 cm),( 8 , 9 , 10 ) highlighting the importance of HCC surveillance at the very early stage.
Current clinical guidelines recommend surveillance with biannual ultrasonography (US) with or without serum alpha‐fetoprotein (AFP) for the early detection of HCC in at‐risk populations.( 11 , 12 ) Nevertheless, the sensitivity of US in detecting early‐stage HCC is only 47% in patients with cirrhosis, as reported by a meta‐analysis.( 13 ) The addition of AFP to US significantly increases the sensitivity in detecting early‐stage HCC from 45% to 63%, which remains suboptimal. Thus, there are urgent unmet clinical needs for new biomarkers that can provide high performance in HCC surveillance.
The molecular heterogeneity of HCC limits its detection by a single biomarker,( 14 ) necessitating combinations of biomarkers (panels) for HCC surveillance. This approach might be particularly valuable when considering recent advances in proteomics, which has enabled the discovery of numerous protein biomarker candidates for HCC. The most significant advantage of multiple reaction monitoring–mass spectrometry (MRM‐MS) is its multiplexing feature, allowing rapid and simultaneous quantification of hundreds of candidate proteins and peptides in a high‐throughput mode.( 15 , 16 , 17 )
In this multicenter study, we analyzed large‐scale serum biomarkers by using MRM‐MS in patients with early‐stage HCC and at‐risk controls to develop a robust multimarker panel that significantly improves the surveillance of HCC compared with AFP.
Materials and Methods
Study Design and Participants
This was a multicenter phase 2 biomarker case‐control study based on the Early Detection Research Network (EDRN) definition.( 18 ) A total of 1,168 patients were enrolled from three tertiary care centers in Korea (Asan Medical Center, Samsung Medical Center, and Seoul National University Hospital). Serum samples were collected from 474 patients with HCC and 694 at‐risk controls with chronic hepatitis B (CHB), chronic hepatitis C (CHC), or liver cirrhosis (LC). All patients gave informed consent before being enrolled. Serum samples were collected between 6:00 and 8:00 am after an overnight fast to limit the differences that were caused by variations in the patients’ diets. The samples were then stored immediately at −80°C and thawed on ice just before analysis. This study was approved by the institutional review board of Asan Medical Center (2015‐1156 and 2017‐1049), Samsung Medical Center (2017‐08‐164), and Seoul National University Hospital (H‐1710‐028‐891).
The detection of HCC was prompted by the presence of suspicious nodules on surveillance images (US, computed tomography [CT], or magnetic resonance imaging [MRI]) or elevation in AFP. HCC was confirmed, based on the results of the histological examination or typical imaging features (nodule > 1 cm with arterial hypervascularity and portal/delayed‐phase washout) by CT or MRI per clinical practice guidelines.( 11 , 12 ) HCC stages were defined using the Barcelona Clinic Liver Cancer (BCLC) system: very early stage (stage 0), single nodule <2 cm; early stage (stage A), single 2‐5 cm or 2‐3 lesions each <3 cm.( 11 , 12 ) Patients with Child‐Pugh class C liver function or any malignancy were excluded. All control patients were confirmed not to have HCC or did not develop HCC for at least 12 months of follow‐up after the collection of the samples, to avoid the possibility of having undiagnosed subclinical HCC. Cirrhosis was clinically or radiologically defined, based on the following criteria: coarse liver echotexture and nodular liver surface on US, clinical features of portal hypertension (e.g., ascites, splenomegaly, varices), or thrombocytopenia (<150 × 1,000/mm3). HBV infection was defined as the persistence of serum hepatitis B surface antigen or HBV DNA for more than 6 months. HCV infection was defined as the persistence of anti‐HCV and HCV RNA for more than 6 months.
A total of 1,168 serum samples were collected from three medical centers and randomly assigned to the training or validation set to avoid possible bias. For the discovery set (which was not randomized, because a homogeneous cohort of HBV‐associated samples was required), 150 serum samples (50 CHB, 50 HBV‐related LC, and 50 HBV‐related HCC) were provided by Asan Medical Center (Fig. 1). The serum samples in the discovery set were collected between June 2009 and April 2010. Sera for the training (HCC, n = 297; controls, n = 416) and validation sets (HCC, n = 127; controls, n = 178) were collected at three participating centers between November 2009 and December 2016. The characteristics of the cases and controls are presented in Table 1.
Fig. 1.
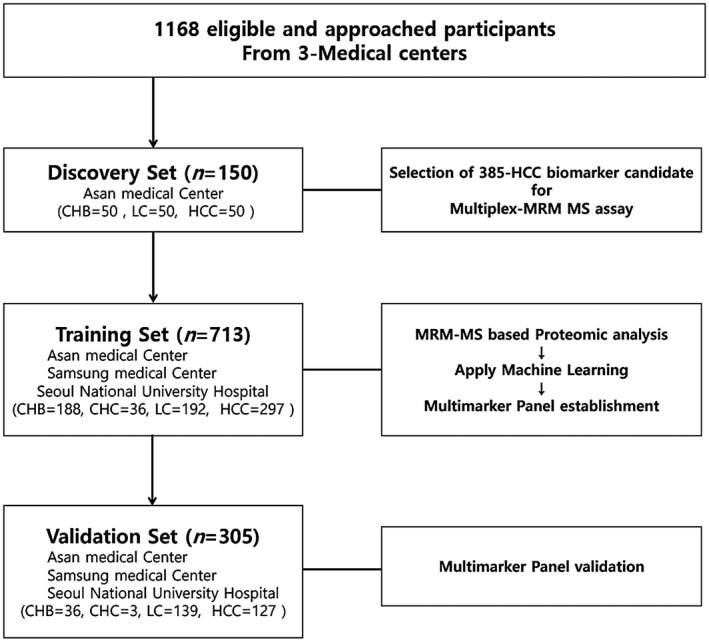
Overview of the workflow. Patients were enrolled in independent discovery, training, and validation sets. The 385 HCC biomarker candidates were identified by an MRM‐MS‐based proteomic method in the discovery set, consisting of serum samples from 50 patients with early HCC and 100 high‐risk controls (50 with CHB and 50 with LC). The multimarker panel that was established in the training set consisted of 713 serum samples, consisting of 297 patients with HCC and 416 high‐risk controls (187 with CHB, 36 with CHC, and 193 with LC) enrolled from three sites. The validation set included 127 patients with HCC and 178 high‐risk controls (36 with CHB, 3 with CHC, and 139 with LC) enrolled from the three aforementioned sites and was used to further evaluate the performance of the multimarker panel.
Table 1.
