

Abstract
Background:
Persons living with HIV (PLWH) are at risk of developing different phenotypes of chronic lung disease, including chronic obstructive pulmonary disease (COPD). Mechanisms underlying these phenotypes are unclear.
Objective:
To identify clusters of peripheral inflammatory mediators associated with pulmonary function to determine inflammatory pathways and phenotypes of COPD in PLWH and HIV-uninfected individuals.
Methods:
Study participants were PLWH and HIV-uninfected individuals enrolled in the Pittsburgh HIV Lung Cohort. Pulmonary function tests (PFTs) were performed for all participants. Chest computed tomographic (CT) scans were performed in a subset of PLWH. Plasma levels of 19 inflammatory mediators were measured by Luminex or ELISA. Clusters were identified based on the expression pattern of inflammatory mediators in PLWH and HIV-uninfected individuals and the relationships among clinical parameters were evaluated within clusters by using cluster and network analyses.
Results:
In PLWH, we identified a distinct cluster with higher levels of Th1, Th2 and Th17 inflammatory mediators with increased complexity of these mediators and inferred presence of pathogenic Th17 cell types. Individuals in this cluster had worse airway obstruction and more radiographic emphysema. In HIV-uninfected individuals, a cluster with high-grade systemic inflammation also had worse diffusing capacity for carbon monoxide.
Conclusions:
Inflammatory pathways associated with pulmonary dysfunction in PLWH suggest multi-faceted immune dysregulation involved in different phenotypes of pulmonary dysfunction with a potential specific contribution of the Th17 pathway to airway obstruction in PLWH. Identification of these associations may help in development of treatments that could alter the course of the disease.
Keywords: HIV infection, systemic inflammation, inflammatory pathways, phenotypes of pulmonary dysfunction, cluster analysis, network analysis
INTRODUCTION
The incidence of pulmonary infections in persons living with HIV (PLWH) has declined with efficacious antiretroviral therapy (ART), but the prevalence of non-infectious pulmonary diseases such as chronic obstructive pulmonary disease (COPD) has not been altered [1–3]. Although a higher proportion of PLWH smoke tobacco, HIV is an independent risk factor for pulmonary diseases including COPD and pulmonary vascular disease [4–9]. Features unique to chronic HIV infection are likely to contribute to differences in pulmonary disease incidence, prevalence, and progression between PLWH and HIV-uninfected individuals. Peripheral inflammatory mediators including interleukin-6 (IL-6), C-reactive protein (CRP) and tumor necrosis factor alpha (TNF-α) are elevated in PLWH including with undetectable plasma levels of HIV RNA. Higher levels of these mediators have been linked to worse pulmonary dysfunction [10, 11]. Endothelin-1 (ET-1) is a marker of endothelial damage and dysfunction [12], and is associated with COPD in HIV [10, 13]. These observations suggest that enhanced systemic inflammation and endothelial damage due to HIV infection could play important roles in pulmonary diseases.
COPD is a complex and heterogeneous disease with a wide range of clinical features, pathophysiology, biology, anatomy and progression [14, 15]. This heterogeneity may reflect differing pathogenic mechanisms that are likely to respond to different therapeutic interventions. Thus, identification of COPD phenotypes may help develop treatments that could alter disease. Cluster analysis is a statistical approach in which individuals are grouped based on multiple similarities in clinical or biologic measures [16]. This approach has been used to characterize COPD [17–19] and asthma [20] phenotypes in the general population, but not in PLWH. In this study, we investigated a panel of peripheral inflammatory mediators with potential relationship to HIV and/or COPD using cluster analysis to determine associations between inflammatory pathways and phenotypes of pulmonary dysfunction in a large cohort of PLWH and HIV-uninfected individuals.
METHODS
Study Participants
We included all participants from the Pittsburgh HIV Lung Cohort [10, 11] who had acceptable baseline pulmonary function testing and concurrent plasma samples collected. These participants had been enrolled in the University of Pittsburgh, the University of California San Francisco, and University of Washington from both HIV outpatient clinics and the ongoing cohort studies of the MACS and WIHS. Individuals were excluded if they had acute or worsening respiratory symptoms, fever, or respiratory infections within four weeks of testing. Demographic and baseline clinical data were collected by standardized participant interview including age, gender, smoking history, current antiretroviral therapy (ART), and history of prior pneumonia. Peripheral CD4+ counts and plasma HIV RNA levels (viral load) were confirmed by chart review or direct testing within 6 months from the current visit. Fifteen individuals did not have available specimens. They were similar in race, gender, and pulmonary function to those who were included, but were older and less likely to be HIV-infected.
Pulmonary Function Testing
All participants performed spirometry before and after bronchodilator administration, including forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) [21, 22] and diffusing capacity for carbon monoxide (DLco). Spirometry and DLco were performed following American Thoracic Society (ATS)/European Respiratory Society (ERS) standards [23]. PFT and DLco measurements that passed quality control criteria were included in the final analysis. Hankinson and Neas equations were used to determine percent predicted values of spirometry and DLco [23, 24], respectively. DLco was corrected for hemoglobin and carboxyhemoglobin [23].
Chest Computed Tomographic Scan
Standardized non-contrast chest computed tomographic (CT) scans were acquired at a moderate radiation exposure (100 mAs) with a breathhold at end-inspiration. Images were reconstructed at an image thickness less than or equal to 0.900 mm. Percentage of lung voxels associated with emphysema was defined as voxels below −950 Hounsfield units (HU) [25]. We used a fully automated computer scheme to detect and quantify airway sections depicted in axial section of the CT examination as previously described [26].
