

Abstract
Yellow fever (YF) is a viral hemorrhagic fever that typically involves the liver. Brazil recently experienced its largest recorded YF outbreak, and the disease was fatal in more than a third of affected individuals, mostly because of acute liver failure. Affected individuals are generally treated only supportively, but during the recent Brazilian outbreak, selected patients were treated with liver transplant. We took advantage of this clinical experience to better characterize the clinical and pathological features of YF‐induced liver failure and to examine the mechanism of hepatocellular injury in YF, to identify targets that would be amenable to therapeutic intervention in preventing progression to liver failure and death. Patients with YF liver failure rapidly developed massive transaminase elevations, with jaundice, coagulopathy, thrombocytopenia, and usually hepatic encephalopathy, along with pathological findings that included microvesicular steatosis and lytic necrosis. Hepatocytes began to express the type 3 isoform of the inositol trisphosphate receptor (ITPR3), an intracellular calcium (Ca2+) channel that is not normally expressed in hepatocytes. Experiments in an animal model, isolated hepatocytes, and liver‐derived cell lines showed that this new expression of ITPR3 was associated with increased nuclear Ca2+ signaling and hepatocyte proliferation, and reduced steatosis and cell death induced by the YF virus. Conclusion: Yellow fever often induces liver failure characterized by massive hepatocellular damage plus steatosis. New expression of ITPR3 also occurs in YF‐infected hepatocytes, which may represent an endogenous protective mechanism that could suggest approaches to treat affected individuals before they progress to liver failure, thereby decreasing the mortality of this disease in a way that does not rely on the costly and limited resource of liver transplantation.

Abbreviations
- 5mC
5‐methyl cytosine
- ANOVA
analysis of variance
- ATP
adenosine triphosphate
- dpi
days post‐infection
- EGF
epidermal grown factor
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- HCC
hepatocellular carcinoma
- H&E
hematoxylin & eosin
- ICG
indocyanine green
- IFN
interferon
- IHC
immunohistochemistry
- ITPR
inositol trisphosphate receptor
- KO
knockout
- MAM
mitochondria‐associated membranes
- MOI
multiplicity of infection
- mRNA
messenger RNA
- PCNA
proliferating cell nuclear antigen
- PCR
polymerase chain reaction
- PFU
plaque‐forming unit
- R3KO
knockout for ITPR3
- RPMI
Roswell Park Memorial Institute
- UFMG
Universidade Federal de Minas Gerais
- YF
yellow fever
- YFV
yellow fever virus
- YFV17DD
YFV 17DD vaccine substrain
- YFVWT
YFV wild‐type strain
- WT
wild type
The yellow fever virus (YFV) is the prototypical member of the Flaviviridae family, which also includes viruses such as hepatitis C virus, dengue, West Nile, Zika, and Japanese encephalitis.( 1 , 2 ) Yellow fever (YF) is endemic in 47 countries across Africa and Central and South America,( 3 ) and over 2,000 cases were reported in the Brazilian outbreak between July 2016 and June 2018.( 4 ) The infection was fatal in more than one‐third of these patients, and the lethal cases were mostly attributed to acute liver failure.( 5 ) Therefore, YF remains a major health problem despite intense study about transmission and prophylaxis of the virus and the availability of an effective vaccine. The pathogenesis of the liver disease caused by YFV is poorly understood; indeed, treatment of infected individuals has typically been limited to supportive measures. During the recent Brazilian outbreak, however, selected patients with liver failure were considered for urgent liver transplant.( 6 , 7 ) This provided the opportunity to more carefully characterize the clinical and pathological features of YF‐induced acute liver failure and to investigate the cell biology of its pathogenesis. Calcium signals in hepatocytes regulate pathological processes that are known to occur in YF, including changes in cell proliferation, lipid droplet formation,( 7 ) and cell death.( 2 , 8 ) Therefore, we examined cellular and molecular alterations in the calcium signaling machinery in livers of patients infected with YFV and in isolated liver cells and an animal model.
Materials and Methods
Human Liver Specimens
Human liver samples and clinical data from patients with YF were obtained under the auspices of protocols approved by the Ethics Committee of Hospital Felicio Rocho (Belo Horizonte, Brazil) number CAAE: 90780318.9.0000.5125. YF specimens were from explanted livers, posttransplant liver biopsy of a patient who presented retroperitoneal bleeding 48 hours after the liver transplant, and autopsies of patients hospitalized at Hospital Felicio Rocho in the 2018 YF outbreak. Healthy liver samples were obtained from biopsies of pretransplant liver grafts. YF diagnosis was confirmed by real‐time polymerase chain reaction (PCR) in all cases. Patient selection for orthotopic liver transplantation was based on Clichy criteria.( 9 )
Cells
HepG2 wild‐type (WT) and inositol trisphosphate receptor (ITPR) 3 knockout (KO) cells were obtained from the laboratory of Dr. M. Nathanson (Yale University), maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, and kept at 37°C in 5% CO2.
Production of Viruses and Plaque Assay
For in vitro and in vivo experiments we used two YFV strains: (1) the 17DD vaccine substrain (YFV17DD), kindly donated by the Reference Laboratory for Flavivirus, Fiocruz, Brazilian Ministry of Health, and (2) The WT strain (YFVWT), which was isolated in our Laboratory from a clinical specimen of serum from a symptomatic patient from the 2018 outbreak in Minas Gerais. 17DD virus stocks were passaged at a multiplicity of infection (MOI) of 0.01 in Vero cells (ATCC CCL81, existing collection in our laboratory from Banco de Células do Rio de Janeiro [BCRJ, access code 0245]) in Roswell Park Memorial Institute (RPMI) medium (Cultilab, Campinas, Brazil) supplemented with 10% segmented filamentous bacteria for 5 days at 37°C. The YFVWT virus strain was propagated in mosquito cells (Aedes albopictus clone C6/36 [ATCC CRL‐1660, existing collection in our laboratory from BCRJ, code 0343]) in L15 medium supplemented with 10% fetal bovine serum (FBS) (Cultilab) and maintained at 28°C. YFVWT isolation was confirmed by real‐time PCR as described subsequently. To concentrate the virus, 50 mL of the cell culture supernatant was loaded onto a VivaCell 100 centrifugal concentrator (Sartorius, Göttingen, Germany) and centrifuged at 2,000g for 10 minutes, and the supernatant remaining in the concentrator was aliquoted and stored at −80°C. For quantification of virus, Vero cells were grown to a confluent monolayer in RPMI 1640 medium with 10% FBS (Cultilab) and 1% penicillin‐streptomycin‐glutamine (Gibco) in 24‐well plates at 37°C. The virus was serially diluted in serum‐free medium and then inoculated into the cells at 37°C with gentle shaking every 15 minutes for 1 hour. Next, the medium was replaced with RPMI 1640 medium containing 2% carboxymethylcellulose and 2% FBS (Cultilab) (overlay medium) and the culture kept at 37°C. After 5 days, the cells were fixed in 3% formalin in phosphate‐buffered saline for 1 hour, washed, and stained with 1% crystal violet in 10% formalin solution for 1 hour. Plates were washed with water, and plaques were counted manually.
