

Abstract
Although chronic obstructive pulmonary disease (COPD) risk is strongly influenced by cigarette smoking, genetic factors are also important determinants of COPD. In addition to Mendelian syndromes such as alpha-1 antitrypsin deficiency, many genomic regions that influence COPD susceptibility have been identified in genome-wide association studies. Similarly, multiple genomic regions associated with COPD-related phenotypes, such as quantitative emphysema measures, have been found. Identifying the functional variants and key genes within these association regions remains a major challenge. However, newly identified COPD susceptibility genes are already providing novel insights into COPD pathogenesis. Network-based approaches that leverage these genetic discoveries have the potential to assist in decoding the complex genetic architecture of COPD.
Keywords: COPD, genetic association, sequencing, network medicine, subtyping
1. INTRODUCTION
Although COPD risk is strongly influenced by cigarette smoking, there has been long-standing interest in identifying genetic determinants of COPD susceptibility. COPD is a complex disease, influenced by genetic and environmental factors acting in a developmental context. Understanding the genetic determinants of COPD could provide an unbiased assessment of key molecular determinants of disease pathobiology, which could lead to important new insights into COPD pathogenesis. Complex diseases such as COPD are not caused by single genetic variants; rather, they develop due to perturbations of biological networks consisting of genes and proteins. COPD genetics could provide critical information to build and refine these biological networks.
Defining the genetic determinants of COPD, and the genes and proteins with which they interact, could provide new targets for drug development. Nelson and colleagues (1) demonstrated that 8% of US Food and Drug Administration (FDA)-approved drugs targeted molecules with genetic support from genome-wide association studies (GWAS) or Mendelian syndromes (from the Online Mendelian Inheritance in Man database). However, only 2% of Phase 1 drugs, which have not yet been subjected to the full gauntlet of drug development, had such genetic support. The higher rate of genetic support in FDA-approved drugs suggests that targeting genes and proteins identified by genetic studies for new drug development may have a higher rate of treatment efficacy.
COPD is a heterogeneous syndrome, with variable contributions of lung parenchymal destruction (emphysema) and airway disease in different COPD patients. In addition, COPD patients vary widely in other clinical manifestations, including the frequency of COPD exacerbations, level of exercise capacity, and the development of cachexia. Genetic determinants may influence COPD heterogeneity as well as susceptibility.
COPD genetic studies over the past 25 years have occurred in a series of scientific waves of varying impact, based on technological advances in the assessment and analysis of genetic variation. Early studies of COPD genetics focused on finding shared regions of the genome among affected relatives using linkage analysis (2). Although these linkage-based approaches were highly successful in monogenic syndromes such as cystic fibrosis, they were much less effective in complex diseases such as COPD. Many candidate genes, selected based on what was thought to be known about COPD pathobiology, were assessed in case-control association studies (3). These candidate gene association studies were largely irreproducible, likely due to small sample sizes and failure to utilize an adequately stringent adjustment for multiple statistical testing. This humbling era of candidate gene association studies also emphasized our limited understanding of COPD pathobiology. More recently, COPD genetics has focused on GWAS, which are a major focus of this article. As the cost of whole exome and whole genome sequencing has fallen, these more comprehensive assessments of rare genetic variation are increasingly being used in COPD genetics research. Ultimately, an integrated approach that includes genetic variants along with other omics data types (e.g., transcriptomics, epigenetics, proteomics, etc.) in a network context will likely be required to provide a comprehensive view of the genetic architecture of COPD.
2. FAMILIAL AGGREGATION OF COPD
Many early studies demonstrated that COPD clusters in families (4). Of course, this does not prove that genes influence COPD risk, since cigarette smoking also clusters in families (likely for both genetic and environmental reasons). In the Boston Early-Onset COPD Study, which enrolled extended pedigrees of individuals with severe COPD at a young age [and without severe alpha-1 antitrypsin (AAT) deficiency], significant familial aggregation of airflow obstruction was identified in response to smoking. Compared to smokers in the general population, smoking first-degree relatives (parents, siblings, and children) of severe, early-onset COPD cases had approximately a threefold increased risk for airflow obstruction (5). However, nonsmoking first-degree relatives of severe, early-onset COPD cases had no increased risk for COPD compared to nonsmokers from the general population. Similar results were found by McCloskey and colleagues (6), suggesting that genetic determinants may interact with cigarette smoking to influence COPD susceptibility.
Twin studies can be utilized to estimate heritability, the fraction of phenotypic variation due to genetic factors, by comparing disease prevalence in monozygotic (identical) twins who share all of their genes, and dizygotic (fraternal) twins who share approximately half of their genes. Ingebrigtsen and colleagues (7) studied 22,422 Danish and 27,668 Swedish twin pairs; they estimated COPD heritability to be approximately 60%.
An alternative approach to estimate heritability is based on the low, but nonzero, levels of relatedness in population-based samples. Using this approach, Zhou et al. (8) estimated the heritability for FEV1 to be approximately 40%, and the heritability for quantitative computed tomography (CT) emphysema to be approximately 30%. Thus, COPD and COPD-related phenotypes are influenced by genetic determinants. The challenge is to identify those genes and the functional variants that influence them.
3. ALPHA-1 ANTITRYPSIN DEFICIENCY
Severe AAT deficiency was the first proven genetic determinant of COPD. It remains the most well-established genetic risk factor for COPD, and it is the only genetic subtype of COPD with a specific treatment (9). AAT, encoded by the SERPINA1 gene, is the major plasma protease inhibitor of leukocyte elastase, a powerful enzyme located in the azurophil granules of neutrophils. The most common cause of severe AAT deficiency is homozygosity for the SERPINA1*Z allele, which results from a single base pair change in the coding sequence of the SERPINA1 gene and leads to a single amino acid substitution that causes AAT polymers to form in the hepatocytes that synthesize most AAT. Reduced circulating AAT levels result from this protein production abnormality. In addition to homozygosity for the Z allele, severe AAT deficiency can be caused by heterozygosity of one Z allele and one null allele (null alleles lead to the absence of AAT production); ZZ and Znull individuals are often referred to as PI Z. Approximately 1 in 3,000 people in the United States inherit severe AAT deficiency, which substantially increases their risk for COPD, as well as for liver disease (including hepatitis, cirrhosis, and hepatocellular carcinoma).
Substantial variability in the development of lung disease in PI Z subjects has been observed (10), with some PI Z individuals living to advanced age without developing significant COPD. Because most PI Z individuals are identified due to COPD or liver disease, determining the natural history of AAT deficiency has been challenging. A newborn screening study performed in Sweden in 1972–1974 identified 129 PI Z subjects; the most recent follow-up report of 41 PI Z subjects from this cohort revealed that some current or ex-smokers revealed evidence for hyperinflation and reduced diffusing capacity at ages 37–39 (11). Genetic modifiers likely influence this variability in COPD risk among PI Z individuals, but they have not yet been definitively identified. Augmentation therapy for AAT deficiency is available in the United States as a weekly intravenous infusion. Randomized controlled trials have demonstrated reduction in the rate of emphysema development in response to AAT augmentation therapy (12).
There has been long-standing controversy regarding the risk of COPD in heterozygotes for the SERPINA1 Z allele (13). Several studies in the past decade have supported increased risk for carriers of one normal SERPINA1 M allele and one SERPINA1 Z allele, who are often referred to as PI MZ (14). Sorheim and colleagues (15) included both COPD cases and controls from the GenKOLS study in Norway and families ascertained through COPD subjects in the International COPD Genetics Network. They found that FEV1/FVC was significantly lower in PI MZ subjects from both studies, but only the GenKOLS study showed a significant association of PI MZ with quantitative CT emphysema. Molloy and colleagues (16) performed a study of families in Ireland ascertained through a PI MZ subject with COPD; after excluding the PI MZ proband subjects, the remaining PI MZ ever-smokers were shown to have reduced lung function and an increased risk for COPD compared to PI MM ever-smokers, with a likely gene-by-environment interaction. Recent work in the COPDGene study demonstrated increased risk for reduced FEV1 and increased emphysema in PI MZ current and ex-smoking individuals (17). Similarly, the SPIROMICS investigators recently showed that PI MZ smokers have significantly reduced FEV1 and FEV1/FVC (18). Thus, the evidence is now compelling that PI MZ smokers are at increased risk for COPD compared to PI MM smokers. It remains unclear whether PI MZ nonsmokers have any increased risk for COPD.
4. GENOME-WIDE ASSOCIATION STUDIES OF COPD
As shown in Figure 1, GWAS begin by assembling a large study population, typically of cases and controls—although population-based and family-based samples are also frequently utilized. Phenotypes for GWAS often include disease affection status (e.g., case versus control), although other quantitative or categorical disease-related phenotypes can also be analyzed. Standardized genome-wide single nucleotide polymorphism (SNP) genotyping of panels, including hundreds of thousands of genetic variants, has become commoditized. Quality control is performed at the level of the study subject (excluding subjects with high rates of missing genotypes, suggesting low-quality DNA samples, or gender inconsistencies, suggesting possible sample mix-ups) and the level of the genetic marker (excluding markers with high rates of missing data, deviations from expected genotype distributions in control subjects based on Hardy-Weinberg equilibrium, etc.). Genetic association analysis is performed with regression analysis (e.g., logistic regression for categorical phenotypes and multiple regression for quantitative phenotypes). Mixed regression models can be used to adjust for subject relatedness. The genotyped SNPs can be utilized to impute likely genotypes at other SNPs with which they are correlated (termed linkage disequilibrium) by using statistical imputation approaches with standard reference panels such as the 1,000 Genomes Project or Haplotype Reference Consortium. Due to the large number of genetic variants tested, stringent adjustment for multiple statistical testing is required, with p-values <5 × 10−8 typically utilized to demonstrate genome-wide significance. Meta-analysis of multiple study populations is often required to achieve statistical significance, and replication of association results substantially increases confidence in the validity of the associations.