Measurement of Plasma Proinflammatory Mediators
Plasma samples were isolated from peripheral blood and stored at −80°C until use. Plasma levels of proinflammatory cytokines and chemokines were measured using a Bio-Plex human cytokine, chemokine 17-plex assay kit (Bio-Rad, Hercules, California, USA) per manufacturer’s protocol utilizing recommended standard curve concentrations. The 17-plex assay kit quantified a panel of cytokines with potential relationship to HIV and/or COPD and included interleukin (IL)-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p70, IL-13, IL-17a, interferon (IFN)-γ, CCL2, CCL4, TNF-α, granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF). Luminex data were analyzed using Bio-Plex Manager software (Bio-Rad). Plasma levels of CRP and ET1 were measured using ELISA assay kits (R&D Systems, Minneapolis, Minnesota, USA) per the manufacturer’s protocol.
Statistical Analysis
Demographic and clinical characteristics including pulmonary function variables and plasma levels of inflammatory mediators were compared between PLWH and HIV-uninfected participants using Student’s t-test (after best transformation) or Chi-square test (and Fisher’s exact test if required). Correlation analyses between inflammatory mediators and FEV1%-predicted, FVC%-predicted and DLco%-predicted were performed using Pearson correlation, and adjusted for pack-years smoking using partial correlation. Cytokine concentrations were standardized in PLWH and HIV-uninfected participants. Our correlation analysis revealed a high correlation between 19 plasma inflammatory markers. To reduce this noise in the clusters from these correlations, we used “clustervarsel” in R to choose 14 biomarkers that significantly impact clustering. This approach finds the optimal subset of variables in our data set that have cluster information and improved clustering partitions [27]. We used “NbCluster” function to select the optimal number of clusters in each group. This approach selects the number of clusters based on 30 dissimilarity indices. We used K-means clustering to cluster participants separately by HIV status. Demographic, clinical, pulmonary function test results, and chest CT scan findings were compared between different clusters in both groups using the Kruskal-Wallis test before adjustment and ANOVA after adjustment for confounders. Statistical analyses were performed in 3.5.0 (R Core Team, 2018), Stata 14.2 (StataCorp., College Station, TX). A P-value < 0.05 was considered statistically significant (two-sided test).
Network Analysis
Network analysis was carried out in three clusters using a modified version of our previously published algorithm for Dynamic Network Analysis [28–30]. The network analyses were performed using different stringency levels (0.7–0.95) with a stringency of 0.8 chosen for comparison of inflammatory networks, as it best illustrated differences in network connectivity, while still maintaining network integrity. Connections (edges) between inflammatory mediators (nodes) were created if the Pearson correlation coefficient between any two nodes (inflammatory mediators) was greater or equal to a threshold of 0.8, as indicated. The “network complexity” for each experimental condition was calculated using the following formula: Sum (N1 + N2 +…+ Nn)/n-1, where N represents the number of connections for each mediator and n is the total number of mediators analyzed. The total number of connections represents the sum of the number of connections in a given experimental sub-group.
Inflammatory Mediator Correlations and Computational Inference of Th17 Cell Subsets
Spearman’s correlation analysis was conducted to measure the strength of the association between pairs of inflammatory mediators in the indicated individual sub-groups using a modified version of a MATLAB®-based toolbox as described previously [31] [32].
Ethics Statement
Written informed consent was obtained from each participant prior to any study procedures. The study protocol was approved by the Institutional Review Boards at the University of Pittsburgh, the University of California San Francisco, and University of Washington.
RESULTS
Characteristics of the Cohort
We included 380 PLWH and 147 participants who were HIV-uninfected (Table 1). The majority of PLWH (93%) received antiretroviral therapy (ART) for 5.6 median years (4.5–6.6) and 73% had undetectable plasma HIV RNA levels. Median age was 51 years, 33% were female, and 44% were Caucasian in PLWH. More than half of the cohort had a history of smoking, with higher cumulative pack-years among PLWH (Table 1).
TABLE 1.
Demographic and pulmonary function characteristics of participants
| PLWH (N=380) | HIV-participants (N=147) | P value | Adjusted P value* | |
|---|---|---|---|---|
| Age (year), median (IQR) | 51 (43–56) | 51 (42–57) | 0.7 | |
| Female, n (%) | 124 (33) | 68 (47) | 0.004 | |
| Race, n (%) | 0.43 | |||
| Caucasian | 168 (44) | 74 (50) | ||
| African American | 195 (51) | 68 (46) | ||
| Other | 17 (5) | 5 (3) | ||
| Smoking status, n (%) | 0.14 | |||
| Never | 115 (30) | 57 (39) | ||
| Former | 96 (25) | 36 (25) | ||
| Current | 169 (45) | 54 (37) | ||
| Total Pack-years, median (IQR) | 8 (0–22) | 5 (0–16) | 0.043 | |
| CD4+ T lymphocyte count (cells/μl), median (IQR) | 615 (404–853) | NA | NA | |
| Plasma HIV RNA <40 copies/ml, n (%) | 252 (73) ^ | NA | NA | |
| Current ART use, n (%) | 352 (93%) | NA | NA | |
| ART duration (year), median (IQR) | 5.6 (4.5–6.6) | NA | NA | |
| Post FEV1%, median (IQR) | 93 (80–106) | 94 (82–106) | 0.5 | >0.9 |
| Post FVC%, median (IQR) | 93 (80–103) | 92 (83–104) | 0.6 | 0.9 |
| Post FEV1/FVC, median (IQR) | 81 (75–84) | 81 (76–84) | 0.9 | 0.7 |
| DLco%, median (IQR) | 79 (68–90) | 86 (77–95) | 0.0001 | 0.002 |
IQR: interquartile range; ART: antiretroviral therapy; Post FEV1%: percent predicted post-bronchodilator (BD) forced expiratory volume in 1 second; Post FVC%: percent predicted post-BD forced vital capacity; DLco%: percent single breath diffusing capacity for carbon monoxide.
Adjusted for age, gender, pack- years.
N=344.
