

Abstract
Based on the evidence that hemochromatosis, an iron-overload disease, drives hepatocellular carcinoma, we hypothesized that chronic exposure to excess iron, either due to genetic or environmental causes, predisposes an individual to cancer. Using pancreatic cancer as our primary focus, we employed cell culture studies to interrogate the connection between excess iron and cancer, and combined in vitro and in vivo studies to explore the connection further. Ferric ammonium citrate was used as an exogenous iron source. Chronic exposure to excess iron induced epithelial-mesenchymal transition (EMT) in normal and cancer cell lines, loss of p53, and suppression of p53 transcriptional activity evidenced from decreased expression of p53 target genes (p21, cyclin D1, Bax, SLC7A11). To further extrapolate our cell culture data, we generated EL-KrasG12D (EL-Kras) mouse (pancreatic neoplastic mouse model) expressing Hfe+/+and Hfe−/− genetic background. p53 target gene expression decreased in EL-Kras/Hfe−/− mouse pancreas compared to EL-Kras/Hfe+/+ mouse pancreas. Interestingly, the incidence of acinar-to-ductal metaplasia and cystic pancreatic neoplasms (CPN) decreased in EL-Kras/Hfe−/− mice, but the CPNs that did develop were larger in these mice than in EL-Kras/Hfe+/+ mice. In conclusion, these in vitro and in vivo studies support a potential role for chronic exposure to excess iron as a promoter of more aggressive disease via p53 loss and SLC7A11 upregulation within pancreatic epithelial cells.
Keywords: SLC7A11, p53, Iron, Heme, Epithelial-mesenchymal transition
Graphical abstract
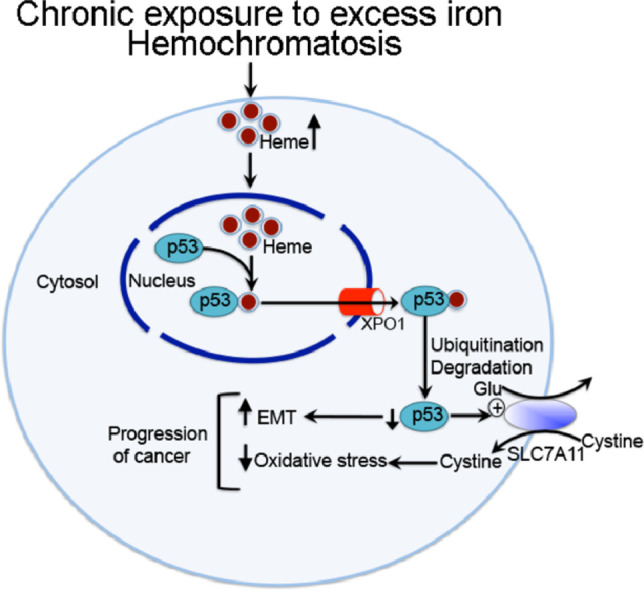
The studies presented here demonstrate that chronic exposure to excess iron leads to heme-mediated export of the tumor suppressor p53 out of the nucleus with subsequent proteasomal degradation, thereby leading to loss of p53 protein and its transcriptional activity. This results in the upregulation of the antioxidant transporter SLC7A11 and in the promotion of EMT. As a consequence, chronic exposure to excessive iron impacts cancer progression and also changes the cancer cells toward EMT-like phenotype, hence also potentially promoting metastasis.
1. Introduction
Iron and heme (Fe2+-Protoporphyrin-IX) are absolutely essential for every cell, but when present in excess, these molecules become toxic. Fe2+ in free form is a potent oxidant; it catalyzes the Fenton reaction to generate hydroxyl radicals (Fe2+ + H2O2 → Fe3+ + OH− + OH⬤), a potent reactive oxygen species [1,2]. Free heme is also toxic as it catalyzes free radical reaction and induces oxidative damage [3,4]. Accumulation of iron and heme to toxic levels occurs in genetic diseases (hemochromatosis, sickle cell disease), pathological conditions (hemolytic anemia, ischemia reperfusion), infections, and clinical/therapeutic conditions (repeated blood transfusion). Excess intake of iron and heme from dietary sources, specifically from red meat with its ∼10-fold higher heme content than white meat, could also lead to iron/heme overload, particularly in the colon. Among these conditions listed, however, hemochromatosis deserves special mention. This is a common recessive disorder in Caucacians and Hispanics, affecting ∼1.5 million people in the United States. The prevalence of homozygosity is ∼1/250. The disease is characterized by increased intestinal absorption of iron and accumulation of iron to toxic levels in almost every organ (e.g., liver, heart, pancreas, kidney, prostate, colon) [5], [6], [7], [8]. The disease is often under-diagnosed or misdiagnosed. In most cases, the patients are treated for the symptoms without being diagnosed as hemochromatosis. This disease is caused by mutations in at least five different proteins, all involved in iron regulation: HFE, hepcidin, hemojuvelin, transferrin receptor 2, and ferroportin [5], [6], [7], [8]. Mutations in HFE constitute ∼85% of the patients, the remaining ∼15% due to mutations in the other four proteins.
The connection between hemochromatosis and liver cancer is well known [5], [6], [7], [8]; excessive iron/heme drives cancer in this tissue. Excess iron in non-hepatic tissues can also pose a cancer risk [9,10]. There is unequivocal evidence for a positive connection between excessive intake of dietary heme in the form of red meat and cancer, not only for colon cancer [11], [12], [13] but also for cancers of pancreas, esophagus, lung, prostate, and mammary gland [4]. These data provide a strong evidence-based rationale for the idea that chronic exposure to excess iron/heme promotes cancer in multiple tissues.
The tumor suppressor protein p53 is a heme-binding protein. When bound to heme, the p53-heme complex becomes a substrate for the nuclear exporter Exportin and is then translocated out of the nucleus for subsequent proteasomal degradation [14]. This could provide the link between chronic exposure to excessive iron/heme and cancer with iron/heme-induced suppression of p53 as the molecular mediator. In this study, we evaluated the influence of chronic exposure to excess iron in vitro in a normal pancreatic epithelial cell line and also in various pancreatic cancer cell lines. We complemented these studies with an in vivo system to interrogate the impact of the iron-overload disease hemochromatosis on pancreatic cancer using the EL-Kras mouse as a model system for Kras-driven pancreatic neoplasia [15] and Hfe−/− mouse as a model for the genetic iron-overload disease hemochromatosis [16]. In general, these in vitro and in vivo studies provide evidence in support of the notion that chronic exposure to excess iron is an important promoter of pancreatic cancer.
