

Abstract
Hyperactivation of sterol regulatory element binding protein 1c (SREBP‐1c), which transcriptionally induces expression of enzymes responsible for de novo lipogenesis and triglyceride (TG) formation, is implicated in nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH) pathogenesis. Posttranslational SREBP‐1c maturation and activation is stimulated by the protein per–arnt–sim kinase (PASK). PASK‐knockout mice are phenotypically normal on a conventional diet but exhibit decreased hypertriglyceridemia, insulin resistance, and hepatic steatosis on a high‐fat diet. We investigated the effects of pharmacologic PASK inhibition using BioE‐1115, a selective and potent oral PASK inhibitor, in Zucker fatty (fa)/fa) rats, a genetic model of obesity, dyslipidemia, and insulin resistance, and in a dietary murine model of NAFLD/NASH. Female Zucker (fa/fa) rats and lean littermate (fa/+) controls received BioE‐1115 (3‐100 mg/kg/day) and/or omega‐3 fatty acids, and blood glucose, hemoglobin A1c, glucose tolerance, insulin, and serum TG were measured. C57BL/6J mice fed a high‐fat/high‐fructose diet (HF‐HFrD) were treated with BioE‐1115 (100 mg/kg/day) or vehicle. Body weight and fasting glucose were measured regularly; serum TG, body and organ weights, and liver TG and histology were assessed at sacrifice. Messenger RNA (mRNA) abundance of SREBP‐1c target genes was measured in both models. In Zucker rats, BioE‐1115 treatment produced significant dose‐dependent reductions in blood glucose, insulin, and TG (all greater than omega‐3 fatty acids) and dose dependently restored insulin sensitivity assessed by glucose tolerance testing. In HF‐HFrD mice, BioE‐1115 reduced body weight, liver weight, fasting blood glucose, serum TGs, hepatic TG, hepatic fibrosis, hepatocyte vacuolization, and bile duct hyperplasia. BioE‐1115 reduced SREBP‐1c target mRNA transcripts in both models. Conclusion: PASK inhibition mitigates many adverse metabolic consequences associated with an HF‐HFrD and reduces hepatic fat content and fibrosis. This suggests that inhibition of PASK is an attractive therapeutic strategy for NAFLD/NASH treatment.
PAS kinase (PASK), a broadly evolutionarily conserved kinase, is nutrient‐responsive metabolic regulator that is required for the proteolytic maturation of sterol regulatory element‐binding protein (SREBP)‐1c, a transcriptional activator of genes that encode the enzymes that catalyze fatty acid and triglyceride production. We have demonstrated in two complementary animal models, a genetic model of obesity, dyslipidemia and insulin resistance (Zucker (fa/fa) rats) and a dietary murine model of NAFLD/NASH, that pharmacological inhibition of PASK mitigates many of adverse metabolic consequences associated with NASH, including hypertriglyceridemia and insulin resistance, and significantly reduces liver fat and fibrosis. Our findings collectively suggest that inhibition of PASK is an attractive therapeutic strategy for NAFLD/NASH treatment.

Abbreviations
- αSMA
alpha‐smooth muscle actin
- ACC
acetyl‐coenzyme A carboxylase
- CoA
coenzyme A
- DNL
de novo lipogenesis
- fa
fatty
- FAE
fatty acid elongase
- GPAT
glycerol‐3‐phosphate acyltransferase
- HFD
high‐fat diet
- HF‐HFrD
high‐fat/high‐fructose diet
- IC50
median inhibitory concentration
- MT
Masson’s trichrome
- mTORC1
mammalian target of rapamycin complex 1
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NCD
normal chow diet
- NMR
nuclear magnetic resonance
- ORO
Oil Red O
- PASK
per–arnt–sim kinase
- PASK−/−
per–arnt–sim kinase knockout
- SCD1
stearoyl‐coenzyme A desaturase1
- SREBP‐1c
sterol regulatory element‐binding protein 1c
- TG
triglyceride
The ability to synthesize and store lipids following feeding may have been critical to the survival of early human hunter–gatherers who had only intermittent access to food. Compared with our hominin ancestors of over a million years ago, however, many humans today are maladapted to an environment in which they have ready and continuous access to food. Nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH), which are emerging as the most important causes of liver disease worldwide,( 1 , 2 , 3 ) are pathologic manifestations of that maladaptation.
Per–arnt–sim kinase (PASK) is a broadly evolutionarily conserved kinase expressed in humans, with orthologs in flies and yeast.( 4 , 5 ) It contains a PAS putative sensory domain as well as a canonical serine/threonine kinase domain( 4 , 5 ) and acts as a nutrient‐responsive metabolic regulator in multiple cell types.( 6 , 7 , 8 , 9 , 10 , 11 , 12 ) PASK is posttranslationally activated in response to nutrient and hormonal signals by phosphorylation by the mammalian target of rapamycin complex 1 (mTORC1) protein kinase.( 13 ) Finally, PASK is also regulated by metabolic status at the level of gene expression.( 7 ) Studies using genetic and pharmacologic approaches have further demonstrated that PASK is required for the proteolytic maturation in mouse and rat liver of sterol regulatory element‐binding protein (SREBP)‐1c, a transcriptional activator of genes that encodes the enzymes that catalyze fatty acid and triglyceride (TG) production.( 14 )
PASK‐knockout (PASK−/−) mice exhibit decreased hepatic expression of the genes encoding enzymes responsible for the de novo synthesis of fatty acids, including acetyl‐coenzyme A (CoA) carboxylase (ACC), stearoyl‐CoA desaturase 1 (SCD1), fatty acid elongase (FAE), and the putative fatty acid transporter cluster of differentiation (CD)36, as well as the enzyme that catalyzes the final step in the synthesis of TG, i.e., esterification of glycerol‐3‐phosphate with long‐chain acyl‐CoA (glycerol‐3‐phosphate acyltransferase [GPAT]).( 10 ) PASK−/− mice are phenotypically normal on a normal chow diet (NCD) but are protected from the increased hepatic lipid accumulation, obesity, hypertriglyceridemia, and insulin resistance observed in wild‐type mice fed a high‐fat diet (HFD).( 10 , 11 )
Collectively, these and other findings suggest that PASK, acting at least partially through SREBP1c‐mediated transcriptional activation, functions as an important physiological regulator of hepatic TG synthesis.( 10 , 11 , 14 ) They further suggest that PASK represents a potentially attractive therapeutic target for NAFLD/NASH in that it is a physiological regulator of early metabolic steps leading to the cascade of events resulting in NAFLD/NASH.( 3 , 15 , 16 , 17 , 18 , 19 )
The studies described herein were designed to test this hypothesis in two different rodent disease models, specifically, Zucker fatty (fa)/fa rats, a genetic model of hyperphagia, obesity, dyslipidemia, and insulin resistance,( 20 ) and a mouse dietary mouse model of NAFLD/NASH.