Figure 1.
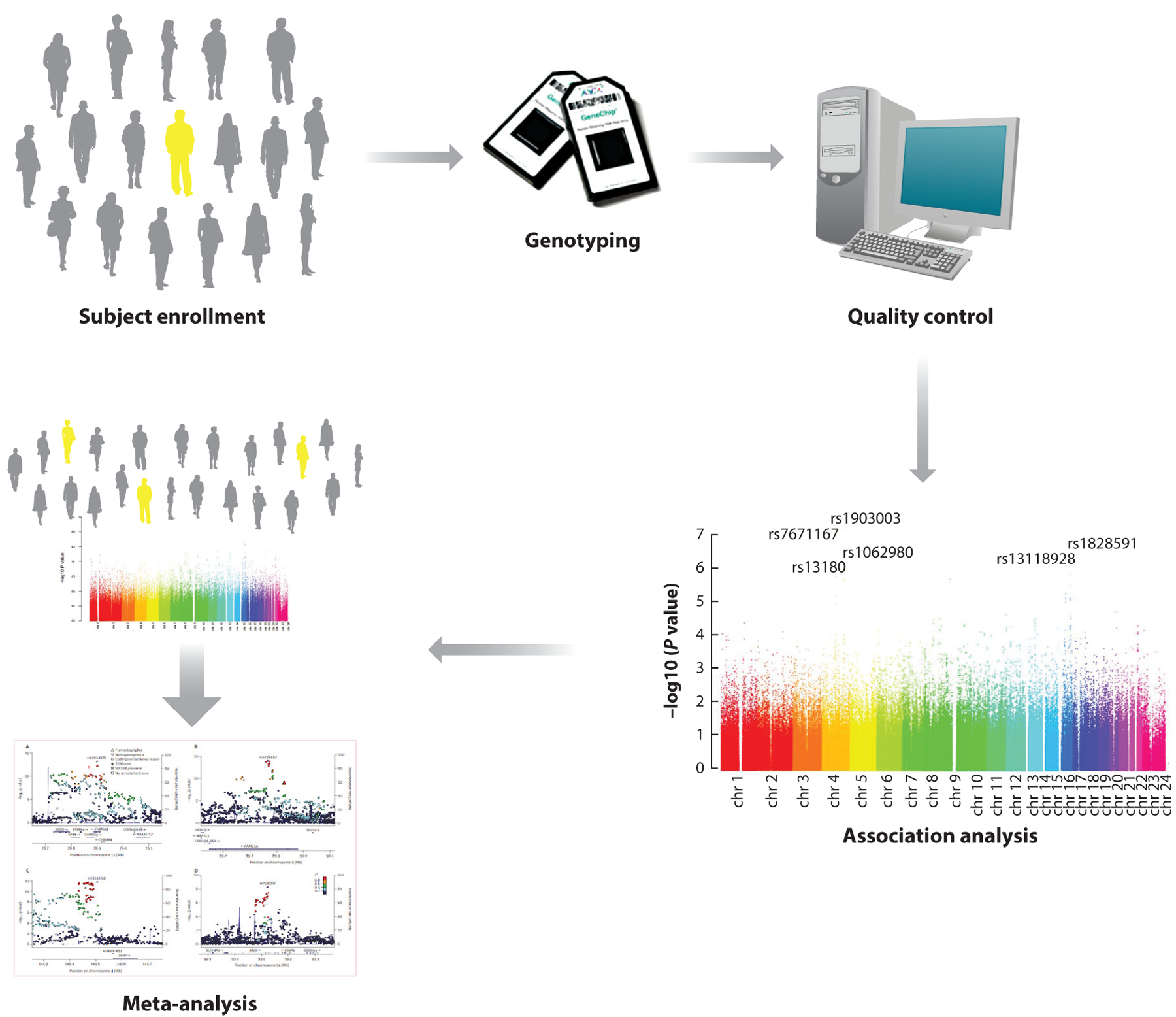
The overall approach for genome-wide association studies of complex diseases. DNA samples and phenotypic information are obtained from cases and controls, families including affected individuals, or subjects from the general population. Subsequently, standard single nucleotide polymorphism (SNP) genotyping panels are tested. Quality control is performed at the level of both the subject and the SNP, and then statistical associations between genotypes and phenotypes are assessed. Adjustment for genetic ancestry is necessary in case-control or population-based studies. To achieve genome-wide statistical significance, meta-analysis of multiple study populations is often required. Adapted with permission from Reference 85.
The first GWAS of COPD were published in 2009; Pillai and colleagues (19) found genome-wide significant associations of the CHRNA3/CHRNA5/IREB2 region on chromosome 15q25 with COPD. In a concurrently published GWAS from the Framingham Heart Study (20), the HHIP region was associated with FEV1/FVC, and this same region nearly reached genome-wide significance with COPD susceptibility in the Pillai et al. study (19). Studies from large general population samples have provided strong support for the association of HHIP SNPs with FEV1/FVC (21, 22). The CHARGE Consortium also found evidence of association of FEV1/FVC with the FAM13A locus (21), which has been strongly associated with COPD susceptibility (23). Subsequently, the COPD GWAS of the full COPDGene cohort of 10,192 smokers was combined in a collaborative meta-analysis with the ECLIPSE, GenKOLS, and NETT-NAS populations (24). In GWAS of moderate-to-severe COPD cases (postbronchodilator FEV1 <80% predicted with FEV1/FVC <0.7) versus control smokers, a novel region near RIN3 was associated with COPD. Additional severe COPD (FEV1 <50% predicted) GWAS regions were found near MMP12 and TGFB2.
The International COPD Genetics Consortium (ICGC) was created to facilitate collaborative research in COPD genetics (25). Using COPD affection status as the phenotype, Hobbs and colleagues (26) performed GWAS in 15,256 COPD cases and 47,936 controls from 26 collaborating studies in the ICGC; they identified 22 genome-wide significant loci for COPD. More recently, a combined COPD GWAS of samples from the ICGC and UK Biobank identified 82 loci associated with COPD at genome-wide significance (Figure 2) (27). Many of these COPD GWAS loci were previously associated with spirometric values in general population samples (see below). Of 35 novel loci (not previously associated with COPD or lung function), 27 loci were at least nominally associated with FEV1 or FEV1/FVC in the SpiroMeta cohort. Interestingly, some of the COPD GWAS loci were more strongly associated with emphysema, while others were more strongly associated with airway phenotypes. Thus, COPD genetic loci may influence the heterogeneous manifestations of COPD.
Figure 2.
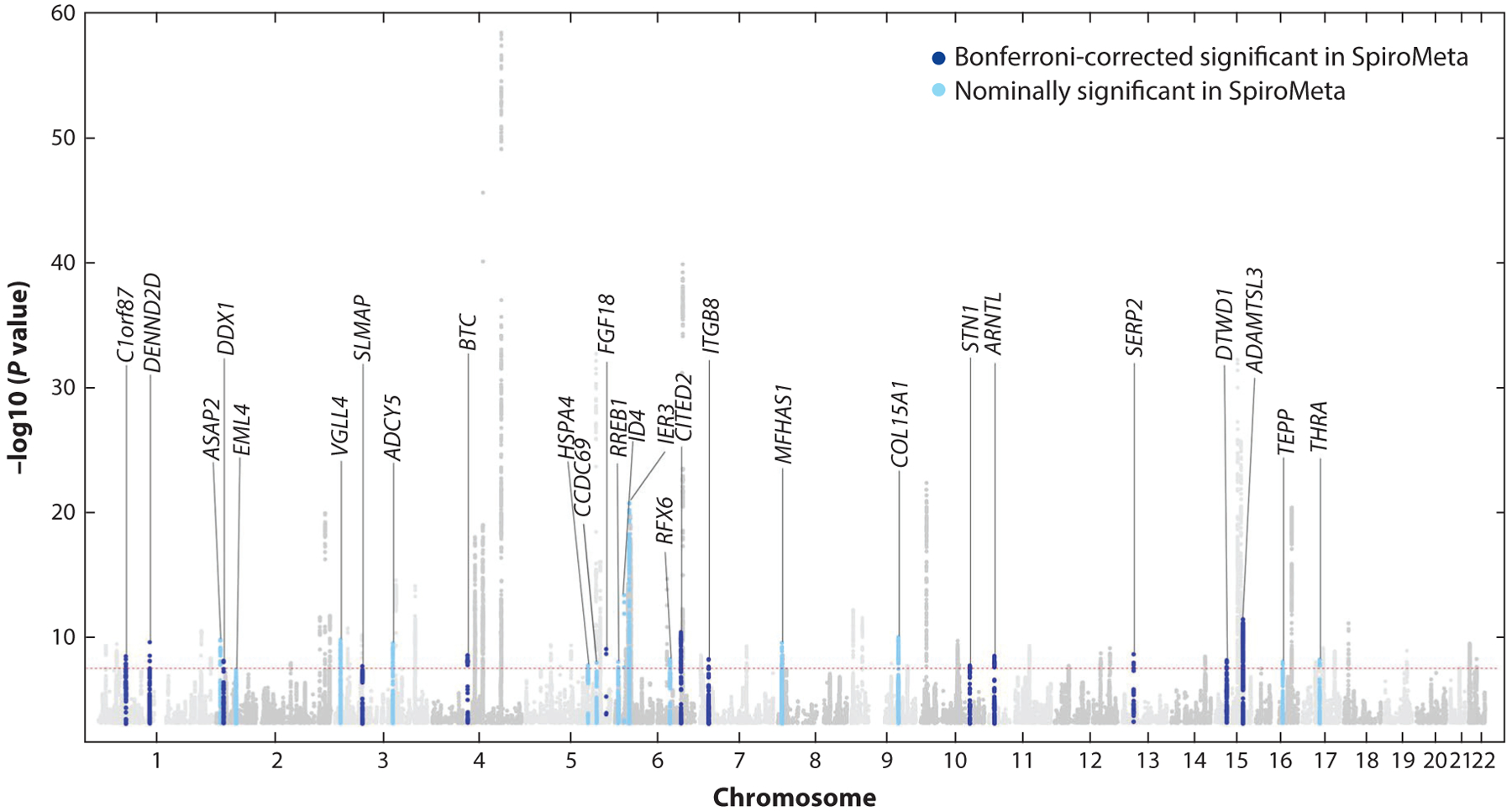
International COPD Genetics Consortium and UK Biobank genome-wide association studies for COPD. Manhattan plot demonstrating 82 genome-wide significant associations to COPD. Novel associations (not previously reported for COPD or lung function) are labeled with the nearest gene, and replication in the SpiroMeta cohort for lung function phenotypes is indicated. Adapted with permission from Reference 27.
