

Abstract
The Fanconi anemia (FA) DNA damage response (DDR) pathway regulate important cellular processes such as DNA replication, cell cycle control and DNA damage repair. Here we show that FANCD2, a key member of the FA DDR pathway, interacts with several important components of the germ-cell-specific Prmt5/piRNA pathways that orchestrate the repression of transposable elements (TEs). By using the Pou5f1-eGFP reporter mice, which marks pure populations of primordial germ cells (PGCs), we demonstrate that FA deficiency results in de-repression of TEs, depletion of PGCs, and defective spermatogenesis and oogenesis. Fancd2–KO PGCs exhibited excessive DNA damage and exacerbated apoptosis. Mechanistically, we observed a significant reduction of PRMT5-catalyzed H2A/H4R3me2s marks on the LINE1 TEs in E10.5 PGCs of Fancd2-KO; Pou5f1-eGFP and Fanca-KO;Pou5f1-eGFP embryos. Furthermore, we utilized the Fancd2-KI model to show that Fancd2 and Prmt5 co-occupied the promoter of LINE1 in WT PGCs, and that this co-occupancy was lost in FA-deficient (Fanca-KO) PGCs. These results suggest that the FA pathway takes part in TE repression in early PGCs, likely through a mechanism involving Fancd2-facilitated, Prmt5-catalyzed repressive H2A/H4R3me2s marks on TEs.
Keywords: DNA damage response (DDR), Fanconi anemia (FA), primordial germ cells (PGCs), Prmt5/piRNA pathways, transposable elements (TEs)
Introduction
Fanconi anemia (FA) is a genetic disease associated with bone marrow failure, increased cancer, and severe germline defects and the FA pathway is known to play a major role in the DNA damage response (DDR) network (Bagby 2003, Tischkowitz & Hodgson 2003, Bagby 2018). FA is genetically heterogeneous, with at least 22 complementation groups (FANCA-FANCW) identified thus far (Dong et al. 2015, Sawyer et al. 2015, Knies et al. 2017). Eight of the FA proteins (FANCA, B, C, E, F, G, L, and M) form the FA core complex that functions as an ubiquitin ligase. In response to DNA damage or DNA replication stress, the FA core complex monoubiquitinates two downstream FA proteins, FANCD2 and FANCI, which then recruit the downstream FA proteins and other DNA repair factors, to nuclear loci containing damaged DNA and consequently influence important cellular processes such as DNA replication, cell-cycle control, and DNA damage response and repair (Kottemann & Smogorzewska, 2013; Deans & West, 2011).
DDR comprises an elaborate network of signaling pathways that enable cells to execute biological responses to genotoxic stress (Jackson & Bartek 2009). Defects in DDR pathways have been shown to be associated with infertility, gamete aneuploidy and diseased risk in offspring (Leduc et al. 2008). As a major DDR pathway, FA has been subjected to extensive study for its role in germline genomic maintenance. In addition to genome instability and cancer susceptibility, FA deficiency in mice has been shown to compromise male and female fertility. Specifically, deletion of the genes for the murine FA proteins Fanca, Fancb, Fancc, Fancl, Fancm and Fancp causes primordial germ cell (PGC) depletion, meiotic defects, and ultimately hypogonadism and infertility (Nadler & Braun 2000, Agoulnik et al. 2002, Wong et al. 2003, Crossan et al. 2011, Kato et al. 2015, Alavattam et al. 2016). More recently, it was shown that hypogonadism of Fancm mutant mice was a result of reduced proliferation of PGCs, likely resulting from elevated DNA damage (Luo et al. 2014). These studies highlight the importance of the FA DDR pathway in germ cell development and function. A recent study demonstrated that the FA pathway is critical for DNA crosslinking repair to safeguard genomic stability of PGCs (Hill RJ & Crossan GP 2019). However, the mechanism by which the FA pathway functions in genome maintenance in the germline, particularly in PGCs, remains largely undefined.
Transposable elements (TEs) induce DNA double-strand breaks (DSBs), insertional mutations, and chromosome rearrangements, leading to genomic instability (Goodier & Kazazian 2008). This genetic consequence is particularly dangerous in germ cells, as genome changes induced by TEs will be transmitted to the next generation. Thus, TE silencing is considered an essential mechanism for defending germline integrity and fertility. There are two key mechanisms for the repression of germline TEs: the H2A/H4R3me2s modification catalyzed by the protein arginine methyltransferase 5 (PRMT5) in early (E8.5-E11.5) PGCs, and the de novo DNA methylation orchestrated by the Piwi-interacting small RNA (piRNA) pathway initiating at approximately E14.5 (Aravin et al. 2008, Kim et al. 2014). Consequently, mice deficient in genes encoding Prmt5 or factors in the piRNA pathway exhibit overexpression of TEs, loss of germ cells and ultimately male and/or female sterility (Saito & Siomi 2010, Siomi et al. 2011, Kim et al. 2014, Weick & Miska 2014, Berrens & Reik 2015). Despite extensive investigation, the mechanisms by which germ cells control TE activity are largely not known and, in particular, the DDR pathways functioning in this critical process have not been identified.
In the current study, we show that Fancd2, a key member of the FA DDR pathway, interacts with several important components of the Prmt5/piRNA pathways essential for TE silencing in primordial germ cells (PGCs). Loss of FANCD2 results in massive de-repression of TEs, depletion of PGCs, defective spermatogenesis and oogenesis, and ultimately infertility. Early FA-deficient PGCs show reduced H2A/H4R3me2s marks on LINE1 transposons. Further, we reveal the co-occupancy of Fancd2 and Prmt5 at the promoter of LINE1 TEs in early PGCs.
Results
Fancd2 interacts with germline-specific Prmt5/piRNA factors
We recently reported that by using a Fancd2 knock-in mouse model and proteomic approach, we unveiled a FANCD2 in vivo interaction network (Zhang et al. 2017). Significantly, analysis of Fancd2-containing complexes from four different tissues (ES cells, E11.5 embryos, testes and spleen) revealed distinct patterns of Fancd2-associated proteins (Fig. 1A), suggesting the presence of developmental and tissue-specific differences in the repertoire of Fancd2 interactors. Specifically in testes, we observed several proteins of the germ-cell-specific Prmt5/piRNA pathways, which orchestrate the repression of transposable elements (TEs) (Saito & Siomi 2010, Siomi et al. 2011, Kim et al. 2014, Weick & Miska 2014, Berrens & Reik 2015). We validated the interaction between the FLAG-tagged Fancd2 and these Prmt5/piRNA factors in Fancd2-KI mice (Fig 1B). We also addressed whether the loss of Fancd2 affected these Prmt5/piRNA factors. Western analysis for Mael and Tdrd8 showed that these proteins were still present in the Fancd2-KO testes (Fig 1C). However, the levels of Prmt5, Mili, Tdrd1 and Tdrd6 were reduced in the Fancd2-KO testes compared to those in WT and Fancd2-KI mice (Fig 1C). These results indicate Fancd2 interacts with germline-specific Prmt5/piRNA factors and suggest that Fancd2 may play a role in the regulation of germline TEs.
Figure 1. Fancd2 interacts with germline-specific Prmt5/piRNA factors.