Materials and Methods
Drug Preparation and Dosing
BioE‐1115, a potent and highly selective PASK inhibitor, was synthesized (Asymchem, Tianjin, China) as described.( 14 ) Prior studies assessing the specificity of BioE‐1115 using a panel of 50 other kinases selected to represent the breadth of the human kinase family demonstrated that the median inhibitory concentration (IC50) for inhibition of PASK of 4 nm is ~2,500 times less than for casein kinase, the second most potently inhibited kinase (IC50 = 10 μm).( 14 ) For the purposes of the present studies, the specificity of BioE‐1115 was further assessed against the CEREP panel (Eurofins CEREP, France), which includes a wide range of transporters, receptors, and enzymes. Specifically, none of the 44 transporters, receptors, or enzymes tested exhibited ≥35% inhibition at 10 μM BioE‐1115.
BioE‐1115 was formulated in 0.5% methylcellulose and 0.025% Tween‐80 (Sigma) in double‐distilled water. Fresh BioE‐1115 suspension was prepared every 6‐7 days, and dosage was calculated based on body weight. The appropriate weight of BioE‐1115 was placed in a 15‐mL glass homogenizer (Kimble Chase) to which vehicle was added. The compound was ground to a fine suspension, transferred to a screw top tube, and the homogenizer rinsed twice with vehicle and then brought to final volume. The drug was stored at 4°C and stirred for an hour before each daily administration.
Zucker (fa/fa) Rats
Female obese Zucker (fa/fa) rats and their lean littermates were obtained at 8‐10 weeks of age (Charles River Laboratories), fed a normal rodent chow diet (Lab Diet; PMI Nutrition International, Brentwood, MO), and housed under standard vivarium conditions (12‐hour light/dark cycle) with water and chow ad libitum. All studies were approved and conducted in accordance with the Institutional Animal Care and Use Committee guidelines of the University of Utah.
Effect of BioE‐1115 on Oral Glucose Tolerance Testing in Zucker Rats
Age‐matched, 10‐week‐old, female Zucker (fa/fa) rats and their lean littermates (fa/+) with an average weight of 432.6 ± 13.9 and 219.8 ± 3.7 g, respectively, were orally dosed once a day for 14 days with 30 or 100 mg/kg BioE‐1115 or vehicle (n = 6‐7 animals/group). Rats underwent retro‐orbital bleeding under isoflurane anesthesia to measure baseline plasma insulin and were fasted for 6 hours before the start of the oral glucose tolerance test. Blood was collected by tail vein puncture just before administration of 2 g/kg glucose by oral gavage and at 0, 15, 30, 45, 60, 90, 120, 180, and 240 minutes after glucose administration. Blood glucose was measured using a glucometer (Breeze 2; Bayer Healthcare, Tarrytown, NY). Animals were euthanized by CO2 asphyxiation, and blood for plasma insulin levels was taken by cardiac puncture. Plasma insulin was measured using a Beckman CX 5Pro (Beckman Coulter, Brea, CA).
Effect of BioE‐1115 Versus Omega‐3 Fatty Acids in Zucker Rats
Zucker (fa/fa) rats were dosed orally once daily with either vehicle, which was a commercially available formulation of omega‐3 fatty acids (Lovaza; 110 mg/kg or 352 mg/kg), or BioE‐1115 (3, 10, 30, or 100 mg/kg). Rats were euthanized, blood was collected by cardiac puncture and centrifuged at 1,200g for 10 minutes, and plasma was collected. Plasma TG, insulin, and glucose were assessed (n = 8‐9 animals/group) using a Beckman CX 5Pro (Beckman Coulter).
Mouse Model of NAFLD/NASH
Eight‐week‐old male C57Bl/6 mice (Jackson Laboratory, Bar Harbor, ME) with an average body weight of 22 g were housed in groups of five per cage in a temperature‐controlled (22°C ± 2°C) vivarium on a 12‐hour light/dark schedule at the University of Utah. Mice were randomly assigned to either an NCD (Harlan‐Teklad; Madison, WI) or a high‐fat/high‐fructose diet (HF‐HFrD).( 21 ) The HF‐HFrD was a combination of HF (D12331, Surwit Diet, 58 kcal% fat with sucrose; Research Diets, New Brunswick, NJ) and drinking water enriched with a high‐fructose corn syrup equivalent, i.e., 42 g/L of fructose (Acros Organics, Morris Plains, NJ) plus sucrose (Sigma‐Aldrich, St. Louis, MO) mixed in drinking water at a ratio of 55% fructose to 45% sucrose by weight. The animals were fed their designated diet ad libitum for 25 weeks, during which body weight was measured weekly and percentage of body fat was measured once at 23 weeks by nuclear magnetic resonance (NMR).
