

Abstract
To date, most insights into the processes shaping vertebrate gut microbiomes have emerged from studies with cross-sectional designs. While this approach has been valuable, emerging time series analyses on vertebrate gut microbiomes show that gut microbial composition can change rapidly from one day to the next, with consequences for host physical functioning, health, and fitness. Hence, the next frontier of microbiome research will require longitudinal perspectives. Here we argue that primatologists, with their traditional focus on tracking the lives of individual animals and familiarity with longitudinal fecal sampling, are well positioned to conduct research at the forefront of gut microbiome dynamics. We begin by reviewing some of the most important ecological processes governing microbiome change over time, and briefly summarizing statistical challenges and approaches to microbiome time series analysis. We then introduce five questions of general interest to microbiome science where we think field-based primate studies are especially well-positioned to fill major gaps: (1) Do early life events shape gut microbiome composition in adulthood? (2) Do shifting social landscapes cause gut microbial change? (3) Are gut microbiome phenotypes heritable across variable environments? (4) Does the gut microbiome show signs of host aging? And (5) do gut microbiome composition and dynamics predict host health and fitness? For all of these questions, we high-light areas where primatologists are uniquely positioned to make substantial contributions. We review preliminary evidence, discuss possible study designs, and suggest future directions.
Keywords: fitness, health, longitudinal, time series, microbial ecology, community dynamics
Introduction
Microbiomes are multilayered, interconnected networks of microbes and their genes, which interact in time and space to produce a well-functioning host (Figure 1). Over the last decade, with the advent of culture-free techniques, researchers have uncovered astonishing diversity in animal microbiomes, especially in the mammalian gut (Bálint et al., 2016). The gut microbiome’s composition and diversity is shaped by many factors, including the host’s evolutionary history, lifestyle, diet, and social interactions (e.g., Moeller, Caro-Quintero, et al., 2016; Moeller, Foerster, et al., 2016; David et al., 2013). Some of these compositional differences may have functional consequences for the services gut microbiomes provide to their hosts, including the host’s ability to digest complex carbohydrates, detoxify plant secondary compounds, and resist infectious diseases (McKenney et al., 2018; Kohl & Dearing, 2016).
Figure 1:
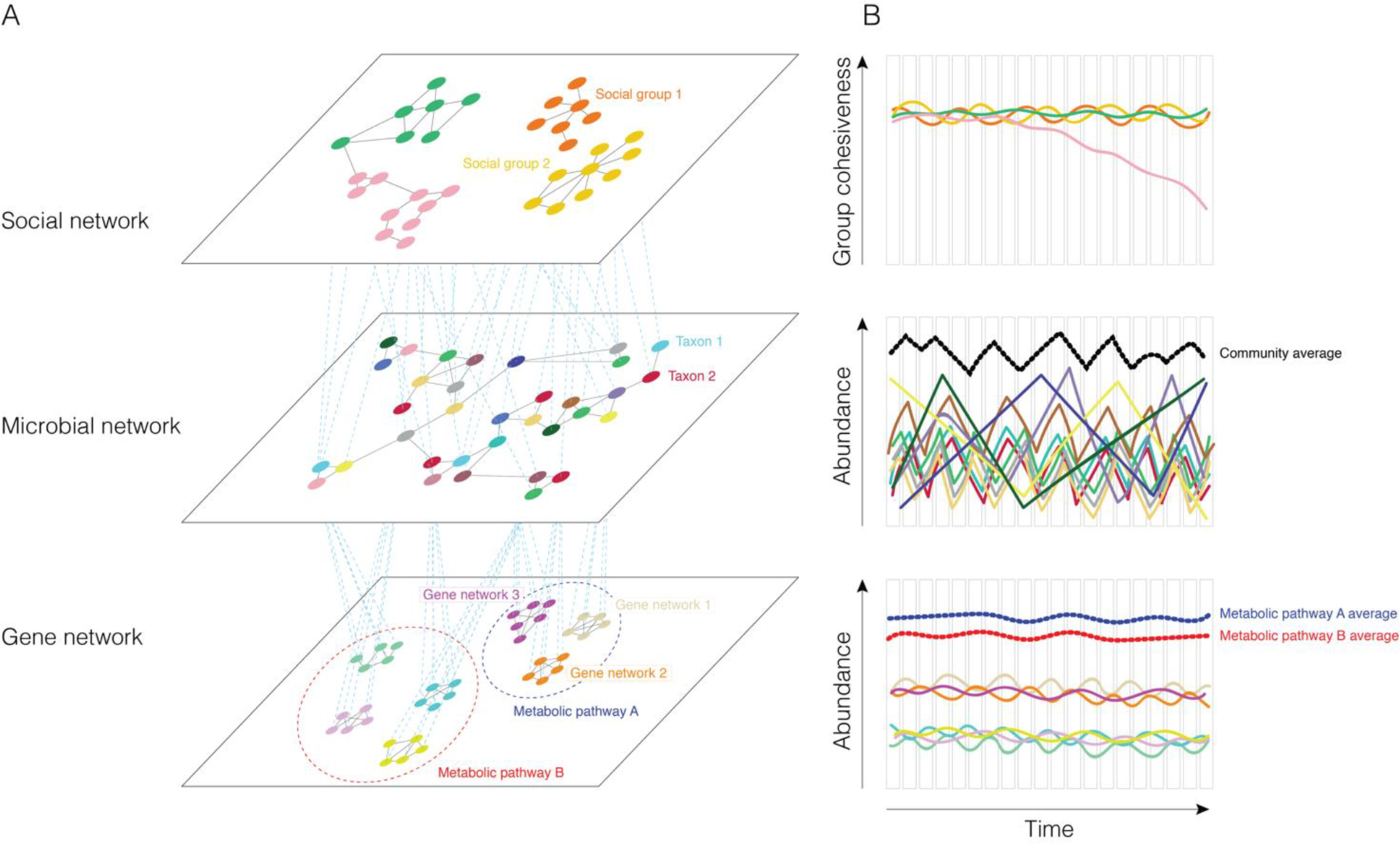
A multilayer representation of hosts and their collective gut microbial communities. (A) In this conceptual figure, the microbiome at time t is represented as a multilayer network where layers are squares, solid black lines are intra-layer edges, and dashed blue lines are inter-layer edges. The first layer is a social network: circles represent individual hosts and edges represent social interactions between hosts living in the same and different social groups, depicted by different colors. The second layer is a gut microbial network: circles represent individual microbial taxa, depicted by different colors, and edges represent positive and negative relationships between taxa. The third layer is a gene network: circles represent gut microbial genes, and edges represent genes that are found in the same gene network, represented by different colors. Different metabolic pathways can be responsible for the same function; the dashed red and blue circles depict functionally redundant metabolic pathways (e.g., the degradation of cellulose or pectin). Multiple metabolic pathways can be present in the same microbe, and multiple microbes can have the same metabolic pathways, but for illustrative purposes, this is not depicted. The inter-layer edges represent different types of associations; edges connect hosts and microbes when that microbe is found in a given host; edges connect microbes and genes when that metabolic pathway is present in a given microbe. (B) Longitudinal time series allow for analyzing how different properties of each layer change over time. Each bar represents a given layer at time t 1, 2 … , T. For instance, in the top plot, cohesiveness among social groups changes over time due to the fission and fusion of social groups. In the second plot, gut microbial taxa change in abundance over time due to similar or different responses to biotic and abiotic factors. Each colored line corresponds to a microbial taxa and the thick black dashed line represents the aggregated fluctuations at the whole microbiome community level. In the last plot, individual metabolic pathways fluctuate over time (depicted by thin colored lines), but due to functional redundancy, the host’s functional capacity is stable over time (red and blue thick dashed lines). See Pilosof et al. (2017) for a review of multilayer networks in ecology.
To date, many of these insights have emerged from cross-sectional studies, which provide only a snapshot perspective on the gut microbiome at a single time point in a host’s life. However, a handful of influential time series analyses (see Box 1 for our glossary) on vertebrate gut microbiomes suggest considerable dynamism: microbial presence and abundance can change considerably from one day to the next, with potential consequences for host physical functioning (e.g., Caporaso et al., 2011; David et al., 2014). Hence, the next frontier of gut microbiome research must consider time: compared to cross-sectional studies, time series analyses of mammalian gut microbiomes from several subjects will yield deeper insights into the drivers of gut microbiome change and the consequences for host health and fitness. Time series analyses will be essential to forecast or predict microbiome change, connect microbiome dynamics to host health and fitness, learn the causal role that host environments and behaviors play in microbiome change, and understand the role of vertical transmission and historical contingency in microbiome assembly. However, time series data on gut microbiomes–especially data sets that span multiple individual hosts–remain rare. Nearly all such data are collected on humans, which can be challenging and expensive study subjects: dense time series and covariates that explain gut microbiome dynamics, such as diet and social interactions, are difficult to collect. As a result, most such data sets have either few subjects (e.g., David et al., 2014; Caporaso et al., 2011), or if they have more subjects, they have relatively few time points per subject, limiting their statistical power (e.g., Flores et al., 2014; Faith et al., 2013; Claesson et al., 2011). These discrepancies can lead to seemingly contradictory results; for example, Caporaso et al. (2011) sampled two human adults daily for 15 months and found that each individual’s gut microbiome exhibited high variability over time. On the other hand, Faith et al. (2013) sampled 37 human adults 2 to 13 times (up to 296 weeks apart) and concluded that individuals’ gut microbiomes were remarkably stable. However, these studies differed in the time scales over which they measured gut microbial dynamics (daily changes versus weekly or monthly changes).
Box 1 |. Glossary.
Alternative stable states. Different possible stable states of composition or function that a community can move to, either following a perturbation or because of different initial conditions (Beisner et al., 2003).
Complex adaptive systems. A system in which many independent agents interact, leading to emergent outcomes that are often difficult or impossible to predict simply by observing individual interactions (Lansing, 2003).
Dispersal. The movement of species across space (Vellend, 2016).
Drift. Random changes in population sizes via stochastic birth and death events (Vellend, 2016).
Dynamical system. The mathematical notion of a dynamical system consists of two parts: the phase space and the dynamics. The phase space of a dynamical system is the collection of all possible states of the system in question. Each state represents a complete snapshot of the system at some moment in time. The dynamics are governed by rules (state variables) that transform the state of the system at time t into a new state at time t+1. For instance, a principal coordinates analysis (PCoA) plot depicting microbiome similarity can be viewed as the phase space (Didier et al., 2018).
Feedback loops. The effect that change in one part of an ecosystem has on another, and how this effect then influences the source of the change by inducing more or less of it. Positive feedback is a circular path of effects that are self-reinforcing. When part of the system increases, another part of the system also changes in a way that makes the first part increase even more. Positive feedbacks are a source of instability and a strong driver of change as they can force the system outside of its normal operating boundaries (Kéfi et al., 2016).
Heritability. A statistic used in the fields of breeding and genetics that estimates the proportion of variation in a phenotypic trait in a population that is due to genetic variation between individuals in that population (Wray & Visscher, 2008).
Keystone species. A species on which other species in an ecosystem largely depend, such that if it was removed the ecosystem would change drastically (Paine, 1969).
Keystone-pathogen. A microbial species that supports and stabilizes disease by instigating inflammation (Hajishengallis et al., 2012).
Priority effects. The initial order and timing at which species disperse and colonize an empty community, which in turn alters how drift, selection, and diversification influence community assembly and succession (Fukami, 2015).
Prospective longitudinal study design. A study design that follows a set of subjects, which differ with respect to factors under study, over time to determine how these factors predict a specific outcome (Diggle et al., 1994).
Selection. In an ecological context, selection occurs when individuals of different species vary in their fitness and niche requirements, producing variation in reproduction and extinction rates between individuals and species (Vellend, 2016).
Speciation. The process by which new species arise in the course of evolution (Vellend, 2016).
Stability. There are many definitions of stability, some measuring the temporal variability of e.g., abundance or biomass over time, or some that measure responses to perturbations (see Box 2).
Steady state. If the state variables that are used to describe the state in a dynamical system are unchanged over time, the system is said to be in a steady state (Didier et al., 2018).
Time series data. A time series is a sequence of data points collected over time (Chatfield, 2013). In the microbiome, time series often represent microbial taxonomic or genic composition from the same host over multiple time points.
Time series analysis. Time series analysis encompasses a wide range of statistical methods for analyzing time series, including tests of temporal autocorrelation, Fourier and wavelet transforms to analyze frequencies, time series decomposition to extract seasonal, trend and noise components, and state space models to retrospectively study the behavior of a system underlying the time series, or to make forecasts beyond the last observation (Chatfield, 2013; M. West & Harrison, 1989).
As such, it is unclear which drivers explain gut microbiome dynamics at different time scales, and which of these dynamics are most relevant to host physiology, health, and fitness.
