

Abstract
Background
Nipah belongs to the genus Henipavirus and the Paramyxoviridae family. It is an endemic most commonly found at South Asia and has first emerged in Malaysia in 1998. Bats are found to be the main reservoir for this virus, causing disease in both humans and animals. The last outbreak has occurred in May 2018 in Kerala. It is characterized by high pathogenicity and fatality rates which varies from 40% to 70% depending on the severity of the disease and on the availability of adequate healthcare facilities. Currently, there are no antiviral drugs available for NiV disease and the treatment is just supportive. Clinical presentations for this virus range from asymptomatic infection to fatal encephalitis.
Objective
This study is aimed at predicting an effective epitope-based vaccine against glycoprotein G of Nipah henipavirus, using immunoinformatics approaches.
Methods and Materials
Glycoprotein G of the Nipah virus sequence was retrieved from NCBI. Different prediction tools were used to analyze the epitopes, namely, BepiPred-2.0: Sequential B Cell Epitope Predictor for B cell and T cell MHC classes II and I. Then, the proposed peptides were docked using Autodock 4.0 software program. Results and Conclusions. The two peptides TVYHCSAVY and FLIDRINWI have showed a very strong binding affinity to MHC class I and MHC class II alleles. Furthermore, considering the conservancy, the affinity, and the population coverage, the peptide FLIDRINWIT is highly suitable to be utilized to formulate a new vaccine against glycoprotein G of Nipah henipavirus. An in vivo study for the proposed peptides is also highly recommended.
1. Introduction
Nipah virus (NiV) is an RNA virus that belongs to the genus Henipavirus within the family Paramyxoviridae and has first emerged in Malaysia in 1998, gaining its name from a village called Sungai Nipah where it was isolated from the cerebrospinal fluid (CSF) of one of the patients [1–4]. NiV is transmitted zoonotically (from bats to humans, or from bats to pigs, and then to humans) as well as human-to-human routes. Its clinical presentation varies from asymptomatic (subclinical) infection to acute respiratory illnesses and fatal encephalitis in most of the patients who has been in direct contact with infected pigs. It has also been found that the virus causes central nervous system illnesses in pigs and respiratory illnesses in horses resulting in a significant economic loss for farmers [1, 5–9]. Large fruit bats of the genus Pteropus seem to act as a natural reservoir of NiV based on the isolation of Hendra virus which has showed the presence of neutralizing antibodies to the Hendra virus on the bats [10, 11]. Although, there are no more cases of NiV in Malaysia, several outbreaks have been frequently occurring in India, Bangladesh, Thailand, and Cambodia [12]. The case fatality rate ranges from 50% to 100%, making it one of the deadliest viruses known to infect humans [3, 13, 14].
Laboratory diagnosis of Nipah virus infection is made using reverse transcriptase polymerase chain reaction (RT-PCR) from throat swabs, cerebrospinal fluid, urine, and blood analysis during acute and convalescent stages of the disease. IgG and IgM antibody detection can be done after recovery to confirm Nipah virus infection. Immunohistochemistry on tissues collected during an autopsy can also confirm the disease [15, 16]. Currently, there are no effective treatments for the Nipah virus infection. Therefore, a few precautions should be followed such as practicing standard infection control, barrier nursing to avoid the spread of the infection from person to person, and the isolation of those suspected to have the infection [7, 8, 17]. Recent computational approaches have provided further information about viruses, including the study conducted by Badawi M et al. on Zika virus, where the envelope glycoprotein was obtained using protein databases. The most immunogenic epitope for the T and B cells involved in cell-mediated immunity was previously analyzed [18]. The main focus of the analysis was the MHC class I potential peptides using in silico analysis techniques [19, 20]. In this study, the same techniques were applied to keep MHC classes I and II along with the world population coverage as our main focus. Furthermore, in this study, we aimed to design an epitope-based peptide vaccine against Nipah virus using peptides of its glycoprotein G as an immunogenic part to stimulate a protective immune response [3].
Nipah virus invades host cells by the fusion of the host cell membranes at an optimal physiological pH for cleavage without requiring viral endocytosis. Cell-cell fusion is a pathological lineament of Nipah virus infections, resulting in a cell-to-cell spread, inflammation, and destruction of endothelial cells and neurons [21]. Both Nipah virus entry and cell-cell fusion require concerted efforts of the attachment of glycoprotein G and fusion (F) glycoprotein. Upon receptor binding, Nipah virus glycoprotein G triggers a conformational cascade in Nipah virus glycoprotein F that executes a viral and/or a cell membrane fusion [22]. Due to the potency of glycoprotein G over F, we have considered this incident to be the target of this study. There are a lot of challenges regarding the development of peptide-based vaccines, and therefore, we have decided to study and propose a new vaccine against the Nipah virus, since they make a helpful alternative strategy that relies on the usage of short peptide fragments to induce immune responses [23–26]. Antigenic epitopes from single proteins may not be really necessary, whereas some of these epitopes may even be detrimental to the induction of protective immunity. This logic has created an interest in peptide vaccines and especially those containing only epitopes that are capable of inducing desirable T cell- and B cell-mediated immune responses. Less than 20 amino acid sequences make up the peptides used in such vaccines, which are then synthesized to form an immunogenic peptide molecule. These molecules represent a specific epitope of an antigen. These vaccines are also capable of inducing immunity against different strains of a specific pathogen by forming noncontiguous and immunodominant epitopes that are usually conserved in the strains of the pathogen [27].
The production of peptide vaccines is extremely safe and cost-effective, especially when they are compared to conventional vaccines. Traditional vaccines that prevent emerging infectious diseases (EIDs) are very difficult to produce because they require the need to culture pathogenic viruses in vitro. However, epitope-based peptide vaccines do not require any means of in vitro culturing which makes them biologically safe, allowing a large scale of bioprocessing to be carried out rapidly and economically. Finally, their selectivity allows a precise activation of the immunological responses by means of selecting immunodominant and conserved epitopes [25, 28]. The complexity of an epitope-based peptide vaccines' design depends largely on the properties of its carrier molecules' reactogenicity as well as its allergenicity [29, 30]. When it comes to the selection of epitopes, it is based on the analysis of the B cells, cytotoxic T cells, and the induction of the helper T cells. Then, it is important to identify the epitopes capable of activating T cells vital for stimulating a protective immunity. One of the issues concerning peptide vaccines representing T cells in a human population and that are highly MHC-heterogeneous is to identify the highly conserved immunodominant epitopes that are considered to be among a broad spectrum of vaccines due to their ability to work against multiple serovars of pathogens [30]. In this study, we have used a variety of bioinformatics tools for the prediction of epitopes along with the population coverage and epitope selection algorithms, including the translocation of peptides into MHC class I and MHC class II.