Importantly, many of the COPD GWAS loci have been replicated by other investigators in other study populations. For loci including HHIP (28–30) and FAM13A (31–33), the same top SNP (or a nearby SNP in linkage disequilibrium) has been replicated with the same direction of effect. The results on chromosome 15q25 have been more complicated, with some evidence supporting the existence of two COPD GWAS loci in that region—one related to nicotine addiction (related to the nicotinic acetylcholine receptor genes such as CHRNA3 and CHRNA5) and one unrelated to nicotine addiction (related to IREB2) (34). Although the effect sizes of COPD GWAS loci are individually modest, their impact is more substantial when combined into a genetic risk score. For example, Busch and colleagues (35) created a simple genetic risk score based on the first seven COPD GWAS loci; they applied this risk score to the International COPD Genetics Network (which had not been used for the GWAS). After adjustment for relevant covariates, each additional COPD risk allele was associated with a 1.9% decrease in FEV1 (as % predicted), which could be clinically significant.
Although COPD GWAS have identified multiple genome-wide significant associations, much of the estimated heritability for COPD remains unexplained. This missing heritability could be related to common variants of very small effect and/or rare variants of either small or large effect. One of the most interesting findings in COPD GWAS is the identification of five genomic regions that are also associated with idiopathic pulmonary fibrosis (IPF), but with opposite directions of effect (the risk allele for COPD protects from IPF and vice versa) (27). Although risk for both of these pulmonary diseases is influenced by cigarette smoking, these genetic clues could point to molecular switches that influence the distinct pathological patterns of COPD and IPF.
5. GENOME-WIDE ASSOCIATION STUDIES OF LUNG FUNCTION
As noted above, several of the initially discovered COPD GWAS loci, including HHIP and FAM13A, were also found to be genetic determinants of spirometric values in general population samples. As the sample sizes for both COPD and lung function GWAS analyses have steadily increased, this overlap has become even more substantial. For example, Wain et al. (36) performed GWAS for FEV1, FVC, and FEV1/FVC in 48,943 subjects selected from the extremes of lung function in the UK BiLEVE cohort, with follow-up genotyping in 95,375 additional subjects. They identified 97 GWAS loci for lung function, many of which showed at least nominal association with COPD. Burkart and colleagues (37) found several novel genome-wide significant regions of association with spirometric measures (including ZSWIM7 and HAL) in the Hispanic Community Health Study/Study of Latinos (HCHS/SOL) cohort, as well as support for previous regions associated with lung function in European populations. Wyss and colleagues (38) studied 90,715 subjects from ethnically diverse cohorts in the CHARGE consortium, including 8,429 African ancestry, 9,959 Asian ancestry, and 11,775 Hispanic ancestry subjects. They found 60 novel loci associated with FEV1, FVC, and/or FEV1/FVC. The studies by Burkart’s and Wyss’s groups demonstrate the importance of assessing genetic associations in multiple ancestries. In the largest general population GWAS of lung function yet reported, Shrine and colleagues (39) analyzed more than 400,000 participants in either the UK Biobank or SpiroMeta Consortium, identifying 279 genome-wide significant loci for spirometric measures. A genetic risk score based on these 279 lung function variants was significantly associated with COPD in independent cohorts, including in non-European ancestries.
Perhaps it is not surprising that genetic determinants of COPD and lung function levels show substantial overlap. After all, COPD is defined based on lung function. COPD is a common disease, and a substantial number of COPD cases are included in general population samples used for lung function GWAS analyses—especially for the UK BiLEVE study, which selected subjects from the extremes of lung function. Because COPD is defined by thresholds of lung function, one could easily envision a lung function variant that caused one person to have FEV1/FVC = 0.69 (who would be diagnosed with COPD), while a carrier of the alternate allele who had FEV1/FVC = 0.71 (who would not be diagnosed with COPD) would be associated with COPD. In addition, there is growing recognition that a sizeable proportion of COPD cases relates to abnormal growth and development rather than rapid decline in lung function (40). However, it remains difficult to understand how the pathological processes that characterize advanced COPD, including emphysema, small airway destruction, and small airway fibrosis, would be influenced by genetic determinants of normal lung growth and development. Further studies to determine if lung function genes are involved in COPD-related lung inflammation, tissue destruction, and inadequate repair are warranted.
Although the search for genetic determinants of lung function levels has been quite fruitful, identifying genetic determinants of lung function decline has been considerably more challenging. Tang and colleagues (41) performed a GWAS meta-analysis of FEV1 change in 27,249 European ancestry subjects from 14 cohorts in the CHARGE and SpiroMeta consortia; no genome-wide significant associations were found. John et al. (42) performed GWAS of longitudinal change in FEV1 and FEV1/FVC in 4,167 subjects; they did not identify any genome-wide significant associations. Moreover, none of the 26 previously identified genetic determinants of lung function level was associated with lung function decline. Genetic determinants of longitudinal change in lung function are quite likely to exist; however, variability in technical assessment of lung function levels over time as well as the possibility that lung function changes are episodic rather than gradual will make it difficult to identify such genetic determinants. Focusing on longitudinal trajectories may provide a more powerful approach to find genetic determinants of lung function decline (43).
6. GENETIC DETERMINANTS OF COPD-RELATED PHENOTYPES
In addition to genetic determinants of COPD and lung function, many other COPD-related phenotypes have been assessed using GWAS. These genetic analyses in the COPDGene study were recently summarized by Ragland and colleagues (44) and are shown in Figure 3. In contrast to GWAS of COPD and lung function, the limited availability of these COPD-related phenotypes has led to smaller sample sizes and/or lack of available cohorts for replication. Some COPD GWAS loci, such as TGFB2, FAM13A, HHIP, CYP2A6, and CHRNA3/IREB2, are associated with multiple COPD-related phenotypes. Other COPD GWAS loci, such as MTCL1 and SFTPD, have only been associated with COPD. Whether these differences reflect the overall strength of association to COPD or the impact on different aspects of COPD pathogenesis remains to be determined.
Figure 3.

Phenogram plot of representative GWAS for COPD susceptibility and COPD-related phenotypes on different chromosomes. Genetic associations are denoted by the nearest gene to the most significantly associated SNP. COPD susceptibility associations (blue) include 22 loci reported by Hobbs et al. (26). Lung function (FEV1, FVC, and FEV1/FVC) associations depicted here (red) include associations from the study conducted by Wain et al. (36). Abbreviations: BDR, bronchodilator response; BMI, body mass index; CB, chronic bronchitis; EMPH.DIST.RATIO, emphysema distribution ratio of upper divided by lower lung fields; FEV1, forced expiratory volume in 1 s; FFMI, fat-free mass index; FVC, forced vital capacity; GWAS, genome-wide association study; LAA-950, percentage of lung density histogram below −950 HU; LHE, local histogram–based emphysema; MOD, moderate centrilobular emphysema on LHE; NORM, normal/nonemphysematous on LHE; OS, resting oxygen saturation; PAE, pulmonary artery enlargement; PAN, panlobular emphysema on LHE; PB, postbronchodilator; PCT.GAS, percent gas-trapping at −856 HU on expiratory computed tomography; PERC15, fifteenth percentile point of the lung density histogram; SEV, severe centrilobular emphysema on LHE; SNP, single nucleotide polymorphism; VIS.EMPH, visual emphysema (presence/absence). Adapted with permission from Reference 44.
