

Abstract
Background
Cystic fibrosis is a life‐limiting autosomal recessive genetic illness. A feeling of shortness of breath is common in cystic fibrosis, especially as the disease progresses. Reversing the underlying cause is the priority when treating breathlessness (dyspnoea), but when it is not feasible, palliation (easing) becomes the primary goal to improve an individual's quality of life. A range of drugs administered by various routes have been used, but no definite guidelines are available. A systematic review is needed to evaluate such treatments.
Objectives
To assess the efficacy and safety of drugs used to ease breathlessness in people with cystic fibrosis.
Search methods
We searched the Cochrane Cystic Fibrosis Trials Register, compiled from electronic database searches and handsearching of journals and conference abstract books. Date of last search: 18 November 2019.
We searched databases (clinicaltrials.gov, the ISRCTN registry, the Clinical Trials Registry India and WHO ICTRP) for ongoing trials. These searches were last run on 06 March 2020.
Selection criteria
We planned to include randomised and quasi‐randomised controlled trials in people with cystic fibrosis (diagnosed by a positive sweat chloride test or genetic testing) who have breathlessness. We considered studies comparing any drugs used for easing breathlessness to another drug administered by any route (inhaled (nebulised), intravenous, oral, subcutaneous, transmucosal (including buccal, sublingual and intra‐nasal) and transdermal).
Data collection and analysis
The authors assessed the search results according to the pre‐defined inclusion criteria.
Main results
The new searches in 2020 yielded two ongoing studies that were not relevant to the review question. Previous searches had found only one study (cross‐over in design), which did not fulfil the inclusion criteria as no data were available from the first treatment period alone.
Authors' conclusions
Due to the lack of available evidence, this review cannot provide any information for clinical practice. The authors call for specific research in this area after taking into account relevant ethical considerations. The research should focus on the efficacy and safety of the drugs with efficacy being measured in terms of improvement in quality of life, dyspnoea scores and hospital stay.
Plain language summary
Drug treatments to ease breathlessness in cystic fibrosis
Review question
What is the evidence that drug treatments for breathlessness (also known as dyspnoea) in people with cystic fibrosis are effective and safe?
Background
Cystic fibrosis is a life‐limiting genetic disease. As the disease progresses, thick sticky mucus is formed in the lungs, pancreas and other organs. This thick mucus increases risk of infection, blocks the airways and causes severe breathlessness. Easing of this breathlessness is an important goal for care of people with cystic fibrosis. Many methods have been suggested to address this problem and many drugs have been reported to ease breathlessness. The drugs can be administered through various routes ‐ they might be inhaled, swallowed, injected under the skin, into muscle or into a vein or be absorbed through the skin (either on the body or through membranes inside the mouth or nose). Despite so many drugs being available, there are no defined guidelines for drug treatments aimed at easing breathlessness in people with cystic fibrosis.
Search date
The evidence from the Cochrane Cystic Fibrosis Trials Register is current to 18 November 2019; the searches of ongoing trial registries are current to 06 March 2020.
Study characteristics
We had planned to include studies comparing different treatments to ease breathlessness in people with cystic fibrosis where the participants were put into the different treatment groups at random. Our search found only one study in which seven people first received either a drug called hydrocodone or a placebo (dummy treatment) and then swapped to the other treatment; but this study did not report results separately for each treatment arm, so we are not able to include it in the review.
Key results
No studies fulfilled the inclusion criteria, therefore it is not possible to comment on the outcomes we wished to report on in this review. Until evidence becomes available, the review authors feel that it is advisable for doctors to follow any local or national guidelines. The authors recommend research in this area should take ethical considerations into account and should focus on the safety of the drugs, improvement in quality of life, breathlessness scores and length of hospital stay.
Background
Description of the condition
Cystic fibrosis (CF) is a life‐limiting autosomal recessive genetic illness. In the past 70 years, the survival of people with CF has improved substantially due to advances in medical care, both in diagnostic techniques and in the pharmacological and non‐pharmacological treatment of the condition (Dasenbrook 2012). Before the introduction of widespread screening programmes, CF was diagnosed by the age of two years in more than 70% of individuals with the condition, but now it can be diagnosed very soon after birth (median age of four months) (CF Foundation 2013; Kleven 2008). The predicted median age of survival for a person with CF has increased progressively; in the UK it was recently reported as being 43.5 years (UK CF Trust 2014), while in the USA it was reported to be over 41 years (CF Foundation 2013). However, according to the WHO, globally the median survival is 33.5 years (WHO 2015). Dodge reported that median survival will be over 50 years of age for those born after 2000 (Dodge 2007). More than 49% of people with CF are aged 18 years or older (CF Foundation 2013) and the UK CF Registry states that more than 58% of people with CF are over 16 years of age (UK CF Trust 2014). The global incidence of CF is varied. It is severely under‐diagnosed in Asia, while in the European Union it is estimated at 1 in 2000 to 3000 births and in the USA it is estimated as 1 in 3500 births (WHO 2015). Approximately 1000 new cases of CF are diagnosed each year in the USA (CF Foundation 2010b; O'Sullivan 2009).