Daily dosing by gavage with 100 mg/kg of BioE‐1115 or vehicle began 1 week after initiation of HF‐HFrD feeding. On the final day at week 25, mice did not receive drug or placebo and were fasted for 12 hours, after which they were anaesthetized with isoflurane and euthanized. Cardiac puncture was performed, and organs, including liver (right and left lobes), heart, muscle, and lung were removed, weighed, and snap frozen in liquid N2 or preserved for histology. Blood collected by cardiac puncture was centrifuged at 1,200g for 10 minutes, and serum was analyzed for TG content. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Utah.
Body, Blood, and Tissue Analyses
Body composition, including lean tissue, fat mass, and fluid, was analyzed using the Bruker Minispec LF50 body composition analyzer and assessed as percentage body weight. Glucose was measured at 8, 16, and 23 weeks following initiation of the HF‐HFrD after a 6‐hour fast, using a Contour glucometer (Bayer, Mishawaka, IN). TG concentration in liver tissue (100 mg of snap‐frozen liver) and serum collected at week 25 was measured using a commercially available Triglyceride Colorimetric Assay Kit (Cayman Chemical, Ann Arbor, MI).
Real‐Time Quantitative Reverse‐Transcription Polymerase Chain Reaction Gene Expression
Total RNA was isolated from liver tissue using Trizol reagent. Samples were homogenized with a TissueLyzer II and purified with RNeasy Mini kit (both from Qiagen, Valencia, CA). First‐strand complementary DNA synthesis was carried out with SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA). Real‐time polymerase chain reaction was performed on a LightCycler480 (Roche Diagnostics, Indianapolis, IN) by using the SYBR Green‐based method, as described.( 22 ) Melting‐curve analysis and a mock reverse‐transcribed control were included to ensure the specificity of the amplicons. Analysis was performed by a calculated relative expression extrapolated from a standard curve for each primer pair that was then normalized to the expression of the housekeeping gene cyclophilin.
Tissue Preparation
Liver and heart samples from HF‐HFrD‐fed mice fixed in 10% neutral‐buffered formalin and/or optimal cutting temperature (OCT)‐embedded frozen liver samples were submitted for histological analyses by HistoTox Labs, Inc. (Boulder, CO) as were formalin‐fixed hepatic tissue with the right and left lobes separately identified. The heart was trimmed into two separate blocks with standardized orientation of the samples in the paraffin blocks. Three slides per paraffin block were sectioned at approximately 5 µm and stained with hematoxylin and eosin (H&E), Masson’s trichrome (MT) without counterstain, and/or by routine immunohistochemistry methods for alpha‐smooth muscle actin (αSMA). One slide per OCT‐embedded frozen block was sectioned and stained with Oil Red O (ORO).
Image Analysis
MT‐, αSMA‐, and ORO‐stained slides were scanned using an Aperio AT2 whole‐slide scanner (Leica Biosystems, Buffalo Grove, IL). Image analysis for each stain was performed on the digital slide images using Visiopharm software (Visiopharm, Hoersholm, Denmark). Whole‐slide images were annotated to delineate regions of interest. Exclusions were applied to remove vascular lumens and tissue artifacts, such as folds and tears, and an algorithm for an area‐based threshold was applied. Positive area for each stain was reported as a percentage of total tissue area. The output images underwent a quality control check after analysis was complete. These data were exported to Microsoft Excel following completion of image analysis.
Histology
H&E‐ and MT‐stained glass slides were independently evaluated using light microscopy by a board‐certified veterinary pathologist. Histologic lesions in heart and liver tissues and increased collagen (fibrosis) in livers were graded for severity (0, absent; 0.5, very minimal; 1, minimal; 2, mild; 3, moderate; 4, marked; 5, severe).
Statistical Analyses
Data are presented as mean ± SEM. Semiquantitative morphologic pathology scores were analyzed by a nonparametric t test (Mann‐Whitney U test). We used t tests for analysis of lipids, collagen, and αSMA immunolabeling. Significance was set at P ≤ 0.05 for all analyses.
Results
Effects of BioE‐1115 on Glucose Tolerance in Zucker Rats
We first evaluated the effect of PASK inhibition by BioE‐1115 in the Zucker rat genetic obesity model. Compared with the other groups, vehicle‐treated Zucker fa/fa rats demonstrated a slightly higher baseline plasma glucose level and, after oral glucose administration, a markedly higher peak plasma glucose level with a slower return toward baseline (Fig. 1A). By contrast, Zucker fa/fa rats treated with 100 mg/kg/day of BioE‐1115 were indistinguishable from Zucker lean +/fa rats, while Zucker fa/fa rats treated with 30 mg/kg/day BioE‐1115 exhibited an intermediate response (Fig. 1A).
Fig. 1.
