

Abstract
Astrocytomas often recur after surgical resection, but the underlying mechanism remains enigmatic. Elucidation of clonal evolution in primary and relapse tumors may provide important information on tumor progression. Here, we examined genetic factors underlying recurrence in a patient with astrocytoma initially diagnosed with World Health Organization (WHO) grade II astrocytoma, who then relapsed with glioblastoma (WHO grade IV) complicated with local anaplastic astrocytoma (WHO grade III). We performed genomic DNA sequencing and data analysis of paired tumor tissue specimens and a peripheral blood sample (control), and used expands software for subclone analysis. A germline NOTCH1 missense mutation was identified in the peripheral blood sample, the primary tumor and the relapse tumor; in addition, we identified a tumor protein p53 (TP53) heterozygous nonsense mutation in the primary tumor and a TP53 homozygous nonsense mutation and an IDH1 heterozygous missense mutation in the relapse tumor. Clonal evolution trees indicated higher heterogeneity in the relapse tumor. Although germline mutations might contribute to the driving force of the primary tumor, aggressive chemotherapy and radiation may apply selective pressure for tumor clonal evolution; furthermore, a total loss of function of gatekeeping genes (TP53) may result in impaired DNA repair and catastrophic chromosomal aberrations.
Keywords: astrocytoma, clonal evolution, glioblastoma, relapse, TP53
Astrocytoma often recurs after surgery. In this study, we examined the clonal structures of a primary astrocytoma and its relapse tumor, a malignant glioblastoma. We observed accelerated carcinogenesis in the relapse tumor after intensive radiation and chemotherapy, suggesting that such treatment may contribute to astrocytoma malignant progression.

Abbreviations
- BWA
burrows‐wheeler aligner
- CNV
copy number variation
- IDH
isocitrate dehydrogenase
- INDELs
insertions and deletions
- SNP
single nucleotide polymorphism
- SP
subpopulation
- SV
structure variation
- TP53
tumor protein p53
- WHO
World Health Organization
Gliomas are the most common central nervous system tumors derived from glial cells. Traditionally, they are classified according to the World Health Organization (WHO) and include astrocytomas, oligodendrogliomas and ependymomas [1]. Like most cancers, gliomas develop because of genetic aberrations that accumulate with tumor progression [2, 3, 4]. Astrocytomas are tumors that arise from astrocyte cells that make up the “glue‐like” or supportive tissues of the brain. Low‐grade astrocytomas are usually localized and grow slowly; high‐grade astrocytomas grow at a rapid pace and require a different course of treatment.
Currently, the treatment options for astrocytoma include surgery, radiation and chemotherapy. Compared with high‐grade astrocytomas, low‐grade astrocytomas are slow growing, but over time they may progress to more malignant tumors after resection, leading to reduced overall survival. The factors associated with recurrence and malignant degeneration vary and include patient age, tumor size, type, location and the extent of resection [5, 6, 7, 8, 9]. The genetic factors were also proved to play an important role in the pathogenicity and development of gliomas disease. Most cases of astrocytoma‐associated mortality are due to tumor recurrence and malignant transformation, which may be associated with clonal evolution at the cytogenetic level [10, 11, 12, 13]. Over about the past 10 years, the number of studies focused on the molecular biology of astrocytomas has increased significantly. Numbers of genetic markers associated with astrocytomas were identified [14, 15, 16, 17]. Studies have shown that low‐grade astrocytomas usually carry mutations of isocitrate dehydrogenase 1/2 (IDH1/2) and tumor protein p53 (TP53) gene [18, 19, 20]. In contrast, studies show that glioblastoma usually arises without IDH mutation [19]. Research data also identified different genetic changes associated with the degree of astrocytomas differentiation [21, 22]. To determine the genetic spectrum associated with relapse and malignant transformation of astrocytoma, we performed whole‐genome sequencing of primary tumor, relapse tumor, and a peripheral blood sample from a patient first diagnosed with grade II astrocytoma who then relapsed with glioblastoma (grade IV) complicated by local anaplastic astrocytoma (grade III).