The abnormal CF gene and its mutated protein lead to thick and sticky mucus in the airways (Torphy 2009). An absence of the cystic fibrosis transmembrane regulator (CFTR) at the surface of airway epithelium decreases chloride secretion and increases sodium absorption, leading to water depletion from the airway surface; this results in impaired mucociliary clearance which favours infections, due to bacterial colonisations like Staphylococcus aureus, Pseudomonas aeruginosa, Burkholderia cepacia complex, etc. (Gautam 2015), and inflammation, thereby causing lung destruction (Dasenbrook 2012). In people with CF, thick, sticky mucus forms in the lungs, pancreas, and other organs. In the lungs, this mucus blocks the airways, making it hard to breathe and leading to serious lung infections and lung damage (CF Foundation 2010a). The natural history of CF is a progressive decline in lung function secondary to chronic airway infection. Respiratory failure is the most common cause of death. In addition to nausea, pain and anxiety, breathlessness (also known as dyspnoea) is a distressing symptom requiring medical management, especially in the terminal phase of CF (Sands 2011). Contemporary theories suggest that breathlessness radiates from multiple sensory inputs in the brain, and evokes a response i.e. sending motor signals or nerve impulses to both the breathing muscles and to the sensory cortex (corollary discharge) (Killian 2005; Peters 2006). The release of these signals or impulses from the respiratory centres in the brainstem represents a complex processing and integration of multiple inputs, which include receiving information regarding central and peripheral chemoreceptor stimulation, forced displacement in the chest wall, lung stretch and information from higher levels in the brain (Killian 2005).
A feeling of shortness of breath is common in people with CF and palliation (easing) becomes the primary goal as the disease progresses. In the later stages of the disease, people with CF also experience increased respiratory exacerbations and oxygen dependence.
Description of the intervention
The easing of breathlessness is a reasonable goal to improve comfort as the disease progresses. Severe breathlessness is one of the most troubling aspects of CF for many clinicians and patients (Robinson 2000). A recent review reported an improvement in the quality of life (QoL) in people with chronic breathlessness using a combination of (mostly) non‐drug and drug interventions. These interventions also improve clinical outcomes and reduce patient suffering and futile use of medical services (Booth 2019). A range of drugs administered by various routes have been used, but no definite guidelines are available. Drugs such as morphine and fentanyl have been tried and proved useful in relieving breathlessness (Booth 2009; Cohen 2002; Graff 2004; Janahi 2000; Kallet 2007), in addition to their use in palliative sedation (Palliative Sedation Protocol 2004; Sands 2011). Intravenous morphine of less than 5 mg/hour has been shown to be sufficient in relieving breathlessness in over 50% of people with CF (Robinson 2000). Anxiolytic agents depress the hypoxic or hypercapnic ventilatory responses and thus relieve breathlessness, but also alter emotional responses related to breathlessness. Other drugs classified as mucolytics (e.g. dornase alfa and nebulised hypertonic saline), inhaled anaesthetics (e.g. lidocaine), inhaled beta‐2 adrenergic agonists and inhaled anti‐cholinergics have also been used for easing breathlessness in CF (ATS 1999; Parshall 2012).
How the intervention might work
Drug treatments for easing breathlessness can be inhaled or administered through intranasal, oral, subcutaneous, transdermal, buccal, intravenous or intra‐muscular routes (Simon 2012).
Opiates and inhaled anaesthetic agents decrease the perceptual responses and thus lessen the intensity of respiratory sensation (Hayes 2010; Jennings 2002). The exact mechanism by which opioids alleviate breathlessness is still not known. However, suggested mechanisms of action include reducing the effects of arterial carbon dioxide (pCO₂) levels and oxygen (pO₂) levels on ventilation, reducing oxygen consumption as well as altering the individual's perception of breathlessness (ATS 1999; Pattinson 2009).
In contrast, anxiolytic agents depress the hypoxic or hypercapnic ventilatory responses and thus relieve breathlessness, but also alter emotional responses related to breathlessness (Tait 2014). These drugs include clonazepam, which has a long half‐life allowing administration as a subcutaneous bolus once or twice a day and midazolam which has a short half‐life and therefore requires a syringe driver or frequent subcutaneous administration to provide the equivalent outcome. Both sublingual and subcutaneous clonazepam have a quick onset of action, making clonazepam suitable for breakthrough symptoms (Tait 2014).
Mucolytics like dornase alfa and N‐acetyl cysteine act by either reducing the viscosity or adhesive properties of the mucus, which helps in easing the breathlessness (Henke 2007). Inhaled hypertonic saline acts by hydrating the thick mucus present and helps to re‐establish the deficient aqueous surface layer which is absent in people with CF (Donaldson 2006). The inhaled anticholinergic agents when administered induce bronchodilation and relieve breathlessness (Weinberger 2002).
Why it is important to do this review
A comparison of the various drug treatments may help in finding better options for the amelioration of the symptoms of breathlessness in people with CF. As there are no defined guidelines for the palliation or easing of breathlessness in CF or the evaluation of such treatments, a systematic review is required to identify the best drug treatment. This is an update of an already published review (Jaiswal 2017).