2. Materials and Methods
2.1. Sequence Retrieval
The amino acid sequences of glycoprotein G (Glycoside hydrolase family) for a total of 21 strains of Nipah virus were retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov/protein) [31] in a FASTA format on July 2018. Different prediction tools of Immune Epitope Database (IEDB) Analysis Resource (http://www.iedb.org/) [32] were then used to analyze the candidate epitopes.
2.2. Conservation Region and Physicochemical Properties
Conservation regions were determined using multiple sequence alignments with the help of Clustal-W in the BioEdit software version 7.2.5 [33]. Epitope conservancy prediction for individual epitopes was then calculated using the IEDB Analysis Resource. Conservancy can be defined as the portion of a protein sequence that restrains in which an epitope is measured at or which that is exceeding a specific level of identity [34]. The physicochemical properties of the retrieved sequence, molecular weight, and amino acid composition were also determined by using BioEdit software.
2.3. B Cell Epitope Prediction Tools
Candidate epitopes were analyzed using several B cell prediction methods to determine their antigenicity, flexibility, hydrophilicity, and surface accessibility. The predicted linear epitopes were obtained from the Immune Epitope Database (http://tools.iedb.org/bcell/result/) [35] using a BepiPred test with a threshold value of 0.149 and a window size of 6.0. Moreover, surface accessible epitopes were predicated with a threshold value of 1.0 and a window size of 6.0 using the Emini surface accessibility prediction tool [35]. Kolaskar and Tongaonkar antigenicity methods (http://tools.iedb.org/bcell/result/) were also proposed to determine the sites of antigenic epitopes with a default threshold value of 1.030 and a window size 6.0 [36].
2.4. T Cell Epitope Prediction Tools
2.4.1. Peptide Binding to MHC Class I Molecules
The binding peptide was assessed by the IEDB MHC I prediction tool at http://tools.iedb.org/mhcI. This tool employs different methods to determine the ability of the submitted sequence to bind to a specific MHC class I molecule. The artificial neural network (ANN) method was used to calculate IC50 values of the peptide binding to MHC class I molecules. For both frequent and nonfrequent alleles, the peptide length was set to 9 amino acids prior to the prediction. The alleles having a binding affinity of IC50 that are equal to or less than 500 nM were considered for further analysis [37].
2.4.2. Peptide Binding to MHC Class II Molecules
To predict the peptide binding to MHC class II molecules, the MHC II prediction tool http://tools.iedb.org/mhcII provided by the Immune Epitope Database (IEDB) Analysis Resource consisting of human allele references sets was used [38]. The artificial neural network prediction method was chosen to identify the binding affinity of MHC II grooves and MHC II binding core epitopes. All epitopes that bind to many alleles at a score equal to or less than 1000, half-maximal inhibitory concentration (IC50), were selected for further analysis.
2.5. Population Coverage
The population coverage of each epitope was calculated by the IEDB population coverage tool at (http://tools.iedb.org/tools/population/iedb_input). This tool was used in order to determine the fraction of individuals predicted to respond to a given set of epitopes, with known MHC restrictions [39]. For every single population coverage, the tool computed the following information: (1) predicted population coverage, (2) HLA combinations recognized by the population, and (3) HLA combinations recognized by 90% of the population (PC90). All the epitopes and their MHC I and MHC II molecules were assessed against the population coverage area selected before submission.
2.6. Homology Modeling
The 3D structure of glycoprotein G of Nipah virus was predicted using the RaptorX web portal (http://raptorx.uchicago.edu/), where the reference sequence was submitted in a FASTA format on 14/9/2018 and the structure was received on 15/9/2018 [40]. This structure was then treated with UCSF Chimera 1.10.2 to visualize the position of the proposed peptides [41].
2.7. In Silico Molecular Docking
2.7.1. Ligand Preparation
In order to estimate the binding affinities between the epitopes and molecular structures of MHC I and MHC II, we have carried out an in silico molecular docking. Sequences of proposed epitopes were then selected from the Nipah virus reference sequence using Chimera 1.10 and saved as a (pdb) file. The obtained files were then optimized and energy minimized. The HLA-A∗02:01 was selected as the macromolecule for docking. Its crystal structure (4UQ3) was downloaded from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do), which was in a complex with an azobenzene-containing peptide [42].
All water molecules and heteroatoms in the retrieved target file 4UQ3 were then removed. The target structure was further optimized and energy minimized using Swiss PDB viewer V.4.1.0 software [43].
Molecular docking was performed using AutoDock 4.0 software, based on the Lamarckian genetic algorithm, which combines energy evaluation through grids of affinity potential to find the suitable binding position for a ligand on a given protein [44, 45]. Polar hydrogen atoms were added to the protein targets, and Kollman united atomic charges were computed. The targets' grid map was calculated and set to 60 × 60 × 60 points with a grid spacing of 0.375 Ǻ. The grid box was then allocated properly in the target to include the active residue in the center. The genetic algorithm and its run were set to 100 as the docking algorithms were set on default. Finally, results were retrieved as binding energies and poses that showed the lowest binding energies in which they were visualized using UCSF Chimera.
3. Results
3.1. Nipah Virus Glycoprotein G Physical and Chemical Parameters
The physicochemical properties of the Nipah virus glycoprotein G protein was assessed using BioEdit software version 7.0.9.0. The protein length was found to be 602 amino acids, and the molecular weight was at 67035.54 Daltons. The amino acids that form the Nipah virus glycoprotein G protein are shown in Table 1 along with their numbers and molar percentages in (Mol%).
Table 1.
Number and Mol% of amino acids that constituted Nipah virus glycoprotein G using BioEdit software version 7.2.5.
| Amino acid | Number | Mol% | Amino acid | Number | Mol% |
|---|---|---|---|---|---|
| Ala A | 23 | 3.82 | Met M | 11 | 1.83 |
| Cys C | 17 | 2.82 | Asn N | 45 | 7.48 |
| Asp D | 27 | 4.49 | Pro P | 36 | 5.98 |
| Glu E | 26 | 4.32 | Gln Q | 25 | 4.15 |
| Phe F | 21 | 3.49 | Arg R | 22 | 3.65 |
| Gly G | 40 | 6.64 | Ser S | 51 | 8.47 |
| His H | 5 | 0.83 | Thr T | 37 | 6.15 |
| Ile I | 55 | 9.14 | Val V | 41 | 6.81 |
| Lys K | 39 | 6.48 | Trp W | 7 | 1.16 |
| Leu L | 49 | 8.14 | Tyr Y | 25 | 4.15 |
3.2. B Cell Epitope Prediction
The ref sequence of the Nipah virus glycoprotein G was subjected to a Bepipred linear epitope prediction. Emini surface accessibility and Kolaskar and Tongaonkar antigenicity methods in IEDB were used to determine bindings to the B cell and in testing its surface and immunogenicity. The results are shown in Figures 1–3.