2. Materials and methods
2.1. Cell culture
Human pancreatic cancer cell lines BxPC-3, Capan-2, and MIA PaCa-2 were procured from American Type Culture Collection (ATCC). HPDE, a human pancreatic ductal epithelial cell line, was kindly provided by Dr. Ming Tsao, Ontario Cancer Institute (Toronto, Canada). Normal epithelial cell lines of prostate (RWPE-1), liver (THLE-2), and colon (CC8841) were also procured from ATCC. The ATCC has done morphological, cytogenetic and DNA profile analyses for characterization of these cell lines. BxPC-3 and CCD841 cells were grown in RPMI-1640 medium, supplemented with 10% FBS and subcultured at a 1:5 ratio. HPDE and RWPE-1 cells were cultured in Keratinocyte Serum Free Media supplemented with epidermal growth factor and bovine pituitary extract and subcultured at a 1:4 ratio. MIA PaCa-2 cells were cultured in DMEM, supplemented with 10% FBS and 2.5% horse serum, and subcultured at a 1:8 ratio. Capan-2 cells were cultured in McCoy's 5A Medium Modified supplemented with 10% FBS and subcultured at a 1:4 ratio. THLE-2 was cultured in BEGM medium supplemented with 5 ng/ml EGF, 70 ng/ml Phosphoethanolamine and 10% FBS, and subcultured at a 1:3 ratio. All media for the above cell lines except HPDE (Fisher Scientific, Waltham, MA, USA) and THLE-2 (Lonza/Clonetics Corporation, Walkersville, MD 21,793) were purchased from Mediatech (Manassas, VA, USA) and were supplemented with 100 units/ml penicillin and 2 µg/ml streptomycin. All these cell lines have been routinely tested for mycoplasma contamination using the Universal Mycoplasma Detection Kit obtained from ATCC. Mycoplasma-free cell lines were used in all our experiments.
2.2. RNA isolation and real-time PCR
RNA isolation and real-time PCR were performed as described [17]. The primers used for the real-time PCR are listed in Table 1A and 1B.
Table 1A.
Human primer sequences used for real-time quantitative RT-PCR.
| Gene name | Orientation | Sequence |
|---|---|---|
| SLC7A11 | Forward | TGG ACG GTG TGT GGG GTC CT |
| SLC7A11 | Reverse | CAG CAG TAG CTG CAG GGC GTA |
| p53 | Forward | GGA GCA CTA AGC GAG CAC TG |
| p53 | Reverse | TCT CGG AAC ATC TCG AAG CG |
| p21 | Forward | TTT CCC TTC AGT ACC CTC TC |
| p21 | Reverse | CTT CTT CTT GTG TGT CCC TTC |
| BAX | Forward | CGG AAC TGA TCA GAA CCA TC |
| BAX | Reverse | CTG GAG ACA GGG ACA TCA |
| Cyclin D1 | Forward | CAC TCC TAC GAT ACG CTA CTA |
| Cyclin D1 | Reverse | CAT CTC ATA AAC AGG TCA CTA CA |
| Vimentin | Forward | CCA GCT AAC CAA CGA CAA A |
| Vimentin | Reverse | TCC TCT CTC TGA AGC ATC TC |
| E-cadherin | Forward | CTC CCT TCA CAG CAG AAC TAA C |
| E-cadherin | Reverse | CCA CCT CTA AGG CCA TCT TTG |
| Zeb1 | Forward | AAG TGG CGG TAG ATG GTA ATG |
| Zeb1 | Reverse | AGG AAG ACT GAT GGC TGA AAT AA |
| Zeb2 | Forward | GAG AAA GTA CCA GCG GAA ACA |
| Zeb2 | Reverse | AGG AGT CGG AGT CTG TCA TAT C |
| Snail | Forward | CCA CGA GGT GTG ACT AAC TAT G |
| Snail | Reverse | ACC AAA CAG GAG GCT GAA ATA |
| Twist | Forward | CGG AGA CCT AGA TGT CAT TGT TT |
| Twist | Reverse | ACG CCC TGT TTC TTT GAA TTT G |
| HPRT | Forward | GCG TCG TGA TTA GCG ATG ATG AAC |
| HPRT | Reverse | CCT CCC ATC TCC TTC ATG ACA TCT |
Table 1B.
Mouse primer sequences used for real-time quantitative RT-PCR.
| Gene name | Orientation | Sequence |
|---|---|---|
| p21 | Forward | TCT CCC AGT CTC CAA ACT TA |
| p21 | Reverse | GCC CTA CCG TCC TAC TAA T |
| BAX | Forward | GAG CTG CAG AGG ATG ATT G |
| BAX | Reverse | CCC AGT TGA AGT TGC CAT |
| Cyclin D1 | Forward | CTA AGA TGA AGG AGA CCA TTC C |
| Cyclin D1 | Reverse | ACC AGA AGC AGT TCC ATT T |
| HPRT | Forward | GCG TCG TGA TTA GCG ATG ATG AAC |
| HPRT | Reverse | CCT CCC ATC TCC TTC ATG ACA TCT |
2.3. Immunofluorescence
Immunofluorescence studies were performed as described [17]. Primary antibody used was obtained from Santa Cruz Biotechnology, Inc. (p53 monoclonal antibody, DO-1). Appropriate secondary antibody (goat anti-mouse IgG) conjugated with Alexa Fluor 488 (Molecular Probes, Eugene, OR, USA) was used. Alexa Fluor 594 Phalloidin was also used for determining their morphology.
2.4. Western blotting
Western blotting was performed according to standard procedures [17]. Primary antibodies were obtained from the following sources: p53 (DO-1) from Santa Cruz Biotechnology, Inc.; E-cadherin (24E10), N-cadherin (13A9), p21 (12D1), xCT/SLC7A11, Snail (L70G2), Slug (C19G7), and Twist1 from Cell Signaling; Vimentin/V6389 and β-actin from Sigma Aldrich. HRP-conjugated secondary antibodies (Bethyl Laboratories Inc.) were used as appropriate.
2.5. Apoptosis assay
Apoptosis assay was performed using FITC Annexin V Apoptosis Detection kit I (BD Biosciences), following manufactures protocol. BxPC-3 and Capan-2 cell lines, cultured in the presence and absence of FAC, were dissociated and washed twice with ice-cold phosphate-buffered saline. The cells were then resuspended in 1X binding buffer at a concentration of 1 × 106 cells/ml. This was followed by addition of 5 µl of FITC annexin V and 5 µl propidium iodide to 100 µl of cells. The cells were gently mixed and incubated for 15 min in dark following which 400 µl of 1X binding buffer was added to each tube and the samples were then analyzed using BD Accuri Flow cytometer.
2.6. Uptake measurements
Uptake experiments were carried out as described previously [18]. Briefly, HPDE, Capan-2, BxPC-3 and MIA PaCa-2 cells, which were chronically exposed to excess iron in the form of ferric ammonium citrate (FAC), were seeded in 24-well culture plates at an initial density of 0.2 × 106 cells/assay and the treatment continued until the uptake measurement. [3H]-glutamate (0.5 µCi/assay) uptake was carried out in N-methyl-D-glucamine (NMDG)-chloride to monitor the function of xCT/SLC7A11.
2.7. EL-KrasG12D/Hfe in vivo mouse tumor model
EL-KrasG12D (EL-Kras) transgenic mice were previously generated by microinjection of FVB/N fertilized mouse embryos and subsequent implantation into pseudo-pregnant females (ref ID 12, 727, 811). In this mouse line, the oncogenic Kras is constitutively active due to the G12D mutation and driven by acinar cell-specific minimal promoter of the elastase gene. Hfe−/− mice were obtained from Jackson Laboratories and have been used for several previously published studies [19,20]. The two mouse lines, both of which are on C57BL/6 background, were crossed to generate mouse lines of the following genotypes: EL-Kras/Hfe+/+ and EL-Kras/Hfe−/−.
2.8. Statistical analysis
Student’s unpaired t-tests were used to compare two independent groups or before and after different treatments as appropriate. P values less than 0.05 were considered to be statistically significant.