Animals and YF Mouse Infection
In vivo experiments were conducted using type I INF receptor deficient mice (A129−/−) on a SV129 background. The A129−/− mice were originally from the Jackson Laboratories (reference 010830) and were obtained from Bioterio de Matrizes da Universidade de Sao Paulo and kept under specific pathogen‐free conditions at the Immunopharmacology Lab at the Universidade Federal de Minas Gerais (UFMG). This mouse model was used because its use has already been described for studying the pathogenesis of YF.( 10 ) In particular, this model recapitulates key features of fatal human YF infection, including death, YFV replication and dissemination, proinflammatory cytokine release, and severe pathology in visceral organs, including the liver.( 10 ) The rationale for using the A129−/− strain is that the resistance of mice to fatal viscerotropic YFV infection is mediated by interferons (IFNs), as it appears to be for dengue virus.( 11 ) Mice were housed in filtered cages with autoclaved food and water available ad libitum on ventilated shelves (Alesco, Monte Mor, Brazil). Mice were housed under standard conditions with controlled temperature (18°C‐23°C), humidity (40%‐60%), and 12/12‐hour dark‐light cycle. Sample sizes for in vivo studies were determined using the G*Power 3.1 software package. The experimental protocol was approved by the Committee on Animal Ethics of the UFMG (permit protocol no. 84/2018). All surgeries were performed under ketamine/xylazine anesthesia. Studies with YFV were conducted under biosafety level 2 containment at Immunopharmacology Lab from the Instituto de Ciencias Biológicas at the UFMG.
For the experiments, adult male and female A129−/− mice (7‐9 weeks old, 20‐22 g) were inoculated with different inoculums of either YFV17DD or YFVWT viruses’ strains through intravenous route/200 μL (tail vein). Control (noninfected) mice received 200 μL of C6/36 cell culture supernatant also through intravenous route (tail vein). Morbidity parameters such as lethality rates and bodyweight loss were evaluated daily. For clinical analysis, a kinetic of infection was performed and analysis was conducted on days 1, 3, and 6 following YFV infection.
Statistical Analysis
Data are presented as arithmetic mean ± SEM unless otherwise indicated. For statistical analysis, means between two groups were compared by Student t test, and comparisons among groups were analyzed by one‐way analysis of variance (ANOVA) followed by Bonferroni’s posttest. A P value of 0.05 or less was considered statistically significance.
Further Methodological Details
Detailed additional materials and methods are available in the Supporting Information.
Results
Clinical, Pathological, and Cell Biological Characteristics of YFV‐Induced Liver Failure
We examined clinical presentations (Table 1) and liver biopsies (Fig. 1 and Supporting Fig. S1) of YFV‐infected patients who were considered for liver transplant at Hospital Felicio Rocho, Belo Horizonte, Brazil, during the 2018 outbreak. Fourteen patients were admitted, most of whom transferred from other hospitals specifically for transplant evaluation. Thirteen patients were male (92.8%) with an average age 47 (22‐67 years), and all patients were exposed to forested areas. All but 1 of the patients were not vaccinated. Eight of the patients were deemed appropriate for listing for liver transplantation. These patients had high serum levels of transaminases at admission (9,000‐27,000 U/mL), with aspartate aminotransferase > alanine aminotransferase; 6 developed acute kidney injury and required hemodialysis. All patients had severe coagulopathy, with thrombocytopenia (18,000‐55,000), elevated international normalized ratio (1.5‐10.0), and low factor V activity (11%‐30%). Serum bilirubin also was elevated in nearly all of the patients, although serum alkaline phosphatase was in the normal range (not shown), suggesting that liver failure was due entirely to hepatocellular injury. Patients developed hepatic encephalopathy between 6 and 11 days after onset of symptoms, and progression from grade I to grade IV (24 hours) and then to death (36 hours) was very fast. After listing, 4 patients (50%) underwent orthotopic liver transplantation, whereas the other 4 died on the waiting list after a median of 24 hours. Two of the 4 transplanted patients died: one 3 hours after liver transplantation due to primary graft nonfunction, and the other 3 days after liver transplantation due to septic shock. The 2 patients who survived received sofosbuvir from 2 days before the surgery until 8 days following transplant. Both of these patients were in good health 18 months later.
Table 1.
Clinical Data of Patients Hospitalized at Hospital Felicio Rocho in 2018 YF Outbreak
| Patient | Age (years) | Gender | Platelets (per mL) | AST (IU/L) | ALT (IU/L) | Bilirubin (mg/dL) | INR | Creatinine (mg/dL) | Lactate (IU/L) | Encephalopathy | Transplantation (days) | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 53 | M | 29,000 | 9,693 | 2,282 | 17.2 | 2.9 | 12.4 | 133 | 4 | N | Dead |
| 2 | 64 | M | 26,000 | 7,900 | 4,648 | 3.54 | 2.3 | 6.48 | 61 | 2 | Y | Alive |
| 3 | 37 | M | 58,000 | 8,151 | 7,206 | 8.16 | >10 | 7.1 | 164 | 3 | Y | Dead |
| 4 | 49 | M | 89,000 | 5,160 | 2,463 | 5.12 | 1.81 | 8.74 | 40 | n | N | Alive |
| 5 | 39 | M | 58,000 | 8,707 | 4,509 | 6.81 | 2.9 | 8.81 | 125 | 4 | N | Dead |
| 6 | 58 | M | 12,000 | 12,590 | 7,446 | 21.9 | 1.65 | 1.17 | 42 | n | N | Alive |
| 7 | 69 | M | 35,000 | 8,572 | 3,987 | 5.9 | >10 | 8.11 | 135 | 3 | Y | Dead |
| 8 | 57 | M | 29,000 | 8,451 | 3,854 | 10.1 | 4.4 | 7.49 | 184 | 4 | N | Dead |
| 9 | 57 | F | 46,000 | 28,266 | 9,680 | 4.47 | >10 | 5.06 | 115 | 4 | N | Dead |
| 10 | 63 | M | 56,000 | 3,650 | 2,215 | 5.41 | 1.12 | 0.77 | 20 | n | N | Alive |
| 11 | 23 | M | 18,000 | 9,471 | 3,926 | 13.47 | 1.7 | 0.92 | 20 | n | N | Alive |
| 12 | 45 | M | 79,000 | 2,790 | 2,126 | 1.02 | 1.1 | 1.2 | 8 | n | N | Alive |
| 13 | 47 | M | 38,000 | 13,207 | 2,992 | 8.15 | 2.9 | 1.46 | 41 | 3 | Y | Alive |
| 14 | 40 | M | 49,000 | 157 | 288 | 0.49 | 1.1 | 0.82 | 15 | n | N | Alive |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; INR, international normalized ratio; N, no; Y, yes.
Fig. 1.