Objectives
To assess the efficacy and safety of drugs used to ease breathlessness in people with CF.
Methods
Criteria for considering studies for this review
Types of studies
In this systematic review, it was planned to include randomised controlled trials (RCTs) and quasi‐RCTs. We had also planned only to include cross‐over studies, if first‐arm data were available.
Types of participants
People with CF (diagnosed by a positive sweat chloride test or genetic testing) of either sex, of any age group and experiencing breathlessness.
Types of interventions
Comparisons of pharmacological interventions for dyspnoea in CF were eligible for inclusion. We planned to compare different drug classes used to treat breathlessness in CF.
In a post hoc change, while extending the review from end‐stage to any stage, we decided to also include studies of pharmacological interventions compared with placebo.
Types of outcome measures
Primary outcomes
Breathlessness (dyspnoea) score (for severity of breathlessness) as measured by a validated scoring system, e.g. modified Borg score (Hommerding 2010), a visual analogue scale (VAS) (Gift 1989), numerical rating scales (NRS), the Chronic Respiratory Disease Questionare (CRQ), Medical Research Council Scale (MRC) (Bausewein 2008)
Number of episodes of breathlessness
Time to next episode of breathlessness
Secondary outcomes
QoL (measured by a validated scoring system e.g. Cystic Fibrosis Questionnaire‐Revised (CFQ‐R) (Quittner 2009))
Need for change of therapy or addition of therapy
-
Hospitalization
number of visits to emergency department
duration of hospital stay
-
Adverse events (e.g. diarrhoea, headache, vomiting, etc.)
mild
severe (e.g. hospitalizations due to adverse drug reaction or stopping of a particular drug due to adverse reaction)
Blood oxygen saturation (SpO₂) levels measured as a continuous variable
Search methods for identification of studies
We searched for all relevant published and unpublished trials without restrictions on language, year or publication status.
Electronic searches
We searched for relevant studies from the Group's Cystic Fibrosis Trials Register using the term: dyspnea.
The Cystic Fibrosis Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of The Cochrane Library), weekly searches of MEDLINE, a search of Embase to 1995 and the prospective handsearching of two journals ‐ Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work is identified by searching the abstract books of three major cystic fibrosis conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the relevant sections of the Cochrane Cystic Fibrosis and Genetic Disorders Group website.
Date of most recent search: 18 November 2019.
We also searched for ongoing trials in the following databases using the terms 'cystic fibrosis' AND 'dyspnoea' AND 'pharmacotherapy' for all years:
Clinical Trials Registry India (CTRI) (ctri.nic.in/Clinicaltrials/login.php);
clinicaltrials.gov (clinicaltrials.gov);
the International Standard Randomized Controlled Trial Number (ISRCTN) registry (www.isrctn.org/);
WHO ICTRP (apps.who.int/trialsearch/).
Date of the most recent search: 06 Mar 2020.
Searching other resources
We had also planned to search the reference lists of the retrieved studies to identify other potentially eligible studies.
Data collection and analysis
Selection of studies
Two authors independently reviewed the abstract of the only study identified by the literature searches. If more studies had been identified, two authors would have separated them as excluded or not excluded following the initial review of title and abstract. Two authors would then have independently screened the full texts (if available) of the studies labelled as not excluded against the inclusion criteria and finally decided on the inclusion of the studies as per the Cochrane Handbook of Systematic Reviews of Interventions (Higgins 2011a). The authors would have resolved discrepancies by discussion with the arbiter (MS).
Data extraction and management
If any studies are included in future updates, the authors will independently extract the data from the included studies on separate study report forms. They will resolve any disagreements through discussion. If needed, they will request further information from the study authors (Higgins 2011b). Authors will then enter the extracted data into Review Manager (RevMan) software for analysis (Review Manager 2014)
Assessment of risk of bias in included studies
If any studies are included in future updates, the review authors will assess these for risks of bias using the Cochrane risk of bias tool (Higgins 2011c). The tool assesses the biases introduced through inadequacies in random sequence generation, allocation concealment, blinding of participants and study personnel, blinding of outcome assessments, reporting of incomplete outcome data and selective reporting. The review authors will judge each of these criteria to have a low, unclear or high risk of bias. The risk of bias assessment will follow the same process as data extraction.
Measures of treatment effect
If, in future, the review authors are able to report results for dichotomous outcomes (adverse events, the need for change of therapy, increased tolerance to activities), they will use the risk ratios (RR) with 95% confidence intervals (CIs) as the effect measure. For continuous outcome data (breathlessness score, duration of period free from breathlessness, QoL scores, number of visits to emergency department, duration of hospital stay, SpO₂ levels), they will calculate the mean difference (MD) with 95% CIs in accordance with the Cochrane Handbook of Systematic Reviews of Intervention (Deeks 2011). The authors only plan to pool the QoL results using the MD with 95% CIs if a standardized scale or the same scale is used across the studies. If this is not the case, they will use the standardized mean difference (SMD) with 95% CIs (Deeks 2011). The authors plan to analyse data for the frequency of attacks and the number of admissions as dichotomous data and report the rate ratios using the generic inverse variance method as described in the Cochrane Handbook of Systematic Reviews of Interventions (Deeks 2011; Higgins 2011a; Higgins 2011b). The authors plan to present data at the final time point reported in the studies (for cross‐over studies, they will present data from the first arm only).