Primatologists can help overcome these barriers. One hallmark of field-based primatology is a focus on the behavior, ecology, physiology, and life histories of known individual animals. Indeed, primate field studies often collect long-term, individual-based data on the phenotypes of many subjects, sometimes from the animal’s birth to death (Bronikowski et al., 2016; Strier et al., 2010; Kappeler & Watts, 2012; Clutton-Brock & Sheldon, 2010a,b). Primate studies have long led the the way in using longitudinal fecal sampling to characterize their subjects’ genes (e.g., Snyder-Mackler et al., 2016; Morin et al., 1993), hormones (e.g., Gesquiere et al., 2018; Whitten et al., 1998), and parasites (e.g., Müller-Klein et al., 2018; Stuart & Strier, 1995; Freeland, 1979). Hence, such projects are well-positioned to pair their wealth of long-term life history data with time series of individual subject’s gut microbiomes. With this combination of data types, primatologists may be able to fill key gaps in microbiome dynamics that may be impossible to fill in humans or other animals, including our understanding of the processes governing microbiome assembly and succession, microbiome temporal dynamics and stability, and how these changes influence microbiome function and host health and fitness.
Our objective in this review is to summarize major unanswered questions about the mammalian gut microbiome that require time series data and where individual-based primate studies are well-positioned to provide answers. We begin by introducing some of the major ecological processes governing gut microbiome assembly, dynamics, and stability. We then briefly summarize statistical challenges and approaches to microbiome time series analysis. We next highlight five diverse questions in microbiome science where primatology is poised to make contributions. Our goal is not to provide a comprehensive review of these questions, but rather to introduce key barriers to progress and explain how primate studies might overcome these barriers. The first four questions are united in that each addresses the factors and processes that drive gut microbial change over time: (1) do early life events shape gut microbiome composition in adulthood? (2) do shifting social landscapes cause gut microbial change? (3) are microbiome phenotypes heritable across variable environments? And (4) does the gut microbiome show signs of host aging? Finally, the fifth question addresses how longitudinal microbiome data sets can be used to understand the functional consequences of gut microbiome change by asking, (5) do gut microbiome composition and dynamics predict host health and fitness? Tackling these questions will greatly improve our understanding of both the processes shaping the gut microbiome over time and the consequences of these changes for primate hosts. Such research is also likely to reveal unanticipated discoveries, raising completely new questions for primatologists, microbiologists, ecologists, and evolutionary biologists. While our review largely focuses on 16S rRNA amplicon sequencing data, the research questions we review remain equally relevant for data generated with other “-omics” approaches such as transcriptomics, proteomics, and metabolomics.
1. What ecological processes contribute to gut microbiome assembly, temporal dynamics, and stability?
To date, a handful of papers have explored the temporal dynamics of primate gut microbiomes, revealing considerable dynamism. For instance over developmental scales, different lemur species exhibit different patterns of infant gut microbial succession, and these successional differences are linked to differences in lemur gut morphology and dietary regimes (McKenney et al., 2015). Over seasonal time scales, the gut microbiomes of wild gorillas (Gorilla gorilla gorilla) and chimpanzees (Pan troglodytes troglodytes) fluctuate with rainfall, mirroring the apes’ switch from fiberrich leaves and bark to succulent fruits (Hicks et al., 2018). Over even shorter time scales, Ren et al. (2016) found considerable differences in baboon (Papio cynocephalus) gut microbiome composition from one day to the next: gut microbiomes sampled from the same baboon a few days apart were just as different as microbiomes sampled from that animal 10 years apart. While these studies suggest that the gut microbiomes of non-human primates change in different ways over different time periods and across life, few studies have used existing ecological frameworks to understand the processes that drive these changes.
Community ecology offers many theories and processes to understand gut microbiome dynamics (e.g., Sprockett et al., 2018; McKenney et al., 2018; Koskella et al., 2017; Costello et al., 2012; Walter & Ley, 2011). Ecological communities are examples of complex adaptive systems where large-scale patterns such as diversity-stability, diversity-productivity, and species-energy relationships emerge from interactions among species (Preston, 1948; Levin, 1998). They are also examples of dynamical systems that result from species interactions unfolding over time to produce complex dynamics such as periodicities, chaos, or alternative stable states (May, 1975, 1977). The basic principles guiding community dynamics can be summarized in four overarching processes that parallel well-known processes in evolution: dispersal, selection, drift, and speciation (Vellend, 2016; Vellend & Agrawal, 2010). In ecological terms, speciation generates new species, while dispersal, drift, and selection shape the relative abundances of those species and their loss from communities over time (Vellend, 2016; Vellend & Agrawal, 2010).
The development of ecological theory has significantly increased our understanding of (i) how these processes affect community composition and dynamics, (ii) when certain processes dominate, and (iii) how they can combine to produce complex interacting effects. For instance, the theory of island biogeography focuses on the balance between dispersal and drift (MacArthur & Wilson, 1967). This theory explains how species richness increases with island size and the distance from the mainland: the size of an island influences the colonization and extinction rates, and thus indirectly biodiversity. Island biogeography combined with the neutral theory of molecular evolution was later used to develop the unified neutral theory of biodiversity, which also aims to explain the diversity and relative abundance of species in ecological communities. Neutral theory makes the simple assumption that ecologically similar species in a community are demographically equivalent, such that individuals interact with and experience each other as though they were exactly the same regardless of traits and adaptations (Hubbell, 2001). Owing to its simplicity, neutral theory has had a tremendous impact on our understanding of the role of dispersal, drift, and speciation versus niche-based differences for determining biodiversity. Finally, building on these theories, metacommunity theory has become a new cornerstone of ecology, aiming to explain how species interactions at different temporal and spatial scales work together with dispersal to shape local and regional community composition and dynamics (Leibold et al., 2004). Metacommunity theory can be divided into four paradigms that can be positioned along a continuum ranging from niche-based to neutral processes: patch-dynamic, species-sorting, mass-effect, and neutral, each capturing different processes affecting metacommunity dynamics. At one extreme, variation in the metacommunity is determined by the responses of different species to environmental gradients; and at the other extreme, individuals are assumed to be identical in their fitness, and variation in community composition is mainly driven by drift (Leibold et al., 2004).
Below, we review cases from the microbiome literature where these processes either independently or in combination have been adopted to explain microbiome change over time.
Dispersal is the movement of individuals across space. The effect of dispersal on community dynamics depends on the number and composition of dispersing individuals relative to the size and composition of the recipient community. In animal microbiomes, microbial dispersal may increase gut microbial similarity between hosts that share habitat or have high rates of physical contact with each other. For instance, in controlled experiments, Burns et al. (2017) found that microbial dispersal between zebrafish homogenized the intestinal microbiome of co-housed host pairs, eliminating microbiome differences linked to host strains. In humans, people are constantly over-writing each other’s microbial fingerprints in the built environment. For example, families that moved to a new house replaced the past owner’s microbial fingerprint with their own within 24 hours (Lax et al., 2014). The effects of dispersal on gut microbiome community dynamics also depend on the spatial scale considered. A study of mammal species across the Americas found that gut microbiome similarity decayed with increasing geographic distance between species, suggesting that dispersal limitation of microbial taxa can lead to diversification of microbial lineages between host populations (Moeller et al., 2017). Similar effects may explain why sympatric gorillas (Gorilla gorilla gorilla) and chimpanzees (Pan troglodytes troglodytes) exhibit similar gut microbiomes (Moeller et al., 2013). On smaller spatial scales, direct contact between hosts facilitates microbial dispersal and homogenization of microbiomes both within and between host species (Amato et al., 2017; Grieneisen et al., 2017; Moeller et al., 2017; Moeller, Foerster, et al., 2016; Tung et al., 2015; Lax et al., 2014; Song et al., 2013).
Ecological selection shapes community composition when different species vary in their fitness and niche requirements, producing species-level variation in reproduction and extinction rates. In animal microbiomes, selective processes may partly explain differences in gut microbiome composition linked to host age, diet, and habitat because all of these factors could contribute to differential survival and reproduction of gut microbial species or strains, either inside or outside of hosts. For instance, outside of a host, differences in the climate, soils, and vegetation affect which microbes survive in the environment, and in turn, which microbes have the opportunity to colonize a primate’s intestinal tract (this process is also known as species sorting; Székely & Langenheder, 2014). Within the host, across developmental time scales, microbial selection is thought to increase from early life to adulthood, partly due to physiochemical maturation of the gut, but also because hosts become more effective at curating their microbiomes (Figure 2; Burns et al., 2015; Dini-Andreote & Raaijmakers, 2018). Dietary regimes and gut morphology also represent strong selective forces (Ley et al., 2008; David et al., 2013; McKenney et al., 2015; Groussin et al., 2017). This is because dietary changes affect gut contents, leading to differential growth and survival among gut microbial species, and thus different gut microbiome compositions (David et al., 2013). Selection, in combination with dispersal, may explain why gut microbial composition differs between captive and wild primates (Clayton et al., 2016). Specifically, changes in diet (e.g., the loss of dietary fiber) and in the environment (e.g., from a forest to a cage) affect both the selective regimes affecting on the gut microbiome, as well as which microbes hosts are exposed to (Clayton et al., 2016).
Figure 2:

Conceptual figure depicting the relative contribution of the four ecological processes that govern community dynamics across primate development. In infants, drift and dispersal determine early colonizing microbes, which create priority effects that are thought to have a large influence on subsequent microbial colonization. As the host develops and matures, selection is expected to increase as the physicochemical conditions of the gut stabilize, and as hosts become more effective at curating their microbiomes. As microbial species persist across host development, the chances for speciation increase; hence speciation may play a stronger role in adult microbiomes as compared to infants and young juveniles. Figure adapted from (Dini-Andreote & Raaijmakers, 2018).
Drift arises from random changes in species relative abundances due to stochastic birth and death events (Hubbell, 2001). In a hypothetical scenario where all individuals live in a closed community (i.e., no immigration and dispersal), and all individuals are demographically identical, drift is the only process affecting community dynamics (Vellend & Agrawal, 2010). Drift is stronger in small populations, thus it represents an important process affecting local species extinctions. For example, rare gut microbes that are perturbed by antibiotics or illness should be more prone to extinction than more abundant microbes (Fukuyama et al., 2017; Dethlefsen & Relman, 2011). The effects of drift on animal microbiomes are not well understood, partly because it is difficult to tease apart drift from selection and dispersal. However, Burns et al. (2015) found in an experimental zebrafish system, that drift and dispersal dominated in newly hatched larvae, but were less important as the hosts developed and matured, perhaps because of increasing host control over the microbiome and hence stronger selective forces (Figure 2).
Speciation is the process by which new species arise in the course of evolution. Speciation can therefore shape the regional distribution of species, and in turn, local community composition and dynamics (Vellend, 2016). Because microbes have short generation times, speciation has the potential to shape animal microbiomes over animal lifespans, especially in long-lived primates (Figure 2). Indeed, Koeppel et al. (2013) found that in lab conditions, microbial speciation can occur over the course of a few days. Speciation may partly explain why allopatric populations of chimpanzees (Pan troglodytes troglodytes) and gorillas (Gorilla gorilla gorilla) harbor divergent gut microbiomes compared to sympatric host populations (Moeller et al., 2013). Specifically, while some of this divergence is likely due to selective processes created by site-specific diets and low rates of microbial dispersal between allopatric host populations, microbes can rapidly diversify and adapt when faced with strong selective pressures Koeppel et al. (2013). Furthermore, when there is strong partner fidelity, microbes and their hosts may speciate in parallel. For example, the bacterial families Bacteroidaceae and Bifidobacteriaceae have diversified in concert with hominid hosts, including humans (Moeller, Caro-Quintero, et al., 2016).
These four processes–dispersal, selection, drift, and speciation–interact to produce complex community patterning across space and time. While it is not possible to review all of the relevant interactions and processes relevant to gut microbiome dynamics, below we summarize a few phenomena that have been especially influential in understanding microbiome dynamics, including priority effects, alternative stable states, feedback loops, and keystone species.
In priority effects, the order and timing of species arrivals determine the composition and dynamics of current ecological communities (Fukami, 2015). A newborn infant, for instance, represents a blank canvas ready to be colonized by microbes via dispersal. However, which microbes arrive first determines the identity and the order that later microbial immigrants are able to colonize (Didier et al., 2018; Fukami, 2015). This is because first-arriving microbes fill particular niches by quickly reaching carrying capacity, while simultaneously modifying the gut in their favor, thereby altering the ability of subsequent microbial immigrants to colonize (Sprockett et al., 2018). Such priority effects can also have long-term consequences for overall microbiome stability (see Box 2 for definitions of ecological stability), and may adversely affect host health and fitness later in life (see Sections 3.1 and 3.5 below). Subtle differences in the arrival order of species may shift communities into alternative stable states (Didier et al., 2018; Fukami, 2015). A switch in diet, for instance, can alter the nutritional niche of the gut, thus favouring the growth of pathogens at the expense of commensal microbes (Chen et al., 2017). However, the gut microbiome can also display resilience (see Box 2) and recover its initial state following a perturbation (see Box 2; Faust et al., 2015). For example, David et al. (2014) found that when one healthy adult human traveled abroad for several weeks and underwent a major dietary change, the ratio between the two dominant groups of bacteria in the gut increased nearly two-fold, but reverted to its initial ratio after returning home. We currently know very little about what makes a microbiome more resilient. What is clear is that resilience is an emergent property that applies in different ways in different subjects and in the various functional layers that make up the microbiome (Figure 1; Bäckhed et al., 2015; Gerber, 2014; Hollister et al., 2014).