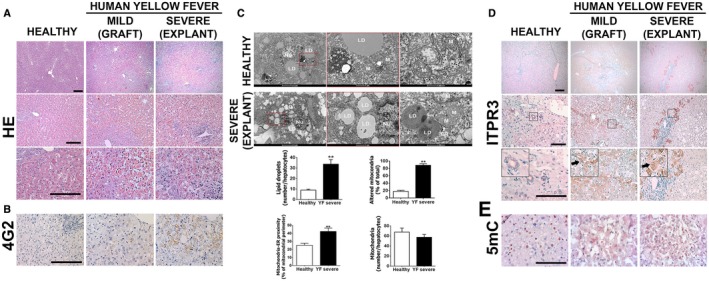
Patients with acute liver failure from YF have steatosis, pan‐lobular cell death, and new expression of ITPR3. (A) H&E‐stained liver sections from healthy patient (left panel), YFV‐infected patient showing mild liver disease (middle panel, biopsy from a liver graft), and a liver explant specimen from a patient with severe hepatic disease due to YFV infection (right panel). Patients were infected in 2018 during the YFV outbreak in Minas Gerais, Brazil. (B) Immunohistochemistry (IHC) staining for flavivirus envelope protein 4G2. (C) TEM images of liver samples from healthy patients (top panels) or YFV‐infected patient displaying severe hepatic disease (bottom panels), showing mitochondria with disorganized or absent crests, phagosomes, and lipid droplets. Graphs show quantification of number of lipid droplets, percentage of altered mitochondria, mitochondria‐endoplasmic reticulum proximity, and mitochondrial number. Bars indicate the average values of samples from at least eight hepatocytes/group. (D) IHC liver slices stained for ITPR3. Arrows indicate ITPR3 localization in the cytoplasm and nucleus in hepatocytes of mild and severe cases of YF. (E) IHC stained for 5mC (scale bar: 50 μm, 100 μm, and 200 μm for H&E and IHC, and 1 μm and 5 μm for TEM). Significance was analyzed using Student t test (**P < 0.01). Abbreviations: LD, lipid droplet; Nu, nucleus; M, mitochondrion; Ph, phagosome.
Histological analysis of liver biopsy specimens showed severe hepatocellular damage throughout the hepatic lobule in the explants, while grafts developed mild injury in the midzone region (Fig. 1A and Supporting Fig. S1A). Pathological changes in explants included steatosis, lytic necrosis, hemorrhage, and inflammatory infiltrates, similar to what was described in a recent case report,( 7 ) and consistent with the clinical presentations of these patients. Pathological changes in midzonal hepatocytes of grafts included swelling, steatosis, apoptosis, and lytic necrosis (Fig. 1A, middle panel). All biopsy specimens also showed positive staining for the flavivirus envelope protein 4G2 (Fig. 1B and Supporting Fig. S1B), confirming the association between liver injury and YFV infection in these patients. Transmission electron microscopy (TEM) analysis demonstrated microvesicular steatosis in the YFV‐infected hepatocytes (Fig. 1C), with a 3‐fold increase in the number of lipid droplets relative to what was observed in normal control hepatocytes. Both steatosis and certain types of cell death have been linked to alterations in mitochondrial calcium signals in hepatocytes,( 12 , 13 , 14 ) which in turn may result from aberrant expression of the ITPRs that conduct calcium from the endoplasmic reticulum (ER) into mitochondria at mitochondria‐associated membranes (MAMs).( 14 ) TEM analysis also showed that YFV‐infected hepatocytes displayed mitochondrial alterations, including dissolved mitochondrial crests and presence of membrane pores, and an increased amount of MAMs, but no change in the total number of mitochondria (Fig. 1C). Transmission of calcium into mitochondria depends on which ITPR isoforms are expressed at the MAM,( 15 , 16 ) which varies among cell types and can change in disease states.( 13 , 14 , 17 , 18 ) Therefore, we examined whether expression of each of the three ITPR isoforms was altered in YF.
ITPR1 and ITPR2 are the isoforms principally expressed in normal hepatocytes,( 19 , 20 ) although hepatocytes begin to express ITPR3 as well in chronic liver diseases and in liver cancer, where it is responsible for certain pathological effects.( 21 , 22 ) Unexpectedly, ITPR3 expression markedly increased in hepatocytes during YF infection (Fig. 1D and Supporting Fig. S1C). Cytoplasmic, perinuclear, and nuclear ITPR3 staining was observed in hepatocytes in both explants and grafts (Fig. 1D). In contrast, there was no change in expression of ITPR1 (Supporting Fig. S1C), which is the isoform that normally localizes to the MAM in hepatocytes,( 14 ) whereas ITPR2 expression was increased but to a much lesser extent than ITPR3 (Supporting Fig. S1C‐E). Demethylation of its promoter can increase ITPR3 expression in hepatocytes,( 22 ) and viral infection can promote DNA demethylation,( 23 , 24 ) so we analyzed the general methylation state in human YFV‐infected tissue by examining tissue staining for 5‐methyl cytosine (5mC), the methylated form of the DNA base cytosine targeted to DNA demethylases (Fig. 1E). Hepatocytes from normal liver were mostly positively stained (Fig. 1E, left panel), while hepatocytes were not labeled in either mild or severe liver injury from YFV (Fig. 1E, middle and right panels, respectively). Together, these findings demonstrate that YF infection induces hepatocytes to express ITPR3 and suggest that this occurs through DNA demethylation.
Mouse Model of YFV Infection Replicates Histological and Functional Characteristics of Liver Involvement in Human Disease
An animal model was used to investigate the mechanism of YF‐induced liver injury and the role of ITPR3 in this. IFN α/β receptor KO SV129 mice were infected with YFV, and the time‐course of ITPR3 expression was monitored. Mice were infected with either the attenuated YFV strain (YFV17DD) used in the YF vaccine (Fig. 2) or with a sylvatic YFV strain (YFVWT) that was isolated from a patient with YFV‐liver failure in February 2018 (Supporting Fig. S2). Both strains caused loss of body weight and death (Fig. 2A and Supporting Fig. S2A). YFV17DD reduced survival by 20% at 4 × 104 plaque‐forming units (PFU) 7 days post‐infection (dpi) (Fig. 2A), while all mice died by 7 dpi with 4 × 104 PFU of YFVWT (Supporting Fig. S2A). Therefore, 4 × 104 PFU injections were used for further studies. Quantitative RT‐PCR showed YFV17DD messenger RNA (mRNA) in the mouse liver at 3 dpi, although the virus was not detected at 1 dpi and was completely eliminated by 6 dpi (Fig. 2B). Kinetics of indocyanine green (ICG) clearance from the blood was used to assess liver function in the YFV17DD‐infected mice (Fig. 2C). There was a significant increase in ICG concentration in the blood after 1, 3, and 6 dpi, reflecting hepatic impairment, although the ICG concentration returned to normal at 10 dpi. Hematoxylin and eosin (H&E) stained liver specimens were microscopically examined. Hepatocytes displayed discrete hydropic degeneration (swollen cells with clear and vacuolated cytoplasm and central nucleus) in response to inoculation with 1 or 3 dpi of YFV17DD (Fig. 2D) or YFVWT (Supporting Fig. S2B); however, at 6 dpi, liver tissues were similar to controls. The frequency of apoptosis, steatosis, necroptosis, and necrosis were analyzed in 300‐nm‐thick sections stained with toluidine blue, and showed a progressive increase following infection, regardless of whether animals were infected with YFV17DD (Fig. 2E) or YFVWT (Supporting Fig. S2C). Hepatocytes isolated from YFV17DD‐infected mice at 3 dpi displayed an increased number of Bodipy‐labeled lipid droplets compared with control hepatocytes (Fig. 2F). As in patients, livers of YFV‐infected mice began to express ITPR3 (Fig. 2G and Supporting Fig. S2D). Western blot analysis demonstrated that expression of ITPR3 was significantly increased 3 dpi and 6 dpi in mice infected with either YFV17DD (Fig. 2G) or YFVWT (Supporting Fig. S2D). Similarly, 5mC staining showed that DNA methylation was decreased at 3 dpi with YFVWT (Supporting Fig. S2E) compared with the noninfected group. Sequencing the ITPR3 promoter region of liver samples from mice at 3 dpi after YFV17DD (Fig. 2H) or YFVWT infection (Supporting Fig. S2F) confirmed demethylation, consistent with this being responsible for the new expression of ITPR3 in infected hepatocytes. Together, these results show that α/β KO SV129 mice infected with YFV display cellular and molecular alterations in the liver that are similar to what is observed in humans, including de novo expression of ITPR3.