(A) List of tissue specific Fancd2-associated proteins. The testis-specific Prmt5/piRNA factors are marked by bold. (B) Validation of the testis-specific Prmt5/piRNA factors by anti-FLAG immunoprecipitation (IP) from testis lysates and detected by antibodies against the indicated proteins. (C) Western analysis of the indicated Prmt5/piRNA factors in the testes of WT, Fancd2-KI and Fancd2-KO mice.
Loss of Fancd2 leads to defective spermatogenesis and oogenesis
The observation that Fancd2 interacts with Prmt5/piRNA factors prompted us to determine whether Fancd2 plays a role in germline development. Consistent with previous report (Houghtaling et al. 2003), we found that Fancd2-KO mice were smaller than WT or Fancd2-KI mice at the embryonic stage (d.p.c 13.5) (Fig. 2A). Second, the size of testes and ovary in 8-week-old-adult Fancd2-KO mice were significantly reduced in comparison to age-matched WT or and Fancd2-KI mice (Fig. 2B). Third, H&E stained testis and ovary of 8-week-old WT, Fancd2-KO and Fancd2-KI mice show severely defective spermatogenesis and oogenesis in Fancd2-KO mice, as compared to WT and Fancd2-KI mice (Fig. 2C). Consequently, loss of Fancd2 results in complete sterility in both males and females (Zhang et al. 2017 and data not shown). Thus, these data confirm the past finding and suggest that Fancd2 is required for germline maintenance.
Figure 2. Loss of Fancd2 leads to hypogonadism.
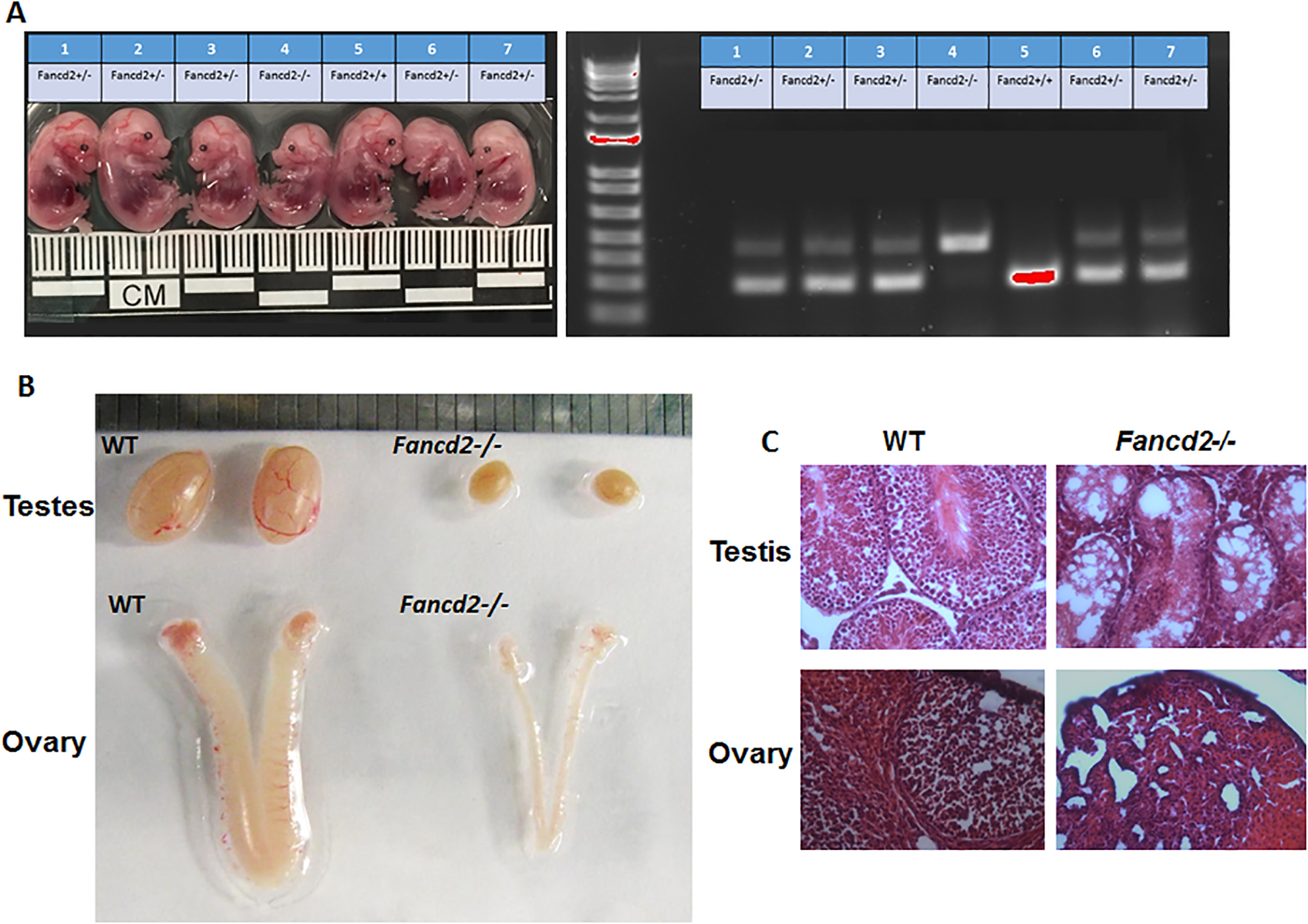
(A) Images of embryos (Left) of the indicated genotypes at E13.5. Genotypes (Right) of the embryos were determined by PCR. (B) Representative photo of the testes and ovary of WT and Fancd2-KO mice. (C) Gonadal defects in Fancd2−/− mice. Shown are H&E stained sections of testes and ovaries from 8-week-old WT and Fancd2−/− mice (200× magnification).
Loss of Fancd2 results in depletion of PGCs during embryonic development
It has been reported that deletion of the mouse FA genes Fanca, Fancb, Fancc, Fancl, Fancm and Fancp causes primordial germ cell (PGC) depletion, hypogonadism and ultimately infertility (Nadler & Braun 2000, Agoulnik et al. 2002, Wong et al. 2003, Crossan et al. 2011, Kato et al. 2015). In addition, it has been shown that hypogonadism in Fancm mutant mice is a result of reduced proliferation of PGCs (Luo et al. 2014). Therefore, we asked whether Fancd2 deficiency would affect PGC maintenance during embryonic development. To facilitate the purification and analysis of PGCs by flow cytometry, we crossed Fancd2+/− mice to Pou5f1 (Oct4)-eGFP (Pou5f1-eGFP hereafter) reporter mice, which have been used to mark and isolate pure populations of PGCs by FACS (Szabo et al. 2002). Using another PGC marker, SSEA1 (Hayashi et al. 2011), we observed a significant reduction of GFP+ SSEA1+ PGCs in Fancd2-KO mice compared to WT controls at both E11.5 (WT 384 ± 76 vs Fancd2-KO 147 ± 49, n =5) and E13.5 (WT 634 ± 61 vs Fancd2-KO 283 ± 44, n =6) (Fig. 3A, 3B). These results indicate that loss of Fancd2 leads to PGC depletion.
Figure 3. Loss of Fancd2 leads to PGC depletion.