Demographics and Clinical Characteristics of the Study Population
| Discovery Set | Training Set | Validation Set | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CHB | LC | HCC | P Value | CHB | CHC | LC | HCC | P Value | CHB | CHC | LC | HCC | P Value | |
| (n = 50) | (n = 50) | (n = 50) | (n = 188) | (n = 36) | (n = 192) | (n = 297) | (n = 36) | (n = 3) | (n = 139) | (n = 127) | ||||
| Age (years) | 45.3 ± 8.0 | 54.2 ± 6.4 | 53.8 ± 8.8 | <0.001 | 48.7 ± 10.4 | 56.3 ± 10.5 | 57.2 ± 8.5 | 58.5 ± 9.4 | <0.001 | 45.8 ± 12.3 | 63.3 ± 4.5 | 56.4 ± 7.8 | 58.2 ± 7.9 | <0.001 |
| Sex, n (%) | 0.878 | <0.001 | <0.001 | |||||||||||
| Female | 15 (30.0%) | 15 (30.0%) | 13 (26.0%) | 49 (26.2%) | 25 (69.4%) | 82 (42.5%) | 64 (21.5%) | 8 (22.2%) | 3 (100.0%) | 62 (44.6%) | 20 (15.7%) | |||
| Male | 35 (70.0%) | 35 (70.0%) | 37 (74.0%) | 138 (73.8%) | 11 (30.6%) | 111 (57.5%) | 233 (78.5%) | 28 (77.8%) | 0 (0.0%) | 77 (55.4%) | 107 (84.3%) | |||
| Body mass index (kg/m2) | 23.9 ± 2.3 | 23.9 ± 2.5 | 25.2 ± 2.8 | 0.029 | 24.6 ± 3.2 | 24.0 ± 2.6 | 24.4 ± 3.2 | 24.5 ± 2.9 | 0.700 | 24.0 ± 1.5 | 24.3 ± 2.5 | 24.3 ± 3.3 | 25.5 ± 3.6 | 0.030 |
| AFP (ng/mL), median [IQR] | 2.8 [2.2‐4.3] | 4.8 [3.2‐9.1] | 29.2 [6.6‐306.8] | <0.001 | 2.7 [1.8‐3.8] | 3.5 [2.3‐5.3] | 2.6 [1.9‐4.0] | 10.1 [3.7‐62.3] | <0.001 | 2.2 [1.6‐3.6] | 4.0 [3.1‐5.0] | 2.8 [1.8‐4.3] | 6.7 [3.2‐24.6] | <0.001 |
| ALT (U/L) | 29.2 ± 12.9 | 34.3 ± 16.4 | 33.8 ± 13.7 | 0.187 | 40.9 ± 89.9 | 57.5 ± 48.9 | 34.8 ± 71.5 | 96.0 ± 124.6 | <0.001 | 34.0 ± 25.3 | 20.3 ± 7.4 | 29.2 ± 23.3 | 101.4 ± 118.2 | <0.001 |
| Albumin (g/dL) | 4.2 ± 0.3 | 3.8 ± 0.5 | 4.0 ± 0.4 | <0.001 | 4.5 ± 0.3 | 4.2 ± 0.3 | 4.1 ± 0.5 | 3.7 ± 0.5 | <0.001 | 4.6 ± 0.2 | 3.9 ± 0.1 | 4.0 ± 0.6 | 3.8 ± 0.4 | <0.001 |
| Bilirubin (mg/dL) | 1.1 ± 0.4 | 1.1 ± 0.3 | 1.0 ± 0.3 | 0.092 | 0.9 ± 0.4 | 0.7 ± 0.3 | 1.3 ± 0.8 | 1.1 ± 0.8 | <0.001 | 0.8 ± 0.4 | 0.8 ± 0.3 | 1.4 ± 0.9 | 1.1 ± 0.7 | 0.001 |
| Platelet (×103/μL) | 176.1 ± 49.3 | 108.1 ± 39.9 | 139.9 ± 50.1 | <0.001 | 206.0 ± 51.7 | 169.9 ± 56.0 | 97.1 ± 50.9 | 149.6 ± 63.0 | <0.001 | 177.1 ± 47.1 | 139.0 ± 78.6 | 97.8 ± 53.2 | 162.3 ± 65.8 | <0.001 |
| Prothrombin time (INR) | 1.0 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.1 | <0.001 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.1 ± 0.1 | 3.2 ± 4.6 | <0.001 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.1 ± 0.2 | 1.1 ± 0.2 | <0.001 |
| Etiology, n (%) | ||||||||||||||
| HBV | 50 (100%) | 50 (100%) | 50 (100%) | 1.000 | 182 (96.8%) | 0 (0.0%) | 125 (65.1%) | 220 (74.1%) | <0.001 | 35 (97.2%) | 0 (0.0%) | 103 (74.1%) | 90 (70.9%) | <0.001 |
| HCV | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 29 (80.6%) | 9 (4.7%) | 22 (7.4%) | 0 (0.0%) | 2 (66.7%) | 5 (3.6%) | 10 (7.9%) | |||
| Others | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 6 (3.2%) | 7 (19.4%) | 58 (30.2%) | 55 (18.5%) | 1 (2.8%) | 1 (33.3%) | 31 (22.3%) | 27 (21.3%) | |||
| Child‐Pugh score, n (%) | 0.029 | <0.001 | 0.004 | |||||||||||
| 5 | 47 (94.0%) | 38 (76.0%) | 47 (94.0%) | 178 (96.7%) | 35 (97.2%) | 148 (76.7%) | 190 (64.0%) | 36 (100.0%) | 3 (100.0%) | 97 (69.8%) | 83 (65.4%) | |||
| 6 | 3 (6.0%) | 10 (20.0%) | 2 (4.0%) | 5 (2.7%) | 1 (2.8%) | 27 (14.0%) | 79 (26.6%) | 19 (13.7%) | 33 (26.0%) | |||||
| 7 | 2 (4.0%) | 1 (2.0%) | 1 (0.5%) | 10 (5.2%) | 23 (7.7%) | 12 (8.6%) | 8 (6.3%) | |||||||
| 8 | 8 (4.1%) | 1 (0.3%) | 7 (5.0%) | 1 (0.8%) | ||||||||||
| 9 | 0 (0.0%) | 4 (1.3%) | 4 (2.9%) | 2 (1.6%) | ||||||||||
| Tumor size (cm) | 2.6 ± 1.1 | 3.1 ± 2.4 | 3.0 ± 2.3 | |||||||||||
| BCLC stage, n (%) | ||||||||||||||
| 0 | 14 (28.0%) | 89 (30.0%) | 35 (27.6%) | |||||||||||
| A | 36 (72.0%) | 189 (63.6%) | 83 (65.4%) | |||||||||||
| B | 0 (0.0%) | 19 (6.4%) | 9 (7.1%) | |||||||||||
| Tumor number, n (%) | ||||||||||||||
| 1 | 45 (90.0%) | 265 (89.2%) | 113 (89.0%) | |||||||||||
| 2 | 5 (10.0%) | 21 (7.1%) | 8 (6.3%) | |||||||||||
| ≥ 3 | 11 (3.7%) | 6 (4.7%) | ||||||||||||
Data are presented as mean ± SD (AFP: median [IQR]). P value for sex, etiology, and Child‐Pugh score were calculated by chi‐squared test. P values for others were calculated by analysis of variance. P values less than 0.001 are shown as “<0.001.”
Abbreviations: HBV, hepatitis B virus; HCV, hepatitis C virus; INR, international normalized ratio; IQR, interquartile ratio.
All clinical data, including the diagnosis of HCC, were blinded to the laboratory technicians and analysts to avoid bias in the measurements. AFP was measured by chemiluminescent microparticle immunoassay (ARCHITECT i2000SR; Abbott, Chicago, IL). This study was approved by the institutional review board of each participating center.
Quantitative MRM‐MS Analysis
The sample preparation methods are detailed in the Supporting Information. The six most abundant proteins (albumin, transferrin, immunoglobulin G, immunoglobulin A, haptoglobin, and α1‐antitrypsin) were depleted on a Multiple Affinity Removal System Human‐6 (MARS Hu‐6, 4.6 × 100 mm; Agilent Technologies, Santa Clara, CA) column that was loaded onto a high‐performance liquid chromatography (HPLC) (Shimadzu Co., Kyoto, Japan). Next, 44 μL of each sample was diluted in 176 μL of buffer A and passed through a 0.22‐μm filter by centrifugation (12,000 g, room temperature). Buffer A was used as a blank, and 200 μL of each sample was injected.
All MRM‐MS assays were performed on an Agilent 6490 triple quadrupole mass spectrometer coupled with a 1260 Infinity HPLC system (Agilent Technologies) with the prepared serum samples. All MRM‐MS raw files were processed in Skyline (McCoss Lab, University of Washington, Seattle, WA).
Data Analysis
Comparisons between two groups were analyzed using Student t test, whereas groups of three or more were analyzed by analysis of variance. Patients’ characteristics were represented in the form of its mean ± SD or percentile (%). A chi‐squared test was performed for categorical variables.
The levels of the 385 candidate markers that were identified in the discovery set were analyzed in the training set. Logistic regression was used to build the multimarker panel, based on the potential biomarkers. Area under the receiver operating characteristic curve (AUROC) analysis was performed to determine the discriminatory performance of each peptide marker combination panel, including its sensitivity and specificity. Youden index( 19 ) was set as the cutoff in the training set and subsequently used to classify patients in the validation set. The predicted probability of being detected as HCC by the multimarker panel was calculated according to the following expression: logit [P = HCC] (Supporting Table S4). In this equation, logit represents the predicted probability of HCC. In cases in which the level exceeded the cutoff of 0.448, the value was replaced by a discrete value and deemed as HCC. The multimarker panel’s ability to diagnose HCC was evaluated using a cutoff of 20 ng/mL AFP. The performance of the combination between multimarker panel and AFP was also assessed. The difference between sensitivity and specificity for multimarker panel and AFP was evaluated by Pearson’s chi‐squared test. All reported P values are two‐sided. P values less than 0.05 were considered significant. All statistical analyses were performed using R (version 3.5.1) and IBM SPSS (version 23.0; IBM, Chicago, IL).