Figure 1.
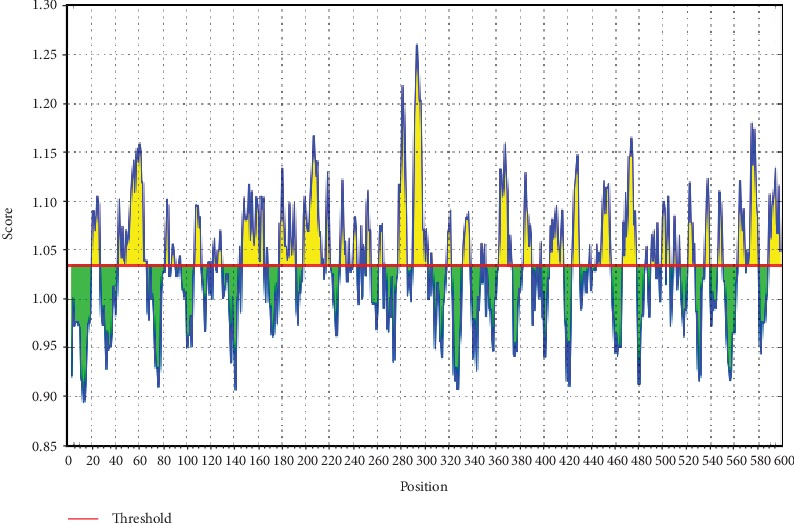
BepiPred linear prediction. Areas above the red line (threshold) are epitopes suggested to be binding to the B cells while the green areas are not.
Figure 2.
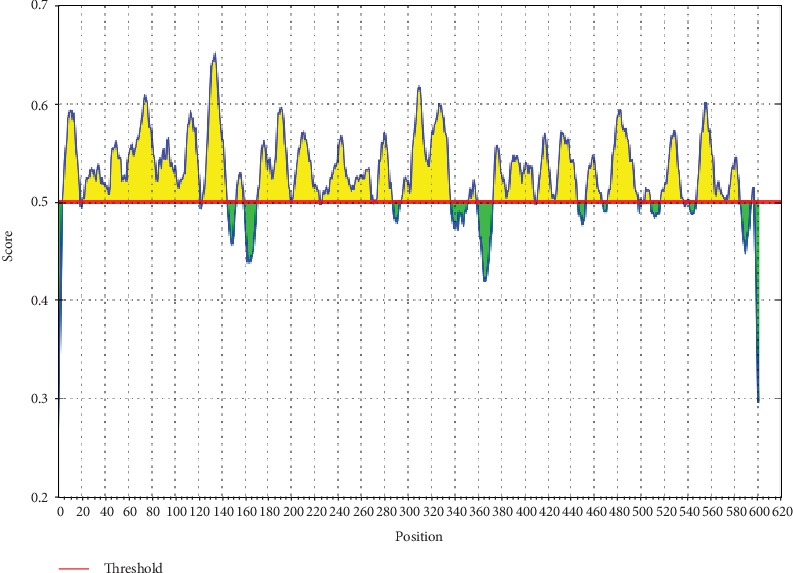
Emini surface accessibility prediction. Areas above the red line (threshold) are epitopes suggested to be binding to the B cells while the green areas are not.
Figure 3.
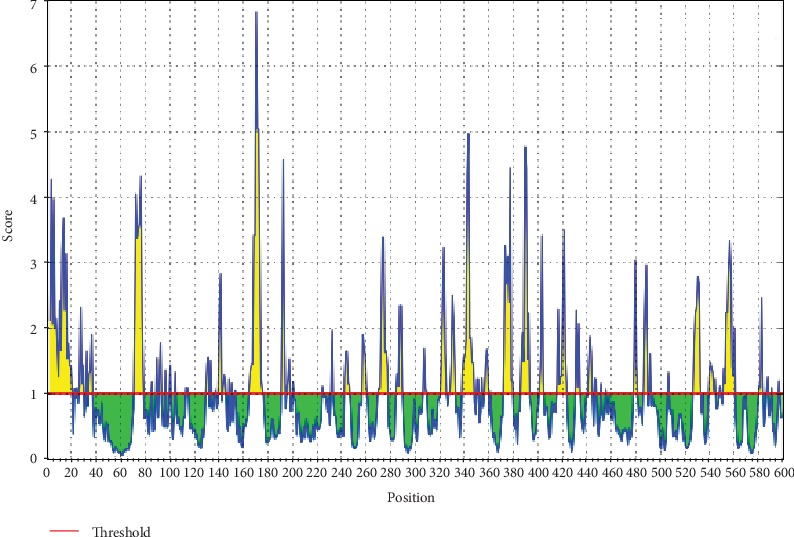
Kolaskar and Tongaonkar antigenicity prediction. Areas above the red line (threshold) are epitopes suggested to be binding to the B cells while the green areas are not.
3.3. Prediction of T Helper Cell Epitopes and Interaction with MHC Class I Alleles
The Nipah virus glycoprotein G sequence was analyzed using the IEDB MHC class I binding prediction tool based on ANN-align with half-maximal inhibitory concentration (IC50) ≤ 500; the least most promising epitopes that had a binding affinity with the class I alleles along with their positions in the Nipah virus glycoprotein G are shown in Table 2.
Table 2.
Most potential T cell epitopes interacting with MHC class I alleles, their positions, IC50, rank, and conservancy.
