

The enhancer of zeste homolog 2 (EZH2) is a histone methyltransferase and functional core subunit of the polycomb repressive complex 2 (PRC2), which is a key epigenetic regulator involved in embryonic development and transcriptional repression of genes by catalyzing the methylation of histone H3 at lysine 27 (H3K27me2/3).1 EZH2 overexpression has been associated with oncogenic activity and worse progression-free survival in several types of cancer including lymphoma, melanoma, and prostate and breast cancer.2,3 Moreover, the detection of recurrent EZH2 mutations, both gain-of-function in lymphomas and loss-of-function, e.g. in medulloblastoma, and bladder and renal cancers, point to a mutual role of EZH2 for disease pathology depending on the distinct type of cancer and indicate the potential of EZH2 as a therapeutic target.4–6 In myeloid malignancies such as myelodysplastic syndromes (MDS), loss of EZH2 activity by inactivating mutations is associated with poor prognosis.7 More recently, EZH2 inactivation by post translational modification was shown to induce chemoresistance in acute myeloid leukemia (AML).8 However, data on the frequency and prognostic role of EZH2 mutations in AML are still scarce and mostly confined to smaller cohorts. To investigate the prevalence and prognostic impact of this alteration in more detail, we analyzed a large cohort of AML patients (n=1,604) for EZH2 mutations.
All patients had newly diagnosed AML, were registered for trials investigating intensive induction chemotherapy of the Study Alliance Leukemia (SAL) (AML96, AML2003 or AML60+, SORAML), and had available material at diagnosis. All analyses were carried out under the auspices of the SAL-bioregistry. Screening for EZH2 mutations and associated alterations was performed using Next-Generation Sequencing (NGS) (TruSight Myeloid Sequencing Panel, Illumina) on an Illumina MiSeq-system using bone marrow (BM) or peripheral blood (PB) (Online Supplementary Appendix). The myeloid gene panel (Illumina) targets 54 genes associated with myeloid neoplasms including full coding exons of EZH2, encoded on the long arm of chromosome 7 (7q36.1). Detection was conducted with a defined cutoff of 5% variant allele frequency (VAF) (median coverage 3,076; range 824-30,565). Patients’ clinical characteristics and co-mutations were analyzed according to the mutational status. Furthermore, multivariate analysis was used to identify the impact of EZH2 mutations on outcome. In addition, a more detailed subgroup analysis of EZH2-mut patients was conducted to integrate clinical outcome with the allelic state of mutations, affected functional domains (CXC-SET), and predicted effects of EZH2 mutations on protein expression.
EZH2 mutations were found in 63 of 1,604 (4%) patients, which is in the range of prevalence (2-13%) typically observed for EZH2 mutations in myeloid malignancies.7,9–13 A detailed list of all detected EZH2 variants is provided in Online Supplementary Table S1. In total, 50 of 63 patients (79%) harbored one single mutation in EZH2, while 13 individuals carried two different EZH2 variants (double mutated). Mutations were detected within several exons (2-6; 8-12; 14-20) and functional domains (D1, D2, CXC and SET), respectively (Figure 1A). In line with previous findings, most EZH2 variants were detected in exons 17 and 18 (28%), comprising the highly conserved SET domain, important for the catalytic activity of the EZH2 protein.7 The majority of detected mutations (67% missense and 33% nonsense/frameshift) were single nucleotide variants (SNV) (86%), followed by small indel mutations. All frameshift and nonsense mutations resulting in a premature stop of transcription were predicted to be “inactivating” for EZH2 protein expression (Figure 1B). One patient harbored a known EZH2 gain-of-function variant at position A682 (A677) recurrently found in large B-cell and non-Hodgkin lymphomas.4 These contrasting observations support recent findings that point to a potential stage-specific role of EZH2 in AML, exerting a tumor suppressor function during early AML induction and an oncogenic function during tumor maintenance.12 Other pathogenic SNV found in two or more patients were detected at residues R25 (COSM53003), G159 (COSM96480), R288 (COSM1000721), R313 (COSM6916439), R502 (COSM87274), N673 (COSM87276), R690 (COSM52980), and E745 (COSM1087033) (highlighted in Figure 1A). The p.Glu745Lys mutation (detected in 2 patients) was previously associated with acute lymphoblastic leukemia and lymphoma in Weaver Syndrome patients.14 Mutations were detected with a median allele burden of 42% (range 6-97%) (Figure 1B). The majority of patients (n=39) were heterozygous for their EZH2 variant, likely representing clonal events. Subclonal EZH2 variants (with significantly lower allelic ratio compared to all other co-mutated driver genes) were detected in nine patients. Patients with EZH2 allelic ratios >70% (n=15) were considered homozygous. EZH2 mutations were previously detected in both monoallelic and biallelic states in MDS/myeloproliferative neoplasms (MPN).7
Figure 1.