Pulmonary Function
Post-bronchodilator FEV1% predicted, FVC% predicted and FEV1/FVC were similar between PLWH and HIV-uninfected participants, but DLco% predicted was significantly lower in PLWH, and the difference remained significant after adjusting for age, gender and pack-years (Table 1). Fifty-four (14.5%) PLWH versus 20 (13.6%) HIV-uninfected individuals had a post-bronchodilator FEV1/FVC <0.70 (P = 0.80), and 193 (51.5%) PLWH had a DLco % predicted <80% versus 50 (34.0%) HIV-uninfected individuals (P <0.001).
Cluster Analyses of Inflammatory Mediators and Pulmonary Function
Fourteen cytokines were selected for K-mean clustering with three cluster solutions in both PLWH and HIV-uninfected participants (Fig. 1A and 1B). In PLWH, cluster 3 had the highest plasma levels of inflammatory mediators and the lowest values of pulmonary function. This cluster also had the highest pack-years and highest CD4+ T lymphocyte counts and frequency of undetectable HIV RNA. Cluster 1 had moderate plasma levels of inflammatory mediators and intermediate measures of pulmonary function, moderate pack-years and the lowest CD4+ T lymphocyte counts. Cluster 2 had the lowest plasma levels of inflammatory mediators with the highest values of pulmonary function, lowest pack-years and intermediate CD4+ T lymphocyte counts (Fig.1C and Table 2). The values of FEV1%, FVC% and DLco% were significantly lower in cluster 3 than in cluster 1 or cluster 2 (Table 2). After adjusting for pack-years, the differences in FEV1% and FVC% between clusters remained significant (Table 2). Additionally, there were not significant differences in smoking status including current smoking status and ART duration among the three clusters. In the HIV-uninfected participants, cluster 3 had the highest plasma levels of inflammatory mediators and the lowest measures of pulmonary function with lower pack-years. Values of DLco% predicted were significantly lower, and there was a trend with lower FVC% than those in cluster 1 or cluster 2 (Fig.1D and Table 3). After adjusting for pack-years, the differences in DLco% between clusters remained significant (Table 3).
Figure 1.
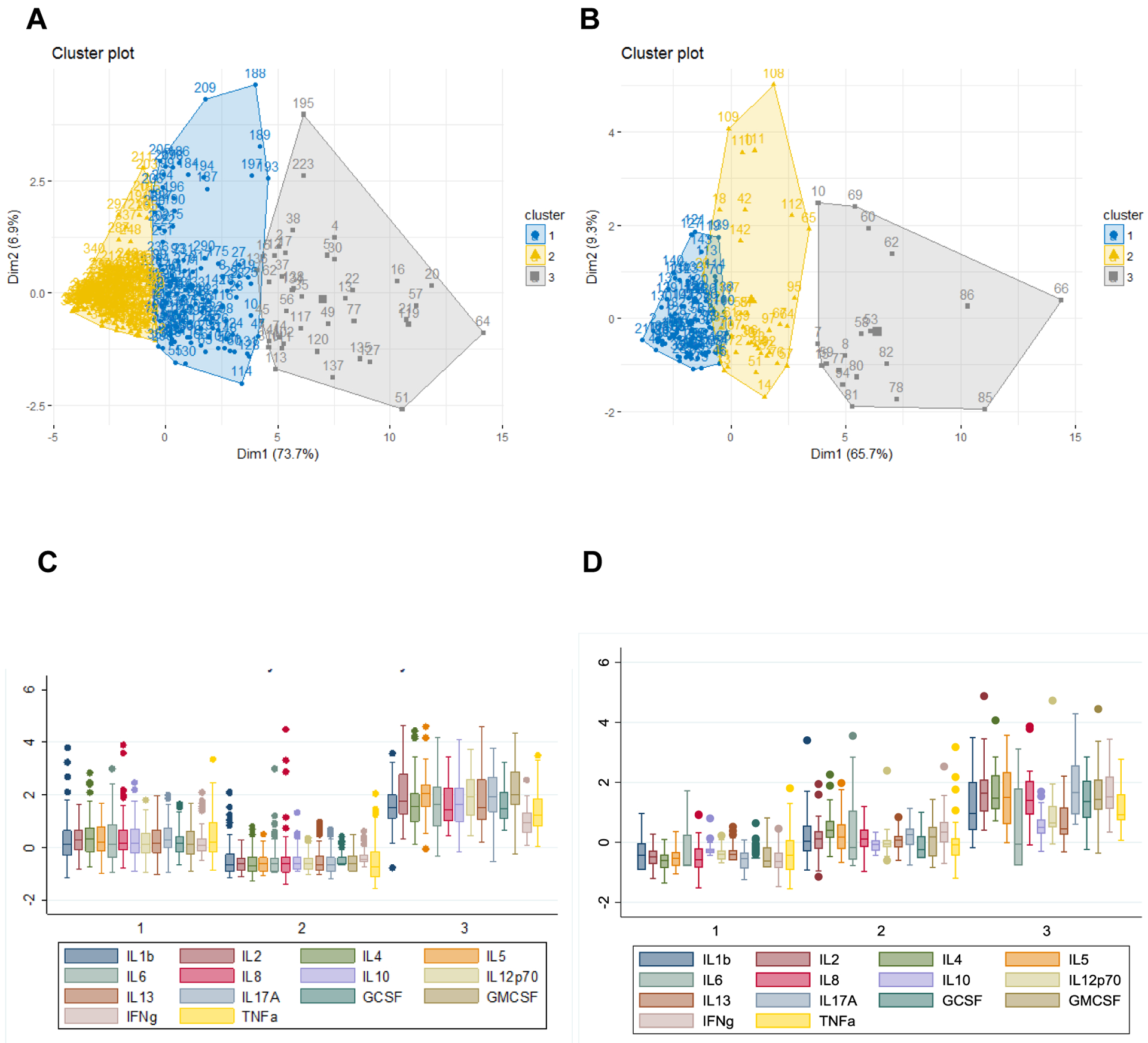
Partitioning of PLWH and HIV-uninfected participants and standardized normal distribution of inflammatory mediators by final clusters. In panel A and B, there were three clusters based on the expression patterns of circulating inflammatory mediators in PLWH (A) and HIV-uninfected participants (B), respectively. The X and Y axis show the first and second Principal Component from cytokines, and each dot represents the location of the observation and the small square represents the centroid of each cluster. In panel C and D, box graphs show the median, interquartile range and range of standardized inflammatory mediators in each cluster in PLWH (C) and HIV-uninfected participants (D), respectively.