Results
Characteristics of Cases and Controls
To define biomarker candidates, 150 serum samples (50 CHB, 50 HBV‐related LC, and 50 HBV‐related HCC) were analyzed in the discovery set. Next, 713 samples (297 HCC and 416 controls with CHB, CHC, or LC) were recruited to test these candidates and establish multimarker panels in the training set. Finally, an independent validation set, consisting of 305 samples (127 HCC and 178 controls), were used to test the performance of the multimarker panels (Fig. 1).
All subjects had Child‐Pugh class A or B liver function, and most (94.1%) of the HCC cases had very‐early stage or early‐stage disease (BCLC stage 0 or A; Table 1). The mean tumor size in the HCC cases was 2.97 cm (±2.2 cm) and was the smallest in the discovery set (2.6 ± 1.1 cm). All cases and controls in the discovery set had compensated liver function, with Child‐Pugh scores ≤ 7 and serum alanine aminotransferase (ALT) levels <80 U/L. Overall, the predominant cause of liver disease and HCC was chronic HBV infection (75.7% in HCC cases and 78.7% in non‐HCC controls).
Development of a MRM‐MS Multimarker Panel
The 385‐multiplex MRM assay was developed to quantify a wide array of HCC‐related blood markers from the discovery set (Supporting Information and Supporting Fig. S1), In total, 713 samples were measured by the 385‐plex MRM assay to generate the training set data, which were then used to develop an optimized multimarker panel for the detection of HCC. Full details of the estimation procedure are described in the Supporting Information.
In the training set, AUROC values were estimated to determine the sensitivity, specificity, and accuracy of various multimarker panels. Four multimarker panels that consisted of 16 to 28 peptides were chosen as models (Supporting Table S3). Among established multimarker panel models, the 28‐biomarker combination performed best (AUROC, 0.976; 95% confidence interval [CI], 0.967‐0.985; Table 2; Fig. 2A; Supporting Table S4). Subsequently, this 28‐biomarker panel was further validated.
Table 2.
Results for the Multimarker Panel and AFP in the Detection of Cases With HCC
| HCC vs. Controls | Very‐Early‐Stage HCC (Single <2 cm) vs. Controls | AFP‐Negative HCC vs. Controls | |||||||
|---|---|---|---|---|---|---|---|---|---|
| AUROC | Sensitivity | Specificity | AUROC | Sensitivity | Specificity | AUROC | Sensitivity | Specificity | |
| (95% CI) | (%) | (%) | (95% CI) | (%) | (%) | (95% CI) | (%) | (%) | |
| Training set | |||||||||
| Multimarker Panel | 0.976 | 91.9 | 92.1 | 0.974 | 92.7 | 92.1 | 0.970 | 80.8 | 96.5 |
| (0.967‐0.985) | (0.949‐0.979) | (0.958‐0.982) | |||||||
| AFP | 0.804 | 40.4 | 98.3 | 0.772 | 38.5 | 98.3 | 0.686 | 0.0 | 100 |
| (0.769‐0.839) | (0.714‐0.823) | (0.636‐0.736) | |||||||
| P value* | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| Multimarker Panel + AFP | 0.981 | 94.6 | 91.5 | 0.981 | 96.3 | 91.5 | 0.971 | 82.5 | 95.7 |
| (0.974‐0.989) | (0.970‐0.992) | (0.963‐0.985) | |||||||
| P value † | 0.374 | 0.253 | 0.882 | 0.432 | 0.385 | 0.853 | 0.691 | 0.783 | 0.691 |
| Validation set | |||||||||
| Multimarker panel | 0.898 | 81.1 | 84.8 | 0.833 | 71.1 | 84.8 | 0.874 | 67.8 | 89.3 |
| (0.863‐0.933) | (0.766‐0.901) | (0.829‐0.918) | |||||||
| AFP | 0.778 | 26.8 | 98.8 | 0.777 | 18.2 | 98.8 | 0.707 | 0.0 | 100 |
| (0.723‐0.833) | (0.697‐0.858) | (0.639‐0.774) | |||||||
| P value* | <0.001 | <0.001 | <0.001 | 0.295 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| Multimarker Panel + AFP | 0.906 | 82.9 | 83.2 | 0.844 | 70.5 | 83.2 | 0.892 | 72.2 | 88.7 |
| (0.723‐0.833) | (0.777‐0.912) | (0.851‐0.933) | |||||||
| P value † | 0.751 | 0.912 | 0.989 | 0.821 | 0.864 | 0.800 | 0.007 | 0.630 | 0.993 |
Note: “Controls” indicates at‐risk populations with HBV, HCV, and LC in the training and the validation sets. “Very‐early‐stage HCC” indicates single tumor size <2 cm HCC. “AFP‐negative HCC” indicates serum AFP <20 ng/mL.
Multimarker panel versus AFP.
Combination of multimarker panel and AFP versus multimarker panel alone. P values less than 0.001 are shown as “<0.001.”
Fig. 2.
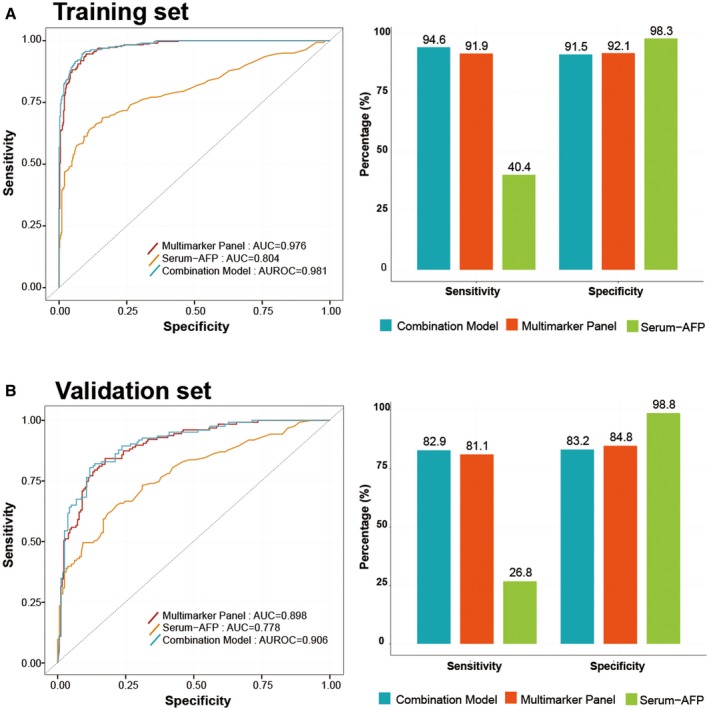
Performance of the multimarker panel and AFP in the diagnosis of HCC. AUROC curve and surveillance ability for the multimarker panel and AFP for all patients with HCC versus high‐risk controls in the training set (A) and validation set (B) (left, AUROC curve; right, bar plot of sensitivity and specificity). The AUROC for the multimarker panel was significantly higher than that of AFP (0.976 vs. 0.804 and 0.898 vs. 0.778 in training and validation sets, respectively). The sensitivities were significantly greater for the multimarker panel than AFP. However, the specificities of the multimarker panel were slightly lower compared with AFP. The AUROC, sensitivity, and specificity of the combination of the multimarker panel with AFP were similar to those of the s panel alone in the training and validation sets. Abbreviation: AUC, area under the curve.
Performance of the 28‐Biomarker Panel in Discriminating HCC and Controls
In the training set, the multimarker panel that was based on this 28‐biomarker combination, yielded a significantly higher AUROC value (0.976 vs. 0.804; P < 0.001) and greater sensitivity (91.9% vs. 40.4%; P < 0.001) compared with AFP (cutoff of 20 ng/mL (Table 2 and Fig. 2A). However, the specificity of this panel was lower than that of AFP (92.1% vs. 98.3%; P < 0.001). Combining AFP with the multimarker panel did not significantly improve the AUROC value compared with the panel alone (0.976 vs. 0.981, P = 0.374; Table 2 and Fig. 2A).