Unit of analysis issues
For any cross‐over studies included in future updates of this review, the authors will use only first‐arm data as participants may not recover to baseline and, in addition to introducing a unit of analysis error, multiple treatment episodes may not be comparable (Elbourne 2002).
Dealing with missing data
If there appear to be any data missing from studies the authors include in future updates of the review, they will contact the study authors for these. If this is not possible, for continuous variables where standard deviations (SDs) are not published, the authors will impute the missing SD using a correlation co‐efficient (Higgins 2011d). Where possible, the authors will perform intention‐to‐treat analyses.
Assessment of heterogeneity
If the authors include studies in future updates of the review, they will assess the forest plot visually for heterogeneity. They will also compute the statistical heterogeneity in the included studies using the Chi² test (considered significant when corresponding P ≤ 0.10) with which a calculation of I² statistic is planned (Higgins 2003). The authors will interpret this value based on thresholds as identified in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011):
0% to 40% as probably not important;
30% to 60% as moderate heterogeneity;
50% to 90% as substantial heterogeneity; and
75% to 100% as considerable heterogeneity.
Assessment of reporting biases
The review authors will contact the authors of any studies included in future updates of the review in an attempt to identify unpublished studies. They also plan to contact the authors of any potentially relevant studies published only in abstract form for additional data. The authors will attempt to identify evidence of outcome reporting biases by comparing the published reports to the study protocols, if these are available. If protocols are not available, the review authors will compare the 'Methods' section to the 'Results' section of the published paper to identify any outcomes that were measured but not reported. If they include and combine sufficient studies (n = 10), they will make further attempts to identify reporting biases by constructing and inspecting funnel plots; they will interpret these in the context of study sizes and methodological rigour (Sterne 2011).
Data synthesis
If the authors include studies in future updates of the review, they will pool the outcome measures using a fixed‐effect method for meta‐analysis. However, if they identify at least substantial heterogeneity, as defined above, they will use the random‐effects method for meta‐analysis.
Subgroup analysis and investigation of heterogeneity
If, in future, the authors are able to combine data from two or more studies and find moderate to considerable heterogeneity between the studies (as defined by I² ranges above (Assessment of heterogeneity)), they will undertake subgroup analyses based on age (less than 18 years versus 18 years and above) and according to the drug used, e.g. nebulized morphine or nebulized dornase alfa. If they find a sufficient number of studies for any given drug (i.e. two or more), they will consider a dose‐specific subgroup analysis.
Sensitivity analysis
If, in the future, the authors are able to combine data from a sufficient number of studies, they will investigate the effect of decisions about missing data and risk of bias judgements made by the review team by undertaking sensitivity analyses of the affected components. If they manage missing data by imputing values, they will investigate the effect of these manipulations by repeating the analyses without these imputations. If they include studies with a high risk of bias in the analysis, they will repeat the analysis using only studies with a low risk of bias.
Results
Description of studies
The searches for the 2020 update found two results for ongoing trials but they were not relevant to the current systematic review. Previous searches had only one study which was excluded as it did not fit the inclusion criteria for the review (Figure 1).
1.
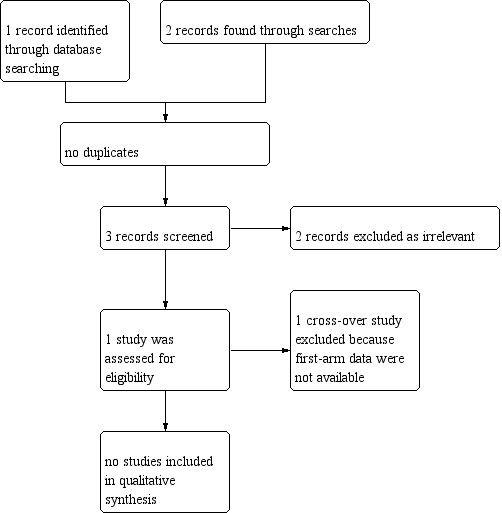
Study flow diagram.
Included studies
No studies were included in the review.
Excluded studies
Only one study was found after searching various databases (Browning 1988). This study was only available as an abstract and was excluded since it was a cross‐over study that did not present separate first‐arm data.
Please also refer to the table 'Characteristics of excluded studies'.
Risk of bias in included studies
There were no studies included in the review.
Effects of interventions
There were no studies included in the review.
Discussion
Summary of main results
There were no randomised controlled trials (RCTs) identified which fulfilled the review's inclusion criteria. The only study found in the searches compared the effect of an active intervention (hydrocodone) to placebo for dyspnoea in adults with cystic fibrosis (CF). The study was cross‐over in design, but did not report data separately for the different treatment arms.