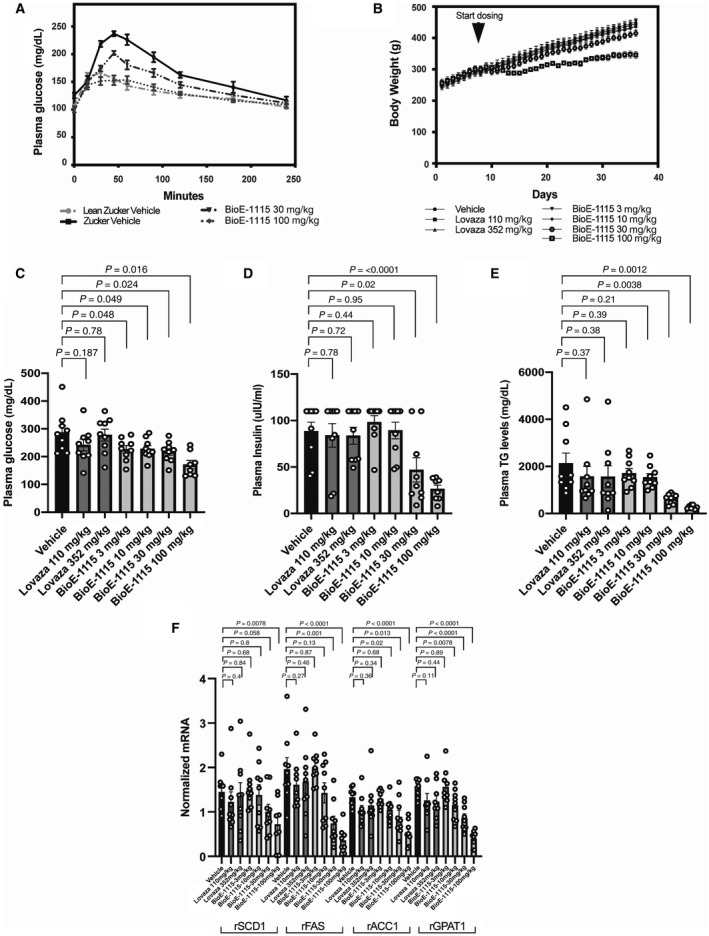
Effect of BioE‐1115 on oral glucose tolerance test, body weight, plasma glucose insulin, and TGs and mRNA transcripts encoding for lipogenic enzymes in Zucker rats. (A) Age‐matched, 10‐week‐old, female Zucker (fa/fa) rats and their lean littermates (fa/+) were orally dosed once a day for 14 days with 30 or 100 mg/kg BioE‐1115 or vehicle (n = 6‐7 animals/group). Plasma glucose was measured following administration of 2 g/kg glucose by oral gavage. In separate longer term studies, Zucker (fa/fa) rats were dosed orally once daily with vehicle, a commercially available formulation of omega‐3 fatty acids (Lovaza; 110 mg/kg or 352 mg/kg) or BioE‐1115 (3, 10, 30, or 100 mg/kg). (B) Weight was measured throughout. Rats were euthanized and (C) plasma glucose, (D) insulin, and (E) TGs were assessed as were (F) liver mRNA transcripts encoding for SCD1, FAE, ACC, and GPAT. All data represent mean ± SEM. Abbreviation: r, rat.
Comparison of BioE‐1115 and Omega‐3 Fatty Acids in Zucker Rats
Weight gain was significantly less in the 100‐mg/kg BioE‐1115 treatment group than the other groups (Fig. 1B), and both the 30‐ and 100‐mg/kg/day BioE‐1115 treatment groups exhibited a temporary decrease in food intake following initiation of drug treatment.
BioE‐1115 produced dose‐dependent decreases in serum glucose, insulin, and TG. The decreases were statistically significant for each of these parameters at the 30 and 100 mg/kg dose but not consistently significant at the 3 or 10 mg/kg/day doses (Fig. 1C‐E). At 100 mg/kg/day, BioE‐1115 decreased TG levels almost to the normal lean level (~87% decrease). By comparison, omega‐3 fatty acids did not significantly affect plasma glucose, insulin, or TG (Fig. 1C‐E).
BioE‐1115 produced dose‐dependent decreases in liver messenger RNA (mRNA) transcripts encoding for SCD1, FAE, ACC, and GPAT, whereas omega‐3 fatty acids had no significant effect on abundance of these transcripts (Fig. 1F).
BioE‐1115 Treated HF‐HFrD Mice Have Lower Body and Liver Weight
We also evaluated the effect of PASK inhibition in a dietary mouse model of NAFLD/NASH.( 21 ) The baseline body weights of vehicle and BioE‐1115‐treated mice were similar (26.9 ± 1.4 g vs. 26.6 ± 1.6 g). By 8 weeks, however, vehicle‐treated mice were significantly heavier (37.9 ± 1.1 g vs. 34.9 ± 1.2 g; p < 0.05). The weights of the vehicle versus BioE‐1115‐treated groups remained statistically significantly different and progressively diverged until death (Fig. 2A), although the BioE‐1115‐treated group remained heavier than chow‐fed controls (Fig. 2B). Food intake was not monitored in this model.
Fig. 2.
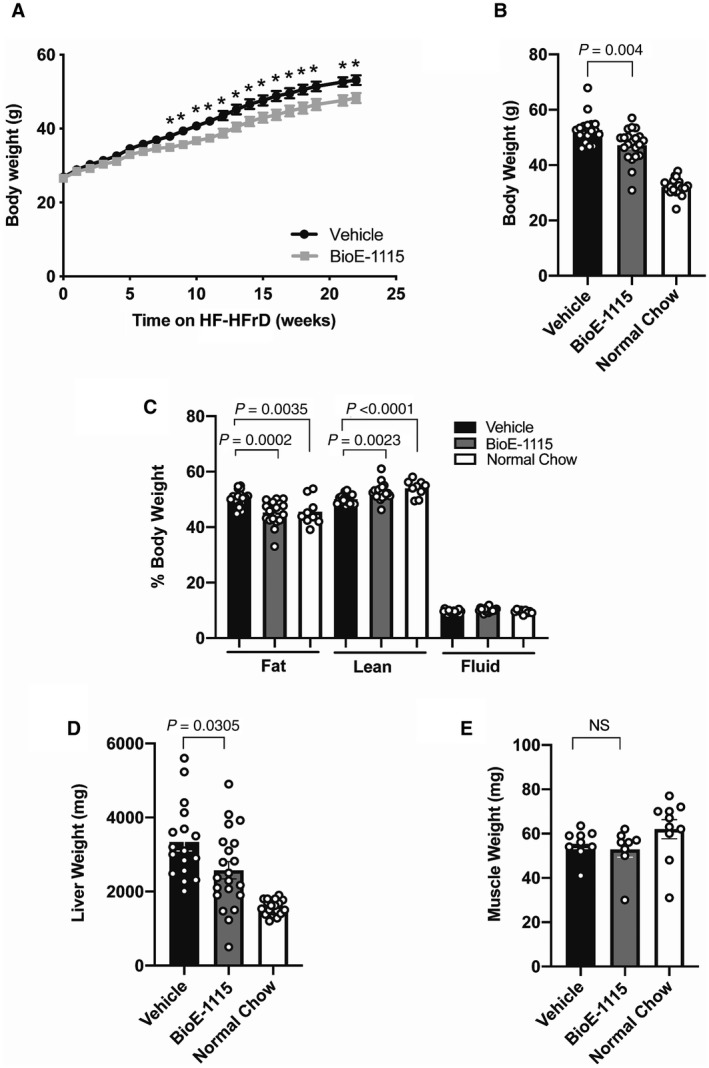
Effect of BioE‐1115 on body weight, body composition, and organ weight in mice fed an HF‐HFrD. (A,B) Body weight of C57Bl/6 mice fed an HF‐HFrD and treated for 25 weeks with vehicle versus BioE‐1115 (A) over time and (B) at the day of sacrifice; the weights of mice fed an NCD were also measured at sacrifice. (C) Body fat, lean, and fluid mass measured by NMR in 23‐week‐dosed C57/Bl6 mice on an HF‐HFrD. (D) Liver and (E) muscle weights at sacrifice. All results represent mean ± SEM for 20‐25 male mice per group. P values are shown. *P < 0.05. Abbreviation: NS, not significant.