Chest CT phenotypes are especially promising assessments to understand COPD heterogeneity, as the presence, severity, distribution, and pattern of emphysema can be determined. Manichaikul and colleagues (45) analyzed quantitative CT emphysema in a multiethnic general population sample of 7,914 subjects, the MESA (Multi-Ethnic Study of Atherosclerosis) Lung Study. They found genome-wide significant associations near SNRPF and PPT2. With additional fine mapping, the most strongly associated SNP in the PPT2 region was located within an intron of the AGER gene. AGER encodes the sRAGE protein biomarker, which has been strongly associated with emphysema (46). Cho and colleagues (47) performed GWAS of chest CT phenotypes in the COPDGene, ECLIPSE, GenKOLS, and NETT studies. Five genome-wide significant associations with quantitative emphysema (percentage of low attenuation areas below −950 HU) were identified, including two previously identified COPD GWAS loci (HHIP and CHRNA3). The AGER region, previously associated with lung function, was also associated with quantitative emphysema, and it was subsequently associated with COPD in the ICGC analysis (26). A region near the SERPINA1 gene was also associated with emphysema, and it appeared to be driven by the Z allele; thus, with a highly specific phenotype (CT emphysema), even rare variants of large effect can be identified in genetic association studies. In addition, a region near DLC1, which has not been associated with lung function levels, was implicated in emphysema. Finding genetic determinants of CT airway wall phenotypes has been more challenging than emphysema phenotypes, potentially because only relatively large airways can be visualized due to the limits of CT resolution.
Emphysema distribution was also studied in GWAS of 11,532 subjects from COPDGene, ECLIPSE, and GenKOLS (48). Five genome-wide significant associations were found, including HHIP and chromosome 15q25 as well as three novel loci (near SOWAHB, TRAPPC9, and KIAA1462). Genetic determinants of emphysema pattern based on local histogram textural analysis in COPDGene were studied by Castaldi et al. (49). In addition to several novel associations (VWA8 with panlobular emphysema and MYO1D with severe centrilobular emphysema), multiple previously identified COPD GWAS loci were associated with textural emphysema patterns: CHRNA3/AGPHD1 (moderate centrilobular, severe centrilobular, and panlobular), TGFB2 (moderate centrilobular), MMP12 (moderate centrilobular), and CYP2A6 (moderate centrilobular). The associations of the chromosome 15q25 region with multiple different emphysema patterns could relate to the impact of nicotine addiction on all emphysema patterns and/or the existence of several COPD-related genetic determinants at that locus.
Studying genetic determinants of COPD-related phenotypes is one approach to understand COPD heterogeneity. An alternative is to define COPD subtypes using machine learning, imaging patterns, or other clinical features, and then to assess genetic associations to those subtypes. Castaldi and colleagues (50) used K-means clustering to define four COPD subtypes based on FEV1 (% predicted), emphysema at −950 HU, emphysema distribution (upper lung field/lower lung field), and segmental airway wall area. A cluster with mild upper lung–predominant emphysema was associated with an SNP near HHIP, while a severe emphysema cluster was most strongly associated with the chromosome 15q25 locus.
7. FINDING FUNCTIONAL VARIANTS IN GENOME-WIDE ASSOCIATION STUDIES LOCI
Although GWAS have successfully identified susceptibility loci for many complex diseases (51), functional variant identification post-GWAS has been quite slow (52, 53), because (a) many functional variants are likely regulatory variants of moderate effect (54) rather than highly penetrant Mendelian variants that often impact protein structure (55); (b) gene transcriptional regulation is complicated by tissue specificity as well as dynamic temporal and spatial controls (56); and (c) the impact of genetic variation on gene expression may escape detection in available tissue samples (57). Importantly, genetic variants associated with a phenotype of interest often do not regulate the closest gene (58, 59). Most GWAS variants are located in noncoding regions, and they can regulate genes based on long-range chromatin interactions. Thus, the discovery of a region of interest using GWAS is just the beginning of an odyssey that includes fine mapping of associated regions, identifying long-range chromatin interactions, and assessing variants for functional activity. An additional challenge is posed by the linkage disequilibrium between genetic variants, which assists in the identification of a GWAS signal but makes the identification of a functional variant more challenging.
Geneticists debate the importance of finding the functional variant in a region of association. Some argue that as long as the key gene is found, the functional variant is not important. However, efforts to identify functional variants can be advantageous for the following reasons. First, confirming the functional variant (or variants) can assist in determining that the right gene (or genes) in a GWAS region has been found. Second, the functional variant can point to the biological mechanism for an association, such as altering an enhancer’s activity, a transcription factor’s binding site, or another gene regulatory event. Finally, the functional variant may regulate multiple genes, and those insights can assist in building biological networks relevant for disease.
In an early example of functional variant identification in a COPD GWAS locus, Zhou and colleagues (29) studied the HHIP GWAS region on chromosome 4q31. Within the chromosome 4q31 COPD GWAS locus, chromosome conformation capture (3C) studies in Beas2B (lung epithelial) and MRC5 (lung fibroblast) cell lines identified a 7-kb region approximately 85 kb upstream from the HHIP gene that showed a long-range interaction with the HHIP promoter (Figure 4). Subsequently, enhancer activity was detected within a 500-bp subset of this genomic region. By resequencing, two common SNPs located inside this enhancer region were found (rs6537296 and rs1542725). Electrophoretic mobility shift assays demonstrated that the COPD-associated allele at rs1542725 binds more avidly to the transcription factor Sp3, likely leading to reduced HHIP gene expression levels (29). Thus, this work identified a common functional variant influencing COPD and demonstrated that it influenced HHIP through long-range chromatin interactions.
Figure 4.
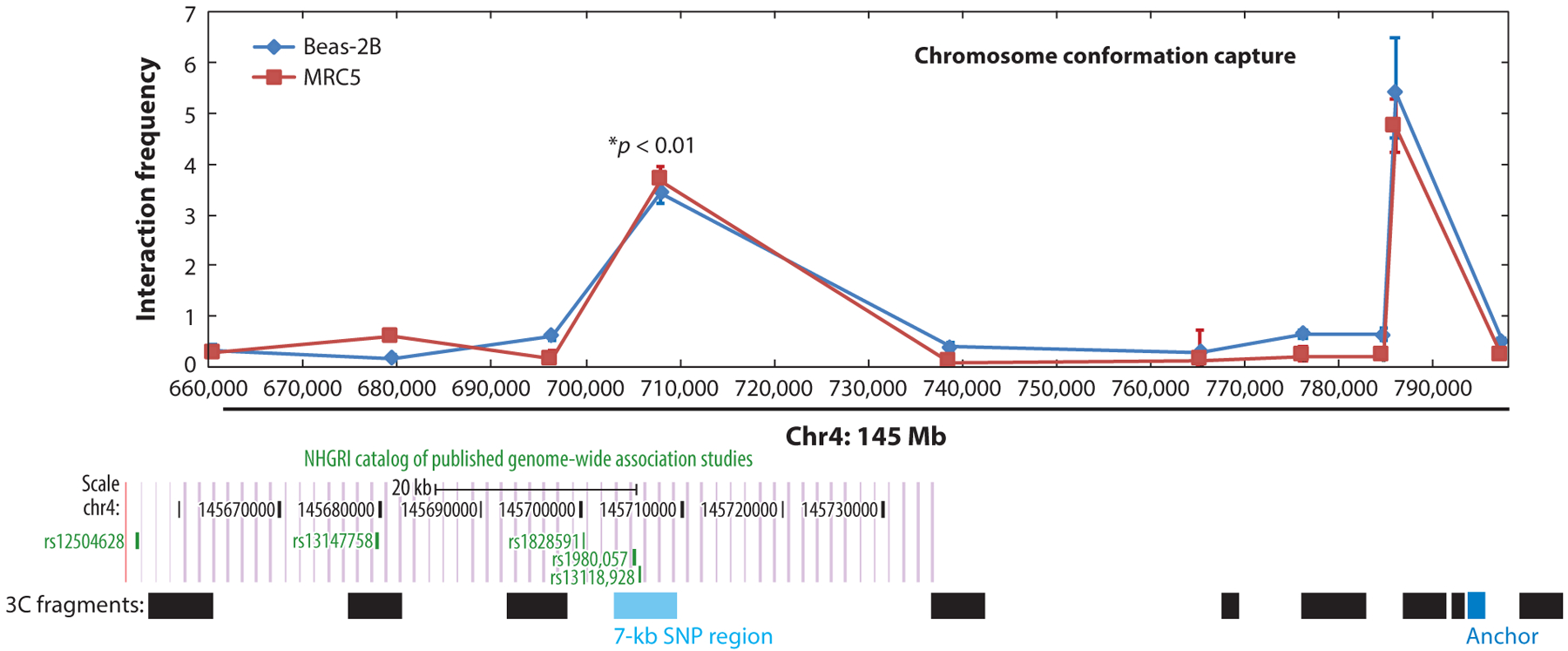
Long-range interaction detected between the COPD GWAS region and HHIP promoter (labeled as Anchor). Chromosome conformation capture assay demonstrated a 7-kb region of interaction upstream from the HHIP gene with the HHIP promoter. This upstream interacting genomic region is located within a frequently replicated COPD GWAS locus. Abbreviation: GWAS, genome-wide association study. Adapted with permission from Reference 29.