Materials and methods
Patient and samples
A 25‐year‐old female patient was diagnosed with grade II astrocytoma with accompanying secondary epilepsy, and surgery was conducted to remove the tumor tissues, followed with radiation (60 Gy total) and chemotherapy (nimustine, 80 mg·m−2, 1 week × 8). Then, after almost 1 year, the tumor recurred, and histopathological examination of the resected specimen revealed that the tumor was glioblastoma (grade IV) complicated by local anaplastic astrocytoma (grade III). The patient was treated by temozolomide (120 mg·m−2, days 1–5 every 28 days for 5 cycles).
Samples of primary (coded as TY‐1) and relapse tumor (TY‐2) and a peripheral blood sample (TY‐NC) were collected, and genomic DNA was extracted from all samples using standard protocols. Then the DNA samples were cryostored for whole‐genome sequencing. The patient gave written informed consent, and the Committee on Studies Involving Human Beings at Army Medical University approved the protocol. The research complied with the Helsinki Declaration and International Ethical Guidelines for Biomedical Research Involving Human Tissue, jointly developed by the WHO and the International Council of Medical Science Organizations.
Whole‐genome sequencing
Adequate amounts of high‐quality DNA of tumor and normal samples were used to construct libraries (TruSeq Library Construction Kit; Illumina, San Diego, CA, USA) according to the manufacturer’s supplied protocols. High qualified DNA samples were randomly broken into 350‐bp fragments using ultrasonicator. Then the fragmented genomic DNA was end repaired and phosphorylated, followed by the addition of A‐tailing, ligated index adaptation, denaturing and amplification for the final product. After genomic DNA library detection, the qualified libraries were sequenced using HiSeq XTen (Illumina). The depth of sequencing was 100× for tumor samples and 30× for the peripheral blood sample.
Bioinformatics analyses of the sequence data
The raw data (sequenced reads) acquired by sequencing were preprocessed by eliminating the low‐quality reads. The sequence coverage for the primary and relapse tumor samples was 98.85% and 98.68%, respectively. The cleaned reads of tumor and normal tissues were then aligned to reference genome (UCSU hg19) [23] using burrows‐wheeler aligner (BWA) software [24] to obtain the initial results, saved in BWA format. The BWA format files were processed by duplication removal, local realignment and base quality recalibration using picard (http://sourceforge.net/projects/picard/), gatk [25] and samtools [26], respectively, to get the final comparison results. Single nucleotide polymorphisms (SNPs) and insertions and deletions (INDELs) were detected using gatk, whereas copy number variations (CNVs) were identified by control‐FREEC [27]. Then variations were annotated using ANNOVAR [28], including genes related to variation, genomic character annotation and function of related genes. Then the somatic SNPs and INDELs were identified using mutect [29] and strelka [30] software, whereas somatic structure variations (SVs), including interchromosomal and intrachromosomal translocations, deletions and insertions, were identified using crest [31], and somatic CNVs were identified by control‐FREEC (Fig. S1). The feature of the identified variants was visualized using the Integrative Genomics Viewer tool [32].
Clonal evolution analysis
Clonal evolution analysis of the primary and the relapse tumor was conducted using expands [33]. Based on the somatic SNPs and CNVs of the tumor, expands predicts the number of subpopulations (SPs) that coexist in a tumor, the size of the SPs in the tumor bulk and the mutations that mark each SP.
Results
SNPs, INDELs, CNVs and chromosome translocations detected in primary and relapse tumor by whole‐genome sequencing
Mutations of primary tumor and relapse tumor were detected through aligning the sequence data to the reference genome (UCSC hg19) (Fig. 1). Compared with the primary tumor, more genetic variants (including the SNPs, INDELs, CNVs and chromosome translocations) were detected in the relapse tumor (Fig. 2). Amino acid–altering SNP annotations were conducted, including the SNPs located in cancer‐related genes. The cancer‐related genes in the peripheral blood sample and primary and relapse tumors are listed in Table 1.
Fig. 1.