TABLE 2.
Demographic characteristics and pulmonary function variables in PLWH by cluster
| Parameter | Cluster 1 N = 125 | Cluster 2 N =193 | Cluster 3 N =40 | P value | Adjusted P value | |
|---|---|---|---|---|---|---|
| Age (year), median (IQR) | 53 (48–57) | 48 (40–55) | 52 (48–57) | 0.0001 | NA | |
| Gender, female, n (%) | 28 (22) | 73 (38) | 12 (30) | 0.015 | NA | |
| Smoking status, n (%) | 0.30 | |||||
| Never | 35 (28) | 62 (32) | 11 (28) | |||
| Former | 33 (26) | 53 925) | 6 (15) | |||
| Current | 57 (46) | 78 (40) | 23 (58) | |||
| Body-mass index [kg/m2], median (IQR) | 26 (23–30) | 26 (23–30) | 26 (23–31) | 0.81 | NA | |
| Pack-years, median (IQR) | 9 (0–30) | 7 (0–17) | 13 (0–33) | 0.04 | NA | |
| Plasma HIV RNA <40 copies/ml, n (%) | 93 (79) * | 127 (70) ** | 32 (82) *** | 0.047 | NA | |
| CD4+ T lymphocytes/μl, median (IQR) | 555 (341–770) | 622 (442–858) | 810 (476–1014) | 0.0059 | NA | |
| ART, n (%) | 118 (94) | 175 (91) | 38 (95) | 0.38 | NA | |
| ART duration (year), median (IQR) | 5.7 (4.9–6.6) | 5.0 (3.9–6.6) | 6.0 (5.2–6.6) | 0.16 | ||
| Post FEV1%, median (IQR) | 0.91 (0.78–1.04) | 0.95 (0.84–1.07) | 0.84 (0.66–0.92) | 0.0013 | 0.021 | |
| Post FVC%, median (IQR) | 0.92 (0.78–1.01) | 0.95 (0.85–1.07) | 0.86 (0.74–0.93) | 0.002 | 0.027 | |
| Post FEV1/FVC, median (IQR) | 0.80 (0.75–0.84) | 0.81 (0.75–0.85) | 0.79 (0.68–0.83) | 0.20 | 0.29 | |
| DLco%, median (IQR) | 0.77 (0.65–0.87) | 0.82 (0.73–0.92) | 0.73 (0.66–0.83) | 0.0052 | 0.30 | |
Adjusted P value: Adjusted for pack-years; IQR: interquartile range; ART: antiretroviral therapy; Post FEV1%: percent predicted post-bronchodilator (BD) forced expiratory volume in 1 second; Post FVC%: percent predicted post-BD forced vital capacity; DLco%: percent single breath diffusing capacity for carbon monoxide.
N = 118,
N = 187,
N = 39.
TABLE 3.
Demographic characteristics and pulmonary function variables in HIV-uninfected participants by cluster
| Parameter | Cluster 1 N = 88 | Cluster 2 N =37 | Cluster 3 N =19 | P value | Adjusted P value | ||
|---|---|---|---|---|---|---|---|
| Age (year) median (IQR) | 48 (41–56) | 54 (50–59) | 53 (43–61) | 0.017 | NA | ||
| Gender, Female, n (%) | 50 (57) | 13 (35) | 3 (16) | 0.001 | NA | ||
| Body-mass index [kg/m2], median (IQR) | 28 (24–33) | 28 (25–32) | 29 (25–32) | 0.92 | NA | ||
| Pack-years, median (IQR) | 5 (0–20) | 7 (0–14) | 0.2 (0–14) | 0.71 | NA | ||
| Post FEV1%, median (IQR) | 0.95 (0.83–1.08) | 0.91 (0.82–1.05) | 0.92 (0.75–1.03) | 0.22 | 0.20 | ||
| Post FVC%, median (IQR) | 0.94 (0.86–1.04) | 0.90 (0.81–0.98) | 0.88 (0.76–1.03) | 0.09 | 0.09 | ||
| Post FEV1/FVC, median (IQR) | 0.81 (0.77–0.84) | 0.80 (0.76–0.84) | 0.77 (0.73–0.83) | 0.40 | 0.35 | ||
| DLco%, median (IQR) | 0.87 (0.78–0.97) | 0.84 (0.77–0.95) | 0.78 (0.64–0.91) | 0.018 | 0.013 | ||
Adjusted P value: Adjusted for pack-years; IQR: interquartile range; Post FEV1%: percent predicted post-bronchodilator (BD) forced expiratory volume in 1 second; Post FVC%: percent predicted post-BD forced vital capacity; DLco%: percent single breath diffusing capacity for carbon monoxide.
Cluster Analyses of Chest CT Scan Variables
In 132 PLWH with chest CT scans, participants in cluster 3 had significantly higher percentages of voxels associated with emphysema (median=0.011%) than those in cluster 1 (median=0.009%) or cluster 2 (median=0.006%) (p=0.022). After adjusting for pack-years, these differences remained statistically significant (p=0.044). However, the airways analysis on the CT imaging did not show significant differences in airway parameters among three clusters (data not shown). We compared the demographic and pulmonary function characteristics among PLWH with or without CT images and found that most of them were similar, but there were still significant differences in age, percentage of HIV RNA<40 copies/ml, ART and FVC%.