The HHIP work described above was laborious and low throughput. One of the key limitations in assessing potential functional regulatory variants has been the low throughput of traditional laboratory assays of gene regulation, such as luciferase reporter assays. Melnikov, Mikkelsen, and colleagues (60, 61) proposed a potential solution to this limitation with the development of massively parallel reporter assays (MPRAs), which enabled the simultaneous assessment of hundreds or even thousands of potential regulatory variants. In MPRAs, oligonucleotides containing potential regulatory sequences are linked to specific sequence tags, cloned into plasmids that include a promoter element (e.g., SV40) and an open reading frame of DNA sequence along with the potential regulatory sequence and sequence tag, and generated into a library of reporter constructs (61). After multiplexed transfection of these constructs into mammalian cells of interest, the expression levels of the potential regulatory sequences are assessed by sequencing both the mRNA of those cells and the library of plasmids. Specific regions are identified by their sequence tags, and the relative expression levels of different regulatory sequences are compared by determining the sequence counts in the plasmid pools and the mRNA. With these data, the expression levels of regulatory constructs containing alternate SNP alleles can be compared. Thus, allele-specific enhancer (and potentially silencer) effects can be assessed.
Castaldi et al. (62) utilized MPRAs to implicate potential regulatory variants in the FAM13A COPD GWAS region. They included all SNPs in the FAM13A GWAS locus associated with COPD at p < 0.02 in the initial GWAS (24); in total, 606 SNPs were tested. They compared the output-to-input ratios of the two alleles for each SNP in the MPRA to identify allele-specific expression differences in Beas2B cells. Using a false discovery rate of 0.05, 45 SNPs demonstrated significant allele-specific regulatory activity in MPRA. Based on COPD GWAS p-values and enhancer/silencer effect size, six SNPs were prioritized for validation in reporter assays. Three (rs2013701, rs7671167, and rs1795739) out of six SNPs demonstrated significant allele-specific activity in 16HBE cells. Using 3C, researchers showed that rs2013701 and rs1795739 interacted with the FAM13A promoter. CRISPR-based studies provided functional support for rs2013701 as at least one of the functional variants in this COPD GWAS region. Although MPRAs have led to important progress in identification of functional variants, there is a clear need for higher-throughput assessments of functional variants.
8. SEQUENCING-BASED APPROACHES TO IDENTIFY COPD GENETIC DETERMINANTS
Although GWAS are highly effective at identifying common genetic determinants of complex diseases such as COPD, they are substantially less useful for the identification of rare genetic determinants. Fortunately, the cost of whole exome and whole genome sequencing studies has fallen substantially, and these assessments are now feasible in large numbers of subjects. Stanley and colleagues (63) utilized whole exome sequencing data in the COPDGene study to demonstrate that rare variants in the TERT gene were associated with emphysema. In small numbers of severe COPD subjects and resistant smokers, Radder et al. (64) performed whole genome sequencing and identified several suggestive associations, including the PTPRO gene.
In the Boston Early-Onset COPD Study, Qiao and colleagues (65) performed whole exome sequencing in extended pedigrees of individuals with severe, early-onset COPD but without AAT deficiency. Although 69 genes had potentially functional variants segregating with COPD in at least two families, there were no genes that had variants segregating in more than three families. Coding variants in a single gene could, at most, only explain a small percentage of severe COPD cases. Thus, if coding variants are the cause of severe, early-onset COPD without AAT deficiency in these families, they are apparently genetically heterogeneous.
Large numbers of COPD subjects are undergoing whole genome sequencing in the National Heart, Lung, and Blood Institute (NHLBI) Trans-Omics for Precision Medicine (TOPMed) program. In 821 severe COPD cases and 973 control smokers with normal spirometry, whole genome sequencing revealed significant associations near the previously reported COPD GWAS locus near HHIP. Several novel suggestive associations to COPD were also observed, including a variant near SERPINA6 in African Americans, which was not explained by the SERPINA1 Z allele. In addition, this study found inconsistent evidence for previously described rare variants in COPD, underscoring the need for further studies in this area.
What can we expect to learn from large-scale whole genome sequencing studies of COPD? Except in other ethnicities, such as African Americans, for whom the genotyping and imputation panels have been suboptimal, additional common variant associations are unlikely. However, statistical fine mapping of COPD GWAS regions will be facilitated (59). Importantly, association of rare variants with COPD and COPD-related phenotypes will be enabled. In addition, these whole genome sequencing data will provide insights into genetic determinants of omics data types collected on the same subjects, which will empower systems and network-based analyses.
9. BIOLOGICAL INSIGHTS FROM COPD GENETICS
Cell-based and animal model studies of genes located within COPD GWAS loci have provided novel insights into COPD pathobiology. Demonstrating a COPD-relevant phenotype in an animal model, such as increased (or decreased) emphysema in gene-targeted mice exposed to chronic cigarette smoke, increases the likelihood that a gene within a COPD GWAS region is functional in COPD pathogenesis. As shown in Figure 5, there are currently approximately 119 genes that demonstrate a COPD-related phenotype in a murine transgenic or knock-out model, and there are currently approximately 84 COPD and/or emphysema loci. The eight genes that are located within a COPD/emphysema GWAS locus and also show a murine COPD phenotype [HHIP (66), FAM13A (67), IREB2 (68), AGER (69), MMP1 (70), MMP12 (71), SFTPD (72), and FBLN5] are likely COPD susceptibility genes that could provide valuable clues to COPD pathogenesis. Some of these biological pathways, such as protease-antiprotease balance (MMP1 and MMP12) and extracellular matrix (FBLN5), have been known for decades. Studies of COPD GWAS genes have revealed novel roles in COPD-related processes, such as FAM13A in WNT/β-catenin signaling (67). However, other biological processes, such as mitochondrial iron (related to IREB2) and hedgehog signaling (related to HHIP, although HHIP may also have other biological functions) (66), were not widely studied before the COPD GWAS era. Surfactant protein D [encoded by SFTPD (73)] and sRAGE (encoded by AGER) are promising blood biomarkers that have been associated with COPD (46).
Figure 5.
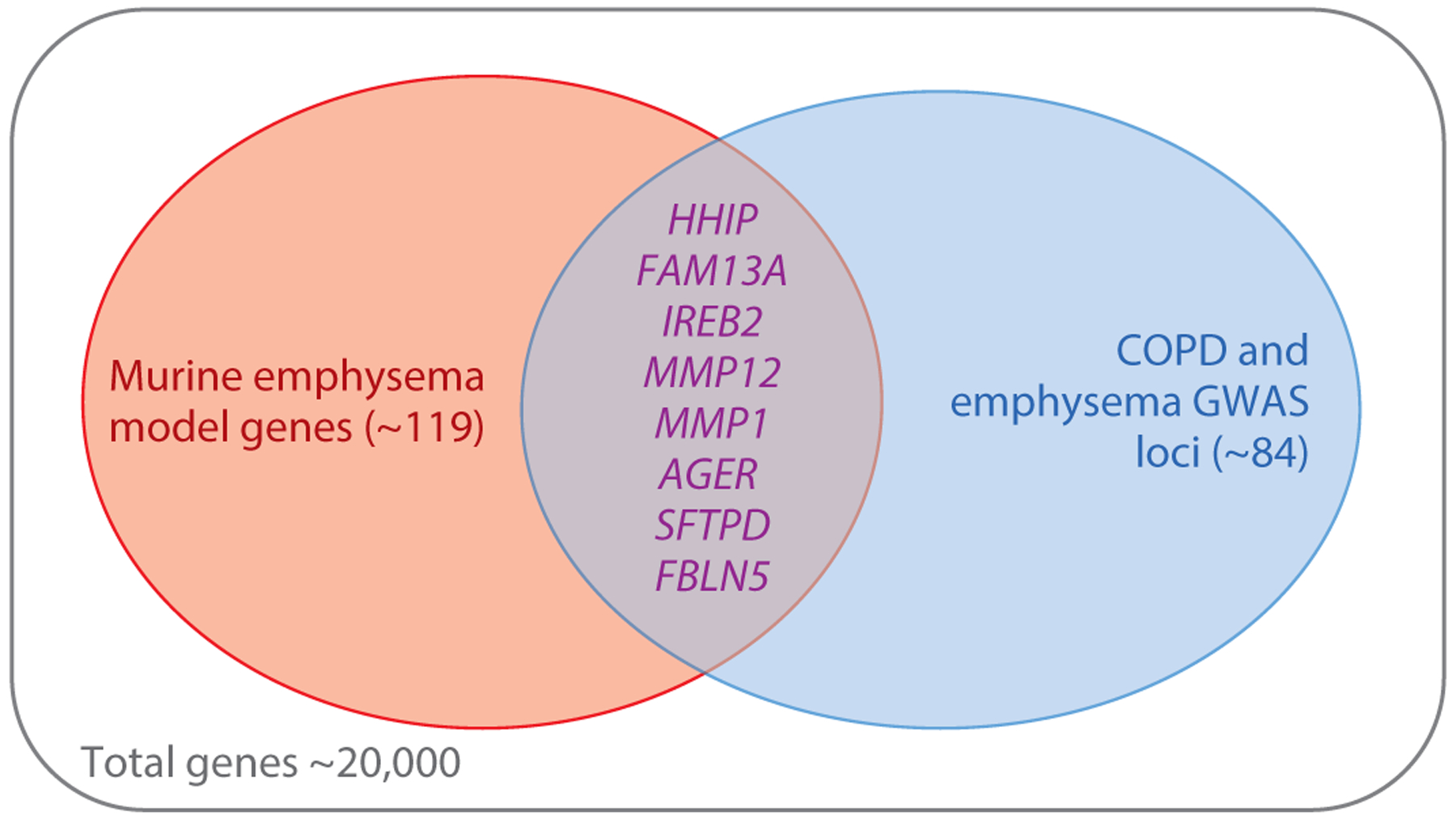
Venn diagram demonstrating the overlap of genes implicated by murine emphysema models and genes located near genome-wide significant associations to COPD. Out of a total of approximately 20,000 mammalian genes, only eight are located in both COPD genome-wide association regions and have been supported by a gene-targeted murine model of emphysema.