Detected EZH2 mutations. (A) Schematic illustration showing the position of acquired EZH2 mutations and the domain structure of EZH2 (ENST00000320356): D1 (domain I), D2 (domain II), CXC (cysteine-rich domain), and SET (suppressor of variegation-enhancer of zeste-trithorax domain). Recurrent alterations are indicated. (B) EZH2 variant allele frequencies (VAF) predicted effect on protein expression and associated co-mutations. Clonality was determined by comparing VAF of EZH2 mutations and co-mutated somatic driver variants. Mutations with uncertain effect on EZH2 protein expression were classified “unknown” (gray). All EZH2 loss-of-function mutations (nonsense SNPs and frameshift INDEL variants) were considered “inactivating” (orange). One patient harbored a p.Ala682Gly (A677) EZH2 variant known to be “activating” for protein expression (green).
Descriptive statistics of clinical parameters and associated co-mutations revealed significant differences between EZH2-mut and wild-type (-wt) patients (Table 1). At diagnosis, patients with EZH2 mutations were significantly older (median age 59 years) than EZH2-wt patients (median age 56 years) (P=0.044). In addition, significantly fewer EZH2-mut patients (71%) were diagnosed with de novo AML compared to EZH2-wt patients (84%) (P=0.036). Accordingly, EZH2-mut patients had a higher rate of secondary AML (sAML) (21%), evolving from prior MDS or after prior chemotherapy (therapy-related AML, tAML) (8%; P=0.036). BM (and PB) blast counts differed between the two groups (EZH2-mut patients had significantly lower BM and PB blast counts; P=0.013), confirming previous reports.13 In contrast, no differences were observed for white blood cell (WBC) counts at diagnosis, karyotype, Eastern Cooperative Oncology Group (ECOG) performance status and European LeukemiaNet (ELN) 2017 risk category compared to EZH2-wt patients. Based on cytogenetics according to the 2017 ELN criteria, 35% of EZH2-mut patients were categorized with favorable risk, 28% had intermediate and 37% adverse risk. Initial studies frequently observed associations of EZH2 mutations in de novo AML with monosomy 7 and deletion 7q (-7/7q-).13 We did not find a significant correlation of the EZH2 mutational status with -7/7q, confirming other recent reports.15 However, as expected, patients with EZH2 allelic ratios >70% (n=15) (considered homozygous) were significantly more often affected by monosomy 7 (27%) compared to patients with heterozygous variants (4.7%) (P=0.002). In addition, short tandem repeat analysis of homozygous EZH2-mut patients without -7/7q showed that other chromosome 7 aberrations such as monoallelic 7q36.1 microdeletions and/or uniparental disomy may be the cause of EZH2 loss of heterozygosity (Online Supplementary Appendix).7,9 In the group of EZH2-mut AML patients, significantly higher rates of co-mutations were detected in RUNX1 (25%), ASXL1 (22%), and NRAS (25%) compared to EZH2-wt patients (with 10%, 8% and 15%, respectively) (Figure 1B and Online Supplementary Table S2). Vice versa, concomitant mutations in NPM1 were (non-significantly) more common in EZH2-wt patients (33%) versus EZH2-mut patients (21%). For other frequently mutated genes in AML, there was no major difference between EZH2-mut and -wt patients, e.g. FLT3ITD (13%), FLT3TKD (10%), CEBPAdm (11%), and CEBPAsm (24%), as well as genes encoding epigenetic modifiers, namely, DNMT3A (21%), IDH1/2 (11/14%), and TET2 (21%). An association of mutations in chromatin modifiers such as EZH2 and ASXL1 with mutations in spliceosome or transcription factors like RUNX1 corresponds to a distinct molecular cluster of co-occurring mutations frequently detected in high-risk MDS.10,11 This is in agreement with the relatively high rate of secondary AML in EZH2-mut patients in our cohort.