Agreements and disagreements with other studies or reviews
The review found that there is lack of studies discussing palliative drug treatments for breathlessness in people with CF. The available literature is in the form of case studies, which need to be assessed when making clinical decisions (Cohen 2002; Graff 2004; Hayes 2010; Janahi 2000). The excluded study was cross‐over in design and compared hydrocodone to placebo (Browning 1988). Results reported at the end of the study showed improvement improvement in exercise performance with hydrocodone, although the sample size was very small. Recent studies have stressed the importance of advance care planning (ACP) (Dellon 2016; Madge 2016). Palliative treatment for breathlessness is therefore more important as it is a part of ACP. There is a need for better palliative care training and education to health professionals involved in caring for people with CF (Goggin 2016; Linnemann 2015). As ACP involves a discussion of various available treatment options, it is very important to have comparative information regarding the drug therapies for breathlessness.
A recently published letter concluded that opioids help in improving treatment of breathlessness (Ekstorm 2017). There are also two other Cochrane Reviews which compare systemic opioids to placebo for treating chronic breathlessness and have shown benefits for relieving breathlessness with low quality evidence (Barnes 2016; Ekstorm 2015). One of these reviews compared systemic opioids to placebo in any advanced disease and in adults with terminal illnesses (Barnes 2016), while the second review discussed the same comparison specifically in people with chronic obstructive pulmonary disease (COPD) (Ekstorm 2015). This Cochrane Review aims to compare not just systemic opioids, but all possible pharmacotherapies that may be used to relieve breathlessness in people with CF.
Authors' conclusions
Implications for practice.
There is no randomised evidence to inform guidelines, although breathlessness is an important symptom that affects the quality of life of people with CF and deserves treatment. Currently the only evidence available to clinicians to inform treatment choices for managing breathlessness in people with CF is to refer to case series and reports and any available national or local recommendations, for example from Nottingham Children's Hospital (UK) (Bhatt 2015).
Implications for research.
The fact that the search could not find any relevant randomised controlled trials for inclusion in this review, highlights the need for research in this area. Relevant ethical considerations should be taken into account when planning a randomised controlled trial.
The research should focus on various drug treatments available for easing breathlessness in people with CF and assess the safety, efficacy and effectiveness in terms of breathlessness score, quality of life, hospital stay and adverse events. This will enable a transparent way of choosing a treatment option and facilitate the involvement of individuals and families in decision making.
What's new
| Date | Event | Description |
|---|---|---|
| 28 April 2020 | New citation required but conclusions have not changed | One author (Kiran K Thumburu) has stepped down from the review team and two new authors have joined the team (Anil Chauhan and Nikita Jaiswal). Since no new trials have been added to the review at this update our conclusions remain the same. |
| 28 April 2020 | New search has been performed | A search of the Cochrane Cystic Fibrosis and Genetic Disorders Cystic Fibrosis Trials Register did not identify any new references potentially eligible for this updated review. Searches of online trials registries yielded two ongoing studies that were not relevant to the review question. |
History
Protocol first published: Issue 9, 2015 Review first published: Issue 8, 2017
Acknowledgements
ICMR Advanced Centre for Evidence‐based Child Health, PGIMER, Chandigarh, India.
The Cochrane Cystic Fibrosis and Genetic Disorders Review Group and Nikki Jahnke (Managing Editor) for helping with and co‐ordinating this review.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Glossary
| Term | Explanation |
| Adrenergic agonist | A drug that stimulates a response from the adrenergic receptors. |
| Adrenergic receptors (or adrenoceptors) | A class of G protein‐coupled receptors that are targets of the catecholamines, especially norepinephrine (noradrenaline) and epinephrine (adrenaline). There are three types of adrenergic receptors: alpha, beta 1, and beta 2. Alpha‐receptors are found in the arteries and when they are stimulated by epinephrine or norepinephrine, the arteries constrict. This increases the blood pressure and the blood flow returning to the heart. Beta 1 receptors are located in the heart and when they are stimulated, they increase the heart rate and increase the heart's strength of contraction or contractility. Beta 2 receptors are found in the lungs and the arteries of the skeletal muscles. When these receptors are stimulated, they increase the diameter of the airways to let more air in and out during breathing and they widen the vessels of the skeletal muscles so they can receive the increased blood flow produced by stimulating the alpha and beta 1 receptors. |
| Afferent | Conducted inwards or towards something. |
| Anti‐cholinergics | A class of drugs that affects the muscles around the bronchi (large airways). When the lungs are irritated, these bands of muscle can tighten, making the bronchi narrower; anticholinergics work by stopping the muscles from tightening. |
| Anxiolytic | Used to reduce anxiety. |
| Bolus | Administration of a discrete amount of medication, drug or other compound in order to raise its concentration in blood to an effective level. |
| Buccal | Relating to cheek |
| Chemoreceptor | A sensory nerve cell or sense organ, as of smell or taste, that responds to chemical stimuli. |