In the validation set, the multimarker panel showed a significantly higher AUROC value than AFP (0.898 [95% CI, 0.863‐0.933] vs. 0.778 [95% CI, 0.723‐0.833]; P < 0.001; Table 2 and Fig. 2B). The sensitivity of this multimarker panel was also significantly higher than that of AFP (81.1% vs. 26.8%; P < 0.001; Table 2 and Fig. 2B). However, specificity was lower in the multimarker panel than in AFP (84.8% vs. 98.8%; P < 0.001; Table 2 and Fig. 2B). Combining AFP with the multimarker panel did not significantly change the AUROC, sensitivity, and specificity (all P > 0.05; Table 2 and Fig. 2B).
Performance of the Multimarker Panel in Cases with Very‐Early‐Stage and AFP‐Negative HCC
For the detection of very‐early‐stage HCC (single lesion <2 cm), the multimarker panel had a significantly higher AUROC value than AFP in the training set (0.974 vs. 0.772; P < 0.001; Table 2 and Fig. 3A); however, the difference in AUROC was insignificant in the validation set (0.833 vs. 0.777; P = 0.295; Table 2 and Fig. 3B). In this subgroup of patients, the multimarker panel was significantly more sensitive than AFP in the training (92.7% vs. 38.5%; P < 0.001) and validation sets (71.1% vs. 18.2%; P < 0.001) but less specific than AFP in these sets (92.1% vs. 98.3% [P < 0.001] and 84.8% vs. 98.8% [P < 0.001], respectively) (Table 2). Combining AFP with the multimarker panel did not significantly improve the AUROC, sensitivity, and specificity in the training and validation sets (all P > 0.05; Table 2 and Fig. 3).
Fig. 3.
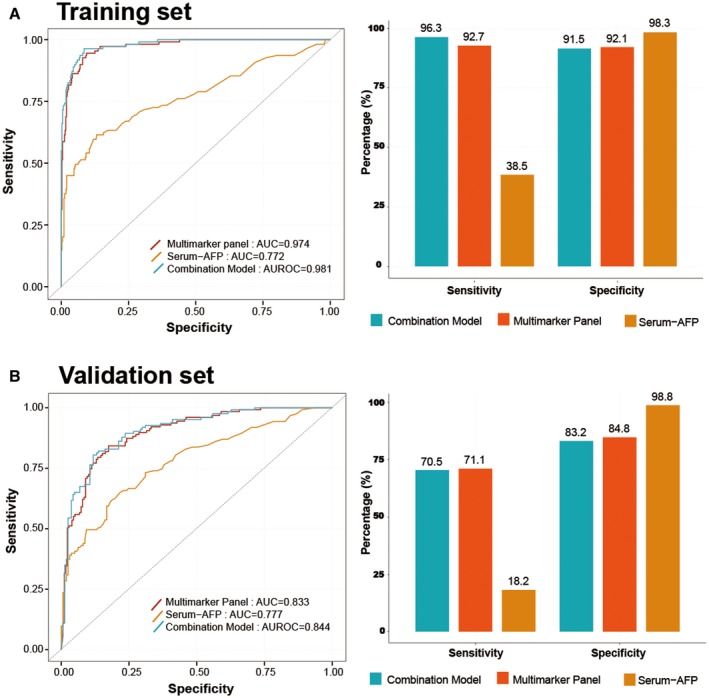
Surveillance ability of the multimarker panel in cases with very‐early‐stage HCC. AUROC curves for the multimarker panel and AFP for very‐early‐stage HCC (single size <2 cm) versus controls in the training set (A) and validation set (B) (left, AUROC curve; right, bar plot of sensitivity and specificity). The multimarker panel outperformed AFP with regard to surveillance of very‐early‐stage HCC (0.974 and 0.833 for multimarker panel vs. 0.772 and 0.777 in training and validation sets, respectively). The AUROC of the combination of multimarker panel and AFP was similar to the panel alone in the training and validation sets. The combined model (combination of multimarker panel and AFP) was more sensitive but less specific than the panel alone. Abbreviation: AUC, area under the curve.
For the detection of AFP‐negative HCC (AFP < 20 ng/mL), the multimarker panel showed a significantly higher AUROC than AFP in the training (0.970 vs. 0.686; P < 0.001; Table 2) and validation sets (0.874 vs. 0.707; P < 0.001; Table 2). The sensitivity of the multimarker panel was 80.8% in the training set and 67.8% in the validation set (Table 2).
Subgroup Analyses
Because most current practice guidelines recommend the surveillance of HCC in patients with CHB and/or LC, subgroup analyses were performed separately for patients with CHB and LC. In HBV‐related HCC cases and CHB controls without cirrhosis, the multimarker panel had a significantly higher AUROC compared with AFP in the training (0.985 vs. 0.810; P < 0.001) and validation sets (0.953 vs. 0.793; P = 0.002; Supporting Table S5 and Supporting Fig. S2). In patients with LC, the multimarker panel had a significantly higher AUROC value than AFP in the training (0.964 vs. 0.801; P < 0.001) and validation sets (0.883 vs. 0.767; P = 0.001; Supporting Table S5 and Supporting Fig. S3).
Diagnostic Performance of the Combination of Multimarker Panel with AFP According to Cutoff in HCC
The putative HCC prediction models consisted of the multimarker panel in conjunction with the two separate cutoffs for AFP levels. In our previously study, we demonstrated that the surveillance performance of AFP when using cutoffs of 5 ng/mL for AFP significantly improved the sensitivity for early detecting of HCC.( 20 ) We compared the ability of the multimarker panel to diagnose HCC with the performance of AFP at two cutoffs: 5 ng/mL (AFP 5) and 20 ng/mL (AFP 20). In this model, subjects were deemed to have HCC if the subject was deemed as HCC by either of the two cutoffs. The predictions of the models were then compared with the actual results.
In the training cohort, the multimarker panel had an AUROC of 0.924 (95% CI, 0.901‐0.942) in discriminating individuals with HCC from controls with a sensitivity of 91.9% and specificity of 92.1% (Table 3 and Fig. 4A). The AUROC for multimarker panel was significantly greater than that of AFP 5 (0.758; P < 0.001), AFP 20 (0.693; P < 0.001), multimarker panel in conjunction with AFP 5 (0.876; P < 0.001), and multimarker panel in conjunction with AFP 20 (0.903, P = 0.003). The 28‐multimarker panel in conjunction with AFP 5 had higher sensitivity compared with the stand‐alone panel (96.6% vs. 91.9%; P = 0.022), but had significantly lower specificity (78.6% vs. 92.1%; P < 0.001; Table 3). However, the specificity of the multimarker panel in conjunction with AFP 5 was significantly lower than that of the multimarker panel in conjunction with AFP 20 (90.1%; Table 3).
Table 3.