NMR analysis of body composition indicated that HF‐HFrD‐fed mice had increased body fat (P = 0.0035) and decreased muscle mass (P = 0.0001) compared with chow‐fed mice. NMR results further indicated that the difference in body weight between HF‐HFrD‐fed mice given vehicle versus BioE‐1115 was completely explained by decreased fat mass (P = 0.0002), whereas lean mass was actually increased (P = 0.0023) in the BioE‐1115‐treated group; there was no difference in body water (Fig. 2C). Among mice fed the HF‐HFrD, liver weight was significantly lower in the BioE‐1115‐treated group compared with vehicle‐treated controls (Fig. 2D). There was no difference in muscle weight (Fig. 2E), heart weight, or lung weight (not shown) among HF‐HFrD mice treated with vehicle or BioE‐1115.
BioE‐1115 Lowers Fasting Glucose, Serum and Liver TG, and Lipogenic Gene Expression
Fasting plasma glucose was significantly (P < 0.001) lower at weeks 8, 16, and 23 among HF‐HFrD‐fed mice treated with BioE‐1115 compared to the vehicle‐treated group (Fig. 3A). Similarly, among mice fed the HF‐HFrD, both serum TG concentration and hepatic TG content were significantly (P ≤ 0.0001) lower after treatment with BioE‐1115 compared to vehicle, and hepatic TGs were very similar in HF‐HFrD‐fed mice treated with BioE‐1115 and NCD‐fed mice. (Fig. 3B,C). The mRNAs encoding for lipogenic enzymes, including GPAT, FAS, ACC, and SCD1, were all increased by the HF‐HFrD compared with the NCD. Among HF‐HFrD‐fed mice, mRNA levels for SCD1 (P = 0.045), GPAT (0.0002), and FAS (0.002) were lower in mice treated with BioE‐1115 compared to vehicle, although the difference for the gene encoding ACC (P = 0.295) was not statistically significant (Fig. 3D).
Fig. 3.
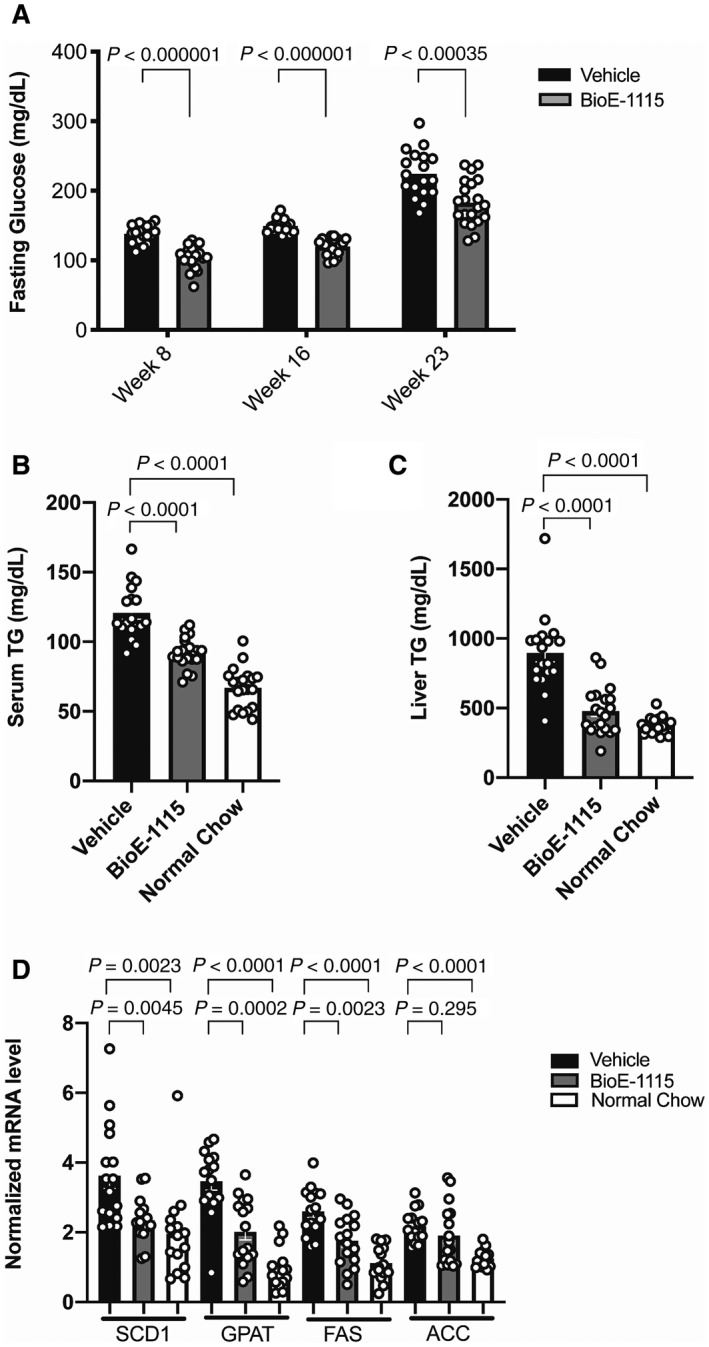
Fasting blood glucose and serum and hepatic TGs in BioE‐1115 versus vehicle‐treated mice fed an HF‐HFrD. (A) Compared with vehicle‐treated mice, fasting glucose was lower among mice treated with 100 mg/kg of BioE‐1115 at each of the time points measured, i.e., 8, 16, and 23 weeks. (B) Serum and (C) hepatic TGs at the end of 25 weeks were lower among mice treated with BioE‐1115 versus vehicle and, in the case of hepatic TGs, very similar to chow‐fed mice. (D) Liver mRNA levels for SCD1, GPAT, FAS, and ACC measured by quantitative reverse‐transcription polymerase chain reaction among mice fed an HF‐HFrD and treated with vehicle or BioE‐1115 or among mice fed an NCD. All results represent mean ± SEM.