3. Results and discussion
3.1. Chronic exposure to excess iron leads to profound morphological changes in pancreatic cell lines
Our first aim was to study whether excessive iron accumulation, due to genetic or environmental causes, impacts cellular features related to cancer in normal and cancer cell lines of the pancreas. We first cultured HPDE, a human pancreatic ductal epithelial cell line, in the presence and absence of ferric ammonium citrate (FAC, 250 µg/ml) for a month, thus chronically exposing the cells to excess iron. The concentration of FAC was selected based on the optimization experiments wherein we found 100 µg/ml of FAC to produce similar effects like 250 µg/ml but less pronounced; concentrations beyond 250 µg/ml were found to be toxic for the cells. FAC is an FDA-approved drug for iron supplementation in children with anemia. It is a physiological form of non-transferrin-bound iron and is ideal as an exogenous iron source to induce iron overload in cell culture since it leads to a time-dependent iron accumulation. Normal iron absorption occurs in the proximal small intestine at a rate of 1–2 mg/d. In people with iron overload conditions like hereditary hemochromatosis, this absorption rate can increase to 4–5 mg/d with progressive accumulation to 15–40 g of iron in the body. As the iron content in FAC is 10%, the maximum iron concentration used in our studies was 25 µg/ml, and morphological changes in cultured cells were evident even at an iron concentration of 10 µg/ml. Hence, the concentration of FAC selected for the current experiments is clinically relevant. It was interesting to note that following prolonged exposure to FAC, HPDE cells transformed into an elongated, spindle-shaped mesenchymal morphology as compared to control cells that appeared rounded with epithelial morphology (Fig. 1). In order to see if a similar phenomenon also occurs in pancreatic cancer cell lines, we cultured Capan-2, BxPC-3, and MIA PaCa-2 in the presence and absence of FAC. Similar results were seen in these cell lines as well. However, the changes were not so pronounced in Capan-2 and BxPC-3 (Fig. 1). Capan-2 and BxPC-3 grow in clumps, therefore making it difficult to detect the epithelial-to-mesenchymal transition (EMT), but we did see loss of cell-to-cell compactness as well as a transition to an elongated and spindle-shaped morphology in cells at the periphery of the clumps. To further determine whether the observed phenomenon of morphological transformation following FAC treatment is only relevant to the pancreatic cells or is a more universal phenomenon, we cultured normal epithelial cell lines of the prostate (RWPE-1), liver (THLE-2), and colon (CCD841) using a similar concentration of FAC. It was interesting to note that these cell lines also exhibited a significant transformation in their morphology, a transition from an epithelial to a mesenchymal morphology (Fig. S1). These results indicate that cell lines in culture undergo significant morphological changes indicative of EMT in response to chronic exposure to excessive iron, and that this phenomenon is universal and occurs irrespective of their tissue origin (pancreas or non-pancreas) or type (normal or cancer).
Fig. 1.

Chronic exposure to excess iron leads to profound morphological changes in pancreatic cell lines. Phase-contrast microscopy demonstrating morphological changes in pancreatic normal (HPDE) and cancer (Capan-2, BxPC-3 and MIA PaCa-2) cells cultured chronically (8 passages) in the absence and presence of FAC (250 µg/ml). Original magnification ×20; Scale bars, 100 µm.
3.2. Chronic exposure to excess iron does not induce apoptosis
Iron is a potent oxidant and excess iron can lead to oxidative damage and cell death. Ferroptosis has been established as a novel form of cell death induced by acute exposure to iron. Here we wanted to interrogate whether chronic exposure to excess iron induces cell death via apoptosis. For this, BxPC-3 and Capan-2 cells cultured in the presence and absence of FAC were subjected with annexin V-FITC/ propidium iodide (PI) dual staining. In this technique, viable cells are FITC annexin V and PI negative, early apoptotic cells are FITC annexin V positive and PI negative, and cells that have undergone apoptotic death versus those that have died as a result of necrotic pathway will stain for both FITC annexin V and PI. It was interesting to note that chronic exposure to FAC did not induce apoptosis in both the cell lines. However, both FAC-treated BxPC-3 and Capan-2 cell lines showed about 15% and 22% dead cell population (Fig. S2), respectively, but these values were not significantly different from untreated control cells.
3.3. Molecular evidence for EMT in pancreatic cells in response to chronic exposure to excessive iron
Our findings that chronic exposure to excessive iron induces profound morphological changes in several pancreatic cell lines suggest that excess iron promotes EMT. To confirm this further at the molecular level, we checked the expression of E-cadherin, vimentin, Zeb1, Zeb2, Snail and Twist in control and FAC-exposed HPDE and Capan-2 cell lines. The two cell lines were selected as model cell lines to represent a normal epithelial cell (HPDE) and a non-metastatic cancer cell line (Capan-2). HPDE, when exposed chronically to FAC, downregulated E-cadherin expression, both at the mRNA (Fig. S3A) and protein level (Fig. S3C) as compared to the control. The decrease in E-cadherin expression was accompanied by an increase in the mRNA and protein expression of vimentin and Snail. There was also an increase in the N-cadherin and Slug protein expression. The expression of Twist remained unchanged but the expression of Zeb1 and Zeb2 was downregulated at least at the mRNA level. In Capan-2, exposure to FAC did not change the expression of E-cadherin but led to a marked increase in vimentin expression (∼150-fold) at the mRNA level (Fig. S3B). The increase in the vimentin mRNA level was accompanied with an increase in vimentin protein level (Fig. S3C). Furthermore, the expression of EMT-related transcription factors were also significantly upregulated; Zeb2 (∼100-fold), and Twist, Snail, and Zeb1 (> 3-fold) in FAC-exposed Capan-2 cells compared to control cells (Fig. S3B). Interestingly, the protein expression of some of the transcription factors, such as Snail and Slug, also showed an increasing trend following FAC treatment. We also checked the protein expression of E-cadherin, N-cadherin, vimentin, Snail, Slug, and Twist in control as well as FAC-treated BxPC-3 and MIA PaCa-2 cells. In BxPC-3, though there was no change in E-cadherin and N-cadherin protein expression, it was accompanied by a significant upregulation of vimentin (Fig. S3C). The expressions of the transcription factors like Snail, Slug and Twist either remained unchanged or was undetected. MIA PaCa-2 is known to express very little E-cadherin and following FAC exposure its expression was further attenuated. The expressions of other transcription factors or N-cadherin either remained unchanged or was not detected. Surprisingly, the expression of Snail was significantly downregulated (Fig. S3C). Additionally, we also checked the mRNA expression of matrix metalloproteases (MMP1, MMP2, MM8, MMP9, MMP13, MMP15, and MMP16) whose increase is indicative of EMT, in all the cell lines. The expression of MMP1, MMP9, and MMP16 significantly increased in FAC-treated Capan-2, BxPC-3 and MIA PaCa-2 but not in HPDE (with the exception of MMP9) (Fig. S4). The rest of the MMPs did not show any significant change. Taken collectively, these data demonstrate that normal epithelial cells, when exposed to excess iron for a prolonged period, transition into a mesenchymal phenotype, which might promote cancer and that cancer cells of epithelial origin that have already undergone genetic and epigenetic changes but are non-metastatic might be transformed into a more invasive tumor phenotype in response to chronic exposure to excessive iron. We speculate that some of the discrepancies in the data among the different cell lines could be due to the variable invasive properties as well as differences in the p53 genotype between the cell lines.