Fig. 2.
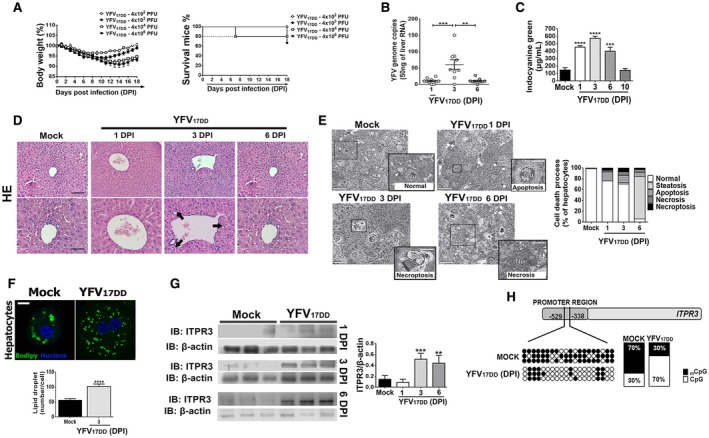
Mouse model replicates histological findings in patients with liver failure from YF infection. IFN‐α/βR−/− SV129 mice (7–9 weeks old) were inoculated intravenously with YFV strain (YFVV17DD) vaccine. (A) Changes in body weight (left panel) and Kaplan‐Meier survival curve (right panel) after inoculums of 4 × 102 to 4 × 106 PFU. (B) Quantitative RT‐PCR analysis of YFV viral load in the mice livers on 1 dpi, 3 dpi, and 6 dpi after inoculation of 4 × 104 PFU. Results are expressed as mean ± SEM (**P < 0.01 and ***P < 0.001 using ANOVA, Bonferroni’s posttest). (C) Liver function measured by the ICG clearance after inoculation of 4 × 104 PFU. Bars indicate mean ± SEM of samples from 3‐6 animals/group (***P < 0.001 and ****P < 0.0001 using ANOVA, Bonferroni’s posttest compared with mock samples). (D) Representative H&E‐stained liver slices of mock and YFV17DD inoculated animals on 1 dpi, 3 dpi, and 6 dpi. Scale bar: 50 μm and 100 μm. Arrows indicate hydropic degeneration characterized by the presence of swollen hepatocytes with clear and vacuolated cytoplasm and central nucleus. (E) Images of high‐resolution light microscopy of 300‐μm‐thick liver sections stained with toluidine blue, and representative images of mock and YFV17DD inoculated mice on 1 dpi (B), 3 dpi (C), and 6 dpi (D) showing hepatocytes with normal aspect (white *) and ongoing different cell death processes (black *). Normal hepatocytes from mock animal with central nucleus, rough cytoplasm with many organelles, and few lipid droplets (arrowheads). Early apoptosis with initial cell and organelle condensation in hepatocyte from 1 dpi of YFV17DD. Necroptosis characterized by pale and swollen nucleus and cytoplasm with plasma membrane integrity in hepatocyte from 3 dpi of YFV17DD. Necrosis, illustrated by empty spaces of cytoplasm in hepatocyte from 6 dpi of YFV17DD (left panel). Arrows indicate sinusoid capillaries; arrowheads indicate lipid droplets (scale bar: 25 μm and 50 μm). Frequency of cell death processes and steatosis analyzed in high‐resolution images of 300‐nm‐thick sections (right panel). (F) Representative immunofluorescence images of isolated hepatocytes stained with Bodipy (green) and 4′,6‐diamidino‐2‐phenylindol (blue) from mock and YFV17DD 3‐dpi inoculated animals (upper panel) (scale bar: 20 μm). Bottom panel shows the quantification of lipid droplets (number/cell). Bars indicate mean ± SEM of samples from three animals/group (****P < 0.0001 using Student t test). (G) ITPR3 expression is increased in liver 3 and 6 days after infection of mice with YFV17DD. Left panel shows representative blots for ITPR3 expression in liver lysates from mock, and YFV17DD 1‐dpi, 3‐dpi, and 6‐dpi inoculated animals. Each lane reflects the blot for the lysate from a separate animal. Right panel shows quantification of ITPR3 expression, normalized by b‐actin. Bars indicate mean ± SEM of samples from 3‐6 animals/group (**P < 0.01 and ***P < 0.001 using ANOVA, Bonferroni’s posttest compared with mock samples). (H) Demethylation sites on cytosine‐guanine dinucleotide (CpG) islands in mouse ITPR3 promoter region after 3 dpi with YFV17DD virus. Black dots represent methylated sites, and white dots represent demethylated sites. Quantification of methylated/demethylated CpG island ratio in mock and YFV‐infected liver samples. Abbreviations: IB, immunoblot; Nu, nucleus.
ITPR3 Expression in Hepatocytes Protects YF‐Infected Liver
To investigate the effects of YF‐induced de novo expression of ITPR3 in hepatocytes, experiments were performed in the liver‐derived HepG2 cell line, which constitutively expresses ITPR3.( 22 , 25 ) These cells were compared with knockout for ITPR3 (R3KO) HepG2 cells, in which CRISPR was used to delete ITPR3 (Fig. 3A).( 22 )
Fig. 3.