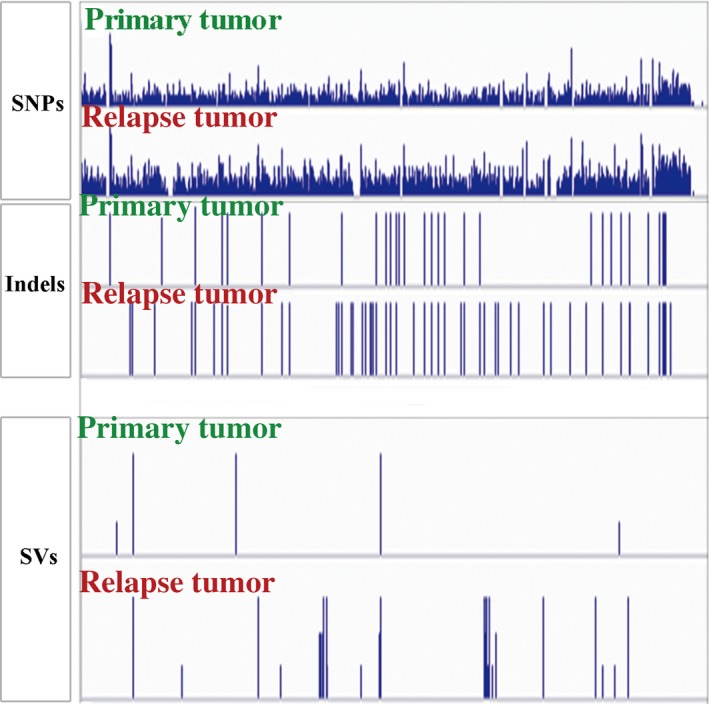
Genetics variants detected in primary and relapse tumor tissues. The identified variants were displayed using the Integrative Genomics Viewer.
Fig. 2.
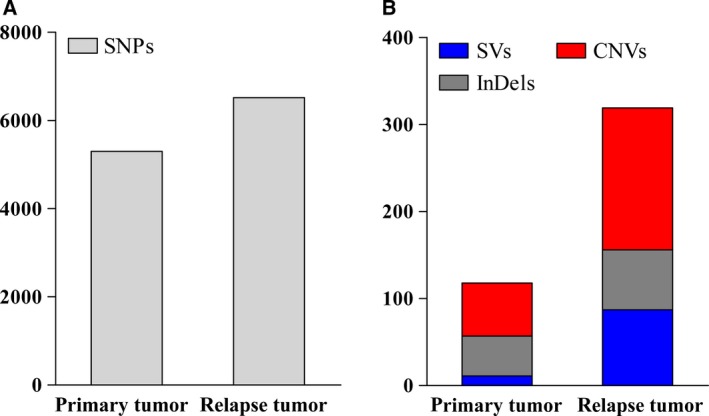
Numbers of SNPs (A), SVs (B), CNVs (B) and insertion and deletions (InDels; B) in the primary and relapse tumor. After filtering the low‐quality reads data, the clean data were used to identify the genetic variants; more variants were detected in the relapse tumor.
Table 1.
Amino acid–altering sequence variants located on cancer‐related genes in the peripheral blood sample, primary tumor and relapse tumor. 0/1, heterozygote; 1/1, homozygote; GT, genotype.
| Sample | Gene | snpID | Variant | GT |
|---|---|---|---|---|
| Peripheral blood | NOTCH1 | – |
NC_000009.11:g.139408966T>C NM_017617:exon13:c.A2203G:p.N735D |
0/1 |
| Primary tumor | ATIC | rs2372536 |
NC_000002.11:g.216190020C>G NM_004044:exon5:c.C347G:p.T116S |
0/1 |
| CIITA | rs7197779 |
NC_000016.9:g.11002927A>G NM_001286403:exon10:c.A947G:p.Q316R |
1/1 | |
| NOTCH1 | – |
NC_000009.11:g.139408966T>C NM_017617:exon13:c.A2203G:p.N735D |
0/1 | |
| KCNJ5 | rs7102584 |
NC_000011.9:g.128782012C>G NM_000890:exon2:c.C844G:p.Q282E |
1/1 | |
| PDE4DIP | rs140993521 |
NC_000001.10:g.144863438G>T NM_001198834:exon37:c.C5965A:p.Q1989K |
0/1 | |
| rs1698605 |
NC_000001.10:g.144871738C>A NM_001198834:exon32:c.G5224T:p.A1742S |
0/1 | ||
| rs145568299 |
NC_000001.10:g.144922593C>T NM_001002811:exon3:c.G1303A:p.A435T |
0/1 | ||
| PER1 | rs2585405 |
NC_000017.10:g.8046772C>G NM_002616:exon19:c.G2884C:p.A962P |
0/1 | |
| ROS1 | rs619203 |
NC_000006.11:g.117622184G>C NM_002944:exon42:c.C6686G:p.S2229C |
0/1 | |
| rs529156 |
NC_000006.11:g.117622188T>G NM_002944:exon42:c.A6682C:p.K2228Q |
0/1 | ||
| rs529038 |
NC_000006.11:g.117622233C>T NM_002944:exon42:c.G6637A:p.D2213N |
0/1 | ||
| TP53 | – |
NC_000017.10:g.7578492C>T NM_001126115:exon1:c.G42A:p.W14X |
0/1 | |
| Relapse tumor | IDH1 | rs121913500 |