Comparison of Diagnostic Performances Across Combinations of the Multimarker Panel and Serum‐AFP Levels at Two Cutoffs
| HCC vs. Controls | Very‐Early‐Stage HCC (Single <2 cm) vs. Controls | AFP‐Negative HCC vs. Controls | |||||||
|---|---|---|---|---|---|---|---|---|---|
| AUROC | Sensitivity | Specificity | AUROC | Sensitivity | Specificity | AUROC | Sensitivity | Specificity | |
| (95% CI) | (%) | (%) | (95% CI) | (%) | (%) | (95% CI) | (%) | (%) | |
| Training set | |||||||||
| Multimarker panel | 0.924 | 91.9 | 92.1 | 0.927 | 92.7 | 92.1 | 0.886 | 80.8 | 96.5 |
| (0.901‐0.942) | (0.901‐0.948) | (0.857‐0.911) | |||||||
| AFP 5 (cutoff = 5 ng/mL) | 0.758 | 67.3 | 84.3 | 0.734 | 62.4 | 84.3 | 0.655 | 45.2 | 85.8 |
| (0.725‐0.790) | (0.693‐0.771) | (0.615‐0.694) | |||||||
| P value* | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| AFP 20 (cutoff = 20 ng/mL) | 0.693 | 40.4 | 98.3 | 0.684 | 38.5 | 98.3 | 0.500 | 0.0 | 100 |
| (0.658‐0.727) | (0.642‐0.724) | (0.458‐0.542) | |||||||
| P value* | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| Multimarker panel + AFP 5 | 0.876 | 96.6 | 78.6 | 0.884 | 98.2 | 78.6 | 0.868 | 90.4 | 83.3 |
| (0.850‐0.899) | (0.853‐0.910) | (0.838‐0.895) | |||||||
| P value † | <0.001 | 0.022 | <0.001 | 0.003 | 0.105 | <0.001 | 0.203 | 0.016 | <0.001 |
| Multimarker panel + AFP 20 | 0.903 | 89.9 | 90.1 | 0.920 | 93.6 | 90.4 | 0.886 | 80.8 | 96.5 |
| (0.879‐0.924) | (0.893‐0.942) | (0.857‐0.911) | |||||||
| P value ‡ | 0.003 | 0.481 | 0.373 | 0.465 | 0.999 | 0.460 | 1.000 | 0.893 | 0.847 |
| Validation set | |||||||||
| Multimarker panel | 0.836 | 81.1 | 84.8 | 0.769 | 71.1 | 84.8 | 0.785 | 67.8 | 89.3 |
| (0.788‐0.877) | (0.708‐0.822) | (0.729‐0.835) | |||||||
| AFP 5 (cutoff = 5 ng/mL) | 0.716 | 61.8 | 81.4 | 0.725 | 63.6 | 81.4 | 0.651 | 47.8 | 82.4 |
| (0.659‐0.767) | (0.658‐0.785) | (0.588‐0.710) | |||||||
| P value* | <0.001 | 0.001 | 0.490 | 0.434 | 0.597 | 0.490 | 0.001 | 0.010 | 0.109 |
| AFP 20 (cutoff = 20 ng/mL) | 0.628 | 26.8 | 98.8 | 0.585 | 18.7 | 98.8 | 0.500 | 0.0 | 100 |
| (0.569‐0.684) | (0.514‐0.653) | (0.436‐0.564) | |||||||
| P value* | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| Multimarker panel + AFP 5 | 0.803 | 89.8 | 70.8 | 0.780 | 86.7 | 70.8 | 0.793 | 84.4 | 74.2 |
| (0.754‐0.846) | (0.717‐0.834) | (0.738‐0.842) | |||||||
| P value † | 0.088 | 0.074 | 0.002 | 0.685 | 0.120 | 0.002 | 0.747 | 0.015 | <0.001 |
| Multimarker panel + AFP 20 | 0.829 | 80.3 | 84.3 | 0.763 | 71.1 | 84.3 | 0.785 | 67.8 | 89.3 |
| (0.780‐0.871) | (0.705‐0.820) | (0.729‐0.835) | |||||||
| P value ‡ | 0.552 | 0.998 | 0.987 | 0.317 | 0.816 | 1.000 | 1.000 | 0.873 | 0.856 |
“Controls” indicates at‐risk populations with CHB, CHC, and LC in the training set and the validation set. “AFP 5” indicates a cutoff value of 5 ng/mL for AFP; “AFP 20” indicates a cutoff value of 20 ng/mL for AFP. “Very‐early‐stage HCC” indicates single tumor size under 2 cm HCC. “AFP‐negative HCC” indicates patients with HCC with AFP level under 20 ng/mL.
Multimarker panel versus AFP.
Combination of multimarker panel and AFP 5 versus multimarker panel alone.
Combination of multimarker panel and AFP 20 versus multimarker panel alone. P values less than 0.001 are shown as “<0.001.”
Fig. 4.
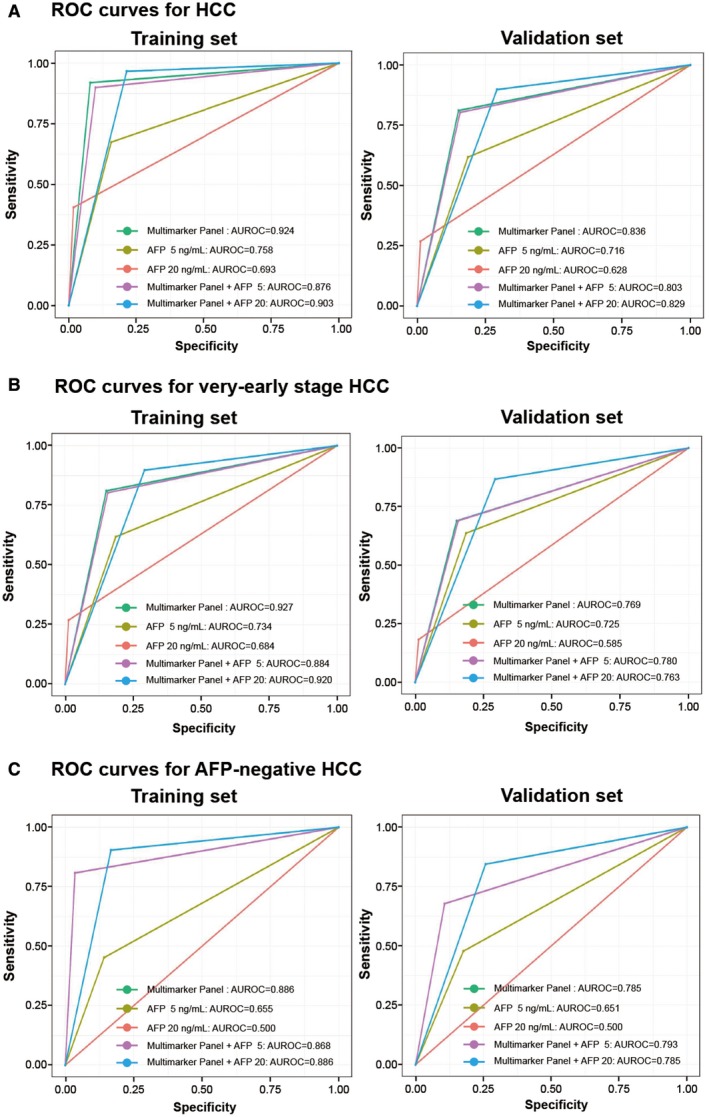
Comparison of the multimarker panel versus combination of multimarker panel and AFP with cutoff values between HCC and controls. AUROC curve and surveillance capability for the multimarker panel, AFP with two cutoff values, and conjunction with AFP 5 or 20 for all HCC versus controls (A), for very‐early‐stage HCC versus controls (B), and AFP‐negative HCC versus controls (C) (left, training set; right, validation set). The AUROC for multimarker panel was significantly higher than serum AFP, and was about the same as the conjunction with AFP 5 or 20 in all sets. Abbreviation: ROC, receiver operating characteristic.
In the validation set, at detection of HCC, the stand‐alone panel displayed the highest AUROC value of 0.836 (95% CI, 0.788‐0.877) in diagnosing HCC than combined with AFP (Table 3 and Fig. 4A), which was combined with AFP 5 and combined with AFP 20. The AUROC for the combination of multimarker panel in conjunction with AFP was not significantly different compared with that of the combination of multimarker panel in conjunction with AFP 5 (0.803; 95% CI, 0.754‐846; P = 0.088), and AFP 20 (0.829; 95% CI, 0.780‐0.871; P = 0.552). The sensitivity of the multimarker panel for HCC was likewise not significantly different from that of the panel in conjunction with AFP 5 (81.1 vs. 89.8%; P = 0.074) and in conjunction with AFP 20 (81.1 vs. 80.3%; P = 0.998). However, the specificity of the multimarker panel for HCC was significantly greater than that of the panel in conjunction with AFP 5 (84.8 vs. 70.8%; P = 0.002), but was not significantly different from that of the panel in conjunction with AFP 20 (84.8% vs. 84.3%; P = 0.987; Table 3 and Fig. 4A). Our results suggest that the multimarker panel is the optimal choice for HCC detection.