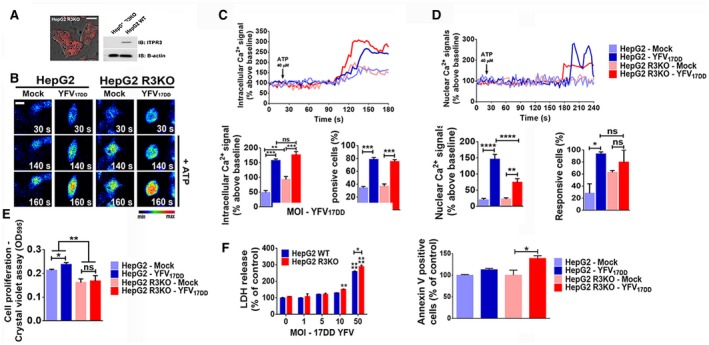
ITPR3 expression is protective in a YF‐infected, liver‐derived cell line. (A) Representative confocal images of HepG2 ITPR3 KO (HepG2 R3KO) cells and western blot for ITPR3 expression in HepG2 and HepG2 R3KO cells. (B) Confocal images of HepG2 and HepG2 R3KO cells loaded with Fluo‐4/AM (6 μM) and stimulated with 40 μM ATP. The cells were infected with 50 MOI of YFV17DD strain 24 hours before the Ca2+ analysis (scale bar: 20 μm). (C) Representative time‐course of total Ca2+ signal (upper panel), quantification of the peak fluorescence following stimulation with ATP (bottom‐left panel), and percentage of responsive cells to increase of cytoplasmic Ca2+ signal (bottom‐right panel). (D) Representative time‐course of nuclear Ca2+ signal in the cells loaded with Fluo‐4/AM (6 μM) (upper graph). Bottom‐left panel graph shows quantification of the peak fluorescence, and bottom‐right graph shows percentage of responsive cells to increase of nuclear Ca2+ signal following stimulation with ATP. (E) Crystal violet proliferation assay 1 dpi with 50 MOI of YFV17DD. (F) YFV17DD cytotoxicity. Cell viability was measured by lactate‐dehydrogenase release by the cells 1 dpi with different YFV17DD inoculums (left panel). Proportion of apoptotic cells at day 1 of YFV17DD 50 MOI infection, measured by flow cytometer analysis for annexin V–positive cells (right panel). Bars indicate the average values of samples from 3‐6 biological replicates (>20 cells/replicate for Ca2+ signaling analysis). Significance was analyzed by the ANOVA, Bonferroni’s posttest (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001) compared with mock samples. Abbreviations: IB, immunoblot; LDH, lactate dehydrogenase; ns, no statistical difference.
Calcium signal indicates the increase of free Ca2+ in a cell compartment relative to the basal level elicited by an agonist that directly or indirectly activates a calcium‐dependent cascade. Cytosolic calcium signals were increased in YFV17DD‐infected cells relative to uninfected controls. The amplitude of the calcium signals was more than 3‐fold greater in infected WT cells, but less than 2‐fold greater in infected R3KO HepG2 cells, each relative to their uninfected controls (Fig. 3B,C). Calcium signaling can be regulated independently in the nucleus,( 25 ) and nuclear calcium signals modulate progression through the cell cycle, hepatocyte proliferation, and liver regeneration,( 26 , 27 , 28 , 29 , 30 ) so we investigated Ca2+ signaling in the nucleus as well (Fig. 3D). YFV17DD infection enhanced nuclear Ca2+ signals in both WT and R3KO cells, relative to uninfected cells. However, the amplitude of the nuclear Ca2+ signal was about 2.2‐fold more pronounced in WT than in R3KO HepG2 cells (Fig. 3D). Consistent with this, a higher proliferation rate was observed in YFV17DD‐infected HepG2 cells compared with mock cells, whereas YFV17DD did not induce proliferation in R3KO HepG2 cells (Fig. 3E). Moreover, cells lacking ITPR3 were more susceptible to the cytotoxic effect of YFV17DD (Fig. 3F). This increased cytotoxic activity may have been due in part to the induction of apoptosis; Annexin V‐positive cells were more prevalent in YFV17DD‐infected R3KO HepG2 cells than in uninfected cells, but not in YFV17DD‐infected WT HepG2 cells relative to their uninfected controls (Fig. 3F). Additionally, lipid droplet formation was higher in R3KO HepG2 cells than in WT cells, and this was even more pronounced after YFV17DD infection (Fig. 4A). This preferential increase in steatosis in R3KO HepG2 was not related to mitochondrial Ca2+ signaling, because the amplitude of mitochondrial Ca2+ signals was increased to a similar extent in R3KO and WT HepG2 cells infected with YFV17DD (Fig. 4B). However, the expression of cpt1b and VLCAD, which are genes related to β‐oxidation, was down‐regulated in YFV17DD‐infected R3KO HepG2 cells, whereas VLCAD was up‐regulated in WT HepG2 cells (Fig. 4C,D). Therefore, the steatosis observed in R3KO HepG2 cells may have been due at least in part to inhibition of β‐oxidation, which is responsible for breakdown of fatty acids derived from lipolysis of lipid droplets.( 31 ) YFV17DD caused down‐regulation of mRNA levels of the lipogenesis genes SREBP1 and FAS in both HepG2 cell lines, but more pronounced in R3KO HepG2 cells than in WT (Fig. 4E,F). These findings are consistent with the idea that ITPR3 stimulates nuclear Ca2+ signaling, cell proliferation, and lipolysis in YFV‐infected hepatocytes, which collectively mitigates the deleterious effects of the infection.
Fig. 4.
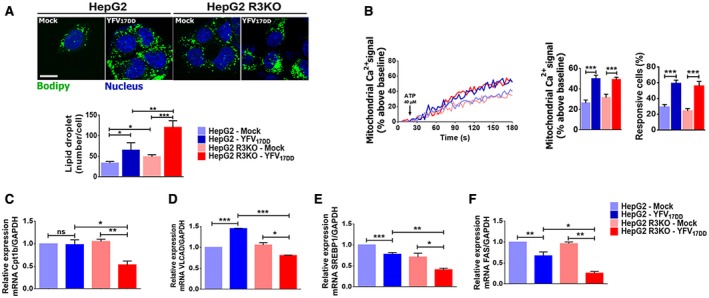
ITPR3 expression decreases steatosis in a YF‐infected, liver‐derived cell line. (A) Immunofluorescence images of HepG2 and HepG2 R3KO cells stained with Bodipy (green) and 4′,6‐diamidino‐2‐phenylindole (blue), both in mock and 1‐dpi YFV17DD cells (upper panel), and quantification of lipid droplets (number/cell) (scale bar: 20 μm) (bottom panel). (B) Representative time‐course of mitochondrial Ca2+ signal (left graph). Cells were transfected with the mitochondrial matrix–targeted Ca2+ indicator inverse‐pericam and stimulated with 40 μM ATP. Graphs show quantification of the peak of fluorescence following stimulation with ATP (middle graph) and percentage of responsive cells to increase of mitochondrial Ca2+ signals (right graph). Quantitative RT‐PCR analysis for mRNA expression of β‐oxidation, Cpt1b (C), VLCAD (D), lipogenesis, and SREBP1 (E) and FAS (F) genes. Bars indicate the average values of samples from three to six biological replicates (>20 cells/replicate for Ca2+ signaling analysis). Significance was analyzed by the ANOVA, Bonferroni’s posttest (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001) compared with mock samples. Abbreviations: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; ns, no statistical difference.