| Peptide | Start | End | Allele | IC50 | Rank | Conservancy |
|---|---|---|---|---|---|---|
| FAYSHLERI | 229 | 237 | HLA-A∗02:01 | 376.44 | 2.5 | |
| 229 | 237 | HLA-A∗02:03 | 19.03 | 0.37 | ||
| 229 | 237 | HLA-A∗02:06 | 23.3 | 0.28 | 76.19 | |
| 229 | 237 | HLA-A∗68:02 | 104.31 | 0.63 | ||
| 229 | 237 | HLA-B∗51:01 | 133.03 | 0.02 | ||
| 229 | 237 | HLA-B∗53:01 | 222.39 | 0.21 | ||
| 229 | 237 | HLA-C∗03:03 | 57.38 | 0.22 | ||
| 229 | 237 | HLA-C∗06:02 | 112.27 | 0.05 | ||
| 229 | 237 | HLA-C∗07:01 | 151.15 | 0.05 | ||
| 229 | 237 | HLA-C∗12:03 | 13.37 | 0.03 | ||
| 229 | 237 | HLA-C∗15:02 | 207.16 | 0.12 | ||
|
| ||||||
| FLIDRINWI | 512 | 520 | HLA-A∗02:01 | 3.65 | 0.02 | |
| 512 | 520 | HLA-A∗02:03 | 2.32 | 0.02 | ||
| 512 | 520 | HLA-A∗02:06 | 4.71 | 0.04 | 100 | |
| 512 | 520 | HLA-A∗68:02 | 488.4 | 1.8 | ||
| 512 | 520 | HLA-C∗03:03 | 366.48 | 0.63 | ||
| 512 | 520 | HLA-C∗06:02 | 163.19 | 0.07 | ||
| 512 | 520 | HLA-C∗07:01 | 416.5 | 0.12 | ||
| 512 | 520 | HLA-C∗12:03 | 55.27 | 0.13 | ||
|
| ||||||
| FSWDTMIKF | 458 | 466 | HLA-A∗02:06 | 131.35 | 1.3 | |
| 458 | 466 | HLA-A∗29:02 | 328.77 | 1.2 | ||
| 458 | 466 | HLA-B∗35:01 | 70.16 | 0.24 | 100 | |
| 458 | 466 | HLA-B∗46:01 | 470.72 | 0.09 | ||
| 458 | 466 | HLA-B∗53:01 | 95.8 | 0.12 | ||
| 458 | 466 | HLA-B∗57:01 | 380.38 | 0.93 | ||
| 458 | 466 | HLA-B∗58:01 | 313.77 | 0.77 | ||
| 458 | 466 | HLA-C∗12:03 | 35.09 | 0.09 | ||
|
| ||||||
| KLISYTLPV | 201 | 209 | HLA-A∗02:01 | 2.36 | 0.02 | |
| 201 | 209 | HLA-A∗02:03 | 2.4 | 0.02 | ||
| 201 | 209 | HLA-A∗02:06 | 3.77 | 0.02 | 100 | |
| 201 | 209 | HLA-A∗30:01 | 146.34 | 0.44 | ||
| 201 | 209 | HLA-A∗32:01 | 21.39 | 0.04 | ||
| 201 | 209 | HLA-B∗15:01 | 303.74 | 1.3 | ||
| 201 | 209 | HLA-C∗14:02 | 187.05 | 0.28 | ||
| 201 | 209 | HLA-C∗15:02 | 364.98 | 0.2 | ||
|
| ||||||
| TVYHCSAVY | 278 | 286 | HLA-A∗03:01 | 84.04 | 0.35 | |
| 278 | 286 | HLA-A∗11:01 | 263.53 | 1.6 | ||
| 278 | 286 | HLA-A∗26:01 | 363.08 | 0.19 | ||
| 278 | 286 | HLA-A∗29:02 | 10.13 | 0.08 | 100 | |
| 278 | 286 | HLA-A∗30:02 | 32.49 | 0.07 | ||
| 278 | 286 | HLA-A∗68:01 | 310.51 | 1.7 | ||
| 278 | 286 | HLA-B∗15:01 | 71.72 | 0.41 | ||
| 278 | 286 | HLA-B∗15:02 | 353.64 | 0.13 | ||
| 278 | 286 | HLA-B∗35:01 | 27.98 | 0.12 | ||
| 278 | 286 | HLA-C∗12:03 | 45.95 | 0.11 | ||
| 278 | 286 | HLA-C∗14:02 | 103.08 | 0.18 | ||
3.4. Prediction of T Helper Cell Epitopes and Interaction with MHC Class II Alleles
The Nipah virus glycoprotein G sequence was analyzed using the IEDB MHC class II binding prediction tool based on NN-align with half-maximal inhibitory concentration (IC50) ≤ 1000. The list of the epitopes and their correspondent bindings to MHC class II alleles, along with their positions in the Nipah virus glycoprotein G, while the list of the most promising epitopes that had a strong binding affinity to MHC class II alleles and depending on the number of their binding alleles is shown in Table 3.
Table 3.
The most potential T cell epitopes (core sequence) and the number of their binding alleles.
| Core sequence | Alleles | Number of alleles |
|---|---|---|
| FLIDRINWI | HLA-DPA1∗01/DPB1∗04:01 | 12 |
| HLA-DPA1∗01:03/DPB1∗02:01 | ||
| HLA-DPA1∗02:01/DPB1∗01:01 | ||
| HLA-DPA1∗02:01/DPB1∗05:01 | ||
| HLA-DPA1∗03:01/DPB1∗04:02 | ||
| HLA-DQA1∗01:01/DQB1∗05:01 | ||
| HLA-DQA1∗05:01/DQB1∗02:01 | ||
| HLA-DRB1∗01:01 | ||
| HLA-DRB1∗03:01 | ||
| HLA-DRB1∗04:01 | ||
| HLA-DRB1∗04:04 | ||
| HLA-DRB1∗04:05 | ||
|
| ||
| FAYSHLERI | HLA-DPA1∗01:03/DPB1∗02:01 | 13 |
| HLA-DPA1∗02:01/DPB1∗01:01 | ||
| HLA-DPA1∗02:01/DPB1∗05:01 | ||
| HLA-DPA1∗03:01/DPB1∗04:02 | ||
| HLA-DQA1∗05:01/DQB1∗02:01 | ||
| HLA-DQA1∗05:01/DQB1∗03:01 | ||
| HLA-DRB1∗01:01 | ||
| HLA-DRB1∗04:04 | ||
| HLA-DRB1∗04:05 | ||
| HLA-DRB1∗07:01 | ||
| HLA-DRB1∗09:01 | ||
| HLA-DRB3∗01:01 | ||
| HLA-DRB5∗01:01 | ||
|
| ||
| FIEISDQRL | HLA-DPA1∗01:03/DPB1∗02:01 | 17 |
| HLA-DPA1∗02:01/DPB1∗01:01 | ||
| HLA-DPA1∗03:01/DPB1∗04:02 | ||
| HLA-DQA1∗05:01/DQB1∗02:01 | ||
| HLA-DRB1∗01:01 | ||
| HLA-DRB1∗04:05 | ||
| HLA-DRB1∗07:01 | ||
| HLA-DRB1∗08:02 | ||
| HLA-DRB1∗13:02 | ||
| HLA-DRB1∗15:01 | ||
| HLA-DRB4∗01:01 | ||
| HLA-DRB5∗01:01 | ||
| HLA-DRB1∗04:01 | ||
| HLA-DRB1∗07:01 | ||
| HLA-DRB1∗09:01 | ||
| HLA-DRB1∗11:01 | ||
| HLA-DRB1∗13:02 | ||
|
| ||
| ILSAFNTVI | HLA-DPA1∗03:01/DPB1∗04:02 | 13 |
| HLA-DQA1∗05:01/DQB1∗03:01 | ||
| HLA-DRB1∗01:01 | ||
| HLA-DRB1∗04:01 | ||
| HLA-DRB1∗04:05 | ||
| HLA-DRB1∗07:01 | ||
| HLA-DRB1∗08:02 | ||
| HLA-DRB1∗09:01 | ||
| HLA-DRB1∗11:01 | ||
| HLA-DRB1∗13:02 | ||
| HLA-DRB1∗15:01 | ||
| HLA-DRB4∗01:01 | ||
| HLA-DRB5∗01:01 | ||
|
| ||
| TVYHCSAVY | HLA-DQA1∗05:01/DQB1∗03:01 | 4 |
| HLA-DRB1∗07:01 | ||
| HLA-DRB1∗13:02 | ||
| HLA-DRB1∗15:01 | ||
3.5. Population Coverage
A population coverage test was performed to detect all the epitopes that bind to MHC class I alleles and MHC class II alleles available in the database in relation to the world, South Asia, Southeast Asia, Sudan, and North Africa.