BioE‐1115 Treatment Decreased Hepatic Steatosis
Based on quantitative grading, HF‐HFrD feeding was associated with histological findings characteristic of NAFLD/NASH, including increased hepatocyte vacuolization/ballooning (Fig. 4A), hepatic fat content (Fig. 4B), and bile duct proliferation (Fig. 4C), all of which were significantly diminished by treatment with BioE‐1115 compared with vehicle. Vacuolization was both microvesicular (small vacuoles without nuclear peripheralization) and macrovesicular (single large vacuole with peripheralization of the nucleus), with centrilobular zones typically more severely affected than periportal zones (Fig. 4D).
Fig. 4.
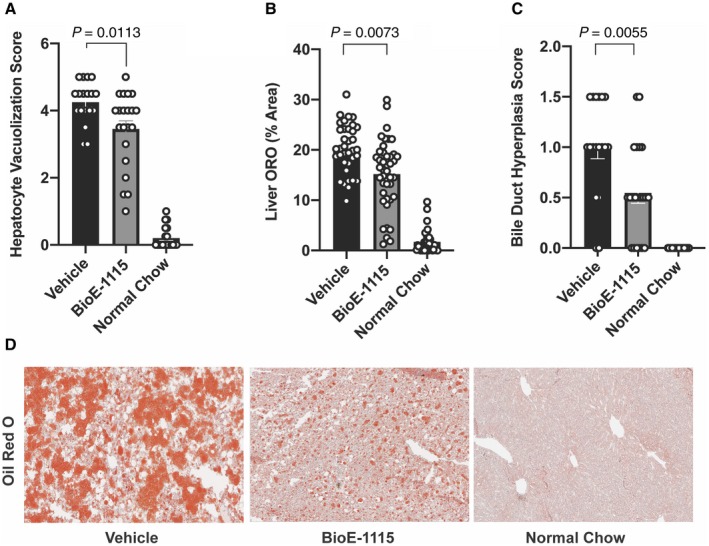
Liver fat and histological findings among treatment groups. Compared with vehicle‐treated mice, mice fed an HF‐HFrD and treated with BioE‐1115 exhibited (A) less fat based on ORO quantification, (B) less hepatocyte vacuolization, and (C) less bile duct hyperplasia. (D) Representative ORO stains from individual mice fed an HF‐HFrD and treated with either vehicle or BioE‐1115 or mice fed an NCD. Magnification ×10. All results represent mean ± SEM.
BioE‐1115 Treatment Decreased Hepatic Fibrosis
HF‐HFrD feeding was also associated with increased levels of liver αSMA (Fig. 5A) and an increased liver fibrosis score (Fig. 5B), both of which were significantly reduced by BioE‐1115‐treated mice compared to vehicle.
Fig. 5.
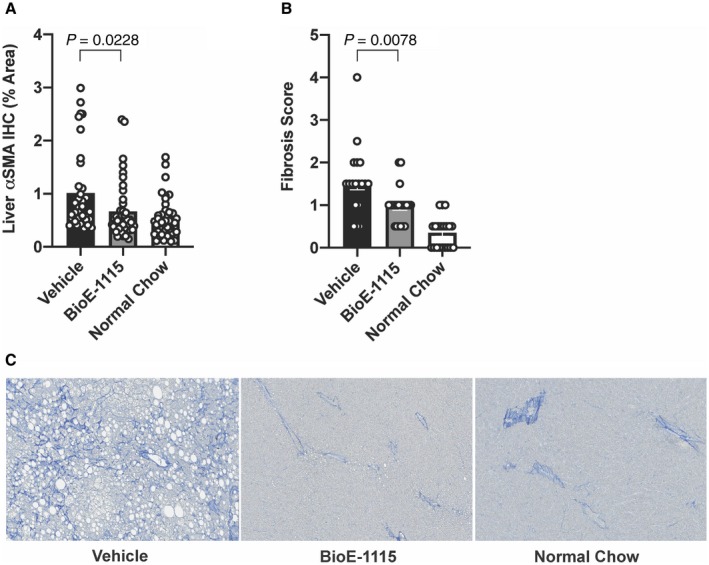
Liver collagen and αSMA among treatment groups. (A) Immunolabeled αSMA as a percentage of total tissue area quantified by image analysis. (B) Fibrosis score assessed as histopathologic quantification of collagen based on MT‐stained slides. Results are depicted as mean ± SEM. (C) Representative MT stains from individual mice fed an HF‐HFrD and treated with vehicle or BioE‐1115, or mice fed an NCD. Magnification ×10. Abbreviation: IHC, immunohistochemistry.
MT‐stained liver sections from mice fed an HF‐HFrD demonstrated significant fibrosis, typically characterized by fine collagen fibers extending out from perivascular zones and along sinusoids and infrequently around individual cells (pericellular fibrosis); significant fibrosis was reduced in HF‐HFrD‐fed mice treated with BioE‐1115 (Fig. 5C).