10. NETWORKS AND COPD GENETICS
Investigators studying complex disease genetics are beginning to recognize the value of network-based approaches in defining complex disease genetic architecture. For example, Boyle and colleagues (74) recently proposed an omnigenic model for complex diseases in which many loci of very small effect influence genes of larger effect through network connections. Morrow et al. (75) found that COPD susceptibility genes typically did not show substantial differences in lung tissue gene expression between COPD cases and controls, but the genes that they interact with (for example, based on protein–protein interactions) are often associated with COPD or lung function. Thus, COPD genetics research would likely benefit from increased used of network-based approaches.
Although genetics, transcriptomics, metabolomics, proteomics, and epigenetics can provide large-scale resources for COPD research, the methods used to analyze these data types need to evolve to take advantage of the multifaceted information available. Standard association analyses assess a single outcome with individual potential predictor variables, using approaches such as linear or logistic regression. Multivariable models can include multiple predictor variables, but integration of multiple omics data remains challenging, and assumptions about linear relationships persist. Interactions are often ignored or analyzed simply with cross-product terms in the regression analysis. The development and application of methods to recognize and quantify nonlinear relationships and interactions will be necessary to analyze Big Data effectively in complex diseases such as COPD, especially in the context of age-related effects.
Network science, based on methods from graph theory, provides approaches that can assist in the analysis of Big Data in complex diseases. Networks provide a useful framework to visualize and analyze relationships—both linear and nonlinear—and interactions between variables of interest. Networks are composed of entities, represented by nodes and edges, which indicate a relationship between specific nodes. For example, in a protein–protein interaction network, the nodes represent individual proteins, and an edge is placed if there is a physical interaction detected between those two proteins (e.g., based on affinity purification/mass spectrometry or yeast 2-hybrid assays). In addition to visualizing relationships between nodes, properties of the network, such as the number of connections to different nodes, can provide important information about network structure and response to perturbations. The multiple interactions encoded within networks can lead to network behaviors that cannot be predicted from studying isolated nodes or pairs of nodes; these complex responses are referred to as emergent properties.
The term network medicine has been used to describe the application of network science to the study of disease (76–78). Network medicine is not limited to a single type of network or a single source of data. Measuring and integrating multiple omics data types are key parts of network medicine. Principled analysis can provide disease-related network models that then can be used to reclassify diseases such as COPD based on their etiology instead of end-stage physiological and pathological manifestations—our current approach for classifying most diseases.
Multiple types of network models can be used in network medicine. Protein–protein interactions within the molecular interactome (79) have been utilized to identify interconnected subsets of interacting proteins related to specific diseases, known as disease network modules. One approach to identify such disease network modules is based on genetic association evidence. Using the dmGWAS method (80), McDonald and colleagues (81) found a consensus module for COPD within the protein–protein interaction network that was enriched in interleukin (IL)-7 pathway members. GWAS genes can also be used as seed genes in random walk approaches within the molecular interactome of protein–protein interactions to define disease network modules; Sharma et al. (82) identified a first-generation COPD disease network module of 163 genes using this approach. Correlation networks based on gene expression levels can identify gene network modules having similar gene expression patterns using approaches such as weighted gene coexpression network analysis (WGCNA) (83). Correlation-based network analysis of COPD and control lung tissue gene expression data demonstrated that B cell–related pathways differentiate COPD cases and controls (75). Gene regulatory networks have been developed based on relating transcription factor binding site information and gene expression levels using ap proaches such as PANDA (84). PANDA analysis using mice heterozygous for deficiency of the COPD GWAS gene Hhip demonstrated network rewiring related to the Klf4 transcription factor (66).
These network medicine modeling approaches can be described as top-down efforts to use Big Data to identify disease-related networks related to COPD pathogenesis. However, bottom-up approaches to build disease networks by identifying the biological mechanisms and molecular interactions of well-supported COPD susceptibility genes such as HHIP and FAM13A can also be used to create disease networks. Ideally, the top-down and bottom-up network-building approaches will synergistically identify biological interactions between key molecules involved in COPD pathobiology, as shown in Figure 6.
Figure 6.
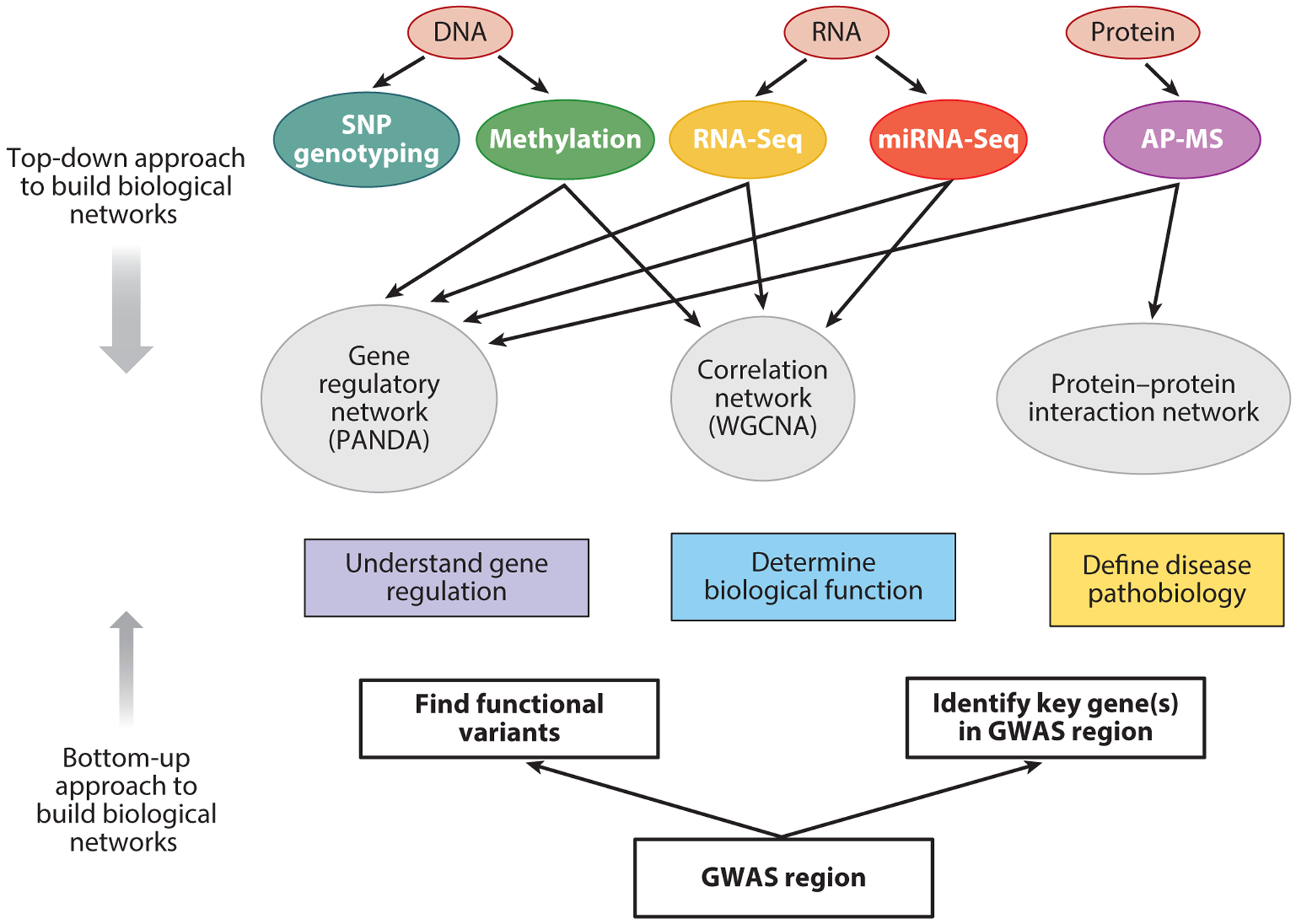
Comparison of top-down and bottom-up approaches to build biological networks. Biological networks can be built from the bottom up, starting with GWAS regions and identifying the key genes and the functional variants that impact those genes. Networks can be extended from those genes using tools that assess binding with other proteins (e.g., tandem affinity purification, coimmunoprecipitation) as well as hypothesis-based molecular biology experiments. Top-down approaches begin with Big Data assessments of key biological molecules such as DNA, RNA, proteins, and metabolites. Various types of networks can be built, including correlation-based networks, gene regulatory networks, and protein–protein interaction networks. Ultimately, bottom-up and top-down approaches may converge to give insights into gene regulation, biological function, and disease pathobiology relevant to COPD. The top-down approach is encompassed by the omics data and network methods shown above the rectangles labeled “Understand gene regulation,” “Determine biological function,” and “Define disease pathobiology,” whereas the bottom-up approach includes the components below those rectangles. Abbreviations: AP-MS, affinity purification–mass spectrometry; GWAS, genome-wide association study; SNP, single nucleotide polymorphism.