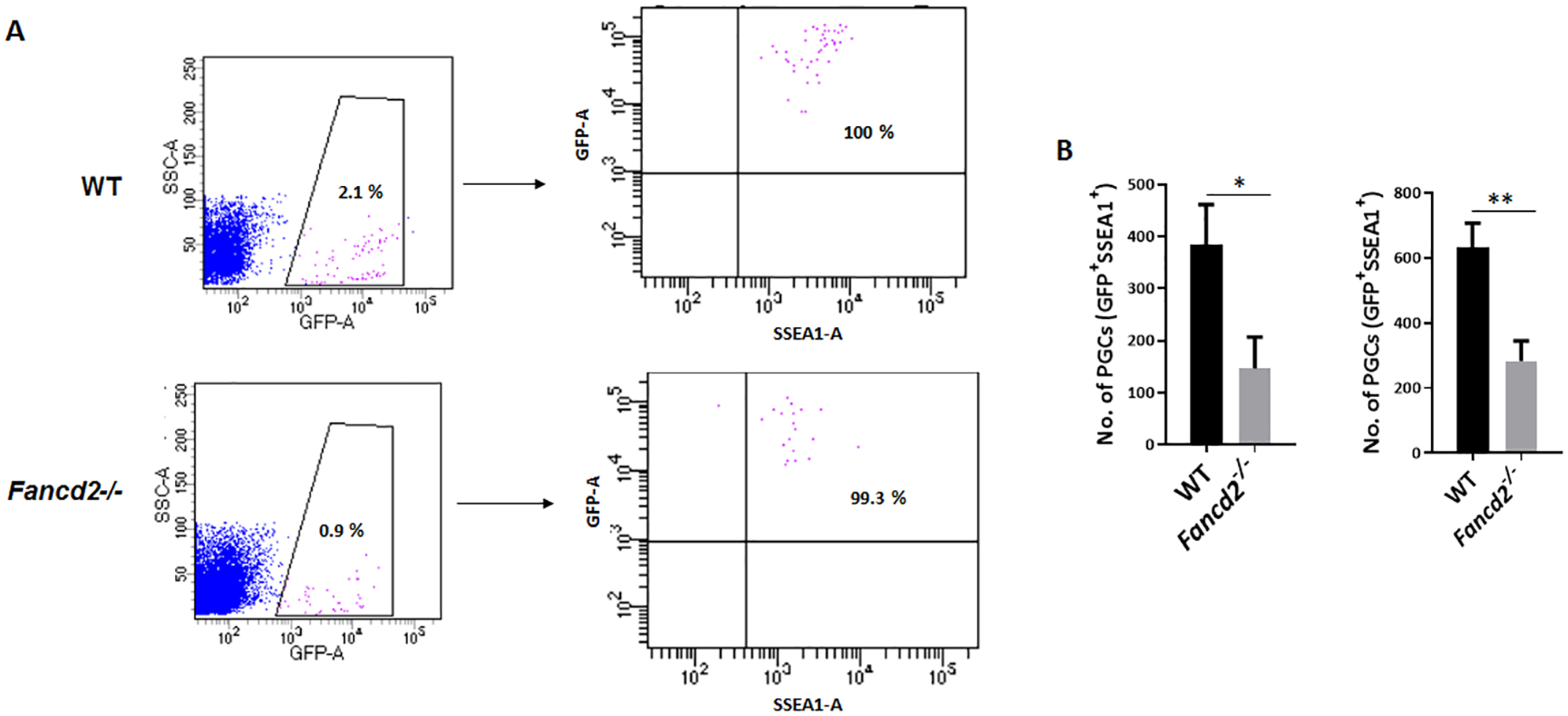
(A) Representative dot plots of flow cytometric analysis of GFP+ SSEA1+ PGCs from E11.5 embryos of WT and Fancd2−/− females expressing the Pou5f1-eGFP reporter gene. (B) The number of PGCs at E11.5 and E13.5 embryos of WT and Fancd2−/− females expressing the Pou5f1-eGFP reporter gene was determined by flow cytometry for GFP+ SSEA1+ PGCs. Results are presented as mean ± SD of three independent experiments. *p < 0.05, **p < 0.01
Reduced proliferation and increased apoptosis in Fancd2-KO PGCs
Since we observed a significant reduction of PGCs in Fancd2-KO embryos, we sought to determine the effect of Fancd2 deficiency on the proliferation and survival of mouse PGCs. To this end, GFP+ PGCs from E11.5 embryos were sorted by FACS and cultured on STO feeder cells for 15 days (Fig. 4A). We found that Fancd2-KO PGCs proliferated significantly slower than WT PGCs in stroma-supported culture (Fig. 4B), suggesting that Fancd2 is essential for proliferation of PGCs. Consistent with these observations, the number of BrdU-positive cells was reduced in Fancd2-KO PGCs (16.2 ± 4.1%, n =5) compared to WT controls (27.8 ± 6.4%, n =5) (Fig. 4C), indicating that Fancd2 is required for proliferation of mouse PGCs. To determine whether Fancd2 is directly involved in the cell survival of mouse PGCs, we analyzed apoptosis using Annexin V staining followed by flow cytometry. Notably, Annexin V-positive cells were up-regulated in Fancd2-KO PGCs (5.40 ± 0.38%, n =5) compared to WT controls (1.73± 0.62%, n =5) (Fig. 4D). These data suggest that Fancd2 plays a role in regulating proliferation and survival of mouse PGCs.
Figure 4. Reduced proliferation and increased apoptosis in Fancd2-KO PGCs.