NC_000002.11:g.209113112C>T NM_001282386:exon4:c.G395A:p.R132H |
0/1 |
| NOTCH2 | rs11810554 |
NC_000001.10:g.120611964G>C NM_001200001:exon1:c.C57G:p.C19W |
0/1 | |
| PDE4DIP | rs2455986 |
NC_000001.10:g.144852390C>T NM_001198834:exon44:c.G7053A:p.W2351X |
0/1 | |
| NOTCH1 | – |
NC_000009.11:g.139408966T>C NM_017617:exon13:c.A2203G:p.N735D |
0/1 | |
| PER1 | rs2585405 |
NC_000017.10:g.8046772C>G NM_002616:exon19:c.G2884C:p.A962P |
1/1 | |
| TP53 | – |
NC_000017.10:g.7578492C>T NM_001126115:exon1:c.G42A:p.W14X |
1/1 |
SPs detected in the primary and the relapse tumor
Somatic mutations are gene mutations that occur in somatic cells after conception, which can lead to a variety of medical issues and are associated with cancers. Based on the somatic SNPs and CNVs of the tumor, four and three SPs were detected in the primary and the relapse tumor, respectively (Fig. 3). Dominant SPs detected in the relapse tumor shared no significant fraction of SNPs with SPs from the primary tumor.
Fig. 3.

Clonal structures of the primary tumor (TY‐1, A) and the relapse tumor (TY‐2, B) constructed by expands. Somatic SNPs and CNVs were used to construct the clonal structures. A “tree‐like” structure was identified in the primary tumor (A) and a “parallel” structure in the relapse tumor (B).
Somatic structural variants shared in the primary and the relapse tumor
Structural variants are large gene fragment variations in the genome that may influence the expression of genes whose biological functions are vital. First, the chromosome aberrations generated by the SVs were identified in the primary and relapse tumor, and then the genes involved in the chromosome variants were annotated. Chromosome aberrations involving the cancer‐related genes and/or DNA repair genes were shown in both primary and relapse tumor tissues (Table 2).
Table 2.
Chromosome aberrations involving the cancer‐related genes and/or DNA repair genes in the primary tumor (astrocytoma, TY‐1) and the relapse tumor (glioblastoma, TY‐2).