Box 2 |. Ecological stability.
Stability is often proposed as a measure of microbiome health, and hence host health (see also Sections 3.4 and 3.5). However, ecological stability is a multifaceted concept that encompasses both variability over time and response to perturbations; it can therefore be measured in multiple ways (see Figure below; Donohue et al. (2016)). Facets of ecological stability that are particularly relevant to gut microbiomes include resilience, resistance, persistence, and temporal stability. While resistance is the degree to which a gut microbiome is able to withstand change following a perturbation (e.g., antibiotics or illness), resilience is the rate at which it returns to the initial steady state, or moves to a new alternative stable state (Pimm, 1984). Microbiomes with faster return times are said to be more resilient than those recovering more slowly. Persistence is the length of time the microbiome maintains the same microbial composition (Pimm, 1991); it can also be defined as a “core” of microbial species persisting beyond some arbitrarily defined threshold of time (Björk, O’Hara, et al., 2018). The most applicable measure of ecological stability that can be directly computed on time series is temporal stability (Si), which is defined as the ratio of the mean abundance (μi) of the i − th microbial species to its standard deviation (σi) calculated across the time series (Tilman, 1999). See Didier et al. (2018); Donohue et al. (2016) for more facets of ecological stability.
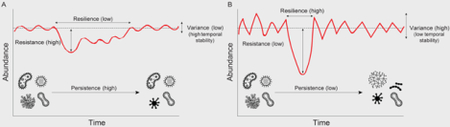
The multifaceted nature of ecological stability. In scenario (A), a gut microbiome shows a high resistance but low resilience following a perturbation. It also has a high persistence, and low variance (i.e., high temporal stability). In scenario (B), another gut microbiome shows the opposite patterns. The horizontal dashed lines depict the steady state, which can be computed as the long-term average abundance prior to any perturbation. The Y-axes show the aggregated abundance of the gut microbiomes, and the X-axes depict time. Figure adapted from Donohue et al. (2016).
At their core, alternative stable states result from positive species interactions and feedback loops (Kéfi et al., 2016). For instance, strong cooperation between microbes may destabilize the gut microbiome because positive interactions induce species coupling and positive feedback: if one microbe decreases in abundance, it will drag other species down with it and cause community collapse (Coyte et al., 2015). In contrast, species that respond differently to biotic and abiotic conditions fluctuate asynchronously over time (Figure 3; Loreau, 2010). These asynchronous fluctuations are expected to increase microbiome stability as one species’ sharp decline is compensated by another’s increase. This is one reason, for example, why community stability tends to increase with species diversity (Figure 3; Loreau, 2010). The above findings further point to the possibility of commensal keystone species, which have a disproportionate negative effect on the microbiome upon their removal (Berry & Widder, 2014; Fisher & Mehta, 2014). However, once the microbiome has been destabilized, alternative stable states associated with dysfunction may be reached by keystone-pathogens–microbes supporting and stabilizing disease by instigating inflammation (Hajishengallis et al., 2012).
Figure 3:
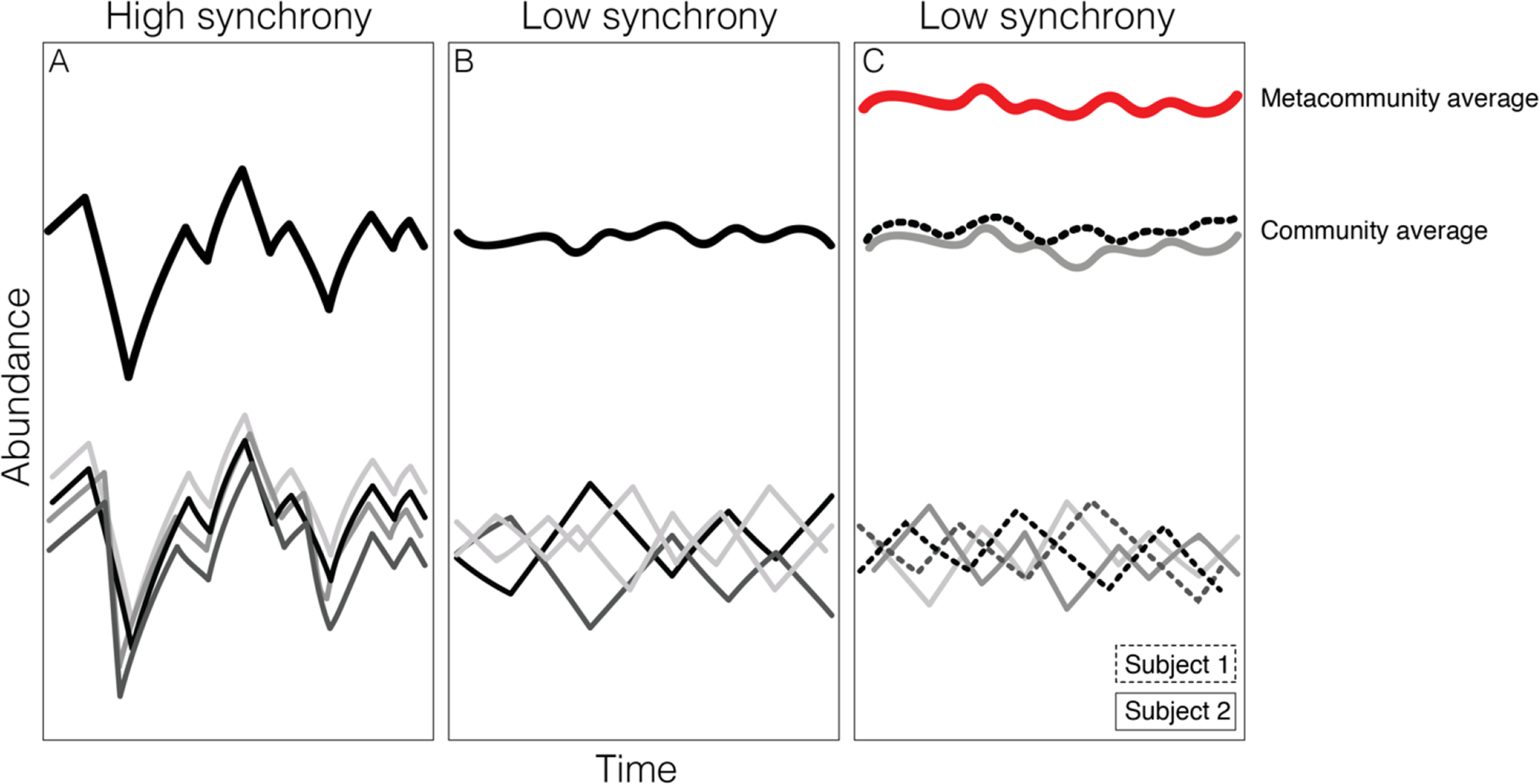
The relationship between species synchrony and stability at higher levels. (A) Depicts a scenario where gut microbes in a single host, indicated by the thin black and grey lines, respond similarly to biotic and abiotic fluctuations in the gut, which leads to higher instability at the microbiome community level (thick black line). (B) Depicts the opposite scenario where gut microbes in a single host respond differently to biotic and abiotic fluctuations in the gut, which in turn, leads to a higher stability at the microbiome community level. (C) is similar to (B), but the plot shows microbes and the microbiome community in two different host subjects (depicted using dashed and solid lines). Again, asynchronous species fluctuations lead to a higher stability at the microbiome level for both hosts, which in turn, lead to a higher stability at the social group level (i.e. metacommunity level; thick red line). The Y-axes show abundance and the X-axes time. Figure adapted from Wilcox et al.
Together, the basic ecological forces–dispersal, selection, drift and speciation–and the processes that emerge from them (e.g. alternative stable states, keystone species etc.) have been important in shaping scientific thinking about the forces underlying microbiome dynamics. However, it is important to note that these ecological frameworks were developed for free-living, macro-communities. Microbial communities, including the gut microbiome, differ from these communities in several important ways: (i) gut microbiome diversity is astonishing, and functional redundancy is probably much more common in microbiomes than in free-living communities; (ii) microbes can disperse both horizontally and vertically, with implications for the evolutionary outcome of host–microbe interactions; (iii) unlike free-living communities, microbiome dynamics are, to some extent governed by the host, introducing a completely new selective force; and (iv) microbes regularly exchange genes via lateral gene transfer; hence microbes can take on functions from their neighbors. These important differences between free-living macro-communities and host-associated microbial communities mean that there is an urgent need to develop ecological theory that is specific to the microbiome. Importantly, microbes’ short generation times and small genomes blur the line between ecological and evolutionary processes and timescales.
2. Time series analysis approaches for microbiome data
Time series analysis encompasses a wide range of statistical methods and models that are often not trivial to apply without some expert knowledge (see e.g., Chatfield, 2013; Faust et al., 2015). Many of the simplest methods require that the time steps between samples are equidistant, which can be challenging to achieve when fecal samples are collected opportunistically. Moreover, microbiome data exhibits several undesirable characteristics that must be considered in the analysis. Most of these characteristics are related to how the data are generated (see e.g., Silverman, Shenhav, et al., 2018). First, high-throughput DNA sequencing produces proportions of counts per operational taxonomic unit (OTU, a placeholder for taxon) per sample that is constrained by an arbitrary sum imposed by the sequencing platform (this characteristic is referred to as compositionality; Gloor et al., 2017). This property limits inference to relative abundances, and introduces uncertainty in the estimates of those relative measurements (Gloor et al., 2017). Second, high-throughput DNA sequencing often produces extremely sparse counts; that is, much of the data consists of zero values that can arise due to multiple processes. For example, an OTU can have zero abundance because it is completely absent from a sample, or because its abundance falls below the detection limit of the sequencing platform (Silverman, Roche, et al., 2018). Third, because microbial diversity is exceptionally high in many biological environments, the generated count table is high-dimensional, with hundreds to thousands of OTUs that can make inference computationally hard or even impossible without additional filtering steps. While it is challenging to account for all these data characteristics in the analysis, failure to do so can lead to spurious correlations between microbes and misleading results (Gloor et al., 2017; Tsilimigras & Fodor, 2016). To tackle the compositional property, data transformations or compositionally robust methods should be used (see e.g., Gloor et al., 2017; Weiss et al., 2016; Lovell et al., 2015).
Owing to the non-trivial nature of time series analysis, and the many undesirable characteristics of microbiome data, there has been a growing interest in developing easy-to-use longitudinal time series methods, such as MetaLonDa (Metagenomics Longitudinal Differential Abundance) to identify significant time intervals of differentially abundant microbial taxa (Metwally et al., 2018), and TIME (Temporal Insights into Microbial Ecology), a web based framework which offers popular time series analyses, including Dickey Fuller tests to calculate time series stationarity, Granger causality to find causal relationships between taxa, and dynamic time warping to measure the displacement between two time series (Baksi et al., 2018). While these methods are valuable to the field, they are limited in their applicability and scope, and many of the questions we introduce in this review will require greater flexibility. In Box 3, we give a brief introduction to state-space models (SSMs), which play a central role in time series analysis. They are typically used to retrospectively study the behavior of a system underlying a series of observations, or to make forecasts beyond the last observation (M. West & Harrison, 1989). Because SSMs model observation error separately from the underlying “state” of the system, they can successfully describe a system’s dynamics and its response to different inputs. An important feature of SSMs is that they do not require time series to be stationary (i.e., statistical properties such as mean, variance, and autocorrelation does not need to be constant over time), and they are therefore not sensitive to nonlinear relationships (M. West & Harrison, 1989). Similar to generalized linear mixed models (GLMMs), SSMs can also handle non-normal data, and account for fixed and random effects. This flexibility means that SSMs are likely to become a cornerstone of longitudinal time series analysis for microbiome data.
Box 3 |. State-space models for microbiomes: future directions.
State-space models (SSMs) differ from e.g., linear mixed models in that they assume there is an unobservable Markov chain called the “state process”, and that the observed time series is an imprecise measurement of that process (see Equations 1 and 2 below). A growing number of SSMs are being developed for microbiome applications, including models that address the technical challenges of microbiome data (e.g., Gibson & Gerber, 2018; Silverman, Durand, et al., 2018; Ridenhour et al., 2017), or infer microbe-microbe interactions (see e.g., Chen et al., 2017; Trosvik & de Muinck, 2015; Fisher & Mehta, 2014). Here we (i) showcase a simple state-space model, and (ii) briefly discuss advancements that are needed in order to answer some of the questions posed in this review. For simplicity, we do not explicitly show how to account for the several undesirable microbiome data characteristics that were mentioned in Section 2 (here we refer interested readers to e.g., Gibson & Gerber, 2018; Silverman, Durand, et al., 2018; Ridenhour et al., 2017; Warton et al., 2015).