To further investigate the effects of YFV infection on ITPR3 expression and calcium signaling, these were examined in freshly isolated primary mouse hepatocytes. Cells were stimulated with either epidermal growth factor (EGF) (Fig. 5A) or adenosine triphosphate (ATP) (Supporting Fig. S3), because both of these stimuli are important for calcium‐mediated hepatocyte proliferation and liver regeneration.( 32 , 33 , 34 , 35 ) The amplitude of both cytosolic and nuclear Ca2+ signals induced by EGF was higher in hepatocytes from YFV17DD‐infected mice than in mock hepatocytes (Fig. 5B,C, respectively). Cytosolic and nuclear Ca2+ signals in YFV17DD‐infected hepatocytes were also more pronounced in cells stimulated with ATP (Supporting Fig. S3A‐C, respectively). ITPR3 expression was increased in the nucleus of YFV17DD‐infected hepatocytes, with its expression peak at 3 dpi (Fig. 5D), and this was associated with a higher number of proliferating cell nuclear antigen (PCNA)‐positive hepatocytes in the YFV17DD‐infected mice as well (Fig. 5E). Specifically, nuclear staining for PCNA, a marker for proliferation, was significantly increased in YFV17DD‐infected mice at 1 dpi, 3 dpi, and 6 dpi (Fig. 5E). Consistent with these findings, a marked increase in PCNA staining was observed in the livers of YFV‐infected patients with either mild (Fig. 5F, middle panel) or severe (Fig. 5F, right panel) liver disease. Together, these results suggest that ITPR3 expression induced by YFV infection triggers a regenerative mechanism through a nuclear Ca2+ increase, in an attempt to protect the liver by stimulating hepatocyte proliferation.
Fig. 5.
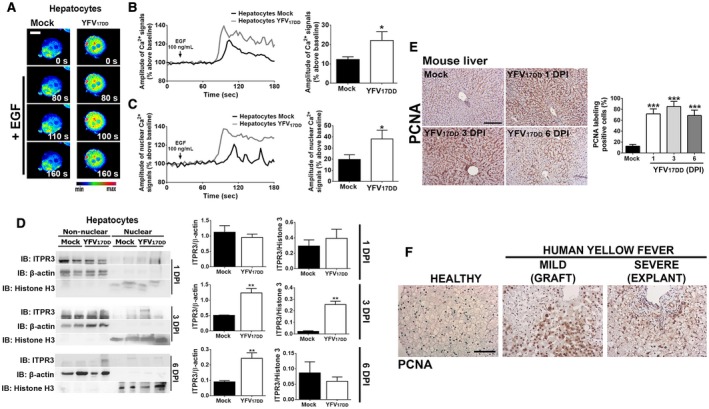
YF infection increases Ca2+ signals in the nucleus and enhances proliferation of hepatocytes. (A) Confocal images of hepatocytes isolated from liver tissue of 3‐dpi YFV17DD‐infected mice. Cells were loaded with Fluo‐4/AM (6 μM) and stimulated with 100 ng/mL EGF (scale bar: 20 μm). (B) Representative time‐course of total Ca2+ signal (left panel) and quantification of the peak fluorescence following stimulation with EGF (right panel). (C) Representative time‐course of nuclear Ca2+ signal (left panel) and quantification of the peak fluorescence of nuclear Ca2+ signal following stimulation with EGF (right panel). (D) Nonnuclear and nuclear protein fractions of liver lysates from mock, 1‐dpi, 3‐dpi, and 6‐dpi YFV17DD‐infected animals were tested for ITPR3 expression by western blot. Left panel shows representative blots; middle and right graphs show quantification of ITPR3 in nonnuclear and nuclear protein fractions. (E) IHC images of liver slices stained for PCNA in mock and YFV17DD‐infected animals on 1 dpi, 3 dpi, and 6 dpi (left panel). Scale bar: 100 μm. Quantification of PCNA‐positive cells is shown in the right graph. (F) PCNA staining of liver slices from a healthy patient (left panel), YFV‐infected hepatic human tissue from a graft biopsy with mild liver disease (middle panel), and an explant sample from a patient with severe hepatic disease (right panel). Scale bar: 50 μm. Bars indicate the average values of samples from three to six biological replicates (>20 cells/replicate for Ca2+ signaling analysis). Significance was analyzed by Student t test or the ANOVA, Bonferroni’s posttest (*P < 0.05, **P < 0.01, and ***P < 0.001) compared with mock samples. Abbreviation: IB, immunoblot.
Discussion
Despite extensive knowledge about YFV transmission and prophylaxis,( 36 ) this infection remains a major threat to human health, and understanding of its pathogenesis remains limited. Here, we characterized the clinical and pathological findings in patients evaluated for liver transplant because they developed acute liver failure from their infection. The current findings complement and extend other recent clinical observations of YF liver failure, including the presence of massive hepatocellular necrosis and microvesicular steatosis, plus the rapid progression of coagulopathy, encephalopathy, and acute kidney injury.( 5 , 6 , 7 , 37 ) Hepatocytes of infected patients also began to heavily express ITPR3, a calcium channel that is normally absent or minimally expressed in hepatocytes.( 19 , 20 , 38 ) This change was observed in a mouse model of YFV infection as well.
New expression of ITPR3 occurs in a variety of chronic liver diseases, and becomes more pronounced in hepatocellular carcinoma (HCC).( 22 ) In that setting, pathological effects such as enhanced proliferation and resistance to apoptosis have been attributed to the ITPR3.( 22 ) We report that ITPR3 can also become expressed in acute liver injury. Some of the ITPR3 in YFV infection localizes to the nucleus, where it is positioned to contribute to the enhanced nuclear calcium signaling observed here. This may have important therapeutic implications, as hepatocyte proliferation is dependent on calcium signals in the nucleus,( 26 ) and the growth factors that are principally responsible for liver regeneration exert their effects by selectively activating nuclear calcium signaling pathways.( 27 , 28 , 29 , 30 , 39 ) It is less clear whether ITPRs that are localized to MAMs play a role in the pathogenesis of YFV liver disease. These ITPRs are responsible for mitochondrial calcium signals, which in turn regulate lipid droplet formation and certain forms of cell death.( 12 , 13 , 14 , 16 ) Normally this is regulated by ITPR1 in hepatocytes,( 14 ) and expression of this isoform was not altered by YFV. ITPR2 can preferentially transmit calcium into mitochondria in some cell types,( 15 , 40 ) and ITPR2 expression was increased in YFV‐infected hepatocytes, but there is disagreement about whether this isoform localizes to the MAM and does not regulate steatosis in hepatocytes.( 14 , 41 ) ITPR3 can preferentially transmit calcium into mitochondria in other cell types( 15 , 16 , 17 , 42 ). Indeed, we found that loss of ITPR3 sensitized YFV‐infected cells to apoptosis; however, effects of ITPR3 on steatosis may instead reflect concomitant changes in expression of enzymes involved in fatty acid oxidation and lipogenesis. The role of ITPR3 in controlling the expression of lipogenic genes is an open question that is just now being investigated. Several calcium signaling genes were recently shown to be important in human and mouse samples of HCC caused by nonalcoholic steatohepatitis. Among them, ITPR3 was aberrantly up‐regulated in this disease,( 21 ) suggesting that this calcium channel could play a role in lipid metabolism. Chronic ITPR3 expression also modulates gene expression, triggering an anti‐apoptotic response in HCC.( 22 ) Together, these observations suggest the possibility of a modulatory effect of ITPR3 on different molecular pathways in YFV‐infected hepatocytes through regulation of gene expression, including genes related to lipid metabolism. Consistent with this, patients who develop symptoms from the YFV vaccine have enhanced ER stress,( 43 ) which typically increases rather than decreases steatosis.( 44 ) Lipid droplets are used by viruses from the Flaviviridae family, including YFV and hepatitis C virus (HCV), as an energy reservoir for replication, a platform for viral assembly( 45 ) and a source of very low density lipoprotein, which is complexed to the HCV virus as a mechanism to evade the immune system.( 46 ) Therefore, the current observation that ITPR3 inhibits steatosis may represent an additional protective role that it serves in YFV‐infected hepatocytes.