Diagnostic Performance of the Combination of Multimarker Panel and AFP Level in Case With Very‐Early‐Stage and AFP‐Negative HCC
We further evaluated the combination of multimarker panel and AFP for detection of very‐early‐stage HCC. Compared with the combination of multimarker panel and AFP, the multimarker panel showed higher AUROC for very‐early‐stage HCC, as shown by the AUCs of 0.927 (95% CI, 0.901‐0.948) in the training set and 0.769 (95% CI, 0.708‐0.822) in the validation set (Table 3 and Fig. 4B). When AFP was used in conjunction with the 28‐multimarker panel, there was no significant improvement in its performance for diagnosing very‐early‐stage HCC. The AUROC for the stand‐alone panel was significantly different from the combined one of multimarker panel with AFP 5 (0.884 [95% CI, 0.853‐0.910; P = 0.003] in the training set and AUROC = 0.780 [95% CI, 0.717‐0.834; P = 0.6850] in the validation set), but was not significantly different than that for combined with multimarker panel with AFP 20 (AUROC = 0.920 [95% CI, 0.897‐0.945; P = 0.465] in the training set and AUROC = 0.763 [95% CI, 0.705‐0.820; P = 0.317] in the validation set). However, the conjunction of multimarker panel with AFP 5 was more sensitive than the stand‐alone panel (98.2% vs. 92.7% [P = 0.104] in the training set, and 86.7% vs. 71.1% [P = 0.120] in the validation set). By contrast, specificity of the conjunction of multimarker panel with AFP 5 was significantly lower than that of the stand‐alone panel (78.6% vs. 92.1% [P < 0.001] in training set, 70.8% vs. 84.8% [P = 0.002] in validation set) and conjunction of multimarker panel with AFP 20 (90.4% vs. 92.1% in training set, 84.3% vs. 84.8% in validation set, respectively; Table 3 and Fig. 4B).
Likewise, for the patients with AFP‐negative HCC (AFP < 20 ng/mL), the multimarker panel was not significantly different in AUROC compared with multimarker panel in conjunction with AFP 5 in the training set (0.886 vs. 0.868; P = 0.203; Table 3 and Fig. 4C) and validation set (0.785 vs. 0.793; P = 0.747; Table 3 and Fig. 4C). However, the conjunction of multimarker panel with AFP 5 identified AFP‐negative HCC with a greater sensitivity than the stand‐alone panel (90.4% vs. 80.8% [P = 0.016] in the training set, and 84.4% vs. 67.8% [P = 0.015] in the validation set), but had lower specificity (83.3% vs. 96.5% [P < 0.001] in the training set and 74.2% vs. 89.3% [P < 0.001] in the validation set; Table 3).
Diagnostic Performance of the Combination of Multimarker Panel and Two AFP Levels in Subgroup
In HBV‐related HCC cases and CHB controls without cirrhosis, the multimarker panel was not significantly different in AUROC (0.920 [95% CI, 0.889‐0.945] in the training set and 0.868 [95% CI, 0.792‐0.923] in the validation set) than the other two combination models. The conjunction of multimarker panel with AFP 5 had higher sensitivity than the stand‐alone panel in the training set (93.2% vs. 86.3% [P = 0.026] in the training set and 86.7% vs. 75.6% [P = 0.087] in the validation set; Supporting Table S6 and Supporting Fig. S4A). The specificity of the conjunction of multimarker panel with AFP 5 was lower than the stand‐alone panel (86.8% vs. 97.3% [P < 0.001] in the training set and 82.9% vs. 94.3% in the validation set). The AUROC, sensitivity, and specificity of the conjunction of multimarker panel with AFP 20 showed similar results to the stand‐alone panel in both the training set and the validation set.
In patients with LC, the multimarker panel showed significantly higher AUROC compared with the multimarker panel in conjunction with AFP 5 in the training set (0.904 vs. 0.860; P = 0.004). Although the AUROC was higher, it was not significant in the validation set (0.820 vs. 0.787; P = 0.071; Supporting Table S6 and Supporting Fig. S4B). The conjunction of multimarker panel with AFP 5 was significantly more sensitive than the stand‐alone panel in both sets (training set: 94.3% vs. 87.2% [P = 0.004]; validation set: 89.8% vs. 81.1% [P = 0.074]), but was less specific (training set: 77.7% vs. 93.3% [P < 0.001]; validation set: 67.6% vs. 82.7% [P = 0.006]; Supporting Table S6). Similar to the HBV‐related HCC cases and CHB control comparison, the AUROC, sensitivity, and specificity of the conjunction of multimarker panel with AFP 20 showed similar diagnostic performance to the stand‐alone panel in both the training set and the validation set when comparing HCC and LC.
Discussion
In this study, 385 candidate biomarkers for HCC were analyzed simultaneously by MRM‐MS in a large cohort of samples from several centers. After comparing the surveillance of these candidates directly, we constructed a multimarker panel that combined 28 peptide markers, improving its performance compared with individual biomarkers. We then performed quantitative proteomics by MRM‐MS, which allowed us to determine broad and systematic changes in the proteome that was associated with liver disease–related proteins. The interassay variation, obtained by analyzing quality control samples over 6 days, was less than 15% for the 28 peptides and ranged from 6.2% to 12.4% (Supporting Information and Supporting Table S7). These results demonstrate that the 28‐peptide panel achieved acceptable reliability by MRM‐MS.
Targeted proteomics approaches that use MRM‐MS are cost‐effective and suitable for multiplex quantitation of hundreds of proteins, with high accuracy and a lower limit of quantitation.( 21 ) In addition, MRM‐MS assays generate consistent and reproducible data sets between laboratories, even in highly complex samples.( 22 ) MRM‐MS has the advantage of automation, as demonstrated in a recent study by Silvia et al.( 23 ) Furthermore, MRM‐MS can simultaneously detect attomole levels of specific peptides in complex biofluids,( 24 , 25 , 26 , 27 ) rendering it useful for validating molecules of interest.( 28 , 29 ) Thus, multimarker panel–based MRM‐MS is a viable method for detecting early‐stage HCC.
The performance of the multimarker panel in distinguishing HCC cases from at‐risk controls was high, with an AUROC value of 0.976, a sensitivity of 91.9%, and a specificity of 92.1% in the training set. The significantly greater AUROC and sensitivity of this panel versus AFP in discriminating HCC cases from controls were observed in the validation set (AUROC, 0.898; sensitivity, 81.1%) and in the subgroups with very‐early‐stage HCC and AFP‐negative HCC. Similar results were replicated for the multimarker panel in the evaluation of subgroup analyses for patients with CHB and LC. The addition of AFP to the multimarker panel did not significantly improve the performance of the multimarker panel alone. These data suggest that the multimarker panel is a valid tool for improving the surveillance for HCC.
Our cohort is unique in that it features patients from multiple centers and that nearly all of the HCC cases used in the discovery, training, and validation sets were in the very‐early or early‐stage disease. The early detection of HCC significantly improves the prognosis of patients and is a key factor in reducing the mortality from HCC.( 30 ) Thus, we aimed to meet at patent clinical need in the management of HCC: biomarker assays that can detect HCC at an early stage in patients who are at risk of developing the cancer. Larger sample sizes increased our ability to identify differentially expressed biomarkers between HCC cases and at‐risk controls. Through MRM‐MS analysis of the large‐scale multicenter samples, we noted distinct proteomic differences between HCC and at‐risk controls in a high‐throughput manner, allowing us to derive a clinically applicable signature that detects early‐stage HCC. Our methodical application of mass spectrometry has improved the likelihood of discovering biomarkers that are most appropriate for surveilling HCC.