3.4. p53 translocation and degradation following chronic exposure to excess iron
It has been unequivocally established that the tumor suppressor protein p53 regulates EMT [21], [22], [23]. Therefore, we wondered if the observed promotion of EMT in response to chronic exposure to excessive iron is related to p53. This idea was prompted by a recent study that showed the binding of heme to p53 with subsequent translocation out of the nucleus followed by proteasomal degradation in the cytoplasm [14]. It is reasonable to think that cells when chronically exposed to excess iron might accumulate heme, which might bind to p53 and promote p53 degradation, thus providing a molecular basis for the iron-induced EMT. In order to validate this idea, immunocytochemical analysis was conducted to monitor the localization of p53 protein in cells with and without chronic exposure to excess iron. Additionally, rhodamine phalloidin staining was also performed in these cells to document the morphological changes following FAC exposure. HPDE, Capan-2 and BxPC-3 were used as model cell lines in the study. HPDE and Capan-2 express wild type p53 whereas BxPC-3 harbors mutant p53. The immunocytochemical data clearly indicated that the p53 protein, which is normally localized in the nucleus, gets translocated into the cytoplasm with consequent decrease in nuclear content of p53 in each of the cell lines when chronically exposed to excessive iron (Fig. 2). In many of the cell lines, the appearance of p53 in the cytoplasm as well as an overall decrease in the cellular content of p53, indicating degradation of the protein, were evident. It was interesting to note that BxPC-3, which expresses mutant p53, also responded in a similar way to excess iron exposure, indicating that the mutation in p53 that occurs in this cell line does not interfere with the binding of heme and subsequent translocation out of the nucleus. Also, the phalloidin staining provided the proof of transformation from an epithelial morphology to a mesenchymal morphology at least in HPDE and BxPC-3 cell lines (Fig. 2). Additionally, to validate our hypothesis that the heme-p53 complex serves as a substrate for XPO1, leading to its translocation out of the nucleus and its subsequent degradation, a reverse experiment was performed using KPT-330, an XPO1 inhibitor. If our hypothesis is right, then inhibition of XPO1 should prevent p53 translocation out of the nucleus and hence prevent its degradation. To test this, we cultured BxPC-3 and Capan-2 cells in the presence and absence of FAC and with and without 1µM KPT-330 for 24 h. Following treatment, immunostaining for p53 protein was performed. It was interesting to observe that the nuclear p53 expression was conserved in the FAC-exposed BxPC-3 and Capan-2 cells following KPT-330 treatment (Fig. 3, Fig. 4), providing support to our hypothesis that excess heme promotes nuclear export of p53 and its subsequent cytoplasmic degradation.
Fig. 2.

Chronic exposure to excess iron promotes p53 translocation out of the nucleus with subsequent degradation in pancreatic cell lines. Immunocytochemical detection of p53 (green) in pancreatic normal epithelial cells (HPDE), and in pancreatic (Capan-2 and BxPC-3), cancer cell lines. Nuclei stained with DAPI are blue. Bar diagrams show p53 fluorescence intensity between control and FAC-treated HPDE, Capan-2, and BxPC-3 cells. *, P < 0.05, **, P < 0.01 compared to corresponding controls. A rhodamine phalloidin (red) demonstrates the morphological differences in control and FAC treated HPDE, Capan-2 and BxPC-3 cells. Original magnification ×100; Scale bar 10 µm (HPDE); Original magnification ×10, Scale bar 100 µm (Capan-2 and BxPC-3).
Fig. 3.

XPO1 inhibitor prevents p53 translocation and its subsequent degradation. Immunocytochemical detection of p53 (green) following KPT-330 treatment in BxPC-3 cells cultured in the presence and absence of FAC. Nuclei stained with DAPI are blue. Bar diagrams show p53 fluorescence intensity of the control and FAC-treated BxPC-3 cells treated with and without KPT-330. *, P < 0.05 compared to corresponding controls. A rhodamine phalloidin (red) demonstrates the morphological differences in control and FAC-treated BxPC-3 cells. Original magnification ×40; Scale bars, 50 µm.
Fig. 4.

XPO1 inhibitor prevents p53 translocation and its subsequent degradation. Immunocytochemical detection of p53 (green) following KPT-330 treatment in Capan-2 cells cultured in the presence and absence of FAC. Nuclei stained with DAPI are blue. Bar diagrams show p53 fluorescence intensity of the control and FAC-treated Capan-2 cells treated with and without KPT-330. *, P < 0.05 compared to corresponding controls. A rhodamine phalloidin (red) demonstrates the morphological differences in control and FAC-treated Capan-2 cells. Original magnification ×40; Scale bars, 50 µm.
3.5. SLC7A11 upregulation following p53 degradation in iron-exposed cells
Recent studies have shown that p53 represses the expression of SLC7A11 [14]. SLC7A11 is a transporter for extracellular cystine (Cys-S-S-Cys) coupled to the efflux of intracellular glutamate [24]. This transporter is a Na+-independent obligatory exchanger. There is increasing evidence that this transporter is a tumor promoter because of its role in maintenance of the cellular antioxidant machinery, promotion of extracellular glutamate signaling, and protection against the cell death pathway ferroptosis [25], [26], [27], [28]. Keeping these facts in mind, we focused on the expression and function of this transporter in control and FAC-exposed cell lines representing the normal pancreatic epithelium and pancreatic cancer. First, we evaluated the levels of SLC7A11 mRNA in control and FAC-exposed cell lines: HPDE, Capan-2, BxPC-3, and MIA PaCa-2. SLC7A11 expression was upregulated in all cancer cell lines when chronically exposed to FAC as compared to the corresponding controls (Fig. 5A–5D). An upregulation was also seen in HPDE, but the magnitude of upregulation was relatively small. We then monitored the transport function of SLC7A11. Even though the transporter mediates the influx of cystine in exchange for glutamate, the transport function is generally measured by the uptake of [3H]-glutamate in a Na+-free uptake medium, which occurs in exchange for cellular glutamate [29]. In all cancer cell lines examined, the transport activity was increased in response to chronic exposure to excess iron (Fig. 5F–5H). Again, the increase in transport activity was evident in HPDE (Fig. 5E), but with a lower magnitude. It was also interesting to note that BxPC-3 and MIA PaCa-2 cells that express mutant p53 were also capable of increasing SLC7A11 expression in response to chronic exposure to excess iron, demonstrating that the mutant p53 present in these two cell lines still retains its ability to suppress the expression of the transporter. Additionally, to further confirm that the increase in SLC7A11 expression and function is regulated by p53, we conducted [3H]-glutamate uptake in mouse embryonic fibroblast cell line, MEFs/p53-WT and MEFs/p53-KO, cultured chronically in the presence and absence of FAC. MEFs/p53-WT showed a significant increase in [3H]-glutamate uptake following FAC exposure, while MEFs/p53-KO exhibited no change in [3H]-glutamate uptake, clearly indicating p53 regulation of SLC7A11 (Fig. S5A).
Fig. 5.