Table 1.
Patients’ characteristics.
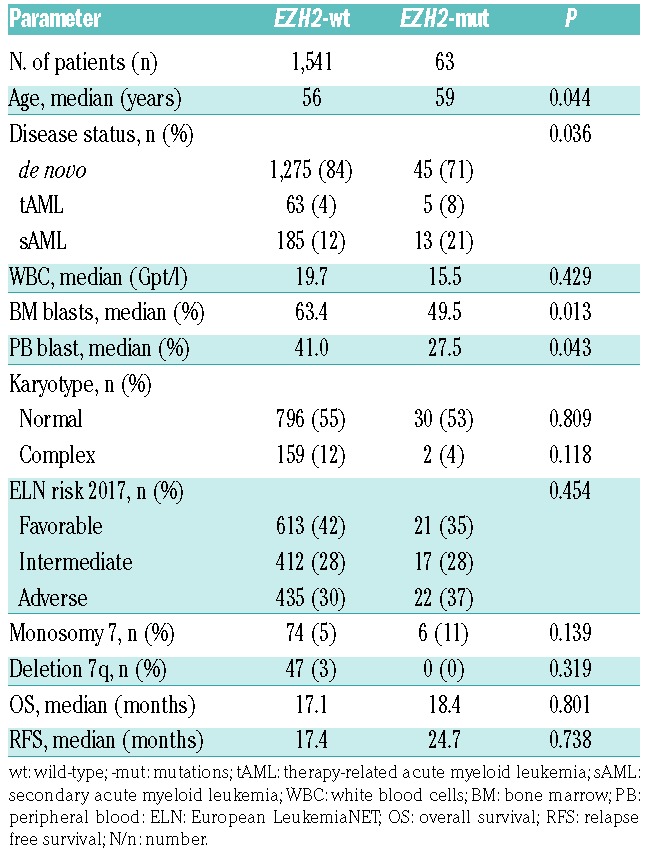
In univariate analyses, the EZH2 mutational status was not associated with the rate of complete remission (CR), relapse-free survival (RFS), or overall survival (OS), with a median OS of 18.4 and 17.1 months for EZH2-mut and –wt patients, respectively (Table 1 and Figure 2A) irrespective of the ELN risk group analyzed. Likewise, multivariate analysis with clinical variables such as age, cytogenetics and WBC using Cox proportional hazard regression revealed that EZH2 mutations were not an independent prognostic factor for OS or RFS. To address the impact of specific EZH2 mutations on clinical outcome in more detail, survival was next analyzed related to the allelic state (monoallelic vs. biallelic) of detected EZH2 variants, the affected functional domains (CXC-SET) and the predicted effect on the catalytic function of the EZH2 protein. There was a trend towards shorter median OS (12.55 vs. 15.61 months) and RFS (8.15 vs. 17.29 months) for patients with homozygous mutations compared to individuals with heterozygous (and subclonal) EZH2 variants, pointing to a potential prognostic impact of high allelic ratio EZH2 mutations in AML (Figure 2B). Likewise, inferior survival was previously associated with the presence of homozygous EZH2 mutations in MDS/MPN.7 Similarly, a slight but statistically not significant effect on OS was observed for patients with mutations in the catalytically active CXC-SET domains (12.4 months) versus patients with variants in other less conserved EZH2 regions (31.7 months), demonstrating an association of the affected functional domain with clinical outcome (Figure 2C). However, in all subgroups, no significant association with survival was observed compared to EZH2-wt patients. Interestingly, also no significant correlation with clinical outcome was observed for patients with EZH2 loss-of-function mutations (nonsense/frameshift) (Figure 2D), which are associated with poor prognosis in MDS/MPN.7 This indicates the importance of other pathogenic mechanisms affecting the epigenetic function of EZH2 in AML, such as the presence or absence of co-occuring driver mutations and/or post-translational modifications of the EZH2 protein.8,11 A more stratified assessment of individual EZH2 variants, patterns of associated co-mutation and functional consequences is warranted to fully understand the prognostic effect of EZH2 mutations in AML. Thus, taken together, EZH2 mutations are recurrent alterations in patients with AML. The association with certain clinical factors and typical mutations such as RUNX1 and ASXL1 points to the fact that EZH2 mutations are associated with sAML. However, in contrast to MDS, where EZH2 mutations are associated with poor prognosis, our data do not indicate that EZH2 mutations represent an independent prognostic factor in AML.