Let Yt,d denote a time series with the number of counts observed for OTU d ∈ {1, … , D} in time point t ∈ {1, … , T }. In the simplest case, samples Yt are assumed to be independent and identically normally distributed as (Yt |θ) ~ N (θ, V), where V reflects the measurement error. However, as the microbiome state θ can change both gradually, for instance with season, or more abruptly, for instance during illness, a time varying state can easily be incorporated, such that
| (1) |
where Ft is a vector of time-varying covariates, with the state vector θ1, … θt, … θT representing the underlying microbiome dynamics in the time series, and νt describing random fluctuations arising from measurement error. The underlying time evolution is further modeled as a simple random walk such that the microbiome state in time t depends on the previous state in time t − 1,
| (2) |
where Gt represents the state matrix describing the dynamics of the microbiome state in the previous time θt−1, and ωt corresponds to unpredictable changes in the dynamics between time t − 1 and t. Which lag to use can be determined by looking at the partial autocorrelation function (see Chatfield, 2013 for more information).
Depending on the exact specification of the system matrices (Ft and Gt) and the covariance matrices (Vt and Wt), different types of models can be specified, such as e.g., static and dynamic mixed effects models, and models with seasonal or polynomial trends (M. West & Harrison, 1989). For simplicity, Equations 1 and 2 only model one time series from one host; however, to answer many of the questions we pose in this review, these equations have to be expanded to model concurrent microbiome time series from multiple hosts (see e.g., Silverman, Durand, et al., 2018). While time-varying covariates such as e.g., diet and rainfall, can already be modeled in Equation 1, new challenges include how to accommodate for more complex covariates in the state-space framework, such as hosts’ social interactions and genetic relatedness. Such covariates can, similar to species phylogenies, be modeled in static linear mixed models (see e.g., Ives & Helmus, 2011; Björk, Hui, et al., 2018). However, to answer questions regarding how e.g., changes in microbiomes over time can be attributed to a shifting social landscape (Section 3.2), or whether microbiome phenotypes are repeatable over time (Section 3.3), time series models, such as state space models are necessary.
3. Key areas of microbiome science where primate studies can contribute
Building on the ecological processes described above, here we briefly review five diverse but fundamental questions in microbiome science that require time series data from multiple hosts, and where individual-based primate studies are especially well-positioned to provide answers. The first four questions address drivers of microbiome change over time. The fifth question addresses the consequences of microbiome change for hosts. Our goal is not to provide a thorough review of each topic; rather we introduce a handful of recent studies in each area, discuss key barriers or gaps to answering these questions, and focus how primate studies can help overcome these barriers and contribute to answering the question.
3.1. Do early life events shape gut microbiome composition in adulthood?
Events early in life can have profound consequences for an animal’s life history, health, and fitness (Lindström, 1999; Chaby, 2016; Berens et al., 2017). An emerging body of work suggests that early life effects may also shape or be mediated by animal microbiomes (Berger et al., 2018; Martínez et al., 2018; Laforest-Lapointe & Arrieta, 2017; Clarke et al., 2014). The suitability of primate systems for understanding these processes stems from primatology’s traditional focus on individual-based research, and its long history of studying early life effects–especially maternal effects (e.g., Bernstein et al., 2012; Altmann & Alberts, 2005; Bailey et al., 2004). Such research has found that diverse maternal effects, ranging from a mother’s rank and cortisol levels to the quality of offspring care and social connectedness, can affect diverse offspring phenotypes, including temperament (Suarez-Jimenez et al., 2013), lifespan (Silk et al., 2009), adult cortisol (Onyango et al., 2008), mating success (Surbeck et al., 2010), and immune function and motor skill acquisition (Berghänel et al., 2016).
Whether these early life effects also shape primate gut microbiomes is unknown, but this possibility is supported by two main lines of evidence. First, as discussed in Section 1, events early in the formation of the gut microbiome may have consequences for subsequent community assembly, dynamics, and stability (Fukami, 2015; Sprockett et al., 2018). In support, there is considerable evidence for priority effects in human microbial development. For example, birth method can determine the initial state of the human gut microbiome and its subsequent assembly (Yassour et al., 2016; Bäckhed et al., 2015; Goedert et al., 2014; Dominguez-Bello et al., 2010). Indeed, Chu et al. (2017) found that infants delivered by cesarean section shared more microbes with their mothers’ skin at the time of birth than infants delivered vaginally. Cesarean infants have also been found to lack the bacterial genera Bacteroides and Bifidobacterium, which are largely responsible for the breakdown of oligosaccharides in breast milk, and these effects can persist for at least 6 to 18 months after delivery (Korpela et al., 2018; Yassour et al., 2016; Bäckhed et al., 2015). Beyond delivery mode, malnutrition can also affect gut microbial development. Acute malnutrition in early life has been found to keep the gut microbiome in a state of persistent immaturity (Subramanian et al., 2014; M. I. Smith et al., 2013). To test whether improved nutrition can rescue an “immature” gut, Subramanian et al. (2014) administered nutritional interventions and found that infants’ guts returned to their initial “immature” state after the intervention. These studies expose the major gaps in the literature–little is known about how long early life effects on the gut microbiome persist, or whether humans are representative of other host-associated microbial systems, such as non-human primates.
The second line of evidence is that the early life effects we already know about in primates could be relevant to gut microbiome assembly and development, including the quality of maternal care, dominance rank, social environment, and harsh conditions such as drought. For instance, in The Gambia, people born during the “hungry season” exhibit much higher mortality in adulthood as compared to those born during the “harvest season” (Moore et al., 1997, 2004). Because many non-human primates also live in seasonal environments, and because seasonal effects have been demonstrated on primate microbiomes (Hicks et al., 2018; Sun et al., 2018; Trosvik et al., 2018), non-human primates may be useful for understanding early life effects on gut microbialy mediated forms of nutritional programming, or the process through which variation in nutrition affects individual development (Langley-Evans, 2009). While long-term effects like these have not yet been shown in non-human primates, there is abundant evidence that factors like stress and rank shape microbiome composition over short time scales. For example, working in captive rhesus macaques (Macaca mulatta), Bailey et al. (2004) demonstrated that maternal stress during pregnancy altered infant gut microbiomes across the first 24 weeks of life. In wild primates, social status is sometimes linked to maternal stress (Murray et al., 2018; Markham et al., 2014); hence, maternal rank may also shape the infant’s gut microbiome in natural populations. In non-human primates, new evidence strongly suggests that diet and social environment are important influences of gut microbial composition (Hale et al., 2018; Grieneisen et al., 2017; Perofsky et al., 2017; Amato et al., 2017; Bennett et al., 2016; Moeller, Foerster, et al., 2016; Tung et al., 2015). However, no studies have yet studied the effects of social isolation or nutrient limitation on the gut microbiome’s of wild primates during early life.
Together, these lines of evidence suggest that early life events could be important in shaping non-human primate gut microbiomes through the juvenile period and into adulthood. Primate hosts with relatively short generation times and lifespans may be particularly useful because of the relative ease of connecting early life events to later-life outcomes in such species. Primate species where it is easy to collect longitudinal samples shortly after birth will also be essential to understand heterogeneity in the initial stages of gut microbial development. Major unanswered questions include: how do hosts, their environments, and ecological processes interact to produce early life effects on gut microbiomes? How long do these effects persist? And, what are the consequences of these effects for host physical functioning in adulthood? These questions are important in a wide variety of systems, and primatologists that are able to collect fecal samples from the same animal in early life and adulthood are well positioned to provide answers.
3.2. Does a shifting social landscape cause gut microbiome change?
To date, cross-sectional research on humans and non-human primates has been at the forefront of understanding how an individual’s social context may shape its gut microbiome (Amato et al., 2017; Perofsky et al., 2017; Bennett et al., 2016; Moeller, Foerster, et al., 2016; Tung et al., 2015; Lax et al., 2014; Song et al., 2013). For instance, recent cross-sectional analyses of wild baboons (Papio cynocephalus), chimpanzees (Pan troglodytes troglodytes), howler monkeys (Alouatta pigra), and lemurs all found that individuals who spend more time in contact or in close proximity to each other have more similar gut microbial compositions than individuals who are not in contact or do not live together (Amato et al., 2017; Perofsky et al., 2017; Moeller, Foerster, et al., 2016; Tung et al., 2015). Taking a longitudinal perspective on this phenomenon will help move the field forward in three important ways. First, longitudinal data coupled with time series analysis will be essential to tease apart the effects of social interactions from other aspects of group living, such as shared environments, similar diets, and host genetic relatedness (see also Section 3.3). Second, the structure of social networks strongly affects the rate at which infectious diseases spread through host populations (Watts & Strogatz, 1998), and microbes likely spread via these same pathways. Primate studies with densely sampled social networks, in combination with microbial strain tracking, could be used to learn how microbial strains, both commensal and pathogenic, propagate through primate societies. Third, taking a metacommunity perspective by treating subjects’ gut microbiomes as local communities, connected by socially-mediated microbial dispersal, will allow primate studies to test current predictions of how of ecological processes contribute towards the stability of each local community, and how this in turn influences the stability at the metacommunity level (e.g., at the social group level).
With respect to the first advance, the multiple, concurrent effects of group living and social interactions that influence gut microbiome composition are difficult to decouple in cross-sectional studies. To date, most such studies attempt isolate the direct effects of microbial transmission between social partners by statistically accounting for correlates and confounds, for instance by controlling for dietary similarity, kinship, and shared environments (Grieneisen et al., 2017; Tung et al., 2015). As a result, there has been a growing interest in developing statistical models to account for complex covariates such as the host species phylogeny (Björk, Hui, et al., 2018) or experimental design (Grantham et al., 2017) in cross-sectional microbiome data sets. While similar statistical models for longitudinal time series data are rare (but see Ridenhour et al., 2017; Laitinen & Lahti, 2018; Silverman, Durand, et al., 2018), time series analyses on multiple, co-resident primates will be essential to decompose variance into sources that directly reflect the myriad contributions of group living and social interactions in each moment of time. This is because different hosts living together in the same group, and even the same host over time, may be influenced by multiple factors at the same time. For instance, while host subject A’s microbiome may, at time t, be largely governed by social interactions because this individual was highly socially connected, host subject B’s microbiome may be largely explained by diet because this individual was socially isolated. Longitudinal data coupled with new statistical models (e.g., modifications of state space models, see Box 3) will be essential to attribute variance to different factors driving microbiome change and to decouple the direct effects of social interactions from the many correlates and confounds of social relationships and group living.
With respect to the second advance, longitudinal analyses offer more direct approaches for testing the role of microbial dispersal between social partners by tracking the spread of microbial strains within socially structured populations over time. Just as disease ecologists use time series data to trace the appearance of new infectious diseases within populations (see e.g., Craft et al., 2010; Eames, 2007), primate studies could combine strain tracking procedures (Smillie et al., 2018; Nayfach et al., 2016; Brito & Alm, 2016; Oh et al., 2014; Morowitz et al., 2011) with information on social networks and events that rewire these networks (e.g., fission or fusion of social groups, births and deaths, and sex-biased dispersal) to identify microbial strains of interest (e.g., commensal and/or pathogenic) and to test whether the network position of an infected host predicts the rate at which those strains colonize other hosts. In non-human primates that live in fission-fusion societies (Aureli et al., 2008), the dynamics of social networks could be leveraged to test predictions about the role of physical contact and proximity in strain sharing. While strain tracking is sometimes attempted with 16S rRNA sequencing data (Knights et al., 2011), these data often lack the phylogenetic resolution to truly trace microbial transmission at the strain level. Hence, we advocate the use population genetics approaches to measure microbial migration or dispersal between hosts. Such approaches rely on identity by common descent and require more extensive genetic data, generated by either shotgun metagenomic sequencing or whole microbial genome sequencing from cultured microbes (Smillie et al., 2018; Asnicar et al., 2017). These methods are becoming increasingly common and affordable and have been used to follow the establishment of microbes in the human gut after fecal transplant (Smillie et al., 2018) or trace vertical microbial transmission between mothers and infants (Smillie et al., 2018; Asnicar et al., 2017).
With respect to the third advance, time series data on multiple hosts will be essential to apply metacommunity theory to understand social effects on primate microbiomes at multiple scales. The microbiomes of individual subjects can be considered local communities connected by microbial dispersal, which is mediated by host social interactions. This perspective could be useful, e.g., to learn how stability at the local level scales up to the metacommunity level (host social groups or populations; Figure 3). Local communities differ in how they respond to environmental fluctuations, such as rain and drought. How much local communities vary with respect to one another determines the level of synchrony at the metacommunity level. Just as low synchrony among species stabilizes local communities (Figure 3A–B; Loreau, 2010), asynchrony among local communities can increase stability at the metacommunity level (Figure 3C; Wilcox et al., 2017). A recent analysis on the colony forming Egyptian fruit bat found that while temporal changes in the fur microbiome were best described at the colony rather than the individual level, this was not the case for the gut microbiome (Kolodny et al., 2019). Perhaps the common finding of strong individual signatures in the gut microbiome of humans and other animals reflects a low degree of synchrony, which may be critical for metacommunity stability and functioning. Overall, how the fluctuations of microbiomes in individual hosts contribute to stability and functioning of the gut microbiome at higher levels, including families, social groups and populations, is completely unknown.