Discussion
Our findings indicate that pharmacologic inhibition of PASK produces metabolically favorable changes in two different and complementary animal disease models. In Zucker (fa/fa) rats, a genetic model of hyperphagia, obesity, insulin resistance, and dyslipidemia, PASK inhibition by BioE‐1115 at doses ranging from 3 to 100 mg/kg, the human equivalent of a total daily dose of ~30 to 1,000 mg for a 60‐kg adult, produced dose‐dependent improvements in insulin sensitivity, as reflected by lower plasma glucose and insulin levels, lowering of serum TG, and decreases in weight gain. In mice fed an HF‐HFrD, a dietary model of NAFLD/NASH, pharmacologic PASK inhibition again reduced hyperglycemia and hypertriglyceridemia, reduced liver fat and fibrosis, and mitigated the signature histological features of NASH, including increased liver fat and ballooning of hepatocytes.( 23 )
The fatty acids that are conjugated with glycerol to form TGs in the liver are either preformed and derived from the diet, adipose tissue, or hepatic extraction of plasma TGs or they are synthesized de novo by the liver from acetyl‐CoA.( 24 , 25 ) The latter, which is referred to as de novo lipogenesis (DNL), has been shown to be increased in patients with NAFLD/NASH.( 26 , 27 ) While the reasons for increased DNL in NAFLD/NASH are incompletely defined, it may reflect selective insulin resistance that is manifested by resistance to the suppression of hepatic gluconeogenesis by insulin but paradoxically preserves insulin sensitivity to stimulate hepatic fatty acid and TG synthesis.( 28 ) This paradoxically preserved sensitivity to insulin stimulation of TG synthesis, coupled with increased circulating insulin to respond to elevated glycemia, is associated with hyperactivation of SREPB‐1c, a transcriptional regulator of the lipogenic pathway.( 29 , 30 , 31 ) It has been proposed that this paradoxical insulin‐responsive activation of SREBP‐1c might be at least partially due to the nutrient‐responsive mTORC1 protein kinase. We recently described that mTORC1 activates PASK by direct phosphorylation. Therefore, the finding that PASK is required for the elevated posttranslational activation of SREBP‐1c under conditions of selective insulin resistance may help to explain the mechanism by which BioE‐1115 mitigates the effect of an HF‐HFrD on hepatic TG accumulation.( 14 )
Although SREBP‐1c‐mediated up‐regulation of DNL is an important contributor to NASH, circulating fatty acids are also increased in patients with NASH,( 32 ) and synthesis from preformed fatty acids may account for half or more of total hepatic TG production.( 24 , 33 ) Inhibition of PASK caused decreased expression of not only the enzymes responsible for DNL but also the enzymes required for esterification of fatty acids with glycerol, like GPAT. Therefore, pharmacologic inhibition of PASK should decrease liver TG synthesis regardless of the source of the fatty acids, and this could help explain the effects of BioE‐1115 to prevent hepatic fat accumulation in animals fed a fatty acid‐rich diet.
While there is no available biomarker to assess PASK target engagement by BioE‐1115 in vivo, several lines of evidence suggest that the observed effects of BioE‐1115 in the present studies are mediated by PASK inhibition. First, the IC50 measured in vitro for inhibition of PASK by BioE‐1115 is several orders of magnitude less than that for other kinases, transporters, receptors, or enzymes.( 14 ) Second, the effects of BioE‐1115 in the two complementary animal models largely mimic the decreased hypertriglyceridemia, insulin resistance, and hepatic steatosis observed in PASK−/− mice fed an HFD.( 10 ) Finally, the lower mRNA levels for SREBP‐1c target genes, including SCD1, ACC, FAS, and GPAT, in animals treated with BioE‐1115 versus vehicle are consistent with inhibition of PASK, activation of which is necessary for proteolytic maturation of SREBP‐1c.( 14 )
One interesting finding in both models is the effect of pharmacologic PASK inhibition on body weight. Specifically, PASK inhibition was associated with less weight gain, which in Zucker (fa/fa) rats was associated with less body fat and increased lean body mass. While this finding might be partly attributable to inhibition of hyperphagia, which our experiments do not directly address, it seems likely that improved insulin sensitivity is also a factor. Indeed, the lesser weight gain among BioE‐1115‐treated Zucker (fa/fa) rats and HF‐HFrD‐fed mice are consistent with our prior findings that PASK−/− mice are phenotypically normal and maintain their weight on an NCD but are resistant to the weight gain, metabolic abnormalities, and increased liver fat observed in wild‐type mice fed an HFD.( 10 ) While PASK activation is linked to SREBP‐1c maturation in liver, it is widely expressed with additional tissue‐specific functions. Collectively, these findings suggest that while the mitigation of hepatic TG accumulation and fibrosis reflect BioE‐1115‐mediated inhibition of PASK in liver, the lesser weight gain and lesser extrahepatic body fat among the BioE‐1115 group might reflect extrahepatic effects of PASK inhibition.
In summary, our findings indicate that PASK inhibition may act to mitigate the pathological increase in hepatic DNL associated with insulin resistance as well as the increased synthesis of TGs from circulating fatty acids. Moreover, pharmacologic inhibition of PASK, a physiological regulator of enzymes responsible for lipogenesis, is conceptually attractive compared with pharmacologic inhibition of individual enzymes in the lipogenic pathway. The findings collectively support the attractiveness of PASK as a therapeutic target for NASH and BioE‐1115 as a candidate for clinical development.
Acknowledgment
We thank Dr. Sarah Fogarty and Dr. Bill Rutter for their assistance and scientific input.
Supported by BioEnergenix, LLC.