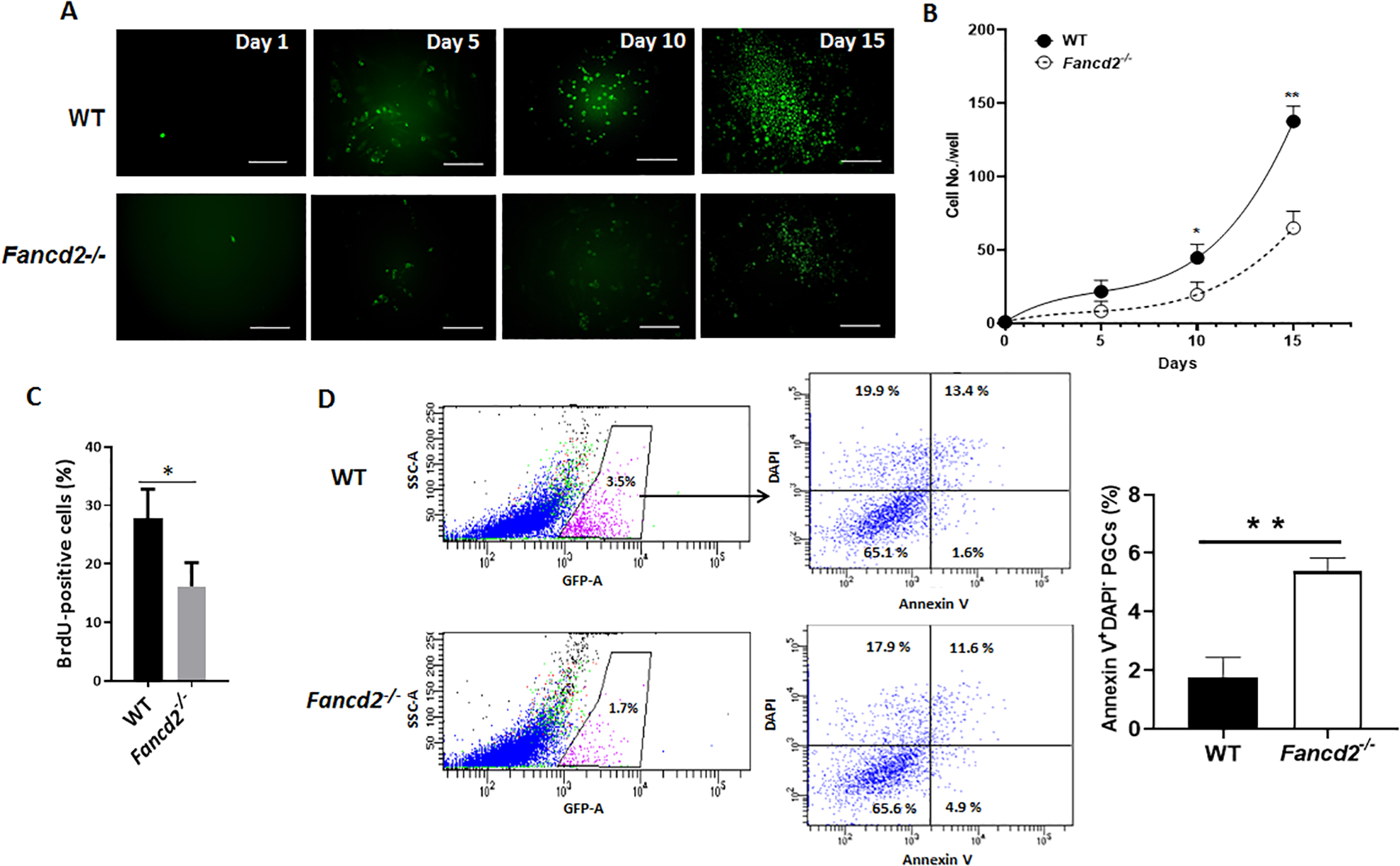
(A) Reduced proliferation in Fancd2-KO PGCs. GFP+ PGCs from E11.5 embryos of WT and Fancd2−/− females expressing the Pou5f1-eGFP reporter gene were sorted by FACS and cultured on STO feeder cells for 15 days. Images were taken at the indicated time points. (B) Cumulative growth curve of PGCs during PGC culture initiation. GFP+ PGCs from E11.5 embryos of WT and Fancd2−/− females expressing the Pou5f1-eGFP reporter gene were sorted by FACS and cultured on STO feeder cells for the indicated days. Values indicate mean ± SD of three independent experiments. *p < 0.05, **p < 0.01. (C) Diminished BrdU incorporation in Fancd2-KO PGCs. GFP+ PGCs from E11.5 embryos of WT and Fancd2−/− females expressing the Pou5f1-eGFP reporter gene were sorted by FACS and subjected to pulse-labeling with BrdU followed by flow cytometric analysis for BrdU-positive cells. Values indicate mean ± SD of three independent experiments. *p < 0.05. (D) Increased apoptosis in Fancd2-KO PGCs. GFP+ PGCs from E11.5 embryos of WT and Fancd2−/− females expressing the Pou5f1-eGFP reporter gene were subjected to flow cytometric analysis for Annexin V-positive, DAPI-negative apoptotic cells. Results are presented as mean ± SD of three independent experiments. **p < 0.01
FA deficiency leads to de-repression of germline TEs
Our proteomic analyses suggested that the FA pathway may play a TE silencing function in vivo during germ cell development. For this reason, we decided to further study the link between the FA pathway and germ cell-specific TE silencing machinery. To determine if the loss of Fancd2 results in TE de-repression in PGCs, we isolated GFP+ SSEA1+ PGCs from WT;Pou5f1-eGFP and Fancd2-KO;Pou5f1-eGFP mice, and determined the expression of two TEs, LINE1 (long interspersed nuclear elements) and IAP (intracisternal A particle), whose unrestrained expression has been linked to infertility (Hayashi et al. 2011), by quantitative qPCR. We observed a 4- to 7-fold increase in LINE1 and a 3- to 5-fold increase in IAP expression in E11.5 Fancd2-KO PGCs compared to WT controls (Fig. 5A). Thus, Fancd2 is essential for transcriptional or posttranscriptional silencing of two TEs known to cause infertility in mice.
Figure 5. FA deficiency leads to de-repression of germline TEs.
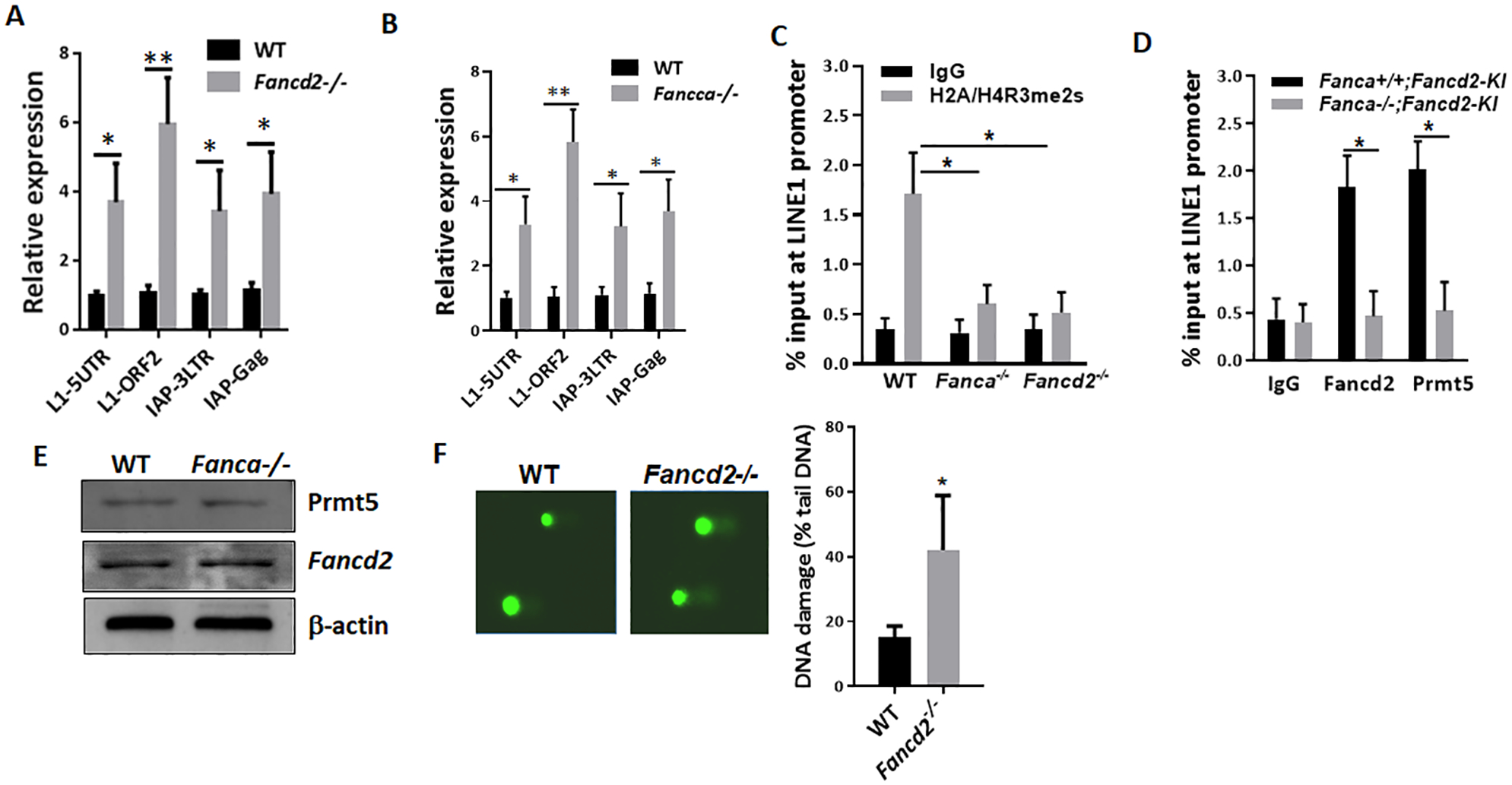
(A) Fancd2 is essential for TE repression in PGCs. qPCR analysis of TEs in FACS-sorted GFP+ SSEA1+ PGCs from E11.5 embryos of WT and Fancd2−/− females expressing the Pou5f1-eGFP reporter gene, using the primer sets described in Materials and Methods. Results are presented as mean ± SD of three independent experiments. *p < 0.05, **p < 0.01. (B) De-repression of germline TEs in Fanca-deficient PGCs. qPCR analysis of TEs in FACS-sorted GFP+ SSEA1+ PGCs from E11.5 embryos of WT and Fanca−/− females expressing the Pou5f1-eGFP reporter gene, using the primer sets described in Materials and Methods. Results are presented as mean ± SD of three independent experiments. *p < 0.05, **p < 0.01. (C) μChIP analysis for H2A/H4R3me2s at the promoter region of LINE1 TEs in FACS-sorted GFP+ SSEA1+ PGCs from E10.5 embryos of WT, Fanca−/− and Fancd2−/− females expressing the Pou5f1-eGFP reporter gene. Percentages of anti- H2A/H4R3me2s immunoprecipitated DNA compared to input are shown. w/o, without primary antibodies. Results are presented as mean ± SD of three independent experiments. *p < 0.05. (D) μChIP analysis for Fancd2 (FLAG) and Prmt5 at the promoter region of LINE1 TEs in FACS-sorted GFP+ SSEA1+ PGCs from E10.5 embryos of WT, Fanca−/− and Fancd2−/− females expressing the Pou5f1-eGFP reporter gene and the Fancd2KI/KI transgene. Percentages of anti-FLAG and anti-Prmt5 immunoprecipitated DNA compared to input are shown. w/o, without primary antibodies. Results are presented