Network Analysis of Inflammatory Mediators and Pulmonary Function
Network analysis was performed for three clusters in both PLWH and HIV-uninfected participants to visualize correlations among inflammatory mediators and pulmonary function. Compared to clusters 1 and 2, both PLWH and HIV-uninfected participants in cluster 3 showed increased network complexity across all network stringency levels (Fig. 2A and 2B). In PLWH of cluster 3, plasma levels of GM-CSF and IL-17A were highly correlated (Fig. 2D), indicating the presence of pathogenic Th17 cell types [33–35]. Similar correlations were not observed in the HIV-uninfected participants of cluster 3 (Fig. 2C).
Figure 2:
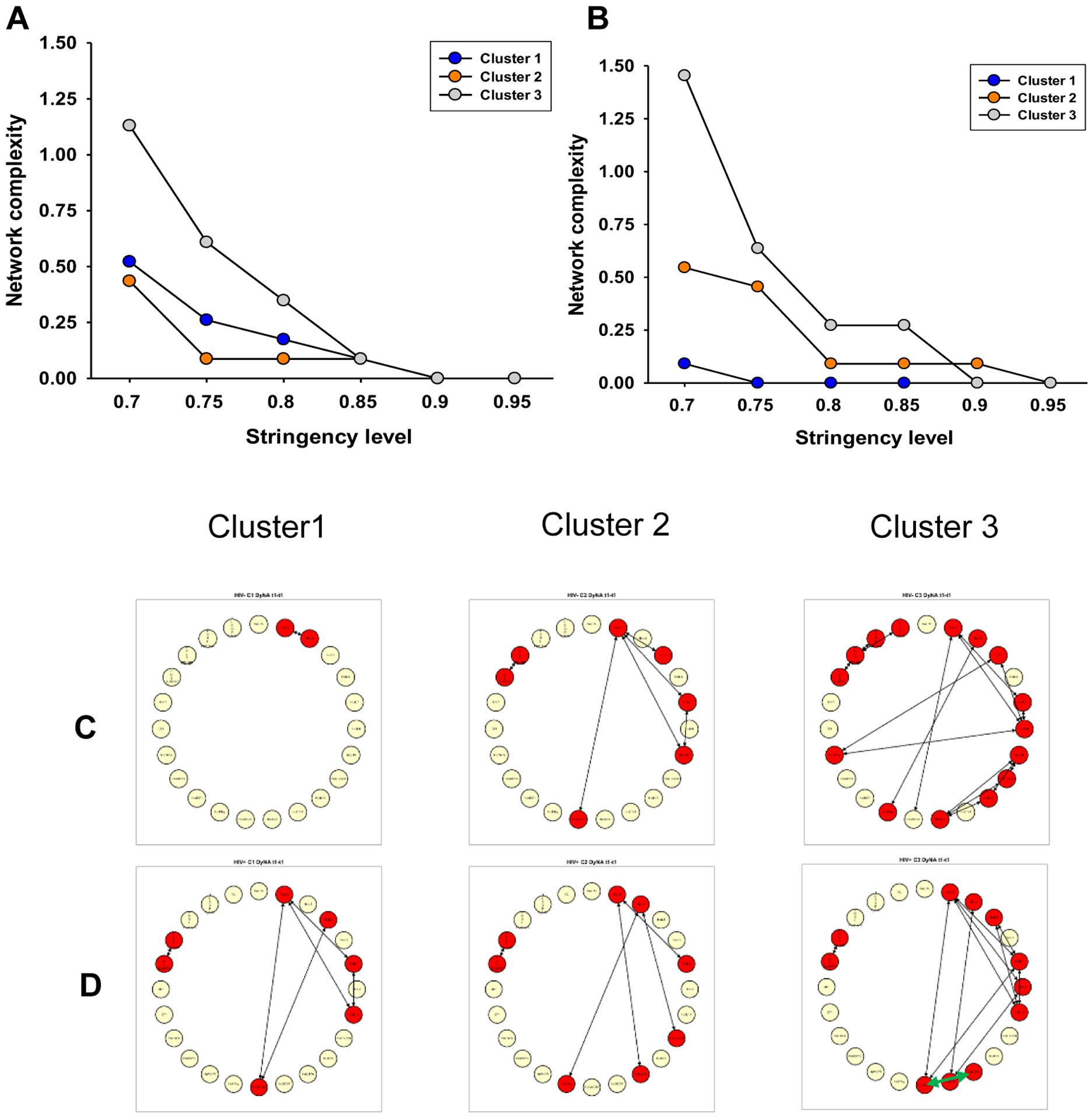
Network analysis of peripheral inflammatory mediators and pulmonary function for three clusters in PLWH and HIV-uninfected participants. The network analyses were performed using different stringency levels (0.7–0.95) as shown in panels A and B, and 0.8 refers to the stringency that was used for the analysis shown in panel C and D. In panels A and B, cluster 3 displays elevated network complexity across all stringency levels in PLWH (A) and HIV-uninfected participants (B). In panels C and D, visualizing inflammatory mediator networks reveals the presence of pathogenic Th17 cell types in PLWH (D) of cluster 3 (green arrow indicates IL-17 and GM-CSF correlation), but similar correlations were not observed in HIV-uninfected participants (C).