A correlation between viral load and worse outcome has been reported in nonhuman primates infected with YFV( 47 ) as well as in patients with YFV,( 5 ) and this relationship was recapitulated in our mouse model. Therefore, even though our findings suggest that de novo expression of ITPR3 in hepatocytes may be an endogenous protective mechanism in YFV infection, there also may be limits to the extent of this beneficial effect. It remains to be determined whether and to what extent the regulation of ITPR3 expression and targeting of this calcium channel to the nucleus can mitigate the liver damage that commonly contributes to the morbidity and mortality of this disease.
Author Contributions
F.O.L. designed and performed experiments, analyzed the data, and wrote the manuscript. A.F., A.C.M.L.F., R.M.F., M.L.S., D.G.M., and G.O.L.R. performed in vitro and in vivo experiments and analyzed the data. G.O.L.R., I.B.S.P., and V.V.C. performed the wild‐type YF virus isolation, YFV propagation, and in vivo experiments, and analyzed the data related to the YF mouse model. F.F.D. processed tissue samples and performed TEM and morphometric analysis in the YF specimens. A.M.F.A, C.X.L., and P.T.V. identified and diagnosed human YF‐infected specimens. M.M.T., V.V.C., M.C.F., M.H.N., and M.F.L. supervised the project, formulated the hypothesis, designed the experiments, and edited the manuscript.
Supporting information
Fig S1
Fig S2
Fig S3
Supplementary Material
Acknowledgments
The authors thank the Center of Microscopy at the Universidade Federal de Minas Gerais (http://www.microscopia.ufmg.br) for providing the equipment and technical support for experiments involving electron microscopy.
Financial support: National Institute of Science and Technology in Dengue (573876/2008); Foundation for the National Institutes of Health (DK112797, DK114041, DK34989, and DK57751); Fundação de Amparo à Pesquisa de Minas Gerais; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior; Fundação de Amparo à Pesquisa do Estado de São Paulo (2018/20014‐0); Conselho Nacional de Desenvolvimento Científico e Tecnológico (159892/2018‐0); and Liver Center at Universidade Federal de Minas Gerais.
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Barrett AD, Monath TP. Epidemiology and ecology of yellow fever virus. Adv Virus Res 2003;61:291‐315. [DOI] [PubMed] [Google Scholar]
- 2. Monath TP. Yellow fever: an update. Lancet Infect Dis 2001;1:11‐20. [DOI] [PubMed] [Google Scholar]
- 3. Klitting R, Gould EA. Paupy C, de Lamballerie X. What does the future hold for yellow fever virus? Genes (Basel) 2018;9. pii: E291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Faria NR, Kraemer MUG, Hill SC, Goes de Jesus J, Aguiar RS, Iani FCM, et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science 2018;361:894‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kallas EG, D'Elia Zanella L, Moreira CHV, Buccheri R, Diniz GBF, Castineiras ACP, et al. Predictors of mortality in patients with yellow fever: an observational cohort study. Lancet Infect Dis 2019;19:750‐758. [DOI] [PubMed] [Google Scholar]
- 6. Duarte‐Neto AN, Cunha MDP, Marcilio I, Song ATW, de Martino RB, Ho YL, et al. Yellow fever and orthotopic liver transplantation: new insights from the autopsy room for an old but reemerging disease. Histopathology 2019;75:638‐648. [DOI] [PubMed] [Google Scholar]
- 7. Song ATW, Abdala E, de Martino RB, Malbouisson LMS, Tanigawa RY, Andrade GM, et al. Liver transplantation for fulminant hepatitis attributed to yellow fever. Hepatology 2019;69:1349‐1352. [DOI] [PubMed] [Google Scholar]
- 8. Quaresma JA, Barros VL, Fernandes ER, Pagliari C, Takakura C, da Costa Vasconcelos PF Jr., et al. Reconsideration of histopathology and ultrastructural aspects of the human liver in yellow fever. Acta Trop 2005;94:116‐127. [DOI] [PubMed] [Google Scholar]
- 9. Bernuau J, Goudeau A, Poynard T, Dubois F, Lesage G, Yvonnet B, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986;6:648‐651. [DOI] [PubMed] [Google Scholar]
- 10. Meier KC, Gardner CL, Khoretonenko MV, Klimstra WB, Ryman KD. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog 2009;5:e1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shresta S, Sharar KL, Prigozhin DM, Beatty PR, Harris E. Murine model for dengue virus‐induced lethal disease with increased vascular permeability. J Virol 2006;80:10208‐10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mansouri A, Gattolliat CH, Asselah T. Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterology 2018;155:629‐647. [DOI] [PubMed] [Google Scholar]
- 13. Feriod CN, Gustavo Oliveira A, Guerra MT, Nguyen L, Mitchell Richards K, Jurczak MJ, et al. Hepatic inositol 1,4,5 trisphosphate receptor type 1 mediates fatty liver. Hepatol Commun 2017;1:23‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arruda AP, Pers BM, Parlakgul G, Guney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum‐mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med 2014;20:1427‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bartok A, Weaver D, Golenar T, Nichtova Z, Katona M, Bansaghi S, et al. IP3 receptor isoforms differently regulate ER‐mitochondrial contacts and local calcium transfer. Nat Commun 2019;10:3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, et al. The type III inositol 1,4,5‐trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem 2005;280:40892‐40900. [DOI] [PubMed] [Google Scholar]
- 17. Kuo IY, Brill AL, Lemos FO, Jiang JY, Falcone JL, Kimmerling EP, et al. Polycystin 2 regulates mitochondrial Ca(2+) signaling, bioenergetics, and dynamics through mitofusin 2. Sci Signal 2019;12. pii: eaat7397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ueasilamongkol P, Khamphaya T, Guerra MT, Rodrigues MA, Gomes DA, Kong Y, et al. Type 3 inositol 1,4,5‐trisphosphate receptor is increased and enhances malignant properties in cholangiocarcinoma. Hepatology 2020;71:583‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirata K, Pusl T, O'Neill AF, Dranoff JA, Nathanson MH. The type II inositol 1,4,5‐trisphosphate receptor can trigger Ca2+ waves in rat hepatocytes. Gastroenterology 2002;122:1088‐1100. [DOI] [PubMed] [Google Scholar]