3.6. 3D Structure
3.7. Molecular Docking
4. Discussion
Traditional vaccination approaches depend on the total amount of pathogens that are either live—constricted or inactivated. Among the significant issues, these vaccines have brought along pivotal security concerns. In light of the fact that they are being utilized for vaccination, this may have caused them to become actuated and may also cause contamination. Additionally, due to the varied hereditary pathogen strains found in the world, vaccines are probably going to lose their viability in various areas or even in certain populations.
However, novel vaccine approaches such as DNA- and epitope-based immunizations may possibly conquer obstructions for this type of immunization approaches, making them increasingly successful, explicit, and long-lasting in vulnerable reactions with insignificant structures and without any undesired impacts [46]. Moreover, many peptide-based vaccines have been effectively proposed through utilizing in silico approaches against Madurella mycetomatis, Mokola rabies virus, Lagos rabies virus, and others [47–52]. Such investigations, in regard to those viruses, have built up immunoinformatics in the computational analysis field.
In our present work, potential peptides were suggested to design an epitope-based vaccine for Nipah virus, using the latest amino acid sequences of glycoprotein G (glycoside hydrolase family) for a total of 21 strains of Nipah virus that were retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov/protein) [31] on July 2018 after the last outbreak at the end of May 2018 in Kerala-India according to the WHO report [53]. Figure 4 summarizes the method of the present work.
Figure 4.
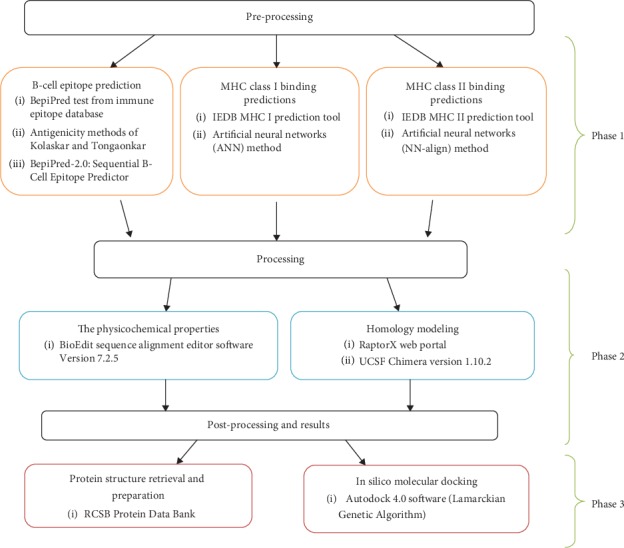
The three phases of Materials and Methods.
Various literatures were surveyed to define the antigenic part of the virus. Glycoprotein G was found to be on the outer surface of the virus which was chosen as our target. Initially, we have evaluated the binding affinity of the virus to MHC alleles. This was done by submitting the protein reference sequence to IEDB MHC, a binding prediction tool, based on the ANN align method with IC50 ≤ 500 [37] for MHC class I molecules. 191 peptides were found to bind to MHC class I with different affinities. It is well known that a better immune response depends on whether or not the recognition of epitopes by HLA molecules with significant affinity is successful. Therefore, a peptide recognized by its highest number of HLA alleles has the best potential to induce a strong immune response, leading us to take into account the only three peptides found with a 100% conservancy. The conserved peptide FLIDRINWI was found to interact with 8 alleles (HLA-A∗02:01, HLA-A∗02:03, HLA-A∗02:06, HLA-A∗68:02, HLA-C∗03:03, HLA-C∗06:02, HLA-C∗07:01, and HLA-C∗12:03), while FSWDTMIKF with 8 alleles (HLA-A∗02:06, HLA-A∗29:02, HLA-B∗35:01, HLA-B∗46:01, HLA-B∗53:01, HLA-B∗57:01, HLA-B∗58:01, and HLA-C∗12:03) and TVYHCSAVY with 11 alleles (HLA-A∗03:01, HLA-A∗11:01, HLA-A∗26:01, HLA-A∗29:02, HLA-A∗30:02, HLA-A∗68:01, HLA-B∗15:01, HLA-B∗15:02, HLA-B∗35:01, HLA-C∗12:03, and HLA-C∗14:02).
The reference sequence of Nipah virus glycoprotein G was reanalyzed using the IEDB MHC II binding prediction tool based on NN-align with half-maximal inhibitory concentration (IC50) ≤ 1000 [38]. The analysis resulted in the prediction of 398 peptides from which FSWDTMIKF, FLIDRINWI, and ILSAFNTVI were potentially proposed according to their high number of binding alleles (15, 12, and 15 alleles, respectively). Additionally, the sequence of Nipah virus glycoprotein G was subjected to BepiPred linear epitope prediction, Emini surface accessibility, and Kolaskar and Tongaonkar antigenicity methods in IEDB. Unfortunately, the peptides with the strongest binding affinities, utilizing the three mentioned tests, were absent.
Population coverage results for the total peptides found and the proposed peptides binding to MHC classes I and II alleles are summarized in Tables 4 and 5. Obtained results from the bindings to MHC I alleles revealed a 99.84% projected population coverage in the world, 98.55% in Southeast Asia, 98.40% in South Asia, 99.23% in North Africa, and 99.36% in Sudan while the population coverage results for the total number of peptides binding to MHC II alleles showed only a 56.84% projected population coverage in the world, 48.63% in Southeast Asia, 56.00% in South Asia, 62.37% in North Africa, and 55.75% in Sudan.
Table 4.