Association between Peripheral Inflammatory Mediators and Pulmonary Dysfunction
In PLWH, plasma levels of all inflammatory mediators except IL-10, CCL4 and CRP were significantly higher compared to HIV-uninfected participants (Supplemental Table 1). Correlation analyses showed that higher plasma levels of Th1 cytokines (IL-2, IL-12p70 and IFN-γ), Th2 cytokines (IL-4, IL-5, IL-6, IL-10 and IL-13), Th17 cytokines (IL-17a), IL-7, IL-8, G-CSF, GM-CSF, CRP and ET1 were correlated with worse FEV1% and FVC% (Supplemental Table 2), and plasma levels of Th1 cytokines (IL-2), Th2 cytokines (IL-5, IL-6 and IL-10), IL-7, IL-8, GM-CSF, CRP and ET1 were negatively correlated with the value of DLco% in PLWH (Supplemental Table 2). After adjusting for pack-years, these correlations remained significant with FEV1% and FVC%, but only IL-8, CRP and ET1 remained significantly correlated with DLco% in PLWH (Supplemental Table 3). However, in HIV-uninfected participants, plasma levels of fewer Th1, Th2 and Th17 cytokines were inversely correlated with DLco%, and fewer Th1 and Th2 cytokine levels were inversely correlated with FEV1% or FVC% (Supplemental Table 2). After adjusting for pack-years, most correlations remained significant (Supplemental Table 3).
DISCUSSION
We investigated peripheral inflammatory mediators associated with pulmonary function to determine relationships between inflammatory pathways and phenotypes of pulmonary dysfunction in PLWH and HIV-uninfected individuals. This is a novel application of cluster and network approaches to determine these associations in PLWH. We identified three clusters of cytokines, and the cluster with the highest levels of inflammatory mediators was also associated with worse measures of pulmonary function in both groups.
A chronic inflammatory state is a hallmark of HIV infection and can lead to premature aging and immune senescence in PLWH [36, 37]. Multiple studies indicate that abnormal T-cell activation and inflammatory mediators do not fully normalize after ART initiation, are associated with pulmonary dysfunction in PLWH receiving ART [10, 11, 38] and may contribute to other non-AIDS co-morbidities [39]. These studies analyzed associations of pulmonary dysfunction with a limited number of individual cytokines in PLWH and did not integrate specific pathways and phenotypes of COPD. In this study, we found a distinct cluster with high levels of Th1, Th2 and Th17 inflammatory mediators and impaired FEV1% and FVC% as well as more radiographic emphysema, suggesting that Th1, Th2 and Th17-related inflammation affects changes in lung structure that correlate with physiologic manifestations of airflow obstruction in HIV-associated COPD. Several prior studies also found that emphysema was independent or overlapping with airflow obstruction in PLWH [40–42]. Correlation analyses also indicated that Th1, Th2 and Th17 inflammatory pathways are involved in airway obstruction, and Th1 and Th2 pathways are related to diffusing capacity impairment in PLWH, suggesting there are both overlapping and unique inflammatory pathways involved in different COPD phenotypes with a potential specific contribution of the Th17 pathway to airway obstruction. In HIV-uninfected individuals, Th1, Th2 and Th17 inflammatory pathways were associated with impairment of diffusing capacity, suggesting that similar inflammatory pathways are involved in different phenotypes of pulmonary dysfunction in PLWH and HIV-uninfected individuals.
We also performed network analyses to visualize the overall immune state of each cluster. The increased network complexity of inflammatory mediators presents among PLWH and HIV-uninfected members of cluster 3 indicates a dysregulated immune state that may be indicative of increased inflammation associated with decreased pulmonary function. Further, the correlation of plasma GM-CSF and IL-17A among the PLWH of cluster 3 suggest the presence of pathogenic Th17 cell types in this cluster, which may represent an additional source of inflammation contributing to increased airway obstruction in PLWH. Th17 cells have been implicated in the pathophysiology of COPD. In a murine model, overexpression of IL-17 in the lung epithelium induced a COPD-like phenotype [43]. Further, IL-17 secretion leads to induction of mucin proteins such as MUC5B and MUC5AC [44] which may be important in COPD. As IL-17 promotes neutrophil and macrophage recruitment to the lung [45], an increased Th17 cell presence in the HIV+ lung could facilitate neutrophilia and worsening lung inflammation. Therefore, Th17 cells may uniquely contribute to the pathophysiology of COPD within PLWH.
Cigarette smoking (CS) induces repetitive inflammation through a chronic activation of immune system accompanied by an abnormal inflammatory response of the airways. Inflammatory mediators including interleukin (IL)-1β, IL-6, IL-8, IL-12, IL-17, TNF-α, MCP-1 and MIP-1α increase in bronchoalveolar lavage fluid of long-term smokers [46, 47]. Additionally, some studies have indicated that CS induces an increase in the numbers of circulating neutrophils, macrophages, and lymphocytes, and elevated levels of a systemic inflammatory mediators such as C-reactive protein (CRP), TNF-α [48]. In this study, we found cluster 3 had the highest plasma levels of inflammatory mediators, highest pack-years and highest CD4+ T lymphocyte counts, suggesting CS could play a role in stimulating inflammatory mediators and CD4+ lymphocytes. The correlations between higher levels of inflammatory mediators and worse PFT parameters remained significant after adjusting for pack-years in PLWH suggesting other factors such as HIV could enhance inflammation. CS damages the pulmonary epithelium and induces inflammation, and is the leading cause of COPD, but HIV also can increase circulating inflammatory mediator levels and lead to lung dysfunction in PLWH. We found that there is still a significant effect in cluster 3 after adjusting for pack-year suggesting inflammatory mediators induced by HIV play an independent role in lung dysfunction in cluster 3 of PLWH.
Strengths of this study are the large size, inclusion of both PLWH and HIV-uninfected participants and assessment of multiple inflammatory markers using novel analytical techniques. However, this study has several limitations. First, only circulating inflammatory mediators were assessed. Evaluation of inflammatory mediators from respiratory samples may identify different relationships that are important in lung dysfunction. Second, the chest CT measures were only available from a subset of the cohort, and the participants with chest CT were a biased group and limits power. Third, causality cannot be determined from the correlations observed. Finally, this is a cross-sectional cohort study and longitudinal investigations could reveal different associations of inflammation and lung function.