11. FUTURE DIRECTIONS FOR COPD GENETICS
Although the rapidly expanding number of genetic loci associated with COPD is encouraging, many areas of investigation will need to be pursued to translate these discoveries into new models of COPD pathogenesis. The key genes and functional variants within these COPD GWAS regions need to be identified. The measurement of omics data could provide important biological clues, but effective multi-omics integration methods will be required. Variable relationships of COPD genetic determinants to COPD-related phenotypes suggest a genetic contribution to COPD heterogeneity, but greater understanding of the mechanisms for those variable associations is needed. Ultimately, we need to perform disease subtyping based on biological mechanisms instead of physiology and pathology. We also need to translate the biological understanding of COPD into new treatments. Treatments tailored to COPD subtype would be ideal, and systems pharmacology approaches that focus on multiple treatment targets, potentially in a dynamic fashion, may be beneficial.
ACKNOWLEDGMENTS
The author thanks Dr. Michael Cho for helpful comments on this article. This work was supported by US National Institutes of Health grants U01 HL089856, R01 HL113264, P01 HL114501, R01 HL133135, and R01 HL137927 to E.K.S.
DISCLOSURE STATEMENT
In the past three years, the author has received grant support from GlaxoSmithKline.
LITERATURE CITED
- 1.Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, et al. 2015. The support of human genetic evidence for approved drug indications. Nat. Genet 47:856–60 [DOI] [PubMed] [Google Scholar]
- 2.Silverman EK, Mosley JD, Palmer LJ, Barth M, Senter JM, et al. 2002. Genome-wide linkage analysis of severe, early-onset chronic obstructive pulmonary disease: airflow obstruction and chronic bronchitis phenotypes. Hum. Mol. Genet 11:623–32 [DOI] [PubMed] [Google Scholar]
- 3.Hersh CP, Demeo DL, Lange C, Litonjua AA, Reilly JJ, et al. 2005. Attempted replication of reported chronic obstructive pulmonary disease candidate gene associations. Am. J. Respir. Cell Mol. Biol 33:71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hersh CP, DeMeo D, Silverman EK. 2005. COPD. In Respiratory Genetics, ed. Silverman EK, Shapiro SD, Lomas DA, Weiss ST, pp. 253–96. London: Hodder Arnold [Google Scholar]
- 5.Silverman EK, Chapman HA, Drazen JM, Weiss ST, Rosner B, et al. 1998. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease: risk to relatives for airflow obstruction and chronic bronchitis. Am. J. Respir. Crit. Care Med 157:1770–78 [DOI] [PubMed] [Google Scholar]
- 6.McCloskey SC, Patel BD, Hinchliffe SJ, Reid ED, Wareham NJ, Lomas DA. 2001. Siblings of patients with severe chronic obstructive pulmonary disease have a significant risk of airflow obstruction. Am. J. Respir. Crit. Care Med 164:1419–24 [DOI] [PubMed] [Google Scholar]
- 7.Ingebrigtsen T, Thomsen SF, Vestbo J, van der Sluis S, Kyvik KO, et al. 2010. Genetic influences on chronic obstructive pulmonary disease—a twin study. Respir. Med 104:1890–95 [DOI] [PubMed] [Google Scholar]
- 8.Zhou JJ, Cho MH, Castaldi PJ, Hersh CP, Silverman EK, Laird NM. 2013. Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am. J. Respir. Crit. Care Med 188:941–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silverman EK, Sandhaus RA. 2009. Alpha1-antitrypsin deficiency. N. Engl. J. Med 360:2749–57 [DOI] [PubMed] [Google Scholar]
- 10.DeMeo DL, Sandhaus RA, Barker AF, Brantly ML, Eden E, et al. 2007. Determinants of airflow obstruction in severe alpha-1-antitrypsin deficiency. Thorax 62:806–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mostafavi B, Diaz S, Piitulainen E, Stoel BC, Wollmer P, Tanash HA. 2018. Lung function and CT lung densitometry in 37- to 39-year-old individuals with alpha-1-antitrypsin deficiency. Int. J. Chron. Obstruct. Pulmon. Dis 13:3689–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapman KR, Burdon JG, Piitulainen E, Sandhaus RA, Seersholm N, et al. 2015. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet 386:360–68 [DOI] [PubMed] [Google Scholar]
- 13.Hersh CP, Dahl M, Ly NP, Berkey CS, Nordestgaard BG, Silverman EK. 2004. Chronic obstructive pulmonary disease in α1-antitrypsin PI MZ heterozygotes: a meta-analysis. Thorax 59:843–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silverman EK. 2016. Risk of lung disease in PI MZ heterozygotes. Current status and future research directions. Ann. Am. Thorac. Soc 13(Suppl. 4):S341–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sorheim IC, Bakke P, Gulsvik A, Pillai SG, Johannessen A, et al. 2010. α;1-Antitrypsin protease inhibitor MZ heterozygosity is associated with airflow obstruction in two large cohorts. Chest 138:1125–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Molloy K, Hersh CP, Morris VB, Carroll TP, O’Connor CA, et al. 2014. Clarification of the risk of chronic obstructive pulmonary disease in α1-antitrypsin deficiency PiMZ heterozygotes. Am. J. Respir. Crit. Care Med 189:419–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foreman MG, Wilson C, DeMeo DL, Hersh CP, Beaty TH, et al. 2017. Alpha-1 antitrypsin PiMZ genotype is associated with chronic obstructive pulmonary disease in two racial groups. Ann. Am. Thorac. Soc 14:1280–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Ortega VE, Ampleford EJ, Graham Barr R, Christenson SA, et al. 2018. Genome-wide association study of lung function and clinical implication in heavy smokers. BMC Med. Genet 19:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pillai SG, Ge D, Zhu G, Kong X, Shianna KV, et al. 2009. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLOS Genet. 5:e1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilk JB, Chen TH, Gottlieb DJ, Walter RE, Nagle MW, et al. 2009. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLOS Genet. 5:e1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hancock DB, Eijgelsheim M, Wilk JB, Gharib SA, Loehr LR, et al. 2010. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat. Genet 42:45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Repapi E, Sayers I, Wain LV, Burton PR, Johnson T, et al. 2010. Genome-wide association study identifies five loci associated with lung function. Nat. Genet 42:36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho MH, Boutaoui N, Klanderman BJ, Sylvia JS, Ziniti JP, et al. 2010. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat. Genet 42:200–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho MH, McDonald ML, Zhou X, Mattheisen M, Castaldi PJ, et al. 2014. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir. Med 2:214–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silverman EK, Vestbo J, Agusti A, Anderson W, Bakke PS, et al. 2011. Opportunities and challenges in the genetics of COPD 2010: an International COPD Genetics Conference report. COPD 8:121–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hobbs BD, de Jong K, Lamontagne M, Bosse Y, Shrine N, et al. 2017. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat. Genet 49:426–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakornsakolpat P, Prokopenko D, Lamontagne M, Reeve NF, Guyatt AL, et al. 2019. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat. Genet 51:494–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Durme YM, Eijgelsheim M, Joos GF, Hofman A, Uitterlinden AG, et al. 2010. Hedgehog-interacting protein is a COPD susceptibility gene: the Rotterdam Study. Eur. Respir. J 36:89–95 [DOI] [PubMed] [Google Scholar]
- 29.Zhou X, Baron RM, Hardin M, Cho MH, Zielinski J, et al. 2012. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum. Mol. Genet 21:1325–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wain LV, Shrine N, Miller S, Jackson VE, Ntalla I, et al. 2015. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir. Med 3:769–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Young RP, Hopkins RJ, Hay BA, Whittington CF, Epton MJ, Gamble GD. 2011. FAM13A locus in COPD is independently associated with lung cancer—evidence of a molecular genetic link between COPD and lung cancer. Appl. Clin. Genet 4:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie J, Wu H, Xu Y, Wu X, Liu X, et al. 2015. Gene susceptibility identification in a longitudinal study confirms new loci in the development of chronic obstructive pulmonary disease and influences lung function decline. Respir. Res 16:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ziółkowska-Suchanek I, Mosor M, Gabryel P, Grabicki M, Zurawek M, et al. 2015. Susceptibility loci in lung cancer and COPD: association of IREB2 and FAM13A with pulmonary diseases. Sci. Rep 5:13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siedlinski M, Tingley D, Lipman PJ, Cho MH, Litonjua AA, et al. 2013. Dissecting direct and indirect genetic effects on chronic obstructive pulmonary disease (COPD) susceptibility. Hum. Genet 132:431–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Busch R, Hobbs BD, Zhou J, Castaldi PJ, McGeachie MJ, et al. 2017. Genetic association and risk scores in a COPD meta-analysis of 16,707 subjects. Am. J. Respir. Cell Mol. Biol 57:35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wain LV, Shrine N, Artigas MS, Erzurumluoglu AM, Noyvert B, et al. 2017. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat. Genet 49:416–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burkart KM, Sofer T, London SJ, Manichaikul A, Hartwig FP, et al. 2018. A genome-wide association study in Hispanics/Latinos identifies novel signals for lung function. The Hispanic Community Health Study/Study of Latinos. Am. J. Respir. Crit. Care Med 198:208–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wyss AB, Sofer T, Lee MK, Terzikhan N, Nguyen JN, et al. 2018. Multiethnic meta-analysis identifies ancestry-specific and cross-ancestry loci for pulmonary function. Nat. Commun 9:2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shrine N, Guyatt AL, Erzurumluoglu AM, Jackson VE, Hobbs BD, et al. 2019. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat. Genet 51:481–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lange P, Celli B, Agusti A, Jensen GB, Divo M, et al. 2015. Lung-function trajectories leading to chronic obstructive pulmonary disease. N. Engl. J. Med 373:111–22 [DOI] [PubMed] [Google Scholar]