- 20. Kruglov EA, Gautam S, Guerra MT, Nathanson MH. Type 2 inositol 1,4,5‐trisphosphate receptor modulates bile salt export pump activity in rat hepatocytes. Hepatology 2011;54:1790‐1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liang JQ, Teoh N, Xu L, Pok S, Li X, Chu ESH, et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat Commun 2018;9:4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guerra MT, Florentino RM, Franca A, Lima Filho AC, dos Santos ML, Fonseca RC, et al. Expression of the type 3 InsP3 receptor is a final common event in the development of hepatocellular carcinoma. Gut 2019:68:1676‐1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gomes AV, de Souza Morais SM, Menezes‐Filho SL, de Almeida LG, Rocha RP, Ferreira JM, et al. Demethylation profile of the TNF‐alpha promoter gene is associated with high expression of this cytokine in Dengue virus patients. J Med Virol 2016;88:1297‐1302. [DOI] [PubMed] [Google Scholar]
- 24. Fan H, Cui Z, Zhang H, Mani SK, Diab A, Lefrancois L, et al. DNA demethylation induces SALL4 gene re‐expression in subgroups of hepatocellular carcinoma associated with Hepatitis B or C virus infection. Oncogene 2017;36:2435‐2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leite MF, Thrower EC, Echevarria W, Koulen P, Hirata K, Bennett AM, et al. Nuclear and cytosolic calcium are regulated independently. Proc Natl Acad Sci U S A 2003;100:2975‐2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rodrigues MA, Gomes DA, Leite MF, Grant W, Zhang L, Lam W, et al. Nucleoplasmic calcium is required for cell proliferation. J Biol Chem 2007;282:17061‐17068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gomes DA, Rodrigues MA, Leite MF, Gomez MV, Varnai P, Balla T, et al. c‐Met must translocate to the nucleus to initiate calcium signals. J Biol Chem 2008;283:4344‐4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Angelis Campos AC, Rodrigues MA, de Andrade C, de Goes AM, Nathanson MH, Gomes DA. Epidermal growth factor receptors destined for the nucleus are internalized via a clathrin‐dependent pathway. Biochem Biophys Res Commun 2011;412:341‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodrigues MA, Gomes DA, Andrade VA, Leite MF, Nathanson MH. Insulin induces calcium signals in the nucleus of rat hepatocytes. Hepatology 2008;48:1621‐1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Amaya MJ, Oliveira AG, Guimaraes ES, Casteluber MC, Carvalho SM, Andrade LM, et al. The insulin receptor translocates to the nucleus to regulate cell proliferation in liver. Hepatology 2014;59:274‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid β‐oxidation. J Inherit Metab Dis 2010;33:469‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Runge DM, Runge D, Dorko K, Pisarov LA, Leckel K, Kostrubsky VE, et al. Epidermal growth factor‐ and hepatocyte growth factor‐receptor activity in serum‐free cultures of human hepatocytes. J Hepatol 1999;30:265‐274. [DOI] [PubMed] [Google Scholar]
- 33. Tackett BC, Sun H, Mei Y, Maynard JP, Cheruvu S, Mani A, et al. P2Y2 purinergic receptor activation is essential for efficient hepatocyte proliferation in response to partial hepatectomy. Am J Physiol Gastrointest Liver Physiol 2014;307:G1073‐G1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Michalopoulos GK, Khan Z. Liver regeneration, growth factors, and amphiregulin. Gastroenterology 2005;128:503‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Besnard A, Gautherot J, Julien B, Tebbi A, Garcin I, Doignon I, et al. The P2X4 purinergic receptor impacts liver regeneration after partial hepatectomy in mice through the regulation of biliary homeostasis. Hepatology 2016;64:941‐953. [DOI] [PubMed] [Google Scholar]
- 36. Klitting R, Fischer C, Drexler JF, Gould EA, Roiz D, Paupy C, de Lamballerie X. What does the future hold for yellow fever virus? Genes (Basel) 2018;9:425‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ribeiro AF, Cavalin RF, Abdul Hamid Suleiman JM, Alves da Costa J, Januaria de Vasconcelos M, Sant'Ana Malaque CM, Sztajnbok J. Yellow fever: factors associated with death in a hospital of reference in infectious diseases, Sao Paulo, Brazil, 2018. Am J Trop Med Hyg 2019;10:180‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dufour JF, Luthi M, Forestier M, Magnino F. Expression of inositol 1,4,5‐trisphosphate receptor isoforms in rat cirrhosis. Hepatology 1999;30:1018‐1026. [DOI] [PubMed] [Google Scholar]
- 39. Garcin I, Tordjmann T. Calcium signalling and liver regeneration. Int J Hepatol 2012;2012:630670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, et al. Binding of FUN14 domain containing 1 with inositol 1,4,5‐trisphosphate receptor in mitochondria‐associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo . Circulation 2017;136:2248‐2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feriod CN, Nguyen L, Jurczak MJ, Kruglov EA, Nathanson MH, Shulman GI, et al. Inositol 1,4,5‐trisphosphate receptor type II (InsP3R‐II) is reduced in obese mice, but metabolic homeostasis is preserved in mice lacking InsP3R‐II. Am J Physiol Endocrinol Metab 2014;307:E1057‐E1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ 2012;19:267‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chan KR, Gan ES, Chan CYY, Liang C, Low JZH, Zhang SL, et al. Metabolic perturbations and cellular stress underpin susceptibility to symptomatic live‐attenuated yellow fever infection. Nat Med 2019;25:1218‐1224. [DOI] [PubMed] [Google Scholar]
- 44. Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, Bailly‐Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non‐alcoholic fatty liver disease. J Hepatol 2018;69:927‐947. [DOI] [PubMed] [Google Scholar]
- 45. Martins AS, Martins IC, Santos NC. Methods for lipid droplet biophysical characterization in flaviviridae infections. Front Microbiol 2018;9:1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Filipe A, McLauchlan J. Hepatitis C virus and lipid droplets: finding a niche. Trends Mol Med 2015;21:34‐42. [DOI] [PubMed] [Google Scholar]
- 47. Engelmann F, Josset L, Girke T, Park B, Barron A, Dewane J, et al. Pathophysiologic and transcriptomic analyses of viscerotropic yellow fever in a rhesus macaque model. PLoS Negl Trop Dis 2014;8:e3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Supplementary Material