Notably, our multimarker panel performed similarly in discriminating very‐early‐stage HCC from controls as with distinguishing all stages of HCC from controls (AUROC, 0.974 vs. 0.772 in the training set; 0.833 vs. 0.777 in the validation set). The sensitivity of this panel in detecting very‐early‐stage HCC varied (92.7% in the training set and 71.1% in the validation set) but far exceeded that of AFP (38.5% and 18.7%, respectively). Although US remains the standard test for HCC surveillance, its sensitivity is unacceptably low in detecting very‐early stage HCC.( 20 , 31 )
Our multimarker panel consisted of 28 proteins that were identified in this study to be associated with HCC (Supporting Table S4). Six AFP surrogate proteins (TTR, SERPINC1, SERPINNF2, APOC3, FABP1, and KNG1) associated independently with HCC, consistent with a previous report.( 32 ) In addition, SERPINA8, a member of the serpin family, suppresses tumor growth and metastasis.( 33 ) C1Q binds DDR1 directly, and activation of this signaling pathway might be involved in the progression of HCC. ( 34 ) CAT has an important function in the development and progression of HCC.( 35 ) CETP is a significant determinant of lipoprotein function, and regulates plasma high‐density lipoprotein (HDL) levels.( 36 , 37 ) HDL has a critical function in the reverse cholesterol transport pathway in the liver.( 36 , 37 ) Complement component 9 is involved in immunity and was recently reported to be down‐regulated in HCV‐infected patients with HCC and overexpressed in various cancers, including familial aggregation HCC.( 38 ) BChE and Factor IX are usually produced in the liver.( 39 , 40 ) Factor IX is a zymogen in the blood coagulation cascade.( 39 ) FCN3 is synthesized in the liver by hepatocytes, bile duct epithelial cells, and type II alveolar and ciliated bronchial epithelial cells. It is secreted into the blood, bile ducts, bronchi, and alveoli. Thus, it might participate in systemic and local innate immune responses.( 41 ) CFHR2 consists of four SCR domains and circulates in human plasma in nonglycosylated and glycosylated forms.( 42 ) FN1 is produced by hepatocytes, and elevated FN1 levels in the liver might accelerate hepatic fibrogenesis and malignant alterations in HCC.( 43 ) Serpina5 is important in many processes beyond hemostasis, including inflammation, innate immunity, fertilization, and carcinogenesis.( 44 , 45 ) ITIH1 is one of the heavy chains of a serine protease inhibitor and is involved in inflammation and carcinogenesis. Decreased ITIH1 levels correlate with the progression of hepatic fibrosis.( 46 ) The expression level of LDHA is critical for metastasis of human HCC, and LDHA is important in tumor proliferation and the growth of HCC.( 47 ) Mannan‐binding lectin (MBL) functions in the innate immune system by protecting against infections. Low serum levels of MBL are associated with an increased chronic HBV and HCV infection.( 48 ) A recent study reported that elevated MBL is associated with the development of HCC.( 49 ) THBS1 is linked to tumor invasiveness and progression in HCC and is a proangiogenic factor that stimulates angiogenesis in HCC.( 50 )
Regardless of the promising outcomes in this study, we have identified several limitations in our study. First, all of our study populations were of Korean ethnicity, and most had chronic HBV infection, which might limit the applicability of our results to the general populations of other ethnicity and etiologies. Despite our strict approach to the ALT levels for the development of 385 candidate biomarkers in the discovery set (ALT < 80 IU/L), some proteins of our multimarker panel might have been associated with liver cell–specific proteins, and the performance of the panel may have varied depending on the activity of HBV replication or level of transaminases. When the correlation between the performance of the multimarker panel and ALT levels was reinvestigated, a subtle yet statistically significant correlation was found in the given data set (r = 0.25 in the training set with P value < 0.0001 [n = 713] and r = 0.35 in the validation set with P value < 0.0001 [n = 305]). Third, whether the detection of preclinical disease by biomarkers can decrease the mortality from HCC remains unknown. However, this aim is beyond the scope of our study, warranting EDRN phase 4 and phase 5 biomarker studies.( 18 ) Last but not least, the lower specificity of our multimarker panel compared with AFP is another limitation. However, unlike in the diagnosis, sensitivity carries more weight than specificity in the surveillance of HCC, as a late diagnosis of HCC would limit the opportunity for survival improvement. Thus, although sensitivity offsets specificity, efforts to enhance the sensitivity should be prioritized in the surveillance of HCC.
In conclusion, using a multiplex MRM‐MS assay, we screened large‐scale serum proteins in a high‐throughput manner in a large cohort of HCC cases and at‐risk controls. The MRM‐MS‐based multimarker panel performed significantly better than AFP in discriminating early‐stage HCC cases from at‐risk controls. Thus, our results suggest that the MRM‐MS multimarker panel—alone or in combination with US and AFP—may have a clinical value in supplementing US, which suffers from low sensitivity, in the surveillance of HCC.
Supporting information
Supplementary Material
Financial Support: Technology Innovation Program of the Republic of Korea (10079271 and 2000134).
Potential conflict of interest: Dr. Young‐Suk Lim consults for, advises, is on the speakers’ bureau for, and received grants from Gilead.
Contributor Information
Young‐Suk Lim, Email: limys@amc.seoul.kr.
Youngsoo Kim, Email: biolab@snu.ac.kr.
References
- 1. Global Burden of Disease Cancer Collaboration , Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability‐adjusted life‐years for 32 cancer groups, 1990 to 2015. JAMA Oncol 2017;3:524‐54 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Choi J, Han S, Kim N, Lim Y‐S. Increasing burden of liver cancer despite extensive use of antiviral agents in a hepatitis B virus‐endemic population. Hepatology 2017;66:1454‐1463. [DOI] [PubMed] [Google Scholar]
- 3. Valery PC, Laversanne M, Clark PJ, Petrick JL, McGlynn KA, Bray F. Projections of primary liver cancer to 2030 in 30 countries worldwide. Hepatology 2018;67:600‐611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. GBD 2013 Mortality and Causes of Death Collaborators . Global, regional, and national age‐sex specific all‐cause and cause‐specific mortality for 240 causes of death, 1990‐2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015;385:117‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Serper M, Taddei TH, Mehta R, D'Addeo K, Dai F, Aytaman A, et al. Association of provider specialty and multidisciplinary care with hepatocellular carcinoma treatment and mortality. Gastroenterology 2017;152:1954‐1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim BH, Lim YS, Kim EY, Kong HJ, Won YJ, Han S, et al. Temporal improvement in survival of patients with hepatocellular carcinoma in a hepatitis B virus‐endemic population. J Gastroenterol Hepatol 2018;33:475‐483. [DOI] [PubMed] [Google Scholar]
- 7. Cadier B, Bulsei J, Nahon P, Seror O, Laurent A, Rosa I, et al. Early detection and curative treatment of hepatocellular carcinoma: a cost‐effectiveness analysis in France and in the United States. Hepatology 2017;65:1237‐1248. [DOI] [PubMed] [Google Scholar]
- 8. Livraghi T, Meloni F, Di Stasi M, Rolle E, Solbiati L, Tinelli C, et al. Sustained complete response and complications rates after radiofrequency ablation of very early hepatocellular carcinoma in cirrhosis: Is resection still the treatment of choice? Hepatology 2008;47:82‐89. [DOI] [PubMed] [Google Scholar]
- 9. Sherman M. Hepatocellular carcinoma: screening and staging. Clin Liver Dis 2011;15:323‐334, vii‐x. [DOI] [PubMed] [Google Scholar]
- 10. Nault JC, Sutter O, Nahon P, Ganne‐Carrie N, Seror O. Percutaneous treatment of hepatocellular carcinoma: state of the art and innovations. J Hepatol 2018;68:783‐797. [DOI] [PubMed] [Google Scholar]
- 11. European Association for the Study of the Liver . EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol 2018;69:182‐236. [DOI] [PubMed] [Google Scholar]