A population coverage for all epitopes that bind to MHC classes I and II alleles from different parts of the world.
| MHC classes | Population | World | South Asia | Southeast Asia | Sudan | North Africa |
|---|---|---|---|---|---|---|
| Class I | Coveragea | 99.84% | 98.40% | 98.55% | 99.36% | 99.23% |
| Average_hitb | 36.62 | 30.60 | 28.42 | 34.02 | 32.43 | |
| PC90c | 16.87 | 9.29 | 8.61 | 13.40 | 12.95 | |
|
| ||||||
| Class II | Coveragea | 56.84% | 56.0% | 48.63% | 55.75% | 62.37% |
| Average_hitb | 54.88 | 50.50 | 36.27 | 36.89 | 50.09 | |
| PC90c | -24.24 | -10.09 | 1.65 | 4.60 | -3.31 | |
aProjected population coverage; baverage number of epitope hits/HLA combinations recognized by the population; cminimum number of epitope hits/HLA combinations recognized by 90% of the population.
Table 5.
Population coverage of the three highly proposed peptides in MHC classes I and II in five different parts of the world.
| Peptide | Population coverage %/area | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| World | Southeast Asia | South Asia | North Africa | Sudan | |||||||||||
| MHC I | MHC II | MHC I & II | MHC I | MHC II | MHC I & II | MHC I | MHC II | MHC I & II | MHC I | MHC II | MHC I & II | MHC I | MHC II | MHC I & II |
|
| FLIDRINWI | 70.4% | 43.7% | 83.3% | 44.5% | 19.8% | 55.5% | 51.4% | 29.8% | 65.9% | 71.0% | 33.8% | 80.8% | 85.4% | 30.7% | 89.9% |
| FAYSHLERI | 74.9% | 40.2% | 85.0% | 50.6% | 31.8% | 66.3% | 60.9% | 40.5% | 76.7% | 77.5% | 35.5% | 85.5% | 89.2% | 19.5% | 91.3% |
| TVYHCSAVY | 61.3% | 40.1% | 76.8% | 54.5% | 18.4% | 62.9% | 63.7% | 45.0% | 80.0% | 50.4% | 43.5% | 72.0% | 50.2% | 20.9% | 60.6% |
The selected peptides were further subjected to both MHC I- and MHC II-based population coverage analysis in the whole world, Southeast Asia, South Asia, North Africa, and Sudan as shown in Table 5. Among the six primarily selected epitopes, the obtained results showed a very strong potential in proposing the epitope FLIDRINWI as a vaccine candidate compared to the rest, taking into consideration its overall epitope conservancy, population coverage, and its affinity for the highest number of HLA molecules. Furthermore, in silico docking was carried out to measure the binding efficacy between the proposed peptides and HLA-A∗02:01, in which it has been specifically chosen in relation to their contribution to several immunological and pathological diseases [54–56], although numerous investigations have shown a relationship between HLA alleles and disease susceptibility, which defines defensive HLA allelic associations that possibly permit a recognizable proof that pathogen epitopes are limited by particular HLA alleles. These epitopes may then be fused into a vaccine design in the expectation that the immunization will be reproduced naturally [55, 56].
Calculations of the root mean square deviation (RMSD) between coordinates of the atoms and formation of clusters based on RMSD values have computed the resemblance of the docked structures. The most favorable docking is considered to be the conformation of the lowest binding energy. The least energy predictions of the peptide FLIDRINWI (-6.95 Kcal/mol) and the 3D structure of the allele and its peptide are shown in Figure 5. Furthermore, the monoisotopic mass, sum formula, and molecular weight of the three highly proposed peptides are shown in Table 6.
Figure 5.
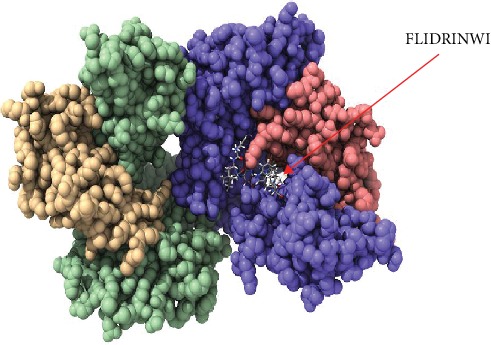
Molecular docking of FLIDRINWI peptide of Nipah virus docked in HLA-A∗02:01 and visualized by UCSF Chimera X version 0.1.0.
Table 6.
Monoisotopic mass, sum formula, and molecular weight of the three highly proposed peptides.
| Sequence (N : H/C : OH) | Sum formula | Monoisot. mass | Mol. weight |
|---|---|---|---|
| FLIDRINWI | C58H88N14O13 | 1188.66551 | 1189.40532 |
| TVYHCSAVY | C47H67N11O14S | 1041.45896 | 1042.16518 |
| FAYSHLERI | C53H78N14O14 | 1134.58218 | 1135.27182 |
As a result of these interesting outcomes, formulating a vaccine using the suggested peptide is highly promising and encouraging to be highly proposed as a universal epitope-based peptide vaccine against Nipah virus.
5. Conclusions
The present study proposed a very promising epitope-based peptide vaccine against glycoprotein G of Nipah virus. It is expected to be highly antigenic with a minimum allergic effect. The proposed peptide FLIDRINWI has a strong binding affinity to both MHC class I and MHC class II alleles. Moreover, it shows an exceptional population coverage result for both MHC class I and MHC class II alleles in the whole world, Southeast Asia, South Asia, North Africa, and Sudan.
Despite having to validate the findings of the current study, an in vivo assessment of the most promising peptides, namely, FLIDRINWI, TVYHCSAVY, and FAYSHLERI, is highly recommended and will serve as the ground data for such work as shown in Figures 5–9.
Figure 6.
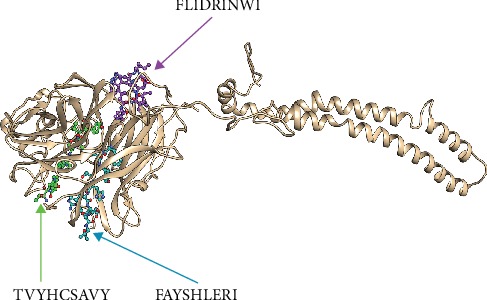
The four potential peptides bound to MHC class I and MHC class II visualized by Chimera X version 0.1.0.
Figure 7.
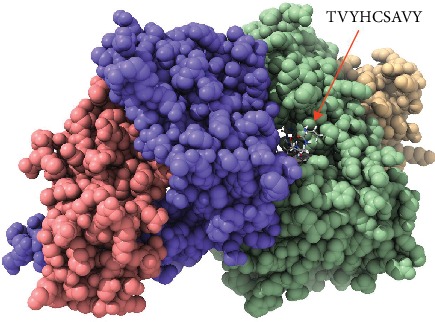
Molecular docking of TVYHCSAVY peptide of Nipah virus docked in HLA-A∗02:01 and visualized by UCSF Chimera X version 0.1.0.
Figure 8.