| Corollary discharge | A copy of a motor command that is sent to the muscles to produce a movement; this copy or corollary does not produce any movement itself but instead is directed to other regions of the brain to inform them of the impending movement. |
| Correlation co‐efficient | This number (between ‐1 & 1) measures the strength of association between two variables. |
| Cross‐over trial | A type of clinical trial comparing two or more treatments where when participants complete the course of one treatment they are switched to another. For example, in a comparison of treatments A and B, half the participants are randomly allocated to receive them in the order A, B and half to receive them in the order B, A. One problem with this design is that the effects of the first treatment may carry over into the period when the second is given. |
| Dichotomous data | Data with two possible categories only, e.g. yes/no, male/female, etc. |
| Dyspnoea | Difficult or laboured breathing. |
| Efferent | Conducted outwards or away from something. |
| Forest plot | A forest plot displays effect estimates and confidence intervals for both individual studies and meta‐analyses |
| Funnel plot | A funnel plot is a simple scatter plot of the intervention effect estimates from individual studies against some measure of each study’s size or precision. |
| Half‐life | The time required for any specified property (e.g. the concentration of a substance in the body) to decrease by half. |
| Hypercapnia | Also known as hypercarbia; a condition where there is an increased amount of carbon dioxide, the waste product of breathing, in the blood. |
| Hypoxia | A deficiency in the amount of oxygen reaching the tissues. |
| Impute | The process of replacing missing data with substituted values |
| Palliation | A kind of care that eases symptoms of disease, even though it can't cure them. |
| Pharmacotherapy | The treatment of diseases with drugs. |
| Quasi‐RCT | A trial using a method of allocating participants to different treatments that is not truly random, for example, allocation by date of birth, day of the week, medical record number, month of the year, or the order in which participants are included in the study. |
| Sublingual | Situated or applied under the tongue. |
Characteristics of studies
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Browning 1988 | This was a small cross‐over study where first‐arm data were not available. |
Differences between protocol and review
The authors have removed the term 'end‐stage' from the title of the review. While this alters the scope of the review which was originally focused on end‐stage disease, we are aware that treatments previously seen as palliative are now used earlier rather than waiting until end of life and can be used simultaneously with therapies designed to prolong life.
It was also clinically relevant to include comparisons of any pharmacological interventions to placebo, hence this possible comparison was also added as a post hoc change.
Following statistical advice, it was decided to present any results for dichotomous outcomes using the risk ratio rather than the odds ratio.
Contributions of authors
| Roles and responsibilities | |
| TASK | WHO WILL UNDERTAKE THE TASK? |
| Protocol stage: draft the protocol | NJ, MS, AA, KKT |
| Review stage: select which trials to include (2 + 1 arbiter) | NJ, KKT, MS |
| Review stage: extract data from trials (2 people) | NJ, AA |
| Review stage: enter data into RevMan | NJ |
| Review stage: carry out the analysis | NJ, MS, AA, KKT |
| Review stage: interpret the analysis | NJ, MS, AA, KKT |
| Review stage: draft the final review | NJ, MS, AA, KKT |
| Update stage: update the review | NJ, MS, AA, AC, NJ |
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
All authors: none known.
New search for studies and content updated (no change to conclusions)
References
References to studies excluded from this review
Browning 1988 {published data only}
- Browning I, D'Alonzo GE, Tobin MJ. Effect of hydrocodone on dyspnea, respiratory drive, and exercise performance in adult patients with cystic fibrosis. American Review of Respiratory Disease 1988;137(Suppl):305. [CFGD REGISTER: CO41] [CN-00208153] [Google Scholar]
Additional references
ATS 1999
- American Thoracic Society. Dyspnoea: mechanisms, assessment and management: a consensus statement. American Journal of Respiratory and Critical Care Medicine 1999;159(1):321-40. [DOI] [PubMed] [Google Scholar]
Barnes 2016
- Barnes H, McDonald J, Smallwood N, Manser R. Opioids for the palliation of refractory breathlessness in adults with advanced disease and terminal illness. Cochrane Database of Systematic Reviews 2016, Issue 3. [DOI: 10.1002/14651858.CD011008.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bausewein 2008
- Bausewein C, Booth S, Higginson IJ. Measurement of dyspnoea in the clinical rather than the research setting. Current Opinion in Supportive and Palliative Care 2008;2(2):95-9. [DOI] [PubMed] [Google Scholar]
Bhatt 2015
- Bhatt J, Smyth A, Bertenshaw C. Nottingham Children's Hospital. Guideline for the management of patients with cystic fibrosis. www.nuh.nhs.uk/handlers/downloads.ashx?id=61166 (accessed prior to 26 June 2017).
Booth 2009
- Booth S, Bausewein C, Higginson I, Moosavi SH. Pharmacological treatment of refractory breathlessness. Expert Review of Respiratory Medicine 2009;3(1):21-36. [DOI] [PubMed] [Google Scholar]
Booth 2019
- Booth S, Johnson MJ. Improving the quality of life of people with advanced respiratory disease and severe breathlessness. Breathe (Sheff) September 2019;15(3):198-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
CF Foundation 2010a
- Cystic Fibrosis Foundation. Hope in our hands. www.cff.org/UploadedFiles/aboutCFFoundation/AnnualReport/2010-Annual-Report.pdf (accessed 16 June 2014).
CF Foundation 2010b
- Cystic Fibrosis Foundation. About cystic fibrosis: what you need to know. www.cff.org/What-is-CF/About-Cystic-Fibrosis/ (accessed 16 June 2014).