Figure 2.

Correlation of EZH2 mutational status with clinical outcome. Kaplan-Meier analysis showing overall survival of patients with (A) EZH2 -mutations (-mut) (n=63) versus wild type (–wt) (n=1,541) acute myeloid leukemia (B) homozygous (n=15) versus heterozygous (n=39) versus subclonal (n=9) EZH2 -mut (C) EZH2 mutations in affected functional (CXC-SET) (n=38) domains versus other EZH2 regions (n=25) and (D) EZH2 loss-of-function mutations (nonsense/frameshift) (n=24) versus missense mutations with unknown functional consequences (n=38).
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Gisslinger H, Gotic M, Holowiecki J, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood. 2013; 121(10):1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gillespie E. Anagrelide: a potent and selective inhibitor of platelet cyclic AMP phosphodiesterase enzyme activity. Biochem Pharmacol. 1988;37(14):2866–2868. [DOI] [PubMed] [Google Scholar]
- 3.Eric M. Mazur AGR, Patricia A. Sohl, Julie L. Newton, Amirthini Narendran. Analysis of the Mechanism of Anagrelide-Induced Thrombocytopenia in Humans. Blood. 1992;79(8):1931–1937. [PubMed] [Google Scholar]
- 4.Solberg LA, Jr, Oles KJ, Tarach JS, Petitt RM, Forstrom LA, Silverstein MN. The effects of anagrelide on human megakaryocytopoiesis. Br J Haematol. 1997;99(1):174–180. [DOI] [PubMed] [Google Scholar]
- 5.Tomer A. Effects of anagrelide on in vivo megakaryocyte proliferation and maturation in essential thrombocythemia. Blood. 2002; 99(5):1602–1609. [DOI] [PubMed] [Google Scholar]
- 6.Takayama N, Nishimura S, Nakamura S, et al. Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. J Exp Med. 2010; 207(13):2817–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamura S, Takayama N, Hirata S, et al. Expandable megakaryocyte cell lines enable clinically applicable generation of platelets from human induced pluripotent stem cells. Cell Stem Cell. 2014; 14(4):535–548. [DOI] [PubMed] [Google Scholar]
- 8.Ito Y, Nakamura S, Sugimoto N, et al. Turbulence Activates Platelet Biogenesis to Enable Clinical Scale Ex Vivo Production. Cell. 2018; 174(3):636–648. [DOI] [PubMed] [Google Scholar]
- 9.Hirata S, Murata T, Suzuki D, et al. Selective Inhibition of ADAM17 efficiently mediates glycoprotein Iba retention during ex vivo generation of human induced pluripotent stem cell-derived platelets. Stem Cells Transl Med. 2017;6(3):720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butcher L, Ahluwalia M, Ord T, et al. Evidence for a role of TRIB3 in the regulation of megakaryocytopoiesis. Sci Rep. 2017;7(1):6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisman R, Surrey S, Ramachandran B, Schwartz E, Poncz M. Structural and Functional Comparison of the Genes for Human Platelet Factor 4 and PF4alt. Blood. 1990;76(2):336–344. [PubMed] [Google Scholar]
- 12.Apostolidis PA, Lindsey S, Miller WM, Papoutsakis ET. Proposed megakaryocytic regulon of p53: the genes engaged to control cell cycle and apoptosis during megakaryocytic differentiation. Physiol Genomics. 2012;44(12):638–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Apostolidis PA, Woulfe DS, Chavez M, Miller WM, Papoutsakis ET. Role of tumor suppressor p53 in megakaryopoiesis and platelet function. Exp Hematol. 2012;40(2):131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou X, Qu M, Fang F, et al. Small Molecule Supplements Improve Cultured Megakaryocyte Polyploidization by Modulating Multiple Cell Cycle Regulators. Biomed Res Int. 2017;2017:2320519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahluwalia M, Butcher L, Donovan H, Killick-Cole C, Jones PM, Erusalimsky JD. The gene expression signature of anagrelide provides an insight into its mechanism of action and uncovers new regulators of megakaryopoiesis. J Thromb Haemost. 2015;13(6):1103–1112. [DOI] [PubMed] [Google Scholar]