3.3. Are gut microbiome phenotypes heritable across dynamic environments?
In humans, host genetic variation predicts microbiome composition, and microbe-by-host genotype associations predict health outcomes (Goodrich et al., 2017; Blekhman et al., 2015). Likewise in non-human primates, host genetic effects may explain the existence of host species-specific gut microbiomes (Amato et al., 2016; Moeller et al., 2014; Yildirim et al., 2010), and the observation that gut microbial similarity recapitulates host phylogenetic relationships (Ochman et al., 2010; Moeller, Caro-Quintero, et al., 2016). However, host and environmental factors, such as diet, social behavior, and season, may also play a strong role in creating these patterns (Yatsunenko et al., 2012; Bailey et al., 2011). Indeed, sympatric primate species have more similar microbiomes than allopatric species (McCord et al., 2014; Moeller et al., 2013), and species-specific microbiomes are absent in at least one primate hybrid zone (Grieneisen et al., 2018). Results like these highlight a key challenge: how do we measure microbiome heritability? That is, how do we disentangle host genetic effects from environmental effects on microbiomes in natural primate populations, especially when environments and microbiomes can both change over time?
To date there is considerable evidence that host genetics can contribute to several microbiome phenotypes, including gut microbial community composition, microbial richness, and the relative abundance of certain gut microbes (Golder et al., 2018; Busby et al., 2017; Blekhman et al., 2015; E. Li et al., 2012; Spor et al., 2011; Turnbaugh et al., 2008). These effects may arise through indirect genetic effects on host behavior and diet, such as lactose tolerance in adulthood, or more directly by host genetic effects on, for example, gut motility, cell-to-cell signaling, the permeability of intestinal epithelial cells, stomach acidity, and insulin secretion (Kreznar et al., 2017; Beasley et al., 2015; Blekhman et al., 2015; Davenport et al., 2015; Zhao et al., 2013; Spor et al., 2011; Rawls et al., 2007). However, measuring the heritability of gut microbiome phenotypes is difficult for at least three reasons. First, genetic and environmental effects are frequently confounded (Wagner et al., 2016). Relatives are often close social partners, and as a result, they may share similar environments and consume similar diets; hence, in addition to being colonized by the same environmental microbes, relatives may also exert similar selective regimes in their guts (Tung et al., 2015; Song et al., 2013). Likewise, if heritability is measured by comparing phenotype similarity in parents and offspring, this can be confounded if parents directly transmit microbes to offspring via vertical transmission (Davenport et al., 2015). Second, the magnitude of environmental effects can swamp host genetic effects. Indeed, a recent study found that the effects of host genetic ancestry were undetectable compared to environmental effects (Rothschild et al., 2018). Third, gut microbiome phenotypes are complex and dynamic, changing with short-term fluctuations in diet (David et al., 2014) and long-term shifts between seasons (Hicks et al., 2018; Amato et al., 2016), and as the host ages (Bennett et al., 2016; Ren et al., 2016; Yatsunenko et al., 2012). As a result, the phenotype in question may change considerably over time within single individuals.
Given the confounds and complexities in detecting heritable microbiome phenotypes, we propose three questions that individual-based primate studies are well-suited to answer. The first question is: which microbiome phenotypes are repeatable within an individual host over time? Prior studies have shown that individual hosts exhibit distinctive, persistent gut microbial communities (Degnan et al., 2012; Caporaso et al., 2011; Turnbaugh et al., 2008). However, many of these studies had durations less than one year, raising the question: how long are these signatures maintained?
By looking over longer time periods, even as long as host lifespans, it is possible test which microbial phenotypes persist through changing social and physical environments. Some primate projects have years of banked fecal samples from known subjects, providing individual-based longitudinal data that are not available in other study systems (Guevara et al., 2017; Moeller, Foerster, et al., 2016; Alberts & Altmann, 2012; Kappeler & Watts, 2012). Establishing the repeatability of a trait (i.e., its consistency within an individual over time) is important because it reflects the upper limit of a trait’s heritability; that is, if variation in a phenotype has low repeatability, it will also have low heritability (Wolak et al., 2011; Boake, 1989; Falconer, 1960). Hence, pinpointing repeatable microbial phenotypes may suggest which phenotypes might be most heritable.
Second, can we detect heritable gut microbial phenotypes, controlling for local environmental conditions? Long-term primate studies possess several advantages in both defining heritability and in quantifying environmental traits. To date, most studies attempting to measure host genetic effects on the microbiome have used either twin studies (Gomez et al., 2017; Goodrich et al., 2016; Turnbaugh et al., 2008), comparisons between populations (Morton et al., 2015; Yatsunenko et al., 2012), or selectively bred livestock lines (Zhao et al., 2013). Primate studies that have pedigree data permit a more powerful approach, as phenotypes can be tracked through families and over multiple generations. In addition, sex-biased dispersal in many non-human primates breaks up tight correlations between genetic relatedness and shared environments. In primates with female-biased dispersal, for instance, maternal half-siblings in different social groups can be compared to identify maternal effects. More generally, longitudinal samples from dispersing individuals may provide insight into heritability across changing environments. Finally, longitudinal data on individual hosts with known birth dates can be used to control for host age. This is important because health-related phenotypes in hosts, such as body weight, blood pressure, and basal metabolic rate, have heritability values that decrease with age because environmental variation swamps genetic effects over time (Ge et al., 2017). By dividing primate datasets into age classes, we can test if microbiome phenotypes likewise demonstrate an age-related decline in heritability, and if this decline could have potential health consequences.
Third and finally, can we determine if heritable microbial species are shared among closely related primate species? Gut microbiome similarity between hosts often reflects host phylogenetic relationships (J. Li et al., 2018; Kropácková et al., 2017; Ochman et al., 2010; Ley et al., 2008); hence, related host species may likewise share more heritable microbial species than expected by chance, especially if heritable microbes are beneficial to hosts. Primates provide a particularly interesting system to test ideas about host-microbe coevolution and heritability because of the variety of environments primates thrive in (Smuts et al., 1987). Humans in particular face different selection pressures than their wild relatives (e.g., live in more artificially constructed environments, consume different diets, etc.), raising the possibility that, counter to the dominant paradigm, humans might not share more heritable microbial species with closely related primates than they do with their more distant relatives. Understanding which gut microbial phenotypes are heritable in wild primates, and which heritable phenotypes are consistent through fluctuating environments and across primate lineages will shed insight on how host selection affects the gut microbiome.
3.4. Does the gut microbiome show signs of host aging?
Across primates, and indeed across the tree of life, most physical systems–from immune function, to memory and muscle mass–decline with age (Hayward et al., 2015; Nussey et al., 2013; Bronikowski et al., 2011; Altmann et al., 2010). However, whether gut microbiomes also senesce is unknown. Healthy microbiomes can change with age, and longitudinal microbiome data are one of the best ways to learn (i) if aging gut microbiomes exhibit systematic and predictable declines in the services microbiomes provide to hosts, (ii) what drives these changes, and (iii) whether those declines are reflected in generalizable markers of ecosystem function, such as gut microbiome diversity, composition, resilience, or stability (Heintz & Mair, 2014; Saraswati & Sitaraman, 2014; Biagi et al., 2012). Answering these questions is important to discover the gut microbiome’s role in age-related changes in human and animal health, to develop gut microbial interventions to improve health in old age, and to learn whether age-related changes in the gut microbiome serve as harbingers of developmental milestones, aging, and mortality (see also Section 3.5). Currently, the vast majority of research on gut microbial senescence is on human subjects (V. J. Martin et al., 2016; Gerber, 2014; Mai & Morris, 2013; Costello et al., 2012; Biagi et al., 2010). However, such research is currently hampered by two barriers that might be overcome by research on non-human primates. The first barrier is the challenge of collecting detailed microbiome time series from a large number of subjects. Time series are essential to understand aging trajectories in individual hosts, learn why some microbiomes age faster than others, measure age-related changes in gut microbial stability and resilience, and test whether gut microbial stability or variation in rates of gut microbial aging predict disease risk or longevity (V. J. Martin et al., 2016; Gerber, 2014; Mai & Morris, 2013; Costello et al., 2012). Nearly all current studies of human gut microbial aging are cross-sectional (Bian et al., 2017; Biagi et al., 2016; Odamaki et al., 2016; Claesson et al., 2011; Biagi et al., 2010). Indeed, to our knowledge, the only longitudinal study on gut microbial aging in humans has just two time points from 26 subjects, collected 3 months apart (Claesson et al., 2011).
A second barrier is the confounds created by human lifestyles, diets, and medications, which affect gut microbial composition and change as human health declines with age (Claesson et al., 2012). These confounds make it difficult to disentangle intrinsic (e.g., immunosenescence, changing gut motility and mucosal barrier function) from extrinsic (e.g. host environments and behaviors) drivers of gut microbial senescence (Saraswati & Sitaraman, 2014). Moreover, these confounds may explain why patterns of gut microbial senescence in humans are often population specific (Bian et al., 2017; Odamaki et al., 2016; Biagi et al., 2013; Claesson et al., 2011; Biagi et al., 2010; Mariat et al., 2009). For example, some studies report rising Bacteroidetes during senescence (Claesson et al., 2011; Mariat et al., 2009), while others report the opposite pattern or no trend (Bian et al., 2017; Biagi et al., 2010). Similarly, while the prevailing view is that gut microbial diversity declines in old age (Voreades et al., 2014; Biagi et al., 2013), several recent studies find that diversity either does not change or increases in the elderly compared to younger adults (Bian et al., 2017; Biagi et al., 2016; Jackson et al., 2016; Kong et al., 2016; Odamaki et al., 2016). This heterogeneity in age-related changes in diversity is especially important because gut microbial diversity is often proposed to be a marker of microbiome functional stability and host health (see Section 3.5; Costello et al., 2012). In support, low gut microbial diversity is frequently linked to high frailty scores in old age (Jackson et al., 2016; Claesson et al., 2012, 2011; van Tongeren et al., 2005). However, the causal pathways connecting host lifestyles, health, frailty, and gut microbial diversity remain completely unknown.
Wild non-human primates may be relatively free from age-related lifestyle confounds. Specifically, wild primates may not exhibit strong changes in behavior or environments as they age; unlike some elderly humans, aging primates do not live in residential care facilities or hospitals. This relative freedom from age-related changes in lifestyle, and primates’ evolutionary and ecological similarity to humans, makes them a useful model to illuminate intrinsic patterns and processes of gut microbial senescence. However, the full potential of this approach has yet to be realized. Most research on age-related differences in primate gut microbiomes compares adults to juveniles (Springer et al., 2017; Aivelo et al., 2016; Su et al., 2016; Amato et al., 2014). More rare are studies that model age as a continuous variable within adults (Mitchell et al., 2017; Ren et al., 2016; Degnan et al., 2012), and only two studies have directly compared gut microbial differences in prime age and senescent adults (Trosvik et al., 2018; Bennett et al., 2016). The picture emerging from these studies is intriguing: unlike in humans, studies on non-human primates have yet to find a relationship between old age and gut microbial diversity; instead, diversity is similar in senescent versus prime age adults (Trosvik et al., 2018; Mitchell et al., 2017; Bennett et al., 2016; Ren et al., 2016). In terms of gut microbial composition, baboons (Papio cynocephalus), vervets (Chlorcebus aethiops sabaeus), and geladas (Theropithecus gelada) all exhibit few or no effects of age on the identity and abundance of gut microbial taxa (Trosvik et al., 2018; Mitchell et al., 2017; Ren et al., 2016). Age-related compositional changes are more pronounced in ring-tailed lemurs (Lemur catta), but the effects of social group and habitat quality are much stronger (Bennett et al., 2016). Interestingly, one study of captive vervet monkeys (Chlorcebus aethiops sabaeus) found that old monkeys did not differ in gut microbial composition from younger adults, despite exhibiting several other physical differences that should affect the gut microbiome, including systemic inflammation, poor intestinal barrier function, and reduced intestinal motility (Mitchell et al., 2017).
Together, these studies provide just a glimpse into primate gut microbial senescence, but their results strongly implicate extrinsic factors unique to humans (e.g., health care, medication, assisted living), and not intrinsic factors fundamental to primate senescence in human gut microbial aging. Moreover, they suggest that studies linking gut microbial diversity to frailty in humans might arise from an indirect path (poor health leads to residential care and medications, which in turn reduce gut microbial diversity) as opposed to a direct causal path between gut microbiome diversity and host health. However, much more research is needed to resolve these causal connections. Researchers studying non-human primates are well positioned to disentangle comparative processes underlying signatures of aging in gut microbiomes. Key questions include: what features of gut microbial senescence, if any, are universal across primates? And what are the underlying processes that generate age-related changes in diversity, stability, and composition? Understanding these processes and resulting patterns is essential to design effective gut microbiome interventions to promote healthy aging, and which signatures of gut microbial senescence are effective markers of host health.