Upregulation of SLC7A11 mRNA and transport function in a normal pancreatic epithelial cell line (HPDE) and pancreatic cancer cell lines (Capan-2, BxPC-3, and MIA PaCa-2) in response to chronic exposure to excess iron (FAC; 250 µg/ml; 8 passages). (mean± SD, n = 3). (A)-(D) Relative SLC7A11 mRNA levels; the expression level in cells cultured in the absence of FAC is taken as 1. (E)-(H) SLC7A11-mediated glutamate uptake in control and FAC-exposed cells. In all cases, the difference between control and FAC-treated cells was significant (P < 0.05).
3.6. Kinetic analysis of SLC7A11 in BxPC-3 and MIA PaCa-2 cells exposed to FAC
The substrate-saturation kinetics of SLC7A11 was analyzed in control and FAC-exposed BxPC-3 and MIA PaCa-2 cells (Fig. S5B). The increase in the transport activity of SLC7A11 observed in FAC-exposed BxPC-3 and MIA PaCa-2 cells as compared to control cells was associated primarily with an increase in the maximal velocity of the transporter with no significant change in the substrate affinity. The maximal velocity (Vmax) of glutamate uptake was ∼5-fold greater in FAC-exposed BxPC-3 cells than in control cells (5146 ± 90 vs 953 ± 56 pmol/106 cells/15 min). FAC-exposed MIA PaCa-2 cells also exhibited a ∼3-fold increase in Vmax compared to control cells (801 ± 75 vs. 274 ± 72 pmol/106 cells/15 min). The Michaelis constant (Kt) for glutamate in control and FAC-exposed cells remained mostly unchanged: 56 ± 17 vs 90 ± 5 µM for control and FAC-exposed BxPC-3 cells; 72 ± 9 vs 100 ± 28 µM for control and FAC-exposed MIA PaCa-2 cells.
3.7. Suppression of p53 transcriptional activity in cells in response to chronic exposure to excess iron
p53 performs a myriad of functions. As a tumor suppressor, the major p53 functions regulate growth arrest and apoptosis [21], [22], [23]. Based on our findings of p53 degradation and loss in normal and cancer cell lines following chronic exposure to excess iron, we wanted to know if the decreased p53 in FAC-exposed cells correlated with a concomitant decrease in the expression of p53 target genes. We specifically checked the expression of Bax, Cyclin D1 and p21. First, we checked p53 expression in HPDE, Capan-2, BxPC-3, and MIA PaCa-2 (control and FAC-exposed) cells. There was a small but significant reduction in the levels of p53 mRNA in cancer cell lines in response to FAC exposure compared to their corresponding controls (Fig. 6A). This effect was however not seen in the normal cell line. The decrease in p53 protein level was however evident in the normal cell line as well as in the cancer cell lines as monitored by western blot (Fig. 6B). As expected, in HPDE cell line that harbors a wildtype p53, a decrease in cellular levels of p53 protein in FAC-exposed cells also led to a significant reduction in the expression of Bax, Cyclin D1, and p21 (Fig. 6C). However, we find that the cancer cell lines that harbor a mutant p53 (eg., MIA PaCa-2) also shows a reduction in p53 protein and a concomitant decrease in the expression of its target genes (Fig. 6D). While this seems counterintuitive, studies have shown that the mutant p53 in spite of losing its tumor suppressor function, is endowed with a gain-of-function abilities and hence, still retains the ability to bind heme/iron and bring about changes in gene expression like the wildtype p53.
Fig. 6.

Decreased levels of p53 protein and downregulation of the expression of p53 target genes in pancreatic cells following chronic exposure to excess iron. (A), relative p53 mRNA expression in pancreatic normal (HPDE), and cancer (Capan-2, BxPC-3, and MIA PaCa-2) cell lines cultured in the absence and presence of FAC (250 µg/ml; 8 passages). (mean ± SD, n = 3); (B) Western blot analysis of p53 protein in whole cell lysates of normal (HPDE), and cancer (Capan-2, BxPC-3, and MIA PaCa-2) cell lines cultured in the presence and absence of FAC. β-actin (42 kDa) was used as a loading control. Densitometric values are indicated for all the control and FAC treated cells; (C) & (D), relative levels of mRNA for Bax, cyclin D1 and p21 in HPDE and MIA PaCa-2 cells cultured in the presence and absence of FAC. (mean ± SD, n = 3); (D) & (E) cellular levels of heme in HPDE and BxPC-3 cells cultured in the absence and presence of FAC; the respective value in control cells (i.e., cells cultured in the absence of FAC) is taken as 100%. * P < 0.05 compared to corresponding controls.
3.8. Elevation of heme levels in FAC-exposed cells
p53 is a heme-binding protein, and heme facilitates the export of this transcription factor out of the nucleus for subsequent ubiquitination and proteasomal degradation [14]. We hypothesized that the decrease in the cellular levels of p53 in FAC-exposed cells was due to increased cellular heme content, which would facilitate p53 degradation. To test this hypothesis, we measured the cellular levels of heme (as hemin, an oxidized form of heme) in control and FAC-exposed cells using the normal pancreatic epithelial cell line HPDE and the pancreatic cancer cell line BxPC-3. The concentration of hemin was increased by ∼20% compared to controls in both cell lines when chronically exposed to excessive iron (Fig. 6E and 6F).
3.9. Impact of chronic iron overload in vivo on p53 protein and its target genes in pancreas in hemochromatosis (Hfe−/−) mouse
Hfe−/− mouse is an animal model for hereditary hemochromatosis [16]. The in vitro experiments described thus far demonstrated that chronic exposure to excessive iron results in decreased cellular levels of p53 protein and hence suppression of p53 transcriptional activity. To determine if a similar phenomenon occurs in vivo, we used Hfe−/− mouse as a model of chronic iron overload. While exposure to FAC represents an non-genetic environmental model of iron overload in cells, the Hfe−/− mouse represents a genetic model of iron overload. We used 12-month-old wild type mice and age-matched Hfe−/− mice for the present study. The pancreatic tissues were collected from these mice and used for isolation of protein lysates as well as RNA. We found that the cellular levels of p53 protein were markedly reduced in pancreatic tissues from Hfe−/− mice compared to pancreatic tissues from control mice (Fig. 7A). This was accompanied with a corresponding decrease in the cellular levels of mRNA for three p53 target genes (Bax, Cyclin D1, and p21) (Fig. 7B). These data show that chronic exposure to excess iron suppresses p53 function both in vitro and in vivo.
Fig. 7.

Deletion of Hfe affects p53 expression and promotes cancer in EL-Kras mice. (A), Western blot analysis of p53 protein in pancreatic tissues from wild type (Hfe+/+) mice and Hfe−/− mice (12-month-old males; 2 mice for each genotype). β-actin (42 kDa) was used as a loading control. Densitometric values are indicated. (B) relative levels of mRNA for the p53 target genes Bax, cyclin D1 and p21 in pancreatic tissues derived from wild type (Hfe+/+) mice and Hfe−/− mice. (mean ± SD, n = 3); *, P < 0.05 compared to corresponding controls). (C) deletion of Hfe in EL-Kras mice decreases the incidence of ADM and CPNs pancreatic lesions but increases the size of CPNs. (D) pancreatic tissue from EL-Kras/Hfe+/+and EL-Kras/Hfe–/– mice (5 and 4 mice in each group respectively) were stained with hematoxylin and eosin to evaluate lesion frequency. Lesions in one section were counted and quantified as lesions per square centimeter at 10×. The surface area of the five greatest lesions was evaluated. The P values were calculated using an unpaired t-test; *, P < 0.05; **, P < 0.01. Original magnification ×10.