as mean ± SD of three independent experiments. *p < 0.05. (E) Western analysis of the Prmt5 and Fancd2 proteins in the in-vitro expanded PGCs from E13.5 WT (WT;Fancd2-KI) and Fanca-deficient (Fanca−/−;Fancd2-KI) embryos, using antibodies against the indicated proteins. (E) Western analysis of the Prmt5 and Fancd2 proteins in the E13.5 PGCs from WT (WT;Fancd2-KI) and Fanca-deficient (Fanca−/−;Fancd2-KI) mice, using antibodies against the indicated proteins. (F) Increased accumulation of DNA damage in Fancd2-deficient PGCs. Genomic DNA was stained with SYBR Gold and imaged after single cell electrophoresis (left). Quantification for the percent of DNA in the comet tail is shown (right). Results are presented as mean ± SD of three independent experiments. *p < 0.05.
To determine whether the depression of LINE1 and IAP TEs in PGCs required the FA core complex, we generated WT; Pou5f1-eGFP and Fanca−/−;Pou5f1-eGFP mice, and analyzed the levels of LINE1 and IAP TEs in E11.5 PGCs. Consistent with the observation in Fancd2-KO PGCs, Fanca deficiency also resulted in de-repression of LINE1 and IAP TEs in PGCs (Fig. 5B). These results indicate that the Fancd2/FA pathway is essential for TE repression during early germline development.
Prmt5-catalyzed repressive H2A/H4R3me2s is essential for TE silencing in PGCs between E8.5–E11.5 (Kim et al. 2014), when these actively dividing PGCs are undergoing global DNA demethylation (Hajkova et al. 2002, Seisenberger et al. 2012). To investigate the mechanistic link between the Fancd2/FA pathway and Prmt5-directed TE silencing in early PGCs, we generated three mouse models: 1) Fancd2-KO;Pou5f1-eGFP for the effect of Fancd2 deficiency on Prmt5-mediated H2A/H4R3me2s repressive chromatin modifications on LINE1 TEs; 2) Fanca-KO;Pou5f1-eGFP for the requirement of the FA core complex for these Prmt5-mediated H2A/H4R3me2s marks on LINE1; and 3) Fancd2-KI;Pou5f1-eGFP in a WT or Fanca-KO background for interaction between Fancd2 and Prmt5 on sites of H2A/H4R3me2s marks in LINE1 chromatin. We chose E10.5 PGCs to investigate the FANCD2-PRMT5 interaction in TE silencing because Prmt5-mediated H2A/H4R3me2s marks on LINE1 are enriched at E10.5-E11.5, which are responsible for TE repression during this specific stage of development when PGCs undergo global demethylation of DNA (Seisenberger et al. 2012, Hajkova et al. 2002). Since LINE1 TEs are silenced via 5′-UTR promoter methylation (Smith et al. 2012), we compared repressive H2A/H4R3me2s marks on the LINE1 5′-UTR promoter in E10.5 PGCs from WT;Pou5f1-eGFP, Fancd2-KO;Pou5f1-eGFP and Fanca-KO; Pou5f1-eGFP embryos by micro-chromatin immunoprecipitation (μChIP) assays (Dahl & Collas 2008). We observed that at E10.5, the LINE1 promoter was strongly enriched with H2A/H4R3me2s marks, indicative of a Prmt5- repressed chromatin state, in WT; Pou5f1-eGFP PGCs (Fig 5C). In contrast, these Prmt5-mediated H2A/H4R3me2s marks were significantly decreased in Fancd2-KO; Pou5f1-eGFP and Fanca-KO;Pou5f1-eGFP PGCs (Fig 5C), consistent with LINE1 transcriptional activation.
Next, we examined the recruitment of Fancd2 and Prmt5 to the promoter of LINE1 in E10.5 PGCs from WT (Fanca+/+;Fancd2-KI) and FA-deficient (Fanca−/−;Fancd2-KI) embryos by μChIP assays using agarose-conjugated antibodies against FLAG (tagged Fancd2) and PRMT5 coupled with protein A magnetic beads. We found that Fancd2 and Prmt5 co-occupied the promoter of LINE1 in WT PGCs, and that this co-occupancy was lost in Fanca−/−;Fancd2-KI PGCs (Fig 5D). Notably, Fanca-KO caused not only complete loss of FLAG-Fancd2 occupancy but also significant reduction of Prmt5 recruitment to the LINE1 promoter, indicating a pattern of Fancd2-dependent Prmt5 occupancy. Together, these results suggest that Fancd2 recruits Prmt5 to the LINE1 promoter to support TE repression.