3.5. Do gut microbiome composition and dynamics predict host fitness and health?
Individual variation in gut microbiome composition has repeatedly been linked to host health (Kostic et al., 2015; Subramanian et al., 2014; Claesson et al., 2012; Greenblum et al., 2012), but the features that separate “unhealthy” from “healthy” microbiomes remain largely unknown. Differentiating healthy from unhealthy microbiomes may be useful for understanding health disparities in humans and animals, but it’s also relevant to ecology and evolution for at least two reasons. First, ecologists and evolutionary biologists are often interested in measuring an animal’s fitness, and animal “health”, including microbiome health, may serve as a useful proxy for fitness. In other words, “healthy” microbiomes may sometimes also be “fit” microbiomes. Second, community ecologists often use concepts such as stability and resilience to define community or ecosystem health, in part because these traits may predict community diversity and productivity (Rapport et al., 1998; Lehman & Tilman, 2000). Because microbiomes are themselves complex communities, stability and resilience may provide useful measures of microbiome health.
In order to test these ideas, many researchers have called for prospective, longitudinal, population-based studies (R. Martin et al., 2016; Mai & Morris, 2013). Such studies would measure microbiome composition and dynamics in a cohort of subjects–ideally at multiple time points–and then follow up with these subjects over time to test whether microbiome markers predict host health or fitness outcomes (Figure 4). Studies like these provide an essential complement to controlled experiments that manipulate microbiome composition in captive animals. This is because what population-based studies lack in experimental control, they make up for in naturalism. Importantly, microbiomes in captive primates differ considerably from their wild relatives (Hird, 2017; Clayton et al., 2016; Amato, 2013). While captive primate microbiomes may be more similar to human microbiomes, it is also challenging to re-create natural environmental, disease, dietary, and social effects in captivity that likely drive microbiome dynamics in the wild.
Figure 4:

Prospective, longitudinal sampling of gut microbial dynamics. Each row represents an individual host, and each circle represents a gut microbiome sample. The color gradient represents changes in microbial composition and/or dynamics that are proposed to be predictive of host death, represented by the skull. Darker colors represent microbiome features that predict death; samples are darkest (least healthy) before the host dies, and the host with the longest lifespan experiences a long period of microbiome health, represented by the series of samples with light colors. Short-lived hosts have samples with consistently dark colors.
Non-human primates are ideal for testing the relevance of microbiome dynamics to host health due to the availability of robust longitudinal and demographic data and their evolutionary relatedness to humans. In addition to tracking individual subjects over time, many primatologists collect data on animal health and performance, including the incidence of illness, parasite infection, observational signs of poor health, fertility, maturational milestones, and lifespan (Alberts & Altmann, 2012; Lonsdorf et al., 2006). Non-human primates also parallel humans in development and lifespan; all primates are long-lived, altricial organisms that have an extended juvenile period and relatively slow life histories. However, compared to humans, non-human primates have much shorter generation times and life spans, making it more feasible to track individuals from birth to death, and even across generations.
Studies that adopt prospective longitudinal study designs to test the relationship between microbiome composition and host health or fitness outcomes are rare. The few studies that have been done focus on health in early life. For example, a small prospective, longitudinal study conducted by Madan et al. (2012) in human infants found that premature infants who maintained high gut microbial diversity did not develop neonatal sepsis during the follow-up period. Additionally, Zhou et al. (2015) found that gut microbial composition in premature infants predicted the onset of necrotizing enterocolitis. To our knowledge, no studies have yet used a prospective, longitudinal design to connect the gut microbiome to fitness components, such as lifespan or reproductive success in a natural non-human primate population. However, experiments in several captive systems suggest there may be links here. In the short-lived killifish (Nothobranchius furzeri), for example, transferring the microbiome from captive 6-week old fish into captive antibiotic-treated 9.5 week old fish increased killifish lifespan significantly (P. Smith et al., 2017). Similarly, male Drosophila inoculated with a commensal species from the genus Lactobacillus experienced longer mating periods and higher offspring production (Morimoto et al., 2017). In contrast, Drosophila inoculated with a Acetobacter species produced daughters with significantly smaller body mass, which may reduce fecundity and other markers of fertility (Morimoto et al., 2017).
Finally, all of the studies reviewed above focus on one-time measures of either gut microbial diversity or composition as markers of healthy microbiomes. No studies in natural systems have yet used longitudinal microbiome data to test the power of gut microbial stability or resilience for predicting host health, fertility, or survival (see Box 2; Sommer et al., 2017). However, experimental studies exploring how the humanized mouse gut microbiome recovers from different types of gastrointestinal infections have found that hosts that fail to return to their original gut community conformation are less healthy than mice that exhibit gut microbial resilience (Schwab et al., 2014; Hsiao et al., 2014; Buffie et al., 2012).
Non-human primates are ideal for connecting the microbiome to host health and fitness, not only because of their evolutionary and physical similarity to humans, but also the broader evolutionary consequences of the microbiome and its role in conservation. Prospective, population-based longitudinal data from non-human primate systems will be essential to answering questions such as: how does the gut microbiome change with long term health? Are gut microbial diversity, stability, or resilience useful markers of host health or fitness? If so, why? If not, “multi-omics” approaches may uncover functional pathways or metabolic products that are useful, specific markers of host health. If the gut microbiome can be leveraged as a noninvasive biomarker of host health, such a marker would be especially powerful as it would encompass the quality of the host’s diet and environment both prior to sample collection and into the future. As such, understanding the role of the gut microbiome in health and fitness could help monitor host diet, habitat, and health noninvasively (Trevelline et al., 2019; A. G. West et al., 2019).
Conclusions and future directions
During the last two decades, major advances have been made in understanding the factors shaping the primate gut microbiome (e.g., Amato et al., 2016; Moeller, Foerster, et al., 2016; Tung et al., 2015; Amato et al., 2015; Moeller et al., 2013). However, for primates, and indeed for most other animals, research on the microbiome is largely cross-sectional or limited to only a few time points. As such, we have not yet captured the highly dynamic nature of vertebrate gut microbiomes. Measuring this dynamism and uncovering its underlying ecological processes, will require dense time series data from multiple hosts. In most vertebrates, the expense and time involved in capturing these time series data is prohibitive. In this review, we have made the argument that field-based primate studies, owing to their focus on the lives of individual animals and the relative ease of collecting longitudinal fecal samples from known animals, are better-positioned to collect these data than studies of humans or many other species. However, analyzing the resulting data will be challenging. Capturing time-evolving (i.e., dynamic) features of microbiomes across multiple subjects is both more statistically challenging and computationally intensive than analyzing cross-sectional data. However, new methods are being developed every day, and state space models may provide a particularly valuable approach (see Section 2 and Box 3).
In the future, we believe that research on microbiome dynamics will benefit from being guided by basic principles of community dynamics, summarized in the four ecological processes of dispersal, selection, drift, and speciation (Vellend, 2016; Vellend & Agrawal, 2010). Interpreting multidimensonal time series data is difficult without appropriate principles and theory. Thus viewing the microbiome through the lens of these overarching processes will facilitate and guide researchers from diverse scientific disciplines in studying primate gut microbiomes. As Koskella et al. (2017) concisely stated: “It is important that we avoid ‘reinventing the wheel’ by ignoring existing theory, but also do not blindly apply theory without understanding the important nuances of host-associated [microbiomes]” (Koskella et al., 2017, p. 1613). While the field certainly benefits from borrowing principles and theory from ecology, there is an urgent need to develop theory specific to the microbiome that incorporates the many complexities and nuances of the microbiome.
Finally, we suggest five questions that we believe are particularly fruitful avenues where longitudinal primate studies are well-positioned to fill key major gaps in microbiome dynamics. While our review largely focuses on 16S rRNA amplicon sequencing, rapid progress may entail longitudinal sampling combined with “multi-omic” approaches, such as transcriptomics, proteomics and metabolomics. We are not aware of any such longitudinal studies to date, but it is certain that such approaches will allow for a considerably higher resolution and more insight into the dynamic interplay between hosts and microbes, including the functional changes that emerge from it.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Aivelo T, Laakkonen J, & Jernvall J (2016). Population- and individual-level dynamics of the intestinal microbiota of a small primate. Applied and Environmental Microbiology, 82(12), 3537–3545. doi: 10.1128/AEM.00559-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts SC, & Altmann J (2012). The amboseli baboon research project: 40 years of continuity and change In Long-term field studies of primates (pp. 261–287). Springer Berlin Heidelberg. doi: 10.1007/978-3-642-22514-7_12 [DOI] [Google Scholar]
- Altmann J, & Alberts SC (2005). Growth rates in a wild primate population: ecological influences and maternal effects. Behavioral Ecology and Sociobiology, 57(5), 490–501. doi: 10.1007/s00265-004-0870-x [DOI] [Google Scholar]
- Altmann J, Gesquiere L, Galbany J, Onyango PO, & Alberts SC (2010). Life history context of reproductive aging in a wild primate model. Ann N Y Acad Sci, 1204, 127–138. doi: 10.1111/j.1749-6632.2010.05531.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato KR (2013). Co-evolution in context: The importance of studying gut microbiomes in wild animals. Microbiome Science and Medicine, 1(1). doi: 10.2478/micsm-2013-0002 [DOI] [Google Scholar]
- Amato KR, Leigh SR, Kent A, Mackie RI, Yeoman CJ, Stumpf RM, … Garber PA (2014). The role of gut microbes in satisfying the nutritional demands of adult and juvenile wild, black howler monkeys (alouatta pigra). American Journal of Physical Anthropology, 155(4), 652–664. doi: 10.1002/ajpa.22621 [DOI] [PubMed] [Google Scholar]
- Amato KR, Martinez-Mota R, Righini N, Raguet-Schofield M, Corcione FP, Marini E, … Leigh SR (2016). Phylogenetic and ecological factors impact the gut microbiota of two neotropical primate species. Oecologia, 180(3), 717–733. doi: 10.1007/s00442-015-3507-z [DOI] [PubMed] [Google Scholar]
- Amato KR, Van Belle S, Di Fiore A, Estrada A, Stumpf R, White B, … Leigh SR(2017). Patterns in gut microbiota similarity associated with degree of sociality among sex classes of a neotropical primate. Microbial Ecology, 74(1), 250–258. doi: 10.1007/s00248-017-0938-6 [DOI] [PubMed] [Google Scholar]
- Amato KR, Yeoman CJ, Cerda GA, Schmitt, C, Cramer JD, Miller MEB, … Leigh SR (2015). Variable responses of human and non-human primate gut microbiomes to a western diet. Microbiome, 3(1), 53. doi: 10.1186/s40168-015-0120-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnicar F, Manara S, Zolfo M, Truong DT, Scholz M, Armanini F, … Segata N (2017). Studying vertical microbiome transmission from mothers to infants by strain-level metagenomic profiling. mSystems, 2(1). doi: 10.1128/mSystems.00164-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aureli F, Schaffner CM, Boesch C, Bearder SK, Call J, Chapman CA, … Schaik C. P. v. (2008). Fission-fusion dynamics: New research frameworks. Current Anthropology, 49(4), 627–654. doi: 10.1086/586708 [DOI] [Google Scholar]
- Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, … Wang J (2015). Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host & Microbe, 17(5), 690–703. doi: 10.1016/j.chom.2015.04.004 [DOI] [PubMed] [Google Scholar]
- Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, & Lyte M (2011). Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain, Behavior, and Immunity, 25(3), 397–407. doi: 10.1016/j.bbi.2010.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MT, Lubach GR, & Coe CL (2004). Prenatal stress alters bacterial colonization of the gut in infant monkeys. Journal of Pediatric Gastroenterology and Nutrition, 38(4). doi: 10.1097/00005176-200404000-00009 [DOI] [PubMed] [Google Scholar]