| Sample | Gene_A | Junction_A | Gene_B | Junction_B | Fusion pair |
|---|---|---|---|---|---|
| Primary tumor | MRPL57 | 13:21750661 | DDX10 | 11:108585751 | MRPL57‐DDX10 |
| MYO5B | 18:47400964 | RAD54B | 8:95409470 | MYO5B‐RAD54B | |
| Relapse tumor | BRIP1 | 17:59932940 | ZNF529 | 19:37039340 | BRIP1‐ZNF529 |
| NSD1 | 5:176672875 | ROBO1 | 3:79308398 | NSD1‐ROBO1 | |
| BCOR | X:39939333 | NPAS3 | 14:33520196 | BCOR‐NPAS3 | |
| PCM1 | 8:17814538 | MLLT11 | 1:151035150 | PCM1‐MLLT11 | |
| SET | 9:131457148 | DPP10 | 2:116376668 | SET‐DPP10 | |
| ABL1 | 9:133751372 | CFAP36 | 2:55760693 | ABL1‐CFAP36 | |
| FOXP1 | 3:71030047 | C8orf37‐AS1 | 8:96526713 | FOXP1‐C8orf37‐AS1 | |
| ACSS2 | 20:33462999 | RNF43 | 17:56455089 | ACSS2‐RNF43 | |
| LAMA1 | 18:7073839 | PTPN11 | 12:112866611 | LAMA1‐PTPN11 | |
| FAM219A | 9:34453196 | NTRK3 | 15:88462594 | FAM219A‐NTRK3 | |
| SKA3 | 13:21750661 | DDX10 | 11:108585748 | SKA3‐DDX10 | |
| PELI2 | 14:56606684 | BRAF | 7:140486815 | PELI2‐BRAF | |
| LOC101927967 | 2:78504999 | BCL11B | 14:99701952 | LOC101927967‐BCL11B | |
| EPB41L4B | 9:111981592 | ALK | 2:29974050 | EPB41L4B‐ALK | |
| RABGAP1L | 1:174333695 | MLH3 | 14:75499567 | RABGAP1L‐MLH3 |
Discussion
Sequencing the genomic DNA of diverse cancers has revealed that intratumors have spatially and temporally heterogeneous clonal architecture [34, 35, 36, 37], which has clinical implications and contributes to therapy resistance [38, 39, 40]. Astrocytoma is the most common of the gliomas, and low‐grade astrocytomas could progress to malignancy. To reveal the pathogenesis and progression of astrocytomas, more studies have focused on identifying the genetic variants in different stages of the tumor. It is important to clarify the reasons why low‐grade tumors recur and progress to malignancy, because this dramatically shortens patient survival. Here we sequenced the genomic DNA of primary tumor and relapse tumor tissues from a patient with astrocytoma to explore the mutational profile associated with relapse, which allowed us to define clonal evolution patterns at relapse.
In this study, the “baseline” background of the patient was a heterozygous NOTCH‐1 (NC_000009.11:g.139408966T>C) missense mutation (Table 1). Notch‐1 plays an essential role of cell fate determination. It is difficult to determine whether the astrocytoma cells originated from the Notch‐1 mutation, although Notch‐1 mutations are among driver mutations in all astrocytomas and glioblastomas, as well as normal tissues.
IDH1 and IDH2 genes mutations were hallmarks of gliomas [41]. IDH1 gene mutation could be identified in most of the patients with low‐grade (grade II–III) astrocytomas and glioblastomas, whereas the gene mutation was rarely detected in patients with primary glioblastomas [18, 19, 42]. Although the IDH1 mutation has been considered a hallmark of astrocytoma and a good prognosis, an IDH1 mutation was not identified until the late stage (relapse tumor, WHO IV). In this case, the IDH1 mutation is likely a secondary change after the accelerated growth of tumor cells and hypoxia.
TP53 mutations were found in most of the patients with astrocytomas with IDH1 mutations [43]. TP53 mutations could be found in most of the low‐grade astrocytomas and secondary glioblastomas, in which the mutation rate in primary glioblastomas was relatively low [16, 20]. In our study, a heterozygous nonsense mutation of TP53 (NC_000017.10:g.7578492C>T) was found in the astrocytoma (WHO II, TY‐1) sample. One of the key mutations found in the WHO stage IV glioblastoma is the homozygous TP53 nonsense mutation. TP53 is a gatekeeper of cell survival after DNA damage; astrocytoma cells with the TP53 mutation have an advantage under selection from radiation and chemotherapy. Indeed, the clonal structure changed dramatically from a “tree‐like” structure in astrocytoma to a “parallel” structure in glioblastoma, suggesting a multiple clonal origin after the TP53 checkpoint failed.
The relapse of astrocytoma is quite common. Because surgical treatments are unlikely to remove all tumor cells, radiation and chemotherapy are suggested. However, very aggressive radiation (60 Gy) provides selection pressure for TP53 mutation‐free cells: cells with TP53 mutations and impaired TP53 function are more likely to survive after radiation therapy.
Indeed, the number of mutations and structure variants increased significantly in the relapse tumor. The tree‐like structure of subclones in the primary tumor could no longer be found in the relapse tumor, suggesting a much higher heterogeneity. The complete loss of TP53 could result in an impaired repairing of DNA damage; therefore, DNA mutations and chromosomal rearrangement became uncontrollable.