In a large cohort of PLWH and HIV-uninfected individuals, we found novel clusters based on patterns of peripheral inflammation that are associated with impaired pulmonary function and HIV-associated variables. These analyses revealed overlapping as well as unique Th1, Th2 and Th17 inflammatory pathways involved in patterns of pulmonary dysfunction that have been associated with different phenotypes of COPD with a potential specific contribution of the Th17 pathway to airway obstruction in PLWH. The inflammatory pathways associated with pulmonary dysfunction in PLWH suggest that therapies targeting immune activation may be beneficial in HIV-associated lung disease.
Supplementary Material
ACKNOWLEDGEMENTS
A subset of subjects in this cohort was recruited from sites of the Multicenter AIDS Cohort Study (MACS) (Charles R. Rinaldo, Lawrence Kingsley). The MACS is funded by the National Institute of Allergy and Infectious Diseases, with additional supplemental funding from the National Cancer Institute. UO1-AI-35042, UL1-RR025005 (GCRC), UO1-AI-35043, UO1-AI-35039, UO1-AI-35040, UO1-AI-35041. Website located at http://www.statepi.jhsph.edu/macs/macs.html. A subset of subjects in this cohort was recruited from the Connie Wofsy Study Consortium of Northern California (Ruth Greenblatt, Phyllis Tien and Bradley Aouizerat) of the Women’s Interagency HIV Study (WIHS) Collaborative Study Group. The WIHS is funded by the National Institute of Allergy and Infectious Diseases (UO1-AI-35004, UO1-AI-31834, UO1-AI-34994, UO1-AI-34989, UO1-AI-34993, and UO1-AI-42590) and by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (UO1-HD-32632). The WIHS is cofunded by the National Cancer Institute, the National Institute on Drug Abuse, and the National Institute on Deafness and Other Communication Disorders. Funding is also provided by the National Center for Research Resources (UCSF-CTSI Grant Number UL1 RR024131).
Supported by the National Institute of Heath [R01 HL125049, R01 HL120398 and K24 HL123342]. LH was partly supported by K24 HL087713.
Footnotes
The authors have no funding or conflicts of interest to disclose.
REFERENCES
- 1.Gingo MR, Morris A. Pathogenesis of HIV and the lung. Curr HIV/AIDS Rep 2013,10:42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Presti RM, Flores SC, Palmer BE, Atkinson JJ, Lesko CR, Lau B, et al. Mechanisms Underlying HIV-Associated Noninfectious Lung Disease. Chest 2017,152:1053–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gingo MR, George MP, Kessinger CJ, Lucht L, Rissler B, Weinman R, et al. Pulmonary function abnormalities in HIV-infected patients during the current antiretroviral therapy era. Am J Respir Crit Care Med 2010,182:790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crothers K, Butt AA, Gibert CL, Rodriguez-Barradas MC, Crystal S, Justice AC, et al. Increased COPD among HIV-positive compared to HIV-negative veterans. Chest 2006,130:1326–1333. [DOI] [PubMed] [Google Scholar]
- 5.Crothers K, Huang L, Goulet JL, Goetz MB, Brown ST, Rodriguez-Barradas MC, et al. HIV infection and risk for incident pulmonary diseases in the combination antiretroviral therapy era. Am J Respir Crit Care Med 2011,183:388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crothers K, McGinnis K, Kleerup E, Wongtrakool C, Hoo GS, Kim J, et al. HIV infection is associated with reduced pulmonary diffusing capacity. J Acquir Immune Defic Syndr 2013,64:271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morris A, Gingo MR, George MP, Lucht L, Kessinger C, Singh V, et al. Cardiopulmonary function in individuals with HIV infection in the antiretroviral therapy era. AIDS 2012,26:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris AM, Huang L, Bacchetti P, Turner J, Hopewell PC, Wallace JM, et al. Permanent declines in pulmonary function following pneumonia in human immunodeficiency virus-infected persons. The Pulmonary Complications of HIV Infection Study Group. Am J Respir Crit Care Med 2000,162:612–616. [DOI] [PubMed] [Google Scholar]
- 9.Sitbon O, Lascoux-Combe C, Delfraissy JF, Yeni PG, Raffi F, De Zuttere D, et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J Respir Crit Care Med 2008,177:108–113. [DOI] [PubMed] [Google Scholar]
- 10.Fitzpatrick ME, Nouraie M, Gingo MR, Camp D, Kessinger CJ, Sincebaugh JB, et al. Novel relationships of markers of monocyte activation and endothelial dysfunction with pulmonary dysfunction in HIV-infected persons. AIDS 2016,30:1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fitzpatrick ME, Singh V, Bertolet M, Lucht L, Kessinger C, Michel J, et al. Relationships of pulmonary function, inflammation, and T-cell activation and senescence in an HIV-infected cohort. AIDS 2014,28:2505–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bohm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc Res 2007,76:8–18. [DOI] [PubMed] [Google Scholar]
- 13.Parikh RV, Ma Y, Scherzer R, Heringer AS, Macgregor JS, Martin JN, et al. Endothelin-1 Predicts Hemodynamically Assessed Pulmonary Arterial Hypertension in HIV Infection. PLoS One 2016,11:e0146355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agusti A, Calverley PM, Celli B, Coxson HO, Edwards LD, Lomas DA, et al. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res 2010,11:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wedzicha JA. The heterogeneity of chronic obstructive pulmonary disease. Thorax 2000,55:631–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ball GH, Hall DJ. A clustering technique for summarizing multivariate data. Behav Sci 1967,12:153–155. [DOI] [PubMed] [Google Scholar]
- 17.Vanfleteren LE, Spruit MA, Groenen M, Gaffron S, van Empel VP, Bruijnzeel PL, et al. Clusters of comorbidities based on validated objective measurements and systemic inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013,187:728–735. [DOI] [PubMed] [Google Scholar]