- Baksi KD, Kuntal BK, & Mande SS (2018). “time”: A web application for obtaining insights into microbial ecology using longitudinal microbiome data. Frontiers in Microbiology, 9, 36. doi: 10.3389/fmicb.2018.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bálint M, Bahram M, Eren AM, Faust K, Fuhrman JA, Lindahl B, … Tedersoo L (2016). Millions of reads, thousands of taxa: microbial community structure and associations analyzed via marker genes. FEMS Microbiology Reviews, 40(5), 686–700. doi: 10.1093/femsre/fuw017 [DOI] [PubMed] [Google Scholar]
- Beasley DE, Koltz AM, Lambert JE, Fierer N, & Dunn RR (2015). The evolution of stomach acidity and its relevance to the human microbiome. PLOS ONE, 10(7), 1–12. doi: 10.1371/journal.pone.0134116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisner B, Haydon D, & Cuddington K (2003). Alternative stable states in ecology. Frontiers in Ecology and the Environment, 1(7), 376–382. doi: 10.1890/1540-9295(2003)001[0376:ASSIE]2.0.CO;2 [DOI] [Google Scholar]
- Bennett G, Malone M, Sauther ML, Cuozzo FP, White B, Nelson KE, … Amato KR (2016). Host age, social group, and habitat type influence the gut microbiota of wild ring-tailed lemurs (lemur catta). American Journal of Primatology, 78(8), 883–892. doi: 10.1002/ajp.22555 [DOI] [PubMed] [Google Scholar]
- Berens AE, Jensen SKG, & Nelson CA (2017). Biological embedding of childhood adversity: from physiological mechanisms to clinical implications. BMC Medicine, 15(1), 135. doi: 10.1186/s12916-017-0895-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger V, Lemaître J-F, Allainé D, Gaillard J-M, & Cohas A (2018). Early and adult social environments shape sex-specific actuarial senescence patterns in a cooperative breeder. The American Naturalist, 192(4), 525–536. doi: 10.1086/699513 [DOI] [PubMed] [Google Scholar]
- Berghänel A, Heistermann M, Schülke O, & Ostner J (2016). Prenatal stress effects in a wild, long-lived primate: predictive adaptive responses in an unpredictable environment. Proceedings of the Royal Society of London B: Biological Sciences, 283(1839). doi: 10.1098/rspb.2016.1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein RM, Setchell JM, Verrier D, & Knapp LA (2012). Maternal effects and the endocrine regulation of mandrill growth. American Journal of Primatology, 74(10), 890–900. doi: 10.1002/ajp.22038 [DOI] [PubMed] [Google Scholar]
- Berry D, & Widder S (2014). Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Frontiers in Microbiology, 5, 219. doi: 10.3389/fmicb.2014.00219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagi E, Candela M, Fairweather-Tait S, Franceschi C, & Brigidi P (2012). Ageing of the human metaorganism: the microbial counterpart. AGE, 34(1), 247–267. doi: 10.1007/s11357-011-9217-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagi E, Candela M, Turroni S, Garagnani P, Franceschi C, & Brigidi P (2013). Ageing and gut microbes: Perspectives for health maintenance and longevity. Pharmacological Research, 69(1), 11–20. doi: 10.1016/j.phrs.2012.10.005 [DOI] [PubMed] [Google Scholar]
- Biagi E, Franceschi C, Rampelli S, Severgnini M, Ostan R, Turroni S, … Candela M (2016). Gut microbiota and extreme longevity. Current Biology, 26(11), 1480–1485. doi: 10.1016/j.cub.2016.04.016 [DOI] [PubMed] [Google Scholar]
- Biagi E, Nylund L, Candela M, Ostan R, Bucci L, Pini E, … De Vos W (2010). Through ageing, and beyond: Gut microbiota and inflammatory status in seniors and centenarians. PLOS ONE, 5(5), e10667. doi: 10.1371/journal.pone.0010667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian G, Gloor GB, Gong A, Jia C, Zhang W, Hu J, … Li J (2017). The gut microbiota of healthy aged chinese is similar to that of the healthy young. mSphere, 2(5). doi: 10.1128/mSphere.00327-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk JR, Hui FK, O’Hara RB, & Montoya JM (2018). Uncovering the drivers of host-associated microbiota with joint species distribution modelling. Molecular Ecology, 27(12), 2714–2724. doi: 10.1111/mec.14718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk JR, O’Hara RB, Ribes M, Coma R, & Montoya JM (2018). The dynamic core microbiome: Structure, dynamics and stability. bioRxiv. doi: 10.1101/137885 [DOI] [Google Scholar]
- Blekhman R, Goodrich JK, Huang K, Sun Q, Bukowski R, Bell JT, … Clark AG (2015). Host genetic variation impacts microbiome composition across human body sites. Genome Biology, 16(1), 191. doi: 10.1186/s13059-015-0759-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boake CRB (1989). Repeatability: Its role in evolutionary studies of mating behavior. Evolutionary Ecology, 3(2), 173–182. doi: 10.1007/BF02270919 [DOI] [Google Scholar]
- Brito IL, & Alm EJ (2016). Tracking strains in the microbiome: Insights from metagenomics and models. Front Microbiol, 7, 712. doi: 10.3389/fmicb.2016.00712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronikowski AM, Altmann J, Brockman DK, Cords M, Fedigan LM, Pusey A, … Alberts SC (2011). Aging in the natural world: comparative data reveal similar mortality patterns across primates. Science, 331(6022), 1325–1328. doi: 10.1126/science.1201571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronikowski AM, Cords M, Alberts SC, Altmann J, Brockman DK, Fedigan LM, … Morris WF (2016). Female and male life tables for seven wild primate species. Scientific Data, 3, 160006 EP −. doi: 10.1038/sdata.2016.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, … Pamer EG (2012). Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to clostridium difficile-induced colitis. Infection and Immunity, 80(1), 62. doi: 10.1128/IAI.05496-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns AR, Miller E, Agarwal M, Rolig AS, Milligan-Myhre K, Seredick S, … Bohannan BJM (2017). Interhost dispersal alters microbiome assembly and can overwhelm host innate immunity in an experimental zebrafish model. Proceedings of the National Academy of Sciences, 114(42), 11181–11186. doi: 10.1073/pnas.1702511114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns AR, Stephens WZ, Stagaman K, Wong S, Rawls JF, Guillemin K, & Bohannan BJ (2015). Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. The Isme Journal, 10, 655 EP −. doi: 10.1038/ismej.2015.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby PE, Soman C, Wagner MR, Friesen ML, Kremer J, Bennett A, … Dangl JL (2017). Research priorities for harnessing plant microbiomes in sustainable agriculture. PLOS Biology, 15(3), 1–14. doi: 10.1371/journal.pbio.2001793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, … Knight R (2011). Moving pictures of the human microbiome. Genome Biology, 12(5), R50. doi: 10.1186/gb-2011-12-5-r50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaby LE (2016). Why are there lasting effects from exposure to stress during development? an analysis of current models of early stress. Physiology & Behavior, 164, 164–181. doi: 10.1016/j.physbeh.2016.05.032 [DOI] [PubMed] [Google Scholar]
- Chatfield C (2013). The analysis of time series: An introduction, sixth edition Taylor & Francis. [Google Scholar]
- Chen T, Long W, Zhang C, Liu S, Zhao L, & Hamaker BR (2017). Fiber-utilizing capacity varies in prevotellaversus bacteroides-dominated gut microbiota. Scientific Reports, 7, 2594. doi: 10.1038/s41598-017-02995-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, & Aagaard KM (2017). Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nature Medicine, 23(3), 314. doi: 10.1038/nm.4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, … O’Toole PW (2011). Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proceedings of the National Academy of Sciences, 108(Supplement 1), 4586–4591. doi: 10.1073/pnas.1000097107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson MJ, Jeffery IB, Conde S, Power SE, O’Connor EM, Cusack S, … O’Toole PW (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature, 488, 178 EP −. doi: 10.1038/nature11319 [DOI] [PubMed] [Google Scholar]
- Clarke G, O’Mahony S, Dinan T, & Cryan J (2014). Priming for health: gut microbiota acquired in early life regulates physiology, brain and behaviour. Acta Paediatrica, 103(8), 812–819. doi: 10.1111/apa.12674 [DOI] [PubMed] [Google Scholar]
- Clayton JB, Vangay P, Huang H, Ward T, Hillmann BM, Al-Ghalith GA, … Knights D(2016). Captivity humanizes the primate microbiome. Proceedings of the National Academy of Sciences, 113(37), 10376–10381. doi: 10.1073/pnas.1521835113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clutton-Brock T, & Sheldon BC (2010a). Individuals and populations: the role of long-term, individual-based studies of animals in ecology and evolutionary biology. Trends in Ecology & Evolution, 25(10), 562–573. doi: 10.1016/j.tree.2010.08.002 [DOI] [PubMed] [Google Scholar]
- Clutton-Brock T, & Sheldon BC (2010b). The seven ages of pan. Science, 327(5970), 1207–1208. doi: 10.1126/science.1187796 [DOI] [PubMed] [Google Scholar]
- Costello EK, Stagaman K, Dethlefsen L, Bohannan BJM, & Relman DA (2012). The application of ecological theory toward an understanding of the human microbiome. Science, 336(6086), 1255–1262. doi: 10.1126/science.1224203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyte KZ, Schluter J, & Foster KR (2015). The ecology of the microbiome: Networks, competition, and stability. Science, 350(6261), 663–666. doi: 10.1126/science.aad2602 [DOI] [PubMed] [Google Scholar]
- Craft ME, Volz E, Packer C, & Meyers LA (2010). Disease transmission in territorial populations: the small-world network of serengeti lions. Journal of The Royal Society Interface. doi: 10.1098/rsif.2010.0511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport ER, Cusanovich DA, Michelini K, Barreiro LB, Ober C, & Gilad Y (2015). Genome-wide association studies of the human gut microbiota. PLOS ONE, 10(11), 1–22. doi: 10.1371/journal.pone.0140301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, … Alm EJ (2014). Host lifestyle affects human microbiota on daily timescales. Genome Biology, 15(7), R89. doi: 10.1186/gb-2014-15-7-r89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, … Turnbaugh PJ (2013). Diet rapidly and reproducibly alters the human gut microbiome. Nature, 505, 559 EP −. doi: 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan PH, Pusey AE, Lonsdorf EV, Goodall J, Wroblewski EE, Wilson ML, … Ochman H (2012). Factors associated with the diversification of the gut microbial communities within chimpanzees from gombe national park. Proceedings of the National Academy of Sciences, 109(32), 13034–13039. doi: 10.1073/pnas.1110994109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, & Relman DA (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proceedings of the National Academy of Sciences, 108(Supplement 1), 4554–4561. doi: 10.1073/pnas.1000087107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didier G, Katharine ZC, Leo L, & Karoline F (2018). Microbial communities as dynamical systems. Current Opinion in Microbiology, 44, 41–49. (Microbiota) doi: 10.1016/j.mib.2018.07.004 [DOI] [PubMed] [Google Scholar]
- Diggle P, Liang K, & Zeger S (1994). Analysis of longitudinal data. Clarendon Press. [Google Scholar]
- Dini-Andreote F, & Raaijmakers JM (2018). Embracing community ecology in plant microbiome research. Trends in Plant Science, 23(6), 467–469. doi: 10.1016/j.tplants.2018.03.013 [DOI] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, & Knight R (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proceedings of the National Academy of Sciences, 107(26), 11971–11975. doi: 10.1073/pnas.1002601107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue I, Hillebrand H, Montoya JM, Petchey OL, Pimm SL, Fowler MS, … Yang Q (2016). Navigating the complexity of ecological stability. Ecology Letters, 19(9), 1172–1185. doi: 10.1111/ele.12648 [DOI] [PubMed] [Google Scholar]
- Eames K (2007). Contact tracing strategies in heterogeneous populations. Epidemiol Infect, 135(3), 443–454. doi: 10.1017/S0950268806006923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, … Gordon JI (2013). The long-term stability of the human gut microbiota. Science, 341(6141). doi: 10.1126/science.1237439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer D (1960). Introduction to quantitative genetics. Ronald Press Company. [Google Scholar]
- Faust K, Lahti L, Gonze D, de Vos M. d. W., & Raes J (2015). Metagenomics meets time series analysis: unraveling microbial community dynamics. Current Opinion in Microbiology, 25, 56–66. doi: 10.1016/j.mib.2015.04.004 [DOI] [PubMed] [Google Scholar]
- Fisher CK, & Mehta P (2014). Identifying keystone species in the human gut microbiome from metagenomic timeseries using sparse linear regression. PLOS ONE, 9(7), 1–10. doi: 10.1371/journal.pone.0102451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores GE, Caporaso JG, Henley JB, Rideout JR, Domogala D, Chase J, … Fierer N(2014). Temporal variability is a personalized feature of the human microbiome. Genome Biol, 15(12), 531. doi: 10.1186/s13059-014-0531-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeland WJ (1979). Primate social groups as biological islands. Ecology, 60(4), 719–728. doi: 10.2307/1936609 [DOI] [Google Scholar]
- Fukami T (2015). Historical contingency in community assembly: Integrating niches, species pools, and priority effects. Annual Review of Ecology, Evolution, and Systematics, 46(1), 1–23. doi: 10.1146/annurev-ecolsys-110411-160340 [DOI] [Google Scholar]