3.10. Effect of Hfe deletion on mutant Kras-driven pancreatic neoplasia
EL-Kras mice carry the spontaneously active mutant KRAS (KRASG12D) under the control of the promoter of the elastase gene [15]. This allows tissue-specific expression of active Kras, specifically in pancreatic acinar cells. These mice develop preinvasive pancreatic neoplasia of ductal phenotype at ≥4 months of age. The results of in vitro and in vivo studies presented here thus far have shown that chronic iron overload suppresses p53 function in cells representing various tissues including the pancreas. Therefore, we hypothesized that deletion of Hfe and consequent development of hemochromatosis in EL-Kras mice would promote pancreatic neoplasia because of the decreased p53 signaling caused by iron overload. As the KrasG12D transgene is integrated into Y-chromosome in the EL-Kras mice, only male mice were used in this study. To evaluate the differences in the pancreatic neoplastic phenotype between EL-Kras mice in two different Hfe genotypes (+/+ and −/−), a group of 5 EL-Kras/Hfe+/+ (10 month-old) was compared with 4 age-matched EL-Kras/Hfe−/− mice for the incidence of acinar-to-ductal metaplasia (ADM) and cystic pancreatic neoplasms (CPN) and for the size of CPNs (Fig. 7C). Histological examination revealed that deletion of Hfe in EL-Kras mice decreased the frequency of ADMs and CPNs; however, CPNs that did develop were significantly larger in EL-Kras/Hfe−/− mice than in EL-Kras/Hfe+/+ mice as evident from increased surface area (Fig. 7D). Furthermore, we also checked the expressions of the p53 target genes and also the EMT proteins in the pancreas of the EL-Kras/Hfe+/+ vs. the EL-Kras/Hfe−/− mice. Interestingly, the expressions of Bax, cyclin D1, and p21 mRNA expressions were low in the knockout mice (Fig. S6A). Our western blot data also showed a significant reduction in p21 expression in the knockouts compared to the wildtypes. At least, 2 out of the 4 knockout mice showed a significant reduction in E-cadherin expression and an increase in the expressions of vimentin, Snail, Slug, Twist, and SLC7A11 (Fig. S6B). These data, despite some inter-animal variations, support the general conclusion that deletion of Hfe in the EL-Kras mice does lead to iron accumulation, which affects p53 and as a result leads to EMT-like changes and eventually promotes cancer in these mice.
3.11. Discussion
Iron is an essential nutrient required by every cell. Its atomic structure gives rise to a number of biochemically important properties, including the useful capacity to both donate and accept electrons, and to reversibly bind to ligands such as oxygen. As such, iron in the form of heme plays a vital role in the transport and storage of oxygen, in oxidative metabolism and in cellular growth and proliferation. On the flip side, excess iron accumulation is implicated in a wide range of diseases, including cardiovascular disease, diabetes, neurological disorders, and most importantly, cancer. Literature evidence has also shown that dietary intake of red meat, which is rich in heme, is associated with a significant increase in cancer risk (4, 11–13). The role of excess iron leading to hepatocellular carcinoma is already well documented (5–8), but little is known on the impact of excess iron on cancers of non-hepatic tissues. Hemochromatosis is not the only genetic disease that affects iron storage. Wilson's disease, also a genetic disease, is associated with excessive copper accumulation and decreased levels of ceruloplasmin in circulation; this is a rare autosomal recessive metabolic disorder caused by mutation in P-type ATPase (ATP7B) [30]. The classical presentations of this disease include hepatic, neurologic, and ophthalmologic manifestations. The relevance of Wilson's disease to iron lies the biologic activity of ceruloplasmin as a ferroxidase. Ferroportin is a transporter that exports iron out of the cells, but this transporter recognizes only the reduced form of iron (Fe2+) as the substrate, and the effluxed iron is immediately oxidized into Fe3+ by ceruloplamin (and also by hephaestin) that is closely associated with the transporter. As a result, decreased levels of ceruloplasmin seen in Wilson's disease lead to increased tissue accumulation of iron even though circulating levels of iron fall. It is of interest to note that even though it was believed until recently that Wilson's disease had a protective effect on liver cancer, recent evidence suggests the opposite. This is not surprising because copper itself is a pro-oxidant and excess iron accumulation in liver that results from ceruloplasmin deficiency also adds to this oxidative process in Wilson's disease. As a result, the risk of liver cancer increases in hemochromatosis as well as in Wilson's disease.
Here we show that chronic exposure to excess iron/heme transforms normal epithelial cell lines into a tumor cell-like phenotype, and also changes the non-metastatic cancer cell line towards EMT that may implicate enhanced ability to metastasize; some, but not all, aspects of in vivo data, including enlarged CPNs, using the EL-Kras mouse model of pancreatic neoplasia show that hemochromatosis resulting from Hfe deletion promotes pancreatic cancer growth and that the phenomenon is associated with loss of p53.
The basic underlying mechanism for the observed phenomenon appears to be loss of p53 initiated by chronic exposure to excessive iron. Of course, iron has many biological functions in addition to its role in the biosynthesis of heme. Free iron is a potent pro-oxidant and facilitates generation of reactive oxygen species; as highly reactive free radicals such as hydroxyl radical have potential to damage lipids, proteins and DNA/RNA, the possibility of iron-induced oxidative stress in the potentiation of tumorigenesis by chronic exposure to excessive iron cannot be discounted. The experiments reported in the present study uncover another aspect of the tumor-promoting role of excess iron. Chronic exposure to excess iron leads to loss of the tumor-suppressor protein p53 and this process is mediated by heme. Even though the relevance of excessive iron to the promotion of pancreatic cancer growth has been specifically highlighted in the present study, the data do implicate a tumor-promoting role for excessive iron in other tissues of epithelial origin such as prostate, liver, and colon.
The present findings have clinical significance in multiple areas of cancer biology. First, we focus on the well-known role of p53 as a tumor suppressor with a regulatory role in cell cycle, cell senescence, apoptosis, metabolism, stem cell maintenance, and EMT [21], [22], [23]. The loss of this tumor suppressor in normal cells when exposed chronically to excess iron is important as a potential cause of tumorigenesis. According to the Knudson's two-hit hypothesis, cancer develops in most cases when two cancer-causing cellular events co-exist; this could be an activation of an oncogene or inactivation of a tumor suppressor gene. As the loss of the tumor suppressor p53 could represent one such event, chronic exposure to excessive iron could cause cancer in the presence of genetic alterations (mutations or change in copy number) that lead to loss of function of tumor suppressors such as PTEN and BRCA1 or gain-of-function in oncogenes such as RAS or growth factor receptors (e.g., EGFR, HER2). Our findings could also be considered significant with regard to incidence and progression of cancer in Li-Fraumeni syndrome, which is caused by heterozygous germline mutations in p53 leading to 50% loss of p53 protein [31]. Chronic exposure to excess iron in patients with this syndrome could synergize with the heterozygous loss of p53 to render more complete loss of p53 protein to promote cancer despite the presence of an intact wild type p53 allele. Second, we emphasize the role p53 plays in cancer metastasis due to the function of the protein as a suppressor of EMT [21], [22], [23]. The loss of p53, induced by chronic exposure to excess iron, could be a trigger for the conversion of cancer cells toward EMT which may implicate enhanced ability to metastasize. The unequivocal evidence from present studies for the promotion of EMT in normal epithelial cell lines as well as non-metastatic cancer cell lines in response to excess iron provides strong support for this notion.