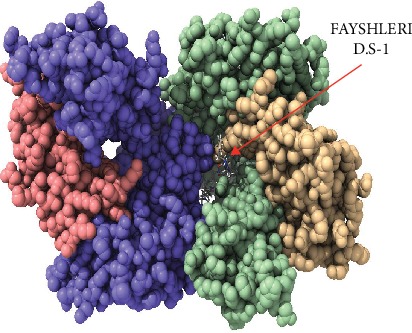
Molecular docking of FAYSHLERI peptide of Nipah virus docked in HLA-A∗02:01 and visualized by UCSF Chimera X version 0.1.0. ∗D.S: Docking Side No.1.
Figure 9.
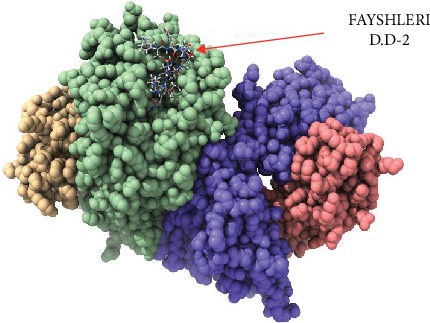
Molecular docking of FAYSHLERI peptide of Nipah virus docked in HLA-A∗02:01 and visualized by UCSF Chimera X version 0.1.0. ∗D.S: Docking Side No.2.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- 1.Chattu V. K., Kumar R., Kumary S., Kajal F., David J. K. Nipah virus epidemic in southern India and emphasizing "One Health" approach to ensure global health security. Journal of Family Medicine and Primary Care. 2018;7(2):275–283. doi: 10.4103/jfmpc.jfmpc_137_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention (CDC) Outbreak of Hendra-like virus--Malaysia and Singapore, 1998-1999. Morbidity and Mortality Weekly Report. 1999;48(13):265–269. [PubMed] [Google Scholar]
- 3.Broder C. C., Xu K., Nikolov D. B., et al. A treatment for and vaccine against the deadly Hendra and Nipah viruses. Antiviral Research. 2013;100(1):8–13. doi: 10.1016/j.antiviral.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breed A. C., Meers J., Sendow I., et al. The distribution of henipaviruses in Southeast Asia and Australasia: is Wallace's line a barrier to Nipah virus? PLoS One. 2013;8(4, article e61316) doi: 10.1371/journal.pone.0061316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weingartl H. M., Berhane Y., Caswell J. L., et al. Recombinant nipah virus vaccines protect pigs against challenge. Journal of Virology. 2006;80(16):7929–7938. doi: 10.1128/JVI.00263-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Field H. E. Bats and emerging zoonoses: henipaviruses and SARS. Zoonoses and Public Health. 2009;56(6-7):278–284. doi: 10.1111/j.1863-2378.2008.01218.x. [DOI] [PubMed] [Google Scholar]
- 7.Luby S. P., Hossain M. J., Gurley E. S., et al. Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001-2007. Emerging Infectious Diseases. 2009;15(8):1229–1235. doi: 10.3201/eid1508.081237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clayton B. A. Nipah virus: transmission of a zoonotic paramyxovirus. Current Opinion in Virology. 2017;22:97–104. doi: 10.1016/j.coviro.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Chua K. B. Epidemiology, surveillance, and control of Nipah virus infections in Malaysia. The Malaysian Journal of Pathology. 2010;32(2):69–73. [PubMed] [Google Scholar]
- 10.Rahman S. A., Hassan L., Epstein J. H., et al. Risk factors for Nipah virus infection among pteropid bats, Peninsular Malaysia. Emerging Infectious Diseases. 2013;19(1):51–60. doi: 10.3201/eid1901.120221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halpin K., Hyatt A. D., Fogarty R., et al. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: a comprehensive experimental study of virus transmission. The American Journal of Tropical Medicine and Hygiene. 2011;85(5):946–951. doi: 10.4269/ajtmh.2011.10-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Majid Z., Majid W. S. NIPAH virus: a new threat to South Asia. Tropical Doctor. 2018;48(4):p. 376. doi: 10.1177/0049475518791327. [DOI] [PubMed] [Google Scholar]
- 13.Luby S. P. The pandemic potential of Nipah virus. Antiviral Research. 2013;100(1):38–43. doi: 10.1016/j.antiviral.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 14.Ramphul K., Mejias S. G., Agumadu V. C., Sombans S., Sonaye R., Lohana P. The killer virus called Nipah: a review. Cureus. 2018;10(8, article e3168) doi: 10.7759/cureus.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goh K. J., Tan C. T., Chew N. K., et al. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. The New England Journal of Medicine. 2000;342(17):1229–1235. doi: 10.1056/NEJM200004273421701. [DOI] [PubMed] [Google Scholar]
- 16.Berhane Y., Berry J. D., Ranadheera C., et al. Production and characterization of monoclonal antibodies against binary ethylenimine inactivated Nipah virus. Journal of Virological Methods. 2006;132(1-2):59–68. doi: 10.1016/j.jviromet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 17.Donaldson H., Lucey D. Enhancing preparation for large Nipah outbreaks beyond Bangladesh: preventing a tragedy like Ebola in West Africa. InternationalJjournal of Infectious Diseases. 2018;72:69–72. doi: 10.1016/j.ijid.2018.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Badawi M. M., Osman M. M., Alla A. A. F., et al. Highly conserved epitopes of ZIKA envelope glycoprotein may act as a novel peptide vaccine with high coverage: immunoinformatics approach. American Journal of Biomedical Research. 2016;4(3):46–60. [Google Scholar]
- 19.Kazi A., Chuah C., Majeed A. B. A., Leow C. H., Lim B. H., Leow C. Y. Current progress of immunoinformatics approach harnessed for cellular- and antibody-dependent vaccine design. Pathogens and Global Health. 2018;112(3):123–131. doi: 10.1080/20477724.2018.1446773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khalili S., Jahangiri A., Borna H., Ahmadi Zanoos K., Amani J. Computational vaccinology and epitope vaccine design by immunoinformatics. Acta Microbiologica et Immunologica. 2014;61(3):285–307. doi: 10.1556/AMicr.61.2014.3.4. [DOI] [PubMed] [Google Scholar]
- 21.Wong K. T., Shieh W.-J., Kumar S., et al. Nipah virus infection: pathology and pathogenesis of an emerging paramyxoviral zoonosis. The American Journal of Pathology. 2002;161(6):2153–2167. doi: 10.1016/S0002-9440(10)64493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Q., Bradel-Tretheway B., Monreal A. I., et al. Nipah virus attachment glycoprotein stalk C-terminal region links receptor binding to fusion triggering. Journal of Virology. 2015;89:1838–1850. doi: 10.1128/JVI.02277-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oscherwitz J. The promise and challenge of epitope-focused vaccines. Human Vaccines & Immunotherapeutics. 2016;12(8):2113–2116. doi: 10.1080/21645515.2016.1160977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nandy A., Basak S. C. A brief review of computer-assisted approaches to rational design of peptide vaccines. International Journal of Molecular Sciences. 2016;17(5):p. 666. doi: 10.3390/ijms17050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skwarczynski M., Toth I. Peptide-based synthetic vaccines. Chemical Science. 2016;7(2):842–854. doi: 10.1039/c5sc03892h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali M. T., Morshed M. M., Hassan F. A computational approach for designing a universal epitope-based peptide vaccine against Nipah virus. Interdisciplinary Sciences, Computational Life Sciences. 2015;7(2, article 23):177–185. doi: 10.1007/s12539-015-0023-0. [DOI] [PubMed] [Google Scholar]
- 27.Woodland D. L. A focus on vaccine development. Viral Immunology. 2015;28(6):p. 303. doi: 10.1089/vim.2015.28999.dlw. [DOI] [PubMed] [Google Scholar]
- 28.Moyle P. M. Progress in vaccine development. Current protocols in microbiology. 2015;36:18.1.1–18.1.26. doi: 10.1002/9780471729259.mc1801s36. [DOI] [PubMed] [Google Scholar]
- 29.Oyarzun P., Ellis J. J., Gonzalez-Galarza F. F., et al. A bioinformatics tool for epitope-based vaccine design that accounts for human ethnic diversity: application to emerging infectious diseases. Vaccine. 2015;33(10):1267–1273. doi: 10.1016/j.vaccine.2015.01.040. [DOI] [PubMed] [Google Scholar]
- 30.Li W., Joshi M. D., Singhania S., Ramsey K. H., Murthy A. K. Peptide vaccine: progress and challenges. Vaccine. 2014;2(3):515–536. doi: 10.3390/vaccines2030515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.NCBI protein sequence database. 2018, https://www.ncbi.nlm.nih.gov/protein.