- 18.Burgel PR, Roche N, Paillasseur JL, Tillie-Leblond I, Chanez P, Escamilla R, et al. Clinical COPD phenotypes identified by cluster analysis: validation with mortality. Eur Respir J 2012,40:495–496. [DOI] [PubMed] [Google Scholar]
- 19.Burgel PR, Paillasseur JL, Caillaud D, Tillie-Leblond I, Chanez P, Escamilla R, et al. Clinical COPD phenotypes: a novel approach using principal component and cluster analyses. Eur Respir J 2010,36:531–539. [DOI] [PubMed] [Google Scholar]
- 20.Moore WC, Hastie AT, Li X, Li H, Busse WW, Jarjour NN, et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol 2014,133:1557–1563 e1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. Standardisation of spirometry. Eur Respir J 2005,26:319–338. [DOI] [PubMed] [Google Scholar]
- 22.Macintyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J 2005,26:720–735. [DOI] [PubMed] [Google Scholar]
- 23.Neas LM, Schwartz J. The determinants of pulmonary diffusing capacity in a national sample of U.S. adults. Am J Respir Crit Care Med 1996,153:656–664. [DOI] [PubMed] [Google Scholar]
- 24.Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med 1999,159:179–187. [DOI] [PubMed] [Google Scholar]
- 25.Leader JK, Zheng B, Rogers RM, Sciurba FC, Perez A, Chapman BE, et al. Automated lung segmentation in X-ray computed tomography: development and evaluation of a heuristic threshold-based scheme. Acad Radiol 2003,10:1224–1236. [DOI] [PubMed] [Google Scholar]
- 26.Zheng B, Leader JK, McMurray JM, Park SC, Fuhrman CR, Gur D, et al. Automated detection and quantitative assessment of pulmonary airways depicted on CT images. Med Phys 2007,34:2844–2852. [DOI] [PubMed] [Google Scholar]
- 27.Scrucca L, Raftery AE. clustvarsel: A Package Implementing Variable Selection for Gaussian Model-Based Clustering in R. J Stat Softw 2018,84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mi Q, Constantine G, Ziraldo C, Solovyev A, Torres A, Namas R, et al. A dynamic view of trauma/hemorrhage-induced inflammation in mice: principal drivers and networks. PLoS.One 2011,6:e19424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ziraldo C, Vodovotz Y, Namas RA, Almahmoud K, Tapias V, Mi Q, et al. Central role for MCP-1/CCL2 in injury-induced inflammation revealed by in vitro, in silico, and clinical studies. PLoS One 2013,8:e79804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zamora R, Vodovotz Y, Mi Q, Barclay D, Yin J, Horslen S, et al. Data-Driven Modeling for Precision Medicine in Pediatric Acute Liver Failure. Mol Med 2016,22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pernet CR, Wilcox R, Rousselet GA. Robust correlation analyses: false positive and power validation using a new open source matlab toolbox. Front Psychol 2012,3:606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abboud A, Namas RA, Ramadan M, Mi Q, Almahmoud K, Abdul-Malak O, et al. Computational Analysis Supports an Early, Type 17 Cell-Associated Divergence of Blunt Trauma Survival and Mortality. Critical care medicine 2016,44:e1074–e1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.González-Amaro R, Marazuela M. T regulatory (Treg) and T helper 17 (Th17) lymphocytes in thyroid autoimmunity. Endocrine 2016,52:30–38. [DOI] [PubMed] [Google Scholar]
- 34.Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 2011,12:560–567. [DOI] [PubMed] [Google Scholar]
- 35.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nature immunology 2011,12:568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desai S, Landay A. Early immune senescence in HIV disease. Curr HIV/AIDS Rep 2010,7:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao W, Jamieson BD, Hultin LE, Hultin PM, Effros RB, Detels R. Premature aging of T cells is associated with faster HIV-1 disease progression. J Acquir Immune Defic Syndr 2009,50:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H, et al. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis 2003,187:1534–1543. [DOI] [PubMed] [Google Scholar]
- 39.Giorgi JV, Hultin LE, McKeating JA, Johnson TD, Owens B, Jacobson LP, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis 1999,179:859–870. [DOI] [PubMed] [Google Scholar]
- 40.Triplette M, Attia E, Akgun K, Campo M, Rodriguez-Barradas M, Pipavath S, et al. The Differential Impact of Emphysema on Respiratory Symptoms and 6-Minute Walk Distance in HIV Infection. J Acquir Immune Defic Syndr 2017,74:e23–e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samperiz G, Guerrero D, Lopez M, Valera JL, Iglesias A, Rios A, et al. Prevalence of and risk factors for pulmonary abnormalities in HIV-infected patients treated with antiretroviral therapy. HIV Med 2014,15:321–329. [DOI] [PubMed] [Google Scholar]
- 42.Guaraldi G, Besutti G, Scaglioni R, Santoro A, Zona S, Guido L, et al. The burden of image based emphysema and bronchiolitis in HIV-infected individuals on antiretroviral therapy. PLoS One 2014,9:e109027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005,6:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem 2003,278:17036–17043. [DOI] [PubMed] [Google Scholar]
- 45.Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol 2010,72:495–516. [DOI] [PubMed] [Google Scholar]
- 46.Crotty Alexander LE, Shin S, Hwang JH. Inflammatory Diseases of the Lung Induced by Conventional Cigarette Smoke: A Review. Chest 2015,148:1307–1322. [DOI] [PubMed] [Google Scholar]
- 47.Kou YR, Kwong K, Lee LY. Airway inflammation and hypersensitivity induced by chronic smoking. Respir Physiol Neurobiol 2011,178:395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Madani A, Alack K, Richter MJ, Kruger K. Immune-regulating effects of exercise on cigarette smoke-induced inflammation. J Inflamm Res 2018,11:155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.