- 41.Tang W, Kowgier M, Loth DW, Artigas MS, Joubert BR, et al. 2014. Large-scale genome-wide association studies and meta-analyses of longitudinal change in adult lung function. PLOS ONE 9:e100776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.John C, Artigas MS, Hui J, Nielsen SF, Rafaels N, et al. 2017. Genetic variants affecting cross-sectional lung function in adults show little or no effect on longitudinal lung function decline. Thorax 72:400–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ross JC, Castaldi PJ, Cho MH, Chen J, Chang Y, et al. 2017. A Bayesian nonparametric model for disease subtyping: application to emphysema phenotypes. IEEE Trans. Med. Imaging 36:343–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ragland MF, Benway CJ, Lutz SM, Bowler RP, Hecker J, et al. 2019. Genetic advances in COPD: insights from COPDGene. Am. J. Respir. Crit. Care Med 200:677–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manichaikul A, Hoffman EA, Smolonska J, Gao W, Cho MH, et al. 2014. Genome-wide study of percent emphysema on computed tomography in the general population. The Multi-Ethnic Study of Atherosclerosis Lung/SNP Health Association Resource Study. Am. J. Respir. Crit. Care Med 189:408–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yonchuk JG, Silverman EK, Bowler RP, Agusti A, Lomas DA, et al. 2015. Circulating soluble receptor for advanced glycation end products (sRAGE) as a biomarker of emphysema and the RAGE axis in the lung. Am. J. Respir. Crit. Care Med 192:785–92 [DOI] [PubMed] [Google Scholar]
- 47.Cho MH, Castaldi PJ, Hersh CP, Hobbs BD, Barr RG, et al. 2015. A genome-wide association study of emphysema and airway quantitative imaging phenotypes. Am. J. Respir. Crit. Care Med 192:559–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boueiz A, Lutz SM, Cho MH, Hersh CP, Bowler RP, et al. 2017. Genome-wide association study of the genetic determinants of emphysema distribution. Am. J. Respir. Crit. Care Med 195:757–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castaldi PJ, San Jose Estepar R, Mendoza CS, Hersh CP, Laird N, et al. 2013. Distinct quantitative computed tomography emphysema patterns are associated with physiology and function in smokers. Am. J. Respir. Crit. Care Med 188:1083–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castaldi PJ, Dy J, Ross J, Chang Y, Washko GR, et al. 2014. Cluster analysis in the COPDGene study identifies subtypes of smokers with distinct patterns of airway disease and emphysema. Thorax 69:415–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, et al. 2009. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. PNAS 106:9362–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Freedman ML, Monteiro AN, Gayther SA, Coetzee GA, Risch A, et al. 2011. Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet 43:513–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Juran BD, Lazaridis KN. 2011. Genomics in the post-GWAS era. Semin. Liver Dis 31:215–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cooper GM, Shendure J. 2011. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet 12:628–40 [DOI] [PubMed] [Google Scholar]
- 55.Gibson G 2011. Rare and common variants: twenty arguments. Nat. Rev. Genet 13:135–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanyal A, Lajoie BR, Jain G, Dekker J. 2012. The long-range interaction landscape of gene promoters. Nature 489:109–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, et al. 2009. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet 41:882–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baxter JS, Leavy OC, Dryden NH, Maguire S, Johnson N, et al. 2018. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nat. Commun 9:1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schaid DJ, Chen W, Larson NB. 2018. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet 19:491–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, et al. 2012. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol 30:271–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Melnikov A, Zhang X, Rogov P, Wang L, Mikkelsen TS. 2014. Massively parallel reporter assays in cultured mammalian cells. J. Vis. Exp 90:e51719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Castaldi PJ, Guo F, Qiao D, Du F, Naing ZZC, et al. 2019. Identification of functional variants in the FAM13A chronic obstructive pulmonary disease genome-wide association study locus by massively parallel reporter assays. Am. J. Respir. Crit. Care Med 199:52–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stanley SE, Chen JJ, Podlevsky JD, Alder JK, Hansel NN, et al. 2015. Telomerase mutations in smokers with severe emphysema. J. Clin. Investig 125:563–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Radder JE, Zhang Y, Gregory AD, Yu S, Kelly NJ, et al. 2017. Extreme trait whole-genome sequencing identifies PTPRO as a novel candidate gene in emphysema with severe airflow obstruction. Am. J. Respir. Crit. Care Med 196:159–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qiao D, Lange C, Beaty TH, Crapo JD, Barnes KC, et al. 2016. Exome sequencing analysis in severe, early-onset chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med 193:1353–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lao T, Glass K, Qiu W, Polverino F, Gupta K, et al. 2015. Haploinsufficiency of Hedgehog interacting protein causes increased emphysema induced by cigarette smoke through network rewiring. Genome Med. 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang Z, Lao T, Qiu W, Polverino F, Gupta K, et al. 2016. A chronic obstructive pulmonary disease susceptibility gene, FAM13A, regulates protein stability of β-catenin. Am. J. Respir. Crit. Care Med 194:185–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cloonan SM, Glass K, Laucho-Contreras M, Bhashyam AR, Cervo M, et al. 2016. Mitochondrial iron as a therapeutic target for IRP2-regulated cigarette smoke-induced bronchitis and emphysema. Nat. Med 22:163–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sambamurthy N, Leme AS, Oury TD, Shapiro SD. 2015. The receptor for advanced glycation end products (RAGE) contributes to the progression of emphysema in mice. PLOS ONE 10:e0118979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.D’Armiento J, Dalal SS, Okada Y, Berg RA, Chada K. 1992. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell 71:955–61 [DOI] [PubMed] [Google Scholar]
- 71.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. 1997. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 277:2002–4 [DOI] [PubMed] [Google Scholar]
- 72.Wert SE, Yoshida M, LeVine AM, Ikegami M, Jones T, et al. 2000. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. PNAS 97:5972–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lomas DA, Silverman EK, Edwards LD, Locantore NW, Miller BE, et al. 2009. Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. Eur. Resp. J 34:95–102 [DOI] [PubMed] [Google Scholar]
- 74.Boyle EA, Li YI, Pritchard JK. 2017. An expanded view of complex traits: from polygenic to omnigenic. Cell 169:1177–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morrow J, Zhou X, Lao T, Jiang Z, Demeo DL, et al. 2017. Functional interactors of three genome-wide association study genes are differentially expressed in severe chronic obstructive pulmonary disease lung tissue. Sci. Rep 7:44232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barabasi AL. 2007. Network medicine—from obesity to the “diseasome.” N. Engl. J. Med 357:404–7 [DOI] [PubMed] [Google Scholar]
- 77.Barabasi AL, Gulbahce N, Loscalzo J. 2011. Network medicine: a network-based approach to human disease. Nat. Rev. Genet 12:56–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loscalzo J, Barabasi AL, Silverman EK, eds. 2017. Network Medicine: Complex Systems in Human Disease and Therapeutics. Cambridge, MA: Harvard Univ. Press [Google Scholar]
- 79.Vidal M, Cusick ME, Barabasi AL. 2011. Interactome networks and human disease. Cell 144:986–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jia P, Zheng S, Long J, Zheng W, Zhao Z. 2011. dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics 27:95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McDonald ML, Mattheisen M, Cho M, Liu Y-Y, Harshfield B, et al. 2014. Beyond GWAS in COPD: probing the landscape between gene-set associations, genome-wide associations and protein-protein interaction networks. Hum. Heredity 78:131–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sharma A, Kitsak M, Cho MH, Ameli A, Zhou X, et al. 2018. Integration of molecular interactome and targeted interaction analysis to identify a COPD disease network module. Sci. Rep 8:14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Langfelder P, Horvath S. 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Glass K, Huttenhower C, Quackenbush J, Yuan GC. 2013. Passing messages between biological networks to refine predicted interactions. PLOS ONE 8:e64832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hardin M, Silverman EK. 2014. Chronic obstructive pulmonary disease genetics: a review of the past and a look into the future. J. COPD Found 1:33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]