- 32.Vita R., Overton J. A., Greenbaum J. A., et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Research. 2015;43(D1):D405–D412. doi: 10.1093/nar/gku938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oni O. O., Owoade A. A., Adeyefa C. A. O. Design and evaluation of primer pairs for efficient detection of avian rotavirus. Tropical Animal Health and Production. 2018;50(2):267–273. doi: 10.1007/s11250-017-1425-2. [DOI] [PubMed] [Google Scholar]
- 34.Bui H. H., Sidney J., Li W., Fusseder N., Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics. 2007;8(1):p. 361. doi: 10.1186/1471-2105-8-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larsen J. E., Lund O., Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Research. 2006;2(1):p. 2. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolaskar A. S., Tongaonkar P. C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Letters. 1990;276(1-2):172–174. doi: 10.1016/0014-5793(90)80535-q. [DOI] [PubMed] [Google Scholar]
- 37.Kim Y., Ponomarenko J., Zhu Z., et al. Immune epitope database analysis resource. Nucleic Acids Research. 2012;40(W1):W525–W530. doi: 10.1093/nar/gks438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Q., Wang P., Kim Y., et al. Immune epitope database analysis resource (IEDB-AR) Nucleic Acids Research. 2008;36(Supplement 2):W513–W518. doi: 10.1093/nar/gkn254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bui H. H., Sidney J., Dinh K., Southwood S., Newman M. J., Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics. 2006;7(1):p. 153. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S., Li W., Liu S., Xu J. RaptorX-Property: a web server for protein structure property prediction. Nucleic Acids Research. 2016;44(W1):W430–W435. doi: 10.1093/nar/gkw306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pettersen E. F., Goddard T. D., Huang C. C., et al. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 42.Choo J. A., Thong S. Y., Yap J., et al. Bioorthogonal cleavage and exchange of major histocompatibility complex ligands by employing azobenzene-containing peptides. Angewandte Chemie (International ed in English) 2014;53(49):13390–13394. doi: 10.1002/anie.201406295. [DOI] [PubMed] [Google Scholar]
- 43.Guex N., Peitsch M. C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 44.Morris G. M., Huey R., Lindstrom W., et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morris G. M., Goodsell D. S., Halliday R. S., et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19(14):1639–1662. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [DOI] [Google Scholar]
- 46.Arnon R. A novel approach to vaccine design—epitope-based vaccines. The FEBS Journal. 2006;273:33–34. [Google Scholar]
- 47.Mohammed A. A., ALnaby A. M. H., Sabeel S. M., et al. Epitope-based peptide vaccine against fructose-bisphosphate aldolase ofMadurella mycetomatisUsing immunoinformatics approaches. Bioinformatics and Biology Insights. 2018;12 doi: 10.1177/1177932218809703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohammed A. A., Hashim O., Elrahman K. A. A., Hamdi A., Hassan M. A. Epitope-based peptide vaccine design against Mokola rabies virus glycoprotein G utilizing in silico approaches. Immunome Research. 2017;13(3) doi: 10.4172/1745-7580.1000144. [DOI] [Google Scholar]
- 49.Ahmed O. H., Abdelhalim A., Obi S., Abd elrahman K. A., Hamdi A., Hassan M. A. Immunoinformatic approach for epitope-based peptide vaccine against Lagos rabies virus glycoprotein G. Immunome Research. 2017;13(3) doi: 10.4172/1745-7580.1000137. [DOI] [Google Scholar]
- 50.Idrees S., Ashfaq U. A. Structural analysis and epitope prediction of HCV E1 protein isolated in Pakistan: an in-silico approach. Virology Journal. 2013;10(1, article 113) doi: 10.1186/1743-422X-10-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hasan M. A., Hossain M., Alam M. J. A computational assay to design an epitope-based peptide vaccine against Saint Louis encephalitis virus. Bioinformatics and Biology Insights. 2013;7:347–355. doi: 10.4137/BBI.S13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharmin I. A highly conserved WDYPKCDRA epitope in the RNA directed RNA polymerase of human coronaviruses can be used as epitope-based universal vaccine design. BMC Bioinformatics. 2014;15(1, article 161) doi: 10.1186/1471-2105-15-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. https://www.who.int/csr/don/07-august-2018-nipah-virus-india/en/
- 54.Burton P. R., Tobin M. D., Hopper J. L. Key concepts in genetic epidemiology. The Lancet. 2005;366(9489):941–951. doi: 10.1016/S0140-6736(05)67322-9. [DOI] [PubMed] [Google Scholar]
- 55.Calafell F., Malats N. Basic molecular genetics for epidemiologists. Journal of Epidemiology & Community Health. 2003;57(6):398–400. doi: 10.1136/jech.57.6.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davenport M. P., Hill A. V. Reverse immunogenetics: from HLA-disease associations to vaccine candidates. Molecular Medicine Today. 1996;2(1):38–45. doi: 10.1016/1357-4310(96)88757-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.