- 12. Heimbach JK, Kulik LM, Finn RS, Sirlin CB, Abecassis MM, Roberts LR, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018;67:358‐380. [DOI] [PubMed] [Google Scholar]
- 13. Tzartzeva K, Obi J, Rich NE, Parikh ND, Marrero JA, Yopp A, et al. Surveillance imaging and alpha fetoprotein for early detection of hepatocellular carcinoma in patients with cirrhosis: a meta‐analysis. Gastroenterology 2018;154:1706‐1718.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chaiteerakij R, Addissie BD, Roberts LR. Update on biomarkers of hepatocellular carcinoma. Clin Gastroenterol Hepatol 2015;13:237‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanash SM, Pitteri SJ, Faca VM. Mining the plasma proteome for cancer biomarkers. Nature 2008;452:571‐579. [DOI] [PubMed] [Google Scholar]
- 16. Whiteaker JR, Lin C, Kennedy J, Hou L, Trute M, Sokal I, et al. A targeted proteomics‐based pipeline for verification of biomarkers in plasma. Nat Biotechnol 2011;29:625‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gillette MA, Carr SA. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods 2013;10:28‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pepe MS, Etzioni R, Feng Z, Potter JD, Thompson ML, Thornquist M, et al. Phases of biomarker development for early detection of cancer. J Natl Cancer Inst 2001;93:1054‐1061. [DOI] [PubMed] [Google Scholar]
- 19. Youden WJ. Index for rating diagnostic tests. Cancer 1950;3:32‐35. [DOI] [PubMed] [Google Scholar]
- 20. Choi J, Kim GA, Han S, Lee W, Chun S, Lim YS. Longitudinal assessment of three serum biomarkers to detect very early‐stage hepatocellular carcinoma. Hepatology 2019;69:1983‐1994. [DOI] [PubMed] [Google Scholar]
- 21. Domanski D, Percy AJ, Yang J, Chambers AG, Hill JS, Freue GV, et al. MRM‐based multiplexed quantitation of 67 putative cardiovascular disease biomarkers in human plasma. Proteomics 2012;12:1222‐1243. [DOI] [PubMed] [Google Scholar]
- 22. Kennedy JJ, Abbatiello SE, Kim K, Yan P, Whiteaker JR, Lin C, et al. Demonstrating the feasibility of large‐scale development of standardized assays to quantify human proteins. Nat Methods 2014;11:149‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Surinova S, Huttenhain R, Chang CY, Espona L, Vitek O, Aebersold R. Automated selected reaction monitoring data analysis workflow for large‐scale targeted proteomic studies. Nat Protoc 2013;8:1602‐1619. [DOI] [PubMed] [Google Scholar]
- 24. Percy AJ, Chambers AG, Yang J, Jackson AM, Domanski D, Burkhart J, et al. Method and platform standardization in MRM‐based quantitative plasma proteomics. J Proteomics 2013;95:66‐76. [DOI] [PubMed] [Google Scholar]
- 25. Rai AJ, Gelfand CA, Haywood BC, Warunek DJ, Yi J, Schuchard MD, et al. HUPO plasma proteome project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics 2005;5:3262‐3277. [DOI] [PubMed] [Google Scholar]
- 26. Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, et al. Multi‐site assessment of the precision and reproducibility of multiple reaction monitoring‐based measurements of proteins in plasma. Nat Biotechnol 2009;27:633‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chambers AG, Percy AJ, Simon R, Borchers CH. MRM for the verification of cancer biomarker proteins: recent applications to human plasma and serum. Expert Rev Proteomics 2014;11:137‐148. [DOI] [PubMed] [Google Scholar]
- 28. Meng Z, Veenstra TD. Targeted mass spectrometry approaches for protein biomarker verification. J Proteomics 2011;74:2650‐2659. [DOI] [PubMed] [Google Scholar]
- 29. Percy AJ, Chambers AG, Yang J, Hardie DB, Borchers CH. Advances in multiplexed MRM‐based protein biomarker quantitation toward clinical utility. Biochim Biophys Acta 2014;1844:917‐926. [DOI] [PubMed] [Google Scholar]
- 30. Jacques F, Isabelle S, Rajesh D, Sultan E, Colin M, Marise R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 31. Kim SY, An J, Lim YS, Han S, Lee JY, Byun JH, et al. MRI with liver‐specific contrast for surveillance of patients with cirrhosis at high risk of hepatocellular carcinoma. JAMA Oncol 2017;3:456‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saito S, Ojima H, Ichikawa H, Hirohashi S, Kondo T. Molecular background of α‐fetoprotein in liver cancer cells as revealed by global RNA expression analysis. Cancer Sci 2008;99:2402‐2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vincent F, Bonnin P, Clemessy M, Contrerès J‐O, Lamandé N, Gasc J‐M, et al. Angiotensinogen delays angiogenesis and tumor growth of hepatocarcinoma in transgenic mice. Cancer Res 2009;69:2853‐2860. [DOI] [PubMed] [Google Scholar]
- 34. Lee J‐H, Poudel B, Ki H‐H, Nepali S, Lee Y‐M, Shin J‐S, et al. Complement C1q stimulates the progression of hepatocellular tumor through the activation of discoidin domain receptor 1. Sci Rep 2018;8:4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marra M, Sordelli IM, Lombardi A, Lamberti M, Tarantino L, Giudice A, et al. Molecular targets and oxidative stress biomarkers in hepatocellular carcinoma: an overview. J Transl Med 2011;9:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hirano R, Igarashi O, Kondo K, Itakura H, Matsumoto A. Regulation by long‐chain fatty acids of the expression of cholesteryl ester transfer protein in HepG2 cells. Lipids 2001;36:401‐406. [DOI] [PubMed] [Google Scholar]
- 37. Jiang JT, Xu N, Wu CP. Metabolism of high density lipoproteins in liver cancer. World J Gastroenterol 2007;13:3159‐3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Joshi V, Shah A, Brown I, Winterford C, Hill M. Complement component C9 as a new biomarker for esophageal adenocarcinoma. J Clin Oncol 2017;35:19. [Google Scholar]
- 39. Orlova NA, Kovnir SV, Vorobiev II, Gabibov AG. Coagulation Factor IX for hemophilia B therapy. Acta Naturae 2012;4:62‐73. [PMC free article] [PubMed] [Google Scholar]
- 40. Santarpia L, Grandone I, Contaldo F, Pasanisi F. Butyrylcholinesterase as a prognostic marker: a review of the literature. J Cachexia Sarcopenia Muscle 2013;4:31‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Swierzko A, Lukasiewicz J, Cedzynski M, Maciejewska A, Jachymek W, Niedziela T, et al. New functional ligands for ficolin‐3 among lipopolysaccharides of Hafnia alvei. Glycobiology 2012;22:267‐280. [DOI] [PubMed] [Google Scholar]
- 42. Eberhardt HU, Buhlmann D, Hortschansky P, Chen Q, Bohm S, Kemper MJ, et al. Human factor H‐related protein 2 (CFHR2) regulates complement activation. PLoS One 2013;8:e78617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim H, Park J, Kim Y, Sohn A, Yeo I, Jong YUS, et al. Serum fibronectin distinguishes the early stages of hepatocellular carcinoma. Sci Rep 2017;7:9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hamada T, Kamada H, Hayashi T, Nishioka J, Gabazza EC, Isaji S, et al. Protein C inhibitor regulates hepatocyte growth factor activator‐mediated liver regeneration in mice. Gut 2008;57:365‐373. [DOI] [PubMed] [Google Scholar]
- 45. Jing Y, Jia D, Wong CM, Oi‐Lin Ng I, Zhang Z, Liu L, et al. SERPINA5 inhibits tumor cell migration by modulating the fibronectin‐integrin beta1 signaling pathway in hepatocellular carcinoma. Mol Oncol 2014;8:366‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hamm A, Veeck J, Bektas N, Wild PJ, Hartmann A, Heindrichs U, et al. Frequent expression loss of Inter‐alpha‐trypsin inhibitor heavy chain (ITIH) genes in multiple human solid tumors: a systematic expression analysis. BMC Cancer 2008;8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li X, Lu P, Li B, Yang R, Chu Y, Zhang Z, et al. Sensitization of hepatocellular carcinoma cells to irradiation by miR34a through targeting lactate dehydrogenaseA. Mol Med Rep 2016;13:3661‐3667. [DOI] [PubMed] [Google Scholar]
- 48. Chong WP, To YF, Ip WK, Yuen MF, Poon TP, Wong WHS, et al. Mannose‐binding lectin in chronic hepatitis B virus infection. Hepatology 2005;42:1037‐1045. [DOI] [PubMed] [Google Scholar]
- 49. Jalal PJ, King BJ, Saeed A, Adedeji Y, Mason CP, Ball JK, et al. Elevated serum activity of MBL and ficolin‐2 as biomarkers for progression to hepatocellular carcinoma in chronic HCV infection. Virology 2019;530:99‐106. [DOI] [PubMed] [Google Scholar]
- 50. Poon RT, Chung KK, Cheung ST, Lau CP, Tong SW, Leung KL, et al. Clinical significance of thrombospondin 1 expression in hepatocellular carcinoma. Clin Cancer Res 2004;10:4150‐4157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material