The requirement of the FA core component Fanca for repression of LINE1 and IAP TEs, the establishment of the Prmt5-mediated H2A/H4R3me2s marks and the recruitment of Fancd2 and Prmt5 to the LINE1 promoter in PGCs prompted us to analyze the levels of Prmt5 and Fancd2 proteins in in-vitro culture-expanded PGCs from WT (WT;Fancd2-KI) and Fanca-deficient (Fanca−/−;Fancd2-KI) mice. We did not observe significant alteration in the protein levels of either Prmt5 or Fancd2 in Fanca−/−;Fancd2-KI mice compared to the WT;Fancd2-KI controls (Fig 5E). These results indicate that, while Fanca is not required for the stability of the Fancd2 and Prmt5 proteins, a functional FA core complex is essential for Fancd2-dependent and Prmt5-mediated germline TE repression.
We also investigated whether the observed increase in the expression of TEs could lead to an increase in the accumulation of DNA damage in Fancd2-deficient PGCs. To this end, we performed the Comet assay and revealed that the Fancd2−/− PGCs exhibited significantly more DNA damage (% comet tail DNA) (Figure 5F) than their wild‐type counterparts. This result demonstrates excessive DNA damage in the Fancd2−/− PGCs, and suggests that de-repression of germline TEs could cause apoptosis of germ cells resulting from DNA damage.
Discussion
The FA DDR pathway plays critical roles in the maintenance of germline integrity; however, the underlying mechanisms are largely unknown. In this study, we show that (1) Fancd2 interacts with several important Prmt5/piRNA factors essential for germline TE silencing. (2) Loss of Fancd2 results in de-repression of TEs, depletion of PGCs, defective spermatogenesis and oogenesis, and infertility. (3) FA deficiency (Fancd2-KO and Fanca-KO) causes a significant reduction of Prmt5-catalyzed H2A/H4R3me2s marks on LINE1 TEs in early PGCs. And (4) Fancd2 and Prmt5 co-occupy the promoter of LINE1 in PGCs, which is lost in FA-deficient (Fanca-KO) PGCs. These results indicate a mechanistic link between the FA DDR pathway and germline maintenance, likely through regulation of TE expression in early PGCs.
De-regulated TE expression causes germ cell loss leading to infertility and induces germline genomic instability compromising the viability and health of offspring. Despite intensive investigation, the mechanisms by which germ cells control TE activity are largely not known and the DDR and repair pathway(s) functioning in this critical process have not been identified. The conventional model suggests that as a major DDR pathway involved in germline genomic maintenance, the FA pathway primarily engages in the processing and enzymatic repair of DNA damage during meiotic recombination or replication (Nadler & Braun 2000, Agoulnik et al. 2002, Wong et al. 2003, Crossan et al. 2011, Luo et al. 2014, Kato et al. 2015). Our results show that Fancd2 is required for Prmt5-catalyzed H2A/H4R3me2s marks, and loss of Fancd2 causes TE de-repression in early PGCs. These studies suggest that Fancd2/FA pathway protects germline genome from TE-induced DNA damage through regulating Prmt5-catalyzed H2A/H4R3me2s marks in TE repression. Our results are also consistent with a recent elegant study, in which the authors employed CRISPR-Cas9 screening strategies to identify genes in the control of the LINE-1 TE and showed that several FA proteins including FANCB, FANCI and FANCL, play a role in LINE-1 repression (Liu et al. 2018).
One novel finding of the current study is the link between the FA pathway and Prmt5-mediatedH2A/H4R3me2s repressive chromatin modification in PGC TE silencing. Prmt5-catalyzed repressive H2A/H4R3me2s chromatin modification is essential for TE silencing in PGCs between E8.5–E11.5, when actively dividing PGCs are undergoing global DNA demethylation (Hajkova et al. 2002, Hajkova et al. 2008, Seisenberger et al. 2012). We proposed that the FA pathway play a crucial role in TE repression through a mechanism that involves Prmt5-mediated repressive H2A/H4R3me2s marks on TEs. Indeed, we observed a significant reduction of H2A/H4R3me2s marks on LINE1 transposons in E10.5 PGCs of Fancd2-KO; Pou5f1-eGFP and Fanca-KO; Pou5f1-eGFP embryos compared to WT PGCs (Fig 5C). Our current study shows that the FA DDR pathway is required for establishing the repressive H2A/H4R3me2s marks catalyzed by Prmt5. These findings are consistent with previous reports that the repressive H2A/H4R3me2s marks is vital for TE silencing for early PGCs at a critical time when these cells reprogram their epigenome via a nearly complete erasure of DNA methylation (Reik et al. 2001, Hajkova et al. 2002, Lees-Murdock et al. 2003, Gulbert et al. 2012). Furthermore, we utilized our newly developed Fancd2-KI mouse model to show that Fancd2 and Prmt5 co-occupied the promoter of LINE1 in WT PGCs, and that this co-occupancy was lost in Fanca−/−;Fancd2-KI PGCs (Fig 5D). These studies established a potential mechanistic link between the Fancd2-Prmt5 interaction and Prmt5-catalyzed repressive H2A/H4R3me2s chromatin modification.
It is possible that the PRMT5 pathway is not the only Fancd2-dependent mechanism in germline TE repression. Another germline TE silencing machinery is the piRNA pathway (Weick & Miska 2014), which functions in a very strict development-stage- and cell-compartment-specific manner in the context of TE repression (Aravin et al. 2008). How these critical processes are regulated is unknown. Using our Fancd2-KI mouse model, we established biochemical interaction between Fancd2 and several components (Mili, Tdrd1, Tdrd6, Tdrd8, Mael) in the piRNA pathways (Fig. 1). In addition, Fancd2 is localized in both cytoplasm and the nucleus (Zhang et al. 2017). Loss of Fancd2 leads to TE overexpression at E11.5 and PGC depletion at E13.5 (Fig. 4–5). These data argue a functional link between Fancd2 and the piRNA pathway in the germline TE repression.
Genome maintenance in the germline is crucial for fertility and health of the next generation. Although DDR and DNA repair have been well characterized in meiocytes, the DDR pathway(s) that function in PGCs, which are the embryonic precursors to sperm and eggs, are less well defined. The current study unveils a novel interaction between a major DDR (FA) pathway and germ cell-specific TE silencing machinery in safeguarding the germline genome. From a clinical standpoint, TE silencing in the germline has a vital role in both parental fertility and offspring health. Therefore, defining the molecular collaboration between the FA DDR pathway and Prmt5/piRNA pathways in the context of germline TE repression will open up a new avenue of research designed to target these interacting pathways for developing innovative therapeutic strategies for reproductive diseases such as infertility and birth defects.