Third, the upregulation of the transporter SLC7A11 in response to iron-induced p53 loss in cancer cell lines and, to a smaller extent, also in normal cell lines has significant relevance to cancer. This transporter is obligatory for the maintenance of antioxidant machinery in cells due to its role as a supplier of cysteine for the synthesis of glutathione [25]. While robust antioxidant mechanisms could deter tumorigenesis in cells via protection against DNA damage by pro-oxidants, the same mechanisms could also promote tumor growth by protecting cancer cells against oxidant-induced cell death. Recent studies have identified a novel form of cell death, known as ferroptosis, which is dependent on free iron as a producer of reactive oxygen species necessary for the induction of this particular cell death process [28]. Ferroptosis is also dependent on p53 as a repressor of SLC7A11, thereby leading to defective antioxidant machinery in cells and hence facilitating the cell death process promoted by iron-induced reactive oxygen species [28]. Even though ferroptosis is an iron-induced cell death process, chronic exposure to excess iron is likely to protect against ferroptosis because of the loss of p53 and consequent increase in the expression of SLC7A11. There is strong evidence that tumor cells modify various components of iron homeostasis in such a way to increase iron levels as a means to promote their survival and growth [9,10]. We postulate that protection against iron-induced ferroptosis is at least one of the underlying mechanisms associated with the tumor-promoting role of chronic exposure to excess iron. This is supported by the recent findings that the expression of SLC7A11 is upregulated in multiple cancers, including breast cancer, prostate cancer, and pancreatic cancer [25,32]. The upregulation of SLC7A11 in response to chronic exposure to excessive iron also has another connotation with regard to tumor growth. As this transporter is an obligatory exchanger that functions under physiological conditions to mediate the influx of cystine coupled to the efflux of glutamate, the enhanced expression of the transporter in tumor cells results in an increase in extracellular levels of glutamate. This phenomenon is equally important to cancer growth.
Our studies are in line with the previous finding that wild type p53 gets translocated out of the nucleus and subsequently gets degraded in the cytoplasm when bound to heme [14]. But the question is why the p53 mRNA expression changed following FAC exposure. That being said, though there is extensive information about the role of p53 protein, very little is known about the transcriptional regulation of p53. It is said that the p53 gene expression is tightly regulated through a variety of transcription factors, miRNAs, anti-sense RNA Wrap53, the insulator protein CTCF and most likely by other genetic and epigenetic mechanisms. It is possible that excess iron might interact with any of these factors and cause a change in the mRNA expression of p53. Also, what is intriguing in the present study is that BxPC-3 and MIA PaCa-2 cancer cells, which harbor mutant p53, also undergo p53 loss in response to chronic exposure to excessive iron and consequent increase in cellular levels of heme. This demonstrates that at least the mutations that occur in these cell lines do not interfere with the ability of p53 to bind heme and undergo subsequent translocation out of the nucleus and proteasomal degradation. As p53 is a tumor suppressor, it makes sense as to how mutations that lead to loss of function of this protein could promote cancer. However, recent studies have suggested that at least in some cases, cancer-causing mutations in p53 may actually lead to gain of some novel functions that are not inherent to wild type p53 [33]. Also, literature evidence has shown that the mutant p53 still retains the ability to bind heme/iron. Our studies suggest that chronic exposure to excess iron could also interfere with tumor-promoting ability of such gain-of-function mutations in p53 because of the iron/heme-induced depletion of mutant p53. This speculation however warrants further study.
Interestingly, the in vivo studies with Hfe-associated hemochromatosis model of iron overload supported the tumor-promoting role of excess iron in pancreatic neoplasia only in certain aspects; in contrast, in vitro studies with cell lines provided strong evidence in support of the role of excess iron in p53 loss and EMT, suggesting a robust cause-effect relationship between excess iron and tumor promotion. There could be several reasons for this apparent discrepancy. Studies have shown that in Hfe−/− mice, pancreatic β-cells are primarily affected with iron overload and acinar cells are mostly spared [34]. The EL-Kras mouse model of pancreatic neoplasia expresses the oncogenic mutant Kras specifically in acinar cells. The lack of robust iron accumulation in acinar cells in Hfe-associated hemochromatosis could be a contributing factor as to why promotion of neoplasia was evident only in increased lesion size but not lesion frequency in EL-Kras/Hfe−/− mouse pancreas. Another caveat to consider is that global loss of Hfe (Hfe−/−) might impact another cell type that prevents, or targets lesion development as observed in EL-Kras/Tgfbr1+/− mice [35], where Tgfbr1 haploinsufficiency led to a robust cytotoxic immune response [34]. This however does not undermine the importance of chronic iron overload as a promoter of pancreatic cancer. Hfe-associated hemochromatosis represents only one of many clinical conditions of iron overload. It is likely that acinar cells and ductal cells are subject to chronic exposure to excess iron in other conditions associated with iron overload. In fact, published reports have shown that mice with the deletion of other iron-regulatory proteins (hepcidin, ferroportin, and hemojuvelin) do indeed lead to excessive iron accumulation in acinar cells with consequent pancreatitis [36,37]. Similarly, iron overload resulting from frequent blood transfusion or hemoglobin disorders such as thalassemia and sickle cell disease might also lead to chronic exposure of pancreatic acinar cells and ductal cells to excessive iron. Therefore, our data that chronic exposure of pancreatic epithelial cells to excess iron induces p53 loss and EMT has significant relevance to tumor promotion in various clinical conditions associated with iron overload.
4. Conclusion
In summary, the studies presented here demonstrate that chronic exposure to excess iron leads to heme-mediated export of the tumor suppressor p53 out of the nucleus with subsequent proteasomal degradation, thereby leading to loss of p53 protein and its transcriptional activity. This results in the upregulation of the antioxidant transporter SLC7A11 and in the promotion of EMT. As a consequence, chronic exposure to excessive iron impacts cancer progression and also changes the cancer cells toward EMT-like phenotype, hence also potentially promoting metastasis.
Declaration of interest
The authors declare that they have no potential conflicts of interest.
Acknowledgments
Acknowledgments
The authors acknowledge the financial support received from the National Institute of Health (CA223271). The authors would also like to thank Dr. Ming Tsao (Ontario Cancer Institute) for the human pancreatic ductal epithelial (HPDE) cell line.
Ethical approval
All experimental procedures were in compliance with the National Institute of Health guidelines and approved by Texas Tech University Health Sciences Center Institutional Animal Care and Use Committee (IACUC).
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ajps.2020.02.003.