As well known, both radiation and chemotherapy could induce DNA damage and mutations. While killing most of the tumor cells, chemotherapies and radiation may provide selection pressures on some of the tumors. It might be a little premature to blame radiation and chemotherapy for the notorious relapsing of glioblastoma, but aggressive radiation therapy of astrocytoma may need reappraisal.
Conclusions
The progressive development from astrocytoma to malignant glioblastoma remains enigmatic; however, our clonal evolution studies suggest accelerated carcinogenesis in the relapse tumor after intensive radiation and chemotherapy. It may suggest that aggressive treatment may not be a good choice for astrocytoma.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
JC, W‐DL and QH designed the study. FY, QH and W‐DL wrote the manuscript. YZ, QG and QH collected the subject and clinical data. FY and W‐DL analyzed the data.
Supporting information
Fig. S1. The workflow for the experimental processing and data analysis.
Acknowledgements
This work was supported in part by National Key R&D Program of China (Grant 2017YFC1001900), National Natural Science Foundation of China (Grants NSFC 81270605, 30971066 and 81470324), Third Military Medical University Clinical and Science Great Fund Project (Grant 2102XLC03), Chongqing Postgraduate Education Reform Project (Grant yjg123114), Chongqing Natural Science Fund Project (Grant CSTC'2008BA5001) and the Military Emphasis Medical Scientific Research Project Fund (to JC).
Fuhua Yang, Yunding Zou and Qiang Gong contributed equally to this article
Contributor Information
Jieping Chen, Email: chenjpxn@163.com.
Wei‐Dong Li, Email: liweidong98@tmu.edu.cn.
Qilin Huang, Email: hqlxqyy@sina.com.
References
- 1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Suva ML (2014) Genetics and epigenetics of gliomas. Swiss Med Wkly 144, w14018. [DOI] [PubMed] [Google Scholar]
- 3. Yong RL and Tsankova NM (2015) Emerging interplay of genetics and epigenetics in gliomas: a new hope for targeted therapy. Semin Peadiatr Neurol 22, 14–22. [DOI] [PubMed] [Google Scholar]
- 4. Grant R, Kolb L and Moliterno J (2014) Molecular and genetic pathways in gliomas: the future of personalized therapeutics. CNS Oncol 3, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chaichana KL, McGirt MJ, Laterra J, Olivi A and Quinones‐Hinojosa A (2010) Recurrence and malignant degeneration after resection of adult hemispheric low‐grade gliomas. J Neurosurg 112, 10–17. [DOI] [PubMed] [Google Scholar]
- 6. Shafqat S, Hedley‐Whyte ET and Henson JW (1999) Age‐dependent rate of anaplastic transformation in low‐grade astrocytoma. Neurology 52, 867–869. [DOI] [PubMed] [Google Scholar]
- 7. Babu R, Bagley JH, Park JG, Friedman AH and Adamson C (2013) Low‐grade astrocytomas: the prognostic value of fibrillary, gemistocytic, and protoplasmic tumor histology. J Neurosurg 119, 434–441. [DOI] [PubMed] [Google Scholar]
- 8. Loh JK, Lieu AS, Chai CY, Hwang SL, Kwan AL, Wang CJ and Howng SL (2013) Arrested growth and spontaneous tumor regression of partially resected low‐grade cerebellar astrocytomas in children. Childs Nerv Syst 29, 2051–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bagley JH, Babu R, Friedman AH and Adamson C (2013) Improved survival in the largest national cohort of adults with cerebellar versus supratentorial low‐grade astrocytomas. Neurosurg Focus 34, E7. [DOI] [PubMed] [Google Scholar]
- 10. Karnes PS, Tran TN, Cui MY, Raffel C, Gilles FH, Barranger JA and Ying KL (1992) Cytogenetic analysis of 39 pediatric central nervous system tumors. Cancer Genet Cytogenet 59, 12–19. [DOI] [PubMed] [Google Scholar]
- 11. Limon J (1994) Cytogenetics of astrocytomas. Folia Neuropathol 32, 205–207. [PubMed] [Google Scholar]
- 12. Hiniker A, Hagenkord JM, Powers MP, Aghi MK, Prados MD and Perry A (2013) Gliosarcoma arising from an oligodendroglioma (oligosarcoma). Clin Neuropathol 32, 165–170. [DOI] [PubMed] [Google Scholar]
- 13. Alentorn A, Labussiere M, Sanson M, Delattre JY, Hoang‐Xuan K and Idbaih A (2013) Genetics and brain gliomas. Presse Med 42, 806–813. [DOI] [PubMed] [Google Scholar]
- 14. Liu Q, Liu Y, Li W, Wang X, Sawaya R, Lang FF, Yung WK, Chen K, Fuller GN and Zhang W (2015) Genetic, epigenetic, and molecular landscapes of multifocal and multicentric glioblastoma. Acta Neuropathol 130, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones TS and Holland EC (2011) Molecular pathogenesis of malignant glial tumors. Toxicol Pathol 39, 158–166. [DOI] [PubMed] [Google Scholar]