Materials and Methods
Mice
Fancd2-KI mice were generated in our laboratory (Zhang & Pang, et al., 2017). Fancd2+/− and Fanca+/− mice were provided by Dr. Markus Grompe (Oregon Health & Sciences University) (Houghtaling et al. 2003) and Dr. Madeleine Carreau (Laval University) (Wong et al. 2003), respectively. The Fancd2-KI;Pou5f1-eGFP, Fanca-KO;Pou5f1-eGFP or Fancd2-KO; Pou5f1-eGFP mice were generated by crossing the Fancd2-KI, Fanca-KO, or Fancd2-KO mice to the Pou5f1-eGFP reporter mice (B6;CBA-Tg(Pou5f1-EGFP)2Mnn/J; JAX Stock #:004654), respectively. Mice were maintained on C57BL/6J background in the animal barrier facility at Cincinnati Children’s Hospital Medical Center. Animals were kept in accordance with the protocol approved by the CCHMC Institutional Animal Care and Use Committee.
Isolation and culture of primordial germ cells (PGCs)
For isolation of PGCs, genital ridges of E10.5, E11.5 or E13.5 embryos from timed crosses of FA mice and Pou5f1-eGFP reporter mice were dissected, and single PGCs from individual genotyped embryos sorted by GFP expression or by SSEA1-APC (R&D Systems; Cat.# FAB2155A) positive cells using a MoFlo XDP high speed flow sorter (Beckman Coulter). For PGC culture, STO feeder cells (American Type Culture Collection (ATCC), Cat.# CRL-1503) were seeded in a 96 well plate coated with 1% gelatin (Sigma-Aldrich, Cat.# G1393) and cultured in DMEM (Gibco, Cat. # 11-960-044) supplemented with 10% FBS and 5% P/S. Upon confluency, STO cells were incubated with mitomycin C (Sigma-Aldrich, Cat.# M4287) at the concentration of 10 μg/ml for 4 hours. Then the cells were washed 3 times with 10 ml 1x PBS. The sorted PGCs were plated onto the STO feeder layer in PGC culture medium [DMEM supplemented with 1% P/S, 10% FBS, 100 ng/ml SCF (Peprotech, Cat.# 250–03), 25 nM bFGF (Peprotech, Cat.# 100–18B) and 25 nM LiF (Peprotech, Cat.# 250–02)].
Proliferation assay
A cell suspension containing GFP-positive PGCs obtained from E10.5 embryos of WT and Fancd2−/− females expressing the Pou5f1-eGFP reporter gene was prepared as described above. PGCs were subjected to pulse-labeling with Bromodeoxyuridine (BrdU; final concentration 10 μM) for 60 min. We used the reagents from the APC-BrdU Flow Kit (BD Biosciences,Cat. #552598), according to the manufacturer’s instructions.
Analysis of apoptosis
Cell apoptosis was measured by Annexin V (BD Pharmingen, Cat. # 550474) and 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI; BD Pharmingen, Cat. # 564907) staining using flow cytometry according to the manufacturer’s instructions.
Quantitative PCR analysis of TE expression
Total RNA from FACS-sorted (GFP+ SSEA1+) PGCs isolated from E11.5 embryos of WT, Fanca−/− and Fancd2−/− females expressing the Pou5f1-eGFP reporter gene was prepared using the RNeasy kit (Qiagen, Valencia, CA) following the manufacturer’s protocol. Reverse transcription was performed with random hexamers and Superscript II RT (Invitrogen, Grand Island, NY) and was carried out at 42 °C for 60 min and stopped at 95 °C for 5 min. First-strand cDNA was used for qPCR using the following primers: L15UTR-F 5’-GGCGAAAGGCAAACGTAAGA-3’, L15UTR-R 5’-GGAGTGCTGCGTTCTGATGA-3’; L1ORF2-F 5’-GGAGGGACATTTCATTCTCATCA-3’, L1ORF2-R 5’-GCTGCTCTTGTATTTGGAGCATAGA-3’; IAP3LTR-F 5’-GCACATGCG CAGATTATTTGTT-3’, IAP3LTR-R 5’-CCACATTCGCCGTTACAAGAT-3’; IAPGag-F 5’-AACCAATGCTAATTTCACCTTGGT-3’, IAPGag-R 5’-GCCAATCAGCAGGCGTTAGT-3’. Samples were normalized to the level of WT controls, and relative expression levels were determined by the standard curve method.
Preparation of protein extracts and immunoblotting
To prepare protein lysates, testes from 8–10 week old mice or in-vitro expanded PGCs isolated from E13.5 embryos were lysed using a homogenizer on ice in 3 volumes of ice-chilled Lysis buffer with Roche Complete proteinase inhibitor (Roche, Cat. # 04693116001). Lysates were cleared by centrifugation (40,000g, 20 min, at 4°C). Protein concentration was quantified by using Bio-Rad reagent and resolved on SDS-PAGE and transferred onto nitrocellulose membranes. Immunoblots were then incubated with primary antibodies specific for Mili (Cell Signaling, Cat. #2071), Prmt5 (Sigma-Aldrich, Cat. # 07–405), Mael (Santa Cruz, Cat. # sc-398925), Tdrd1 (R&D Systems, Cat. # AF320), Tdrd6 (Sigma-Aldrich, Cat. # ABE231), Tdrd8/STK31 (Abcam, Cat. # ab155172), FLAG (Sigma-Aldrich, Cat. # F3165), β-actin (Sigma-Aldrich, Cat. # A5441).
μChIP analysis
Small-scale ChIP (μChIP) experiments were conducted as described previously (Dahl & Collas, 2008). Briefly, FACS-sorted (GFP+ SSEA1+) PGCs isolated from E10.5 embryos of WT, Fanca-KO or Fancd2-KO females expressing both Fancd2-KI transgene and the Pou5f1-eGFP reporter were crosslinked with 0.5% paraformaldehyde, lysed in radio immunoprecipitation assay (RIPA) buffer containing protease inhibitors, and sonicated to achieve a mean DNA fragment size of around 200–400 base pairs. After centrifugation, the chromatin extracts were then precleared with Dynal Magnetic Beads (Invitrogen) (4°C, 1 hr) followed by centrifugation (2,000 rpm, 30 min). Supernatant (precleared chromatin) was immunoprecipitated overnight with Dynal Magnetic Beads coupled with anti-H4R3me2s (1 μg per ChIP, ab5823, Abcam), anti-FLAG (Sigma-Aldrich, Cat. # F3165), anti-Prmt5 (Sigma-Aldrich, Cat. # 07–405), or normal rabbit serum. After reverse crosslinking (with protease K at 42°C for 2 hr and 68°C for 6 hr), DNA was purified (phenol-chloroform extraction) and used for qPCR analysis. Primers used were as follows: LINE1 5UTR-Forward 5’-GGCGAAAGGCAAACGTAAGA-3’, LINE1 5UTR-Reverse 5’-GGAGTGCTGCGTTCTGATGA-3’.
Comet assay
Comet assay was performed using the Trevigen’s Comet Assay Kit, following the manufacturer’s instructions. In brief, the PGCs suspension was mixed with molten LMAgarose at a ratio of 1:10 (v/v), and transferred onto slides. After incubation in the lysis buffer and alkaline treatment in electrophoresis buffer, electrophoresis (20 min, 25 V and approximately 300 mA) and neutralization were performed. Subsequently, slides were dehydrated with ethanol. For evaluation, the slides were stained with 100 μL of diluted SYBR Gold (1:10 000 in TE buffer, Invitrogen), and the comets were evaluated by image analysis using the software comet IV version 4.2 (Perceptive Instruments, Haverhill, UK). The percentage of tail DNA (% tail DNA) for each comet was calculated. The experiments were performed three times (duplicates, 50 cells per technical replicate).