Appendix. Supplementary materials
References
- 1.Kozlowski H., Kolkowska P., Watly J., Krzywoszynska K., Potocki S. General aspects of metal toxicity. Curr Med Chem. 2014;21(33):3721–3740. doi: 10.2174/0929867321666140716093838. [DOI] [PubMed] [Google Scholar]
- 2.Porter J.B., de Witte T., Cappellini M.D., Gattermann N. New insights into transfusion-related iron toxicity: implications for the oncologist. Crit Rev Oncol Hematol. 2016;99:261–271. doi: 10.1016/j.critrevonc.2015.11.017. [DOI] [PubMed] [Google Scholar]
- 3.Dutra F.F., Bozza M.T. Heme on innate immunity and inflammation. Front Pharmacol. 2014;5:115. doi: 10.3389/fphar.2014.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hooda J., Shah A., Zhang L. Heme, an essential nutrient from dietary proteins, critically impacts diverse physiological and pathological processes. Nutrients. 2014;6(3):1080–1102. doi: 10.3390/nu6031080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babitt J.L., Lin H.Y. The molecular pathogenesis of hereditary hemochromatosis. Semin Liver Dis. 2011;31(3):280–292. doi: 10.1055/s-0031-1286059. [DOI] [PubMed] [Google Scholar]
- 6.Pietrangelo A., Caleffi A., Corradini E. Non-HFE hepatic iron overload. Semin Liver Dis. 2011;31(3):302–318. doi: 10.1055/s-0031-1286061. [DOI] [PubMed] [Google Scholar]
- 7.Ganz T., Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823(9):1434–1443. doi: 10.1016/j.bbamcr.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kew M.C. Hepatic iron overload and hepatocellular carcinoma. Liver Cancer. 2014;3(1):31–40. doi: 10.1159/000343856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torti S.V., Torti F.M. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13:342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bystrom L.M., Rivella S. Cancer cells with irons in the fire. Free Radic Biol Med. 2015;79:337–342. doi: 10.1016/j.freeradbiomed.2014.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cross A.J., Leitzmann M.F., Gail M.H., Hollenbeck A.R., Schatzkin A., Sinha R. A prospective study of red and processed meat intake in relation to cancer risk. PLoS Med. 2007;4(12):e325. doi: 10.1371/journal.pmed.0040325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bastide N.M., Pierre F.H., Corpet D.E. Heme iron from meat and risk of colorectal cancer: a meta-analysis and a review of the mechanisms involved. Cancer Prev Res (Phila) 2011;4(2):177–184. doi: 10.1158/1940-6207.CAPR-10-0113. [DOI] [PubMed] [Google Scholar]
- 13.Qiao L., Feng Y. Intakes of heme iron and zinc and colorectal cancer incidence: a meta-analysis of prospective studies. Cancer Causes Control. 2013;24(6):1175–1183. doi: 10.1007/s10552-013-0197-x. [DOI] [PubMed] [Google Scholar]
- 14.Shen J., Sheng X., Chang Z., Wu Q., Wang S., Xuan Z. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014;7(1):180–193. doi: 10.1016/j.celrep.2014.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grippo P.J., Nowlin P.S., Demeure M.J., Longnecker D.S., Sandgren E.P. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant kras in transgenic mice. Cancer Res. 2003;63(9):2016–2019. [PubMed] [Google Scholar]
- 16.Levy J.E., Montross L.K., Andrews N.C. Genes that modify the hemochromatosis phenotype in mice. J Clin Invest. 2000;105(9):1209–1216. doi: 10.1172/JCI9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coothankandaswamy V., Cao S., Xu Y., Prasad P.D., Singh P.K., Reynolds C.P. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer. Br J Pharmacol. 2016;173(23):3292–3306. doi: 10.1111/bph.13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhutia Y.D., Babu E., Ganapathy V. Interferon-gamma induces a tryptophan-selective amino acid transporter in human colonic epithelial cells and mouse dendritic cells. Biochim Biophys Acta. 2015;1848(2):453–462. doi: 10.1016/j.bbamem.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 19.Gnanaprakasam J.P., Thangaraju M., Liu K., Ha Y., Martin P.M., Smith S.B. Absence of iron-regulatory protein hfe results in hyperproliferation of retinal pigment epithelium: role of cystine/glutamate exchanger. Biochem J. 2009;424(2):243–252. doi: 10.1042/BJ20090424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gnanaprakasam J.P., Veeranan-Karmegam R., Coothankandaswamy V., Reddy S.K., Martin P.M., Thangaraju M. Loss of hfe leads to progression of tumor phenotype in primary retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2013;54(1):63–71. doi: 10.1167/iovs.12-10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menendez D., Inga A., Resnick M.A. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9(10):724–737. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 22.Levine A.J., Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9(10):749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bieging K.T., Mello S.S., Attardi L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fotiadis D., Kanai Y., Palacin M. The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med. 2013;3(2–3):139–158. doi: 10.1016/j.mam.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 25.Lewerenz J., Hewett S.J., Huang Y., Lambros M., Gout P.W., Kalivas P.W. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18(5):522–555. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stepulak A., Rola R., Polberg K., Ikonomidou C. Glutamate and its receptors in cancer. J Neural Transm (Vienna) 2014;121(8):933–944. doi: 10.1007/s00702-014-1182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu H., Takano N., Xiang L., Gilkes D.M., Luo W., Semenza G.L. Hypoxia-inducible factors enhance glutamate signaling in cancer cells. Oncotarget. 2014;5(19):8853–8868. doi: 10.18632/oncotarget.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang W.S., Stockwell B.R. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bridges C.C., Kekuda R., Wang H., Prasad P.D., Mehta P., Huang W. Structure, function, and regulation of human cystine/glutamate transporter in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2001;42(1):47–54. [PubMed] [Google Scholar]
- 30.Xu R., Hajdu C.H. Wilson disease and hepatocellular carcinoma. Gastroenterol Hepatol. 2008;49(6):438–439. [PMC free article] [PubMed] [Google Scholar]
- 31.McBride K.A., Ballinger M.L., Killick E., Kirk J., Tattersall M.H., Eeles R.A. Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat Rev Clin Oncol. 2014;11(5):260–271. doi: 10.1038/nrclinonc.2014.41. [DOI] [PubMed] [Google Scholar]
- 32.Bhutia Y.D., Babu E., Ramachandran S., Ganapathy V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015;75(9):1782–1788. doi: 10.1158/0008-5472.CAN-14-3745. [DOI] [PubMed] [Google Scholar]
- 33.Weisz L., Oren M., Rotter V. Transcription regulation by mutant p53. Oncogene. 2007;26(15):2202–2211. doi: 10.1038/sj.onc.1210294. [DOI] [PubMed] [Google Scholar]
- 34.Cooksey R.C., Jouihan H.A., Ajioka R.S., Hazel M.W., Jones D.L., Kushner J.P. Oxidative stress, beta-cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology. 2004;145(11):5305–5312. doi: 10.1210/en.2004-0392. [DOI] [PubMed] [Google Scholar]
- 35.Principe D.R., DeCant B., Mascarinas E., Wayne E.A., Diaz A.M., Akagi N. TGFβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 2016;76(9):2525–2539. doi: 10.1158/0008-5472.CAN-15-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lunova M., Schwarz P., Nurldeen R., Levada K., Kuscuoglu D., Stutzle M. Hepcidin knockout mice spontaneously develop chronic pancreatitis owing to cytoplasmic iron overload in acinar cells. J Pathol. 2017;241(1):104–114. doi: 10.1002/path.4822. [DOI] [PubMed] [Google Scholar]
- 37.Altamura S., Kessler R., Grone H.J., Gretz N., Hentze M.W., Galy B. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab. 2014;20(2):359–367. doi: 10.1016/j.cmet.2014.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.