CF Foundation 2013
- Cystic Fibrosis Foundation. 2012 Annual Report. More tomorrows today. www.cff.org/About-Us/Assets/2012-Annual-Report/ (accessed 16 June 2014).
Cohen 2002
- Cohen SP, Dawson TC. Nebulized morphine as a treatment for dyspnoea in a child with cystic fibrosis. Pediatrics 2002;110(3):e38. [DOI] [PubMed] [Google Scholar]
Dasenbrook 2012
- Dasenbrook EC, Konstan MW. Inhaled hypertonic saline in infants and young children with cystic fibrosis. JAMA 2012;307(21):2316-7. [DOI: 10.1001/jama.2012.5853] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JP, Altman DG, editor(s) on behalf of the Cochrane Statistical Methods Group. Chapter 9: Analysing data and undertaking meta-analysis. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Dellon 2016
- Dellon EP, Chen E, Goggin J, Homa K, Marshall BC, Sabadosa KA, et al. Advance care planning in cystic fibrosis: current practices, challenges, and opportunities. Journal of Cystic Fibrosis 2016;15(1):96-101. [DOI] [PubMed] [Google Scholar]
Dodge 2007
- Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947-2003. European Respiratory Journal 2007;29(3):522-6. [DOI] [PubMed] [Google Scholar]
Donaldson 2006
- Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. New England Journal of Medicine 2006;354(3):241. [DOI] [PubMed] [Google Scholar]
Ekstorm 2015
- Ekström M, Nilsson F, Abernethy AA, Currow DC. Effects of opioids on breathlessness and exercise capacity in chronic obstructive pulmonary disease. A systematic review. Annals of American Thoracic Society 2015;12(7):1079-92. [DOI] [PubMed] [Google Scholar]
Ekstorm 2017
- Ekström M, Bajwah S, Bland JM, Currow DC, Hussain J, Johnson MJ. One evidence base; three stories: do opioids relieve chronic breathlessness? Thorax 2018;73(1):88-90. [DOI: 10.1136/thoraxjnl-2016-209868] [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta-analyses involving cross-over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140-9. [DOI] [PubMed] [Google Scholar]
Gautam 2015
- Gautam V, Shafiq N, Singh M, Ray P, Singhal L, Jaiswal NP, et al. Clinical and in vitro evidence for the antimicrobial therapy in Burkholderia cepacia complex infections. Expert Review of Anti-infective therapy 2015;13(5):629-63. [DOI] [PubMed] [Google Scholar]
Gift 1989
- Gift AG. Validation of a vertical visual analogue scale as a measure of clinical dyspnea. Rehabilitation Nursing 1989;14(6):323-5. [DOI] [PubMed] [Google Scholar]
Goggin 2016
- Goggin J, Cohen RI. CF healthcare workers feel unprepared in providing suitable end of life care and desire more education: results of a nationwide survey. Journal of Cystic Fibrosis 2016;15(1):85-9. [DOI] [PubMed] [Google Scholar]
Graff 2004
- Graff GR, Stark JM, Grueber R. Nebulized fentanyl for palliation of dyspnoea in a cystic fibrosis patient. Respiration 2004;71(6):646-9. [DOI] [PubMed] [Google Scholar]
Hayes 2010
- Hayes D Jr, Anstead MI, Warner RT, Kuhn RJ, Ballard HO. Inhaled morphine for palliation of dyspnea in end-stage cystic fibrosis. American Journal of Health-system Pharmacy 2010;67(9):737-40. [DOI] [PubMed] [Google Scholar]
Henke 2007
- Henke MO, Ratjen F. Mucolytics in cystic fibrosis. Paediatric Respiratory Reviews 2007;8:24-29. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327(7414):557-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JP, Deeks JJ, editor(s). Chapter 7: Selecting studies and collecting data. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011c
- Higgins JP, Altman DG, Sterne JA, editors(s) on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group. Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011d
- Higgins JP, Deeks JJ, Altman DG, editor(s) on behalf of the Cochrane Statistical Methods Group. Chapter 16: Special topics in statistics. In: Higgins JP, Green S, editor(s). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hommerding 2010
- Hommerding PX, Donadio MV, Paim TF, Marostica PJ. The Borg scale is accurate in children and adolescents older than 9 years with cystic fibrosis. Respiratory Care 2010;55(6):729-33. [PubMed] [Google Scholar]
Janahi 2000
- Janahi IA, Maciejewski SR, Teran JM, Oermann CM. Inhaled morphine to relieve dyspnea in advanced cystic fibrosis lung disease. Pediatric Pulmonology 2000;30(3):257-9. [DOI] [PubMed] [Google Scholar]
Jennings 2002
- Jennings AL, Davies AN, Higgins JP, Gibbs JS, Broadley KE. A systematic review of the use of opioids in the management of dyspnoea. Thorax 2002;57(11):939-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kallet 2007
- Kallet RH. The role of inhaled opioids and furosemide for the treatment of dyspnea. Respiratory Care 2007;52(7):900-10. [PubMed] [Google Scholar]
Killian 2005
- Killian K. Dyspnea: mechanisms, measurement and management. In: Mahler DA, O’Donnell DE, editors(s). History of dyspnoea. 2nd edition. Boca Raton: Taylor & Francis, 2005:1–18.10. [Google Scholar]
Kleven 2008
- Kleven DT, McCudden CR, Willis MS. Cystic fibrosis: newborn screening in America. Medical Laboratory Observer 2008;40(7):16-27. [PubMed] [Google Scholar]
Linnemann 2015
- Linnemann RW, O'Malley PJ, Friedman D, Georgiopoulos AM, Buxton D, Altestien LL, et al. Development and evaluation of a palliative care curriculum for cystic fibrosis healthcare providers. Journal of Cystic Fibrosis 2015;15(1):90-5. [DOI] [PubMed] [Google Scholar]
Madge 2016
- Madge S, Sands D. End of Life: Have we got it right? Journal of Cystic Fibrosis 2016;15(1):2-3. [DOI] [PubMed] [Google Scholar]
O'Sullivan 2009
- O'Sullivan BP, Freedman SD. Cystic fibrosis. Lancet 2009;373(9678):1891-904. [DOI: 10.1016/S0140-6736(09)60327-5] [DOI] [PubMed] [Google Scholar]
Palliative Sedation Protocol 2004
- Hospice and Palliative Care Federation of Massachusetts. Palliative sedation protocol: A report of the Standards and Best Practices Committee Hospice & Palliative Care Federation of Massachusetts April 2004. c.ymcdn.com/sites/www.hospicefed.org/resource/resmgr/hpcfm_pdf_doc/pal_sed_protocol_2004.pdf (accessed 16 June 2014).