- 16. Kanu OO, Hughes B, Di C, Lin N, Fu J, Bigner DD, Yan H and Adamson C (2009) Glioblastoma multiforme oncogenomics and signaling pathways. Clin Med Oncol 3, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ohgaki H and Kleihues P (2005) Population‐based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol 64, 479–489. [DOI] [PubMed] [Google Scholar]
- 18. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic‐Haberle I, Jones S, Riggins GJ et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huse JT and Aldape KD (2014) The evolving role of molecular markers in the diagnosis and management of diffuse glioma. Clin Cancer Res 20, 5601–5611. [DOI] [PubMed] [Google Scholar]
- 20. Khani P, Nasri F, Khani Chamani F, Saeidi F, Sadri Nahand J, Tabibkhooei A and Mirzaei H (2019) Genetic and epigenetic contribution to astrocytic gliomas pathogenesis. J Neurochem 148, 188–203. [DOI] [PubMed] [Google Scholar]
- 21. Rohle D, Popovici‐Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E et al (2013) An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Konovalov NA, Asyutin DS, Shayhaev EG, Kaprovoy SV and Timonin SY (2019) Molecular biomarkers of brain and spinal cord astrocytomas. Acta Naturae 11, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM and Haussler D (2002) The human genome browser at UCSC. Genome Res 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H and Durbin R (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M et al (2011) A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G and Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix‐Lerosey I, Delattre O and Barillot E (2012) Control‐FREEC: a tool for assessing copy number and allelic content using next‐generation sequencing data. Bioinformatics 28, 423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang K, Li M and Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES and Getz G (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ and Cheetham RK (2012) Strelka: accurate somatic small‐variant calling from sequenced tumor‐normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- 31. Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, Rusch MC, Chen K, Harris CC, Ding L et al (2011) CREST maps somatic structural variation in cancer genomes with base‐pair resolution. Nat Methods 8, 652–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G and Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Andor N, Harness JV, Muller S, Mewes HW and Petritsch C (2014) EXPANDS: expanding ploidy and allele frequency on nested subpopulations. Bioinformatics 30, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Caldas C (2012) Cancer sequencing unravels clonal evolution. Nat Biotechnol 30, 408–410. [DOI] [PubMed] [Google Scholar]
- 35. Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang H et al (2014) Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 512, 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Greaves M and Maley CC (2012) Clonal evolution in cancer. Nature 481, 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alderton GK (2014) Tumour evolution: clonal ancestry in lung cancer. Nat Rev Cancer 14, 763. [DOI] [PubMed] [Google Scholar]
- 38. Yu H, Han Z, Wang Y and Xin H (2014) The clonal evolution and therapeutic approaches of lung cancer. Cell Biochem Biophys 70, 63–71. [DOI] [PubMed] [Google Scholar]
- 39. Fisher R, Pusztai L and Swanton C (2013) Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer 108, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma QC, Ennis CA and Aparicio S (2012) Opening Pandora's box – the new biology of driver mutations and clonal evolution in cancer as revealed by next generation sequencing. Curr Opin Genet Dev 22, 3–9. [DOI] [PubMed] [Google Scholar]
- 41. Yang H, Ye D, Guan KL and Xiong Y (2012) IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res 18, 5562–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cohen AL, Holmen SL and Colman H (2013) IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 13, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Watanabe T, Nobusawa S, Kleihues P and Ohgaki H (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174, 1149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The workflow for the experimental processing and data analysis.