Potential conflict of interest: Dr. Parnell and Dr. Rutter own stock in and are consultants to BioEnergenix. Dr. Scharschmidt owns stock in and consults for BioEnergenix. The other authors have nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Rinella M, Charlton M. The globalization of nonalcoholic fatty liver disease: Prevalence and impact on world health. Hepatology 2016;64:19‐22. [DOI] [PubMed] [Google Scholar]
- 2. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 3. Younossi ZM, Loomba R, Rinella ME, Bugianesi E, Marchesini G, Neuschwander‐Tetri BA, et al. Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2018;68:361‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rutter J. Essay: Amersham Biosciences and Science Prize . PAS domains and metabolic status signaling. Science 2002;298:1567‐1568. [DOI] [PubMed] [Google Scholar]
- 5. Rutter J, Michnoff CH, Harper SM, Gardner KH, McKnight SL. PAS kinase: an evolutionarily conserved PAS domain‐regulated serine/threonine kinase. Proc Natl Acad Sci U S A 2001;98:8991‐8996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Amezcua CA, Harper SM, Rutter J, Gardner KH. Structure and interactions of PAS kinase N‐terminal PAS domain: model for intramolecular kinase regulation. Structure 2002;10:1349‐1361. [DOI] [PubMed] [Google Scholar]
- 7. da Silva XG, Rutter J, Rutter GA. Involvement of Per‐Arnt‐Sim (PAS) kinase in the stimulation of preproinsulin and pancreatic duodenum homeobox 1 gene expression by glucose. Proc Natl Acad Sci U S A 2004;101:8319‐8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grose JH, Rutter J. The role of PAS kinase in PASsing the glucose signal. Sensors (Basel) 2010;10:5668‐5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grose JH, Smith TL, Sabic H, Rutter J. Yeast PAS kinase coordinates glucose partitioning in response to metabolic and cell integrity signaling. EMBO J 2007;26:4824‐4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hao HX, Cardon CM, Swiatek W, Cooksey RC, Smith TL, Wilde J, et al. PAS kinase is required for normal cellular energy balance. Proc Natl Acad Sci U S A 2007;104:15466‐15471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hao HX, Rutter J. The role of PAS kinase in regulating energy metabolism. IUBMB Life 2008;60:204‐209. [DOI] [PubMed] [Google Scholar]
- 12. Lindsley JE, Rutter J. Nutrient sensing and metabolic decisions. Comp Biochem Physiol B Biochem Mol Biol 2004;139:543‐559. [DOI] [PubMed] [Google Scholar]
- 13. Kikani CK, Wu X, Fogarty S, Kang SAW, Dephoure N, Gygi SP, et al. Activation of PASK by mTORC1 is required for the onset of the terminal differentiation program. Proc Natl Acad Sci U S A 2019;116:10382‐10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu X, Romero D, Swiatek WI, Dorweiler I, Kikani CK, Sabic H, et al. PAS kinase drives lipogenesis through SREBP‐1 maturation. Cell Rep 2014;8:242‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bain G, Shannon KE, Huang F, Darlington J, Goulet L, Prodanovich P, et al. Selective inhibition of autotaxin is efficacious in mouse models of liver fibrosis. J Pharmacol Exp Ther 2017;360:1‐13. [DOI] [PubMed] [Google Scholar]
- 16. Francque S, Vonghia L. Pharmacological treatment for non‐alcoholic fatty liver disease. Adv Ther 2019;36:1052‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kelly MJ, Pietranico‐Cole S, Larigan JD, Haynes NE, Reynolds CH, Scott N, et al. Discovery of 2‐[3,5‐dichloro‐4‐(5‐isopropyl‐6‐oxo‐1,6‐dihydropyridazin‐3‐yloxy)phenyl]‐3,5‐dio xo‐2,3,4,5‐tetrahydro[1,2,4]triazine‐6‐carbonitrile (MGL‐3196), a highly selective thyroid hormone receptor beta agonist in clinical trials for the treatment of dyslipidemia. J Med Chem 2014;57:3912‐3923. [DOI] [PubMed] [Google Scholar]
- 18. Lawitz EJ, Coste A, Poordad F, Alkhouri N, Loo N, McColgan BJ, et al. Acetyl‐CoA carboxylase inhibitor GS‐0976 for 12 weeks reduces hepatic de novo lipogenesis and steatosis in patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol 2018;16:1983‐1991.e1983. [DOI] [PubMed] [Google Scholar]
- 19. Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, et al. GS‐0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology 2018;155:1463‐1473.e1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zucker LM, Antoniades HN. Insulin and obesity in the Zucker genetically obese rat “fatty”. Endocrinology 1972;90:1320‐1330. [DOI] [PubMed] [Google Scholar]
- 21. Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, et al. High‐fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010;52:934‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cooksey RC, de Waard JH, Yakrus MA, Rivera I, Chopite M, Toney SR, et al. Mycobacterium cosmeticum sp. nov., a novel rapidly growing species isolated from a cosmetic infection and from a nail salon. Int J Syst Evol Microbiol 2004;54:2385‐2391. [DOI] [PubMed] [Google Scholar]
- 23. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 24. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343‐1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci U S A 2009;106:15430‐15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ameer F, Scandiuzzi L, Hasnain S, Kalbacher H, Zaidi N. De novo lipogenesis in health and disease. Metabolism 2014;63:895‐902. [DOI] [PubMed] [Google Scholar]
- 27. Lambert JE, Ramos‐Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146:726‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 2008;7:95‐96. [DOI] [PubMed] [Google Scholar]
- 29. Kohjima M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, et al. SREBP‐1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int J Mol Med 2008;21:507‐511. [PubMed] [Google Scholar]
- 30. Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS‐2 and increased SREBP‐1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell 2000;6:77‐86. [PubMed] [Google Scholar]
- 31. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A 2010;107:3441‐3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009;50:1827‐1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vatner DF, Majumdar SK, Kumashiro N, Petersen MC, Rahimi Y, Gattu AK, et al. Insulin‐independent regulation of hepatic triglyceride synthesis by fatty acids. Proc Natl Acad Sci U S A 2015;112:1143‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]