Parshall 2012
- Parshall MB, Schwartzstein RM, Adams L, Banzett RB, Manning HL, Bourbeau J, et al. An official American Thoracic Society statement: update on the mechanisms, assessment, and management of dyspnoea. American Journal of Respiratory and Critical Care Medicine 2012;185(4):435-52. [DOI: 10.1164/rccm.201111-2042ST] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pattinson 2009
- Pattinson KT, Governo RJ, MacIntosh BJ, Russell EC, Corfield DR, Tracey I, et al. Opioids depress cortical centers responsible for the volitional control of respiration. Journal of Neuroscience 2009;29(25):8177-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Peters 2006
- Peters MM, O'Donnell DE. Dyspnoea in chronic obstructive pulmonary disorder. In: Booth S, Dudgeon D, editors(s). Dyspnoea in advanced disease:a guide to clinical management. Vol. 74. Oxford: Oxford University Press, 2006. [Google Scholar]
Quittner 2009
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135(6):1610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Robinson 2000
- Robinson W. Palliative care in cystic fibrosis. Journal of Palliative Medicine 2000;3(2):187-92. [DOI] [PubMed] [Google Scholar]
Sands 2011
- Sands D, Repetto T, Dupont LJ, Eksterowicz K, Catastini P, Madge S. End of life care for patients with cystic fibrosis. Journal of Cystic Fibrosis 2011;10(Suppl 2):S37-S44. [DOI] [PubMed] [Google Scholar]
Simon 2012
- Simon ST, Niemand AM, Benalia H, Voltz R, Higginson IJ, Bausewein C. Acceptability and preferences of six different routes of drug application for acute breathlessness: a comparison study between the United Kingdom and Germany. Journal of Palliative Medicine 2012;15(12):1374-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JA, Egger M, Moher D, editor(s) on behalf of the Cochrane Bias Methods Group. Chapter 10: Addressing reporting biases. In: Higgins JP, Green S, editor(s). Cochrane Handbook forSystematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Tait 2014
- Tait P, Morris B, To T. Core palliative medicines: meeting the needs of non-complex community patients. Australian Family Physician 2014;43(1):29-32. [PubMed] [Google Scholar]
Torphy 2009
- Torpy JM, Lynm C, Glass RM. Cystic fibrosis. JAMA 2009;302(10):1130. [DOI: 10.1001/jama.302.10.1130.] [DOI] [PubMed] [Google Scholar]
UK CF Trust 2014
- UK Cystic Fibrosis Registry. Annual data report 2013. www.cysticfibrosis.org.uk/media/598466/annual-data-report-2013-jul14.pdf (accessed 31 July 2015).
Weinberger 2002
- Weinberger M. Airways reactivity in patients with CF. Clinical Reviews in Allergy and Immunology 2002;23(1):77. [DOI] [PubMed] [Google Scholar]
WHO 2015
- WHO Genomic Resource Centre. Genes and human diseases: cystic fibrosis. www.who.int/genomics/public/geneticdiseases/en/index2.html (accessed prior to 06 April 2017).
References to other published versions of this review
Jaiswal 2015
- Jaiswal N, Singh M, Agarwal A, Thumburu KK. Drug treatments for breathlessness in end-stage cystic fibrosis. Cochrane Database of Systematic Reviews 2015, Issue 9. [DOI: 10.1002/14651858.CD011855] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jaiswal 2017
- Jaiswal N, Singh M, Agarwal A, Thumburu KK. Palliative drug treatments for breathlessness in cystic fibrosis. Cochrane Database of Systematic Reviews 2017, Issue 8. [DOI: 10.1002/14651858.CD011855.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]