Statistical analysis
Student’s t-test was performed using GraphPad Prism v6 (GrapPad software). Comparison of more than 2 groups was analyzed by one-way ANOVA test. Values of p<0.05 were considered statistically significant. Results are presented as mean ± SD. * indicates p<0.05; **, p<0.01; ***, p<0.001.
Acknowledgements
We Dr. Markus Grompe (Oregon Health & Sciences University) for the Fancd2+/−mice, and Dr. Madeleine Carreau (Laval University) for the Fanca+/− mice. This study was partially supported by NIH grants R01 HL076712 and R01 HD089932. Q.P. was supported by a Leukemia and Lymphoma Scholar award.
Footnotes
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
All animal procedures were approved by the Institutional Animal Care and Use Committee of Cincinnati Children’s Hospital Medical Center prior to study initiation (IACUC protocol # 2013–0159).
Competing interests
The authors declare no competing interests.
References
- Agoulnik AI, Lu B, Zhu Q, Truong C, Ty MT, Arango N, Chada KK, Bishop CE 2002. A novel gene, Pog, is necessary for primordial germ cell proliferation in the mouse and underlies the germ cell deficient mutation, gcd. Hum Mol Genet 11 3047–53. [DOI] [PubMed] [Google Scholar]
- Alavattam KG, Kato Y, Sin HS, Maezawa S, Kowalski IJ, Zhang F, Pang Q, Andreassen PR, Namekawa SH 2016. Elucidation of the Fanconi Anemia Protein Network in Meoisis and Its Function in the Regulation of Histone Modifications. Cell Rep. 17 1141–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Bourc’his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ 2008. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell. 31 785–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagby GC 2003. Genetic basis of Fanconi anemia. Curr Opin Hematol 10 68–76. [DOI] [PubMed] [Google Scholar]
- Bagby G 2018. Recent advances in understanding hematopoiesis in Fanconi Anemia. F1000Res. 7 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrens RV, Reik W 2015. Prmt5: a guardian of the germline protects future generations. EMBO J. 34 689–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard PHL, McIntyre RE, Gallagher F, Kettunen MI, Adams DJ, Patel KJ, et al. 2011. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet 43 147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JA, Collas P 2008. A rapid micro chromatin immunoprecipitation assay (ChIP). Nature Protocols 3 1032–1045. [DOI] [PubMed] [Google Scholar]
- Deans AJ & West SC 2011. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 11 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V 2015. Update of the human and mouse Fanconi anemia genes. Hum Genomics 9 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, Kazazian HH Jr. 2008. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135 23–35. [DOI] [PubMed] [Google Scholar]
- Guibert S, Forne T, Weber M 2012. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res 22 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, Cesari F, Lee C, Almouzni G, Schneider R, Surani MA 2008. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 452 877–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA 2002. Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev 117 15–23. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Ohta H, Kurimoto K, Aramaki S, Saitou M 2011. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146 519–32. [DOI] [PubMed] [Google Scholar]
- Hill RJ & Crossan GP 2019. DNA cross-link repair safeguards genomic stability during premeiotic germ cell development. Nat Genet. 51 1283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M 2003. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev 17 2021–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J 2009. The DNA-damage response in human biology and disease. Nature 461 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Alavattam KG, Sin HS, Meetei AR, Pang Q, Andreassen PR, Namekawa SH 2015. FANCB is essential in the male germline and regulates H3K9 methylation on the sex chromosomes during meiosis. Hum Mol Genet. 24 5234–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Günesdogan U, Zylicz JJ, Hackett JA, Cougot D, Bao S, Lee C, Dietmann S, Sengupta R, Surani MA, et al. 2014. PRMT5 protects genomic integrity during global DNA demethylation in primordial germ cells and preimplantation embryos. Mol Cell. 56 564–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knies K, Inano S, Ramírez MJ, Ishiai M, Surrallés J, Takata M, Schindler D 2017. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Invest 127 3013–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann MC & Smogorzewska A 2013. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Murdock DJ, De Felici M, Walsh CP 2003. Methylation dynamics of repetitive DNA elements in the mouse germ cell lineage. Genomics 82 230–237. [DOI] [PubMed] [Google Scholar]
- Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik MC, Wysocka J 2018. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature. 553 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Hartford SA, Zeng R, Southard TL, Shima N, Schimenti JC 2014. Hypersensitivity of Primordial Germ Cells to Compromised Replication-Associated DNA Repair Involves ATM-p53-p21 Signaling. PLoS Genet 10(7): e1004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler JJ, Braun RE 2000. Fanconi anemia complementation group C is required for proliferation of murine primordial germ cells. Genesis 27 117–23. [DOI] [PubMed] [Google Scholar]
- Prudhomme S, Bonnaud B, Mallet F 2005. Endogenous retroviruses and animal reproduction. Cytogenet Genome Res. 110 353–64. [DOI] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J 2001. Epigenetic reprogramming in mammalian development. Science 293 1089–1093. [DOI] [PubMed] [Google Scholar]
- Russell SJ, Stalker L, LaMarre J 2017. PIWIs, piRNAs and Retrotransposons: Complex battles during reprogramming in gametes and early embryos. Reprod Domest Anim. 4 28–38. [DOI] [PubMed] [Google Scholar]
- Saito K, Siomi MC 2010. Small RNA-mediated quiescence of transposable elements in animals. Dev Cell. 19 687–97. [DOI] [PubMed] [Google Scholar]
- Sawyer SL, Tian L, Kähkönen M, Schwartzentruber J, Kircher M 2015. University of Washington Centre for Mendelian Genomics et al. Biallelic Mutations in BRCA1 Cause a New Fanconi Anemia Subtype. Cancer Discov. 5 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W, et al. 2012. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell 48 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, Aravin AA 2011. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol. 12 246–58. [DOI] [PubMed] [Google Scholar]
- Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A 2012. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo PE, Hubner K, Scholer H & Mann JR 2002. Allele-specific expression of imprinted genes in mouse migratory primordial germ cells. Mech. Dev 115 157–60. [DOI] [PubMed] [Google Scholar]
- Tischkowitz MD & Hodgson SV 2003. Fanconi anaemia. J Med Genet 40 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick EM, Miska EA 2014. piRNAs: from biogenesis to function. Development. 141 3458–71. [DOI] [PubMed] [Google Scholar]
- Wong JC, Alon N, Mckerlie C, Huang JR, Meyn MS & Buchwald M 2003. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet 12 2063–76 [DOI] [PubMed] [Google Scholar]
- Zhang T, Du W, Wilson AF, Namekawa SH, Andreassen PR, Meetei AR & Pang Q 2017. Fancd2 in vivo interaction network reveals a non-canonical role in mitochondrial function. Sci Rep. 7 45626. [DOI] [PMC free article] [PubMed] [Google Scholar]