- Fukuyama J, Rumker L, Sankaran K, Jeganathan P, Dethlefsen L, Relman DA, & Holmes SP (2017). Multidomain analyses of a longitudinal human microbiome intestinal cleanout perturbation experiment. PLOS Computational Biology, 13(8), 1–29. doi: 10.1371/journal.pcbi.1005706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge T, Chen C-Y, Neale BM, Sabuncu MR, & Smoller JW (2017). Phenome-wide heritability analysis of the uk biobank. PLOS Genetics, 13(4), 1–21. doi: 10.1371/journal.pgen.1006711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber GK (2014). The dynamic microbiome. FEBS Letters, 588(22), 4131–4139. doi: 10.1016/j.febslet.2014.02.037 [DOI] [PubMed] [Google Scholar]
- Gesquiere LR, Altmann J, Archie EA, & Alberts SC (2018). Interbirth intervals in wild baboons: Environmental predictors and hormonal correlates. American Journal of Physical Anthropology, 166, 107–126. doi: 10.1002/ajpa.23407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson TE, & Gerber GK (2018). Robust and scalable models of microbiome dynamics. arXiv. [Google Scholar]
- Gloor GB, Macklaim JM, Pawlowsky-Glahn V, & Egozcue JJ (2017). Microbiome datasets are compositional: And this is not optional. Frontiers in Microbiology, 8, 2224. doi: 10.3389/fmicb.2017.02224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert JJ, Hua X, Yu G, & Shi J (2014). Diversity and composition of the adult fecal microbiome associated with history of cesarean birth or appendectomy: Analysis of the american gut project. EBioMedicine, 1(2), 167–172. doi: 10.1016/j.ebiom.2014.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder HM, Thomson JM, Denman SE, McSweeney CS, & Lean IJ (2018). Genetic markers are associated with the ruminal microbiome and metabolome in grain and sugar challenged dairy heifers. Frontiers in Genetics, 9, 62. doi: 10.3389/fgene.2018.00062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez A, Espinoza JL, Harkins DM, Leong P, Saffery R, Bockmann M, … Nelson KE (2017). Host genetic control of the oral microbiome in health and disease. Cell Host & Microbe, 22(3), 269–278.e3. doi: 10.1016/j.chom.2017.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C, … Ley RE (2016). Genetic determinants of the gut microbiome in uk twins. Cell Host & Microbe, 19(5), 731–743. doi: 10.1016/j.chom.2016.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JK, Davenport ER, Clark AG, & Ley RE (2017). The relationship between the human genome and microbiome comes into view. Annual Review of Genetics, 51(1), 413–433. doi: 10.1146/annurev-genet-110711-155532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham NS, Reich BJ, Borer ET, & Gross K (2017). Mimix: a bayesian mixed-effects model for microbiome data from designed experiments. arXiv. doi: arXiv:1703.07747 [Google Scholar]
- Greenblum S, Turnbaugh PJ, & Borenstein E (2012). Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proceedings of the National Academy of Sciences, 109(2), 594–599. doi: 10.1073/pnas.1116053109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieneisen LE, Charpentier M, Alberts S, Blekhman R, Bradburd G, Tung J, & Archie E (2018). Genes, geology, and germs: gut microbiota across a primate hybrid zone are explained by site soil properties, not host species. In review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieneisen LE, Livermore J, Alberts S, Tung J, & Archie EA (2017). Group living and male dispersal predict the core gut microbiome in wild baboons. Integr Comp Biol, 57(4), 770–785. doi: 10.1093/icb/icx046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groussin M, Mazel F, Sanders JG, Smillie CS, Lavergne S, Thuiller W, & Alm EJ (2017). Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nature Communications, 8, 14319. doi: 10.1038/ncomms14319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guevara EE, Chen-Kraus C, Jacobs RL, & Baden AL (2017). Celebrating fifty years of research at the duke lemur center. Evolutionary Anthropology: Issues, News, and Reviews, 26(2), 47–48. doi: 10.1002/evan.21521 [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Darveau RP, & Curtis MA (2012). The keystone-pathogen hypothesis. Nature Reviews Microbiology, 10, 717 EP −. doi: 10.1038/nrmicro2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale VL, Tan CL, Niu K, Yang Y, Knight R, Zhang Q, … Amato KR (2018). Diet versus phylogeny: a comparison of gut microbiota in captive colobine monkey species. Microbial Ecology, 75(2), 515–527. doi: 10.1007/s00248-017-1041-8 [DOI] [PubMed] [Google Scholar]
- Hayward AD, Moorad J, Regan CE, Berenos C, Pilkington JG, Pemberton JM, & Nussey DH (2015). Asynchrony of senescence among phenotypic traits in a wild mammal population. Exp Gerontol, 71, 56–68. doi: 10.1016/j.exger.2015.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz C, & Mair W (2014). You are what you host: Microbiome modulation of the aging process. Cell, 156(3), 408–411. doi: 10.1016/j.cell.2014.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks AL, Lee KJ, Couto-Rodriguez M, Patel J, Sinha R, Guo C, … Williams BL (2018). Gut microbiomes of wild great apes fluctuate seasonally in response to diet. Nature Communications, 9(1), 1786. doi: 10.1038/s41467-018-04204-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hird SM (2017). Evolutionary biology needs wild microbiomes. Front Microbiol, 8, 725. doi: 10.3389/fmicb.2017.00725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister EB, Gao C, & Versalovic J (2014). Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology, 146(6), 1449–1458. doi: 10.1053/j.gastro.2014.01.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao A, Ahmed AMS, Subramanian S, Griffin NW, Drewry LL, Petri WA, … Gordon JI (2014). Members of the human gut microbiota involved in recovery from vibrio cholerae infection. Nature, 515, 423 EP −. doi: 10.1038/nature13738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell S (2001). The unified neutral theory of biodiversity and biogeography. Princeton University Press. [DOI] [PubMed] [Google Scholar]
- Ives AR, & Helmus MR (2011). Generalized linear mixed models for phylogenetic analyses of community structure. Ecological Monographs, 81(3), 511–525. doi: 10.1890/10-1264.1 [DOI] [Google Scholar]
- Jackson MA, Jeffery IB, Beaumont M, Bell JT, Clark AG, Ley RE, … Steves CJ (2016). Signatures of early frailty in the gut microbiota. Genome Medicine, 8(1), 8. doi: 10.1186/s13073-016-0262-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappeler P, & Watts D (2012). Long-term field studies of primates. Springer Berlin Heidelberg. [Google Scholar]
- Kéfi S, Holmgren M, & Scheffer M (2016). When can positive interactions cause alternative stable states in ecosystems? Functional Ecology, 30(1), 88–97. doi: 10.1111/1365-2435.12601 [DOI] [Google Scholar]
- Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, … Kelley ST (2011). Bayesian community-wide culture-independent microbial source tracking. Nature Methods, 8, 761 EP −. doi: 10.1038/nmeth.1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppel AF, Wertheim JO, Barone L, Gentile N, Krizanc D, & Cohan FM (2013). Speedy speciation in a bacterial microcosm: new species can arise as frequently as adaptations within a species. The Isme Journal, 7, 1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl KD, & Dearing MD (2016). The woodrat gut microbiota as an experimental system for understanding microbial metabolism of dietary toxins. Front Microbiol, 7, 1165. doi: 10.3389/fmicb.2016.01165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodny O, Weinberg M, Reshef L, Harten L, Hefetz A, Gophna U, … Yovel Y (2019). Coordinated change at the colony level in fruit bat fur microbiomes through time. Nature Ecology & Evolution, 3(1), 116–124. doi: 10.1038/s41559-018-0731-z [DOI] [PubMed] [Google Scholar]
- Kong F, Hua Y, Zeng B, Ning R, Li Y, & Zhao J (2016). Gut microbiota signatures of longevity. Current Biology, 26(18), R832–R833. doi: 10.1016/j.cub.2016.08.015 [DOI] [PubMed] [Google Scholar]
- Korpela K, Costea P, Coelho LP, Kandels-Lewis S, Willemsen G, Boomsma DI, … Bork P (2018). Selective maternal seeding and environment shape the human gut microbiome. Genome Res, 28(4), 561–568. doi: 10.1101/gr.233940.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskella B, Hall LJ, & Metcalf CJE (2017). The microbiome beyond the horizon of ecological and evolutionary theory. Nature Ecology & Evolution, 1(11), 1606–1615. doi: 10.1038/s41559-017-0340-2 [DOI] [PubMed] [Google Scholar]
- Kostic AD, Gevers D, Siljander H, Vatanen T, Hyötyläinen T, Hämäläinen A-M, … Xavier RJ(2015). The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host & Microbe, 17(2), 260–273. doi: 10.1016/j.chom.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreznar JH, Keller MP, Traeger LL, Rabaglia ME, Schueler KL, Stapleton DS, … Rey FE (2017). Host genotype and gut microbiome modulate insulin secretion and diet-induced metabolic phenotypes. Cell Reports, 18(7), 1739–1750. doi: 10.1016/j.celrep.2017.01.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropácková L, Tesický M, Albrecht T, Kubovciak J, Cízková D, Tomásek O, … Kreisinger J (2017). Codiversification of gastrointestinal microbiota and phylogeny in passerines is not explained by ecological divergence. Molecular Ecology, 26(19), 5292–5304. doi: 10.1111/mec.14144 [DOI] [PubMed] [Google Scholar]
- Laforest-Lapointe I, & Arrieta M-C (2017). Patterns of early-life gut microbial colonization during human immune development: An ecological perspective. Front Immunol, 8, 788. doi: 10.3389/fimmu.2017.00788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen V, & Lahti L (2018). A hierarchical ornstein-uhlenbeck model for stochastic time series analysis In Duivesteijn W, Siebes A, & Ukkonen A(Eds.), Advances in intelligent data analysis xvii (pp. 188–199). Springer International Publishing. [Google Scholar]
- Langley-Evans SC (2009). Nutritional programming of disease: unravelling the mechanism. Journal of Anatomy, 215(1), 36–51. doi: 10.1111/j.1469-7580.2008.00977.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansing JS (2003). Complex adaptive systems. Annual Review of Anthropology, 32(1), 183–204. doi: 10.1146/annurev.anthro.32.061002.093440 [DOI] [Google Scholar]
- Lax S, Smith DP, Hampton-Marcell J, Owens SM, Handley KM, Scott NM, … Gilbert JA (2014). Longitudinal analysis of microbial interaction between humans and the indoor environment. Science, 345(6200), 1048–1052. doi: 10.1126/science.1254529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman CL, & Tilman D (2000). Biodiversity, stability, and productivity in competitive communities. The American Naturalist, 156(5), 534–552. doi: 10.1086/303402 [DOI] [PubMed] [Google Scholar]
- Leibold MA, Holyoak M, Mouquet N, Amarasekare P, Chase JM, Hoopes MF, … Gonzalez A (2004). The metacommunity concept: a framework for multi-scale community ecology. Ecology Letters, 7(7), 601–613. doi: 10.1111/j.1461-0248.2004.00608.x [DOI] [Google Scholar]
- Levin SA (1998). Ecosystems and the biosphere as complex adaptive systems. Ecosystems, 1(5), 431–436. doi: 10.1007/s100219900037 [DOI] [Google Scholar]
- Ley RE, Hamady M, Lozupone C, Turnbaugh P, Ramey RR, Bircher JS, … Gordon JI (2008). Evolution of mammals and their gut microbes. Science, 320(5883), 1647–1651. doi: 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Hamm CM, Gulati AS, Sartor RB, Chen H, Wu X, … Frank DN (2012). Inflammatory bowel diseases phenotype, c. difficile and nod2 genotype are associated with shifts in human ileum associated microbial composition. PLOS ONE, 7(6), 1–10. doi: 10.1371/journal.pone.0026284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhan S, Liu X, Lin Q, Jiang J, & Li X (2018). Divergence of fecal microbiota and their associations with host phylogeny in cervinae. Frontiers in Microbiology, 9, 1823. doi: 10.3389/fmicb.2018.01823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström J (1999). Early development and fitness in birds and mammals. Trends in Ecology & Evolution, 14(9), 343–348. doi: 10.1016/S0169-5347(99)01639-0 [DOI] [PubMed] [Google Scholar]
- Lonsdorf EV, Travis D, Pusey AE, & Goodall J (2006). Using retrospective health data from the gombe chimpanzee study to inform future monitoring efforts. American Journal of Primatology, 68(9), 897–908. doi: 10.1002/ajp.20296 [DOI] [PubMed] [Google Scholar]
- Loreau M (2010). From populations to ecosystems: theoretical foundations for a new ecological synthesis. Princeton University Press. [Google Scholar]
- Lovell D, Pawlowsky-Glahn V, Egozcue JJ, Marguerat S, & Bähler J (2015). Proportionality: A valid alternative to correlation for relative data. PLOS Computational Biology, 11(3), e1004075. doi: 10.1371/journal.pcbi.1004075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur R, & Wilson E (1967). The theory of island biogeography. Princeton University Press. [Google Scholar]
- Madan JC, Salari RC, Saxena D, Davidson L, O’Toole GA, Moore JH, … Hibberd PL (2012). Gut microbial colonisation in premature neonates predicts neonatal sepsis. Archives of Disease in Childhood - Fetal and Neonatal Edition, 97(6), F456. doi: 10.1136/fetalneonatal-2011-301373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai V, & Morris JG Jr. (2013). Need for prospective cohort studies to establish human gut microbiome contributions to disease risk. JNCI: Journal of the National Cancer Institute, 105(24), 1850–1851. doi: 10.1093/jnci/djt349 [DOI] [PubMed] [Google Scholar]
- Mariat D, Firmesse O, Levenez F, Guimaraes VD, Sokol H, Doré J, … Furet J-P (2009). The firmicutes/bacteroidetes ratio of the human microbiota changes with age. BMC Microbiology, 9(1), 123. doi: 10.1186/1471-2180-9-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham AC, Santymire RM, Lonsdorf EV, Heintz MR, Lipende I, & Murray CM (2014). Rank effects on social stress in lactating chimpanzees. Animal Behaviour, 87, 195–202. doi: 10.1016/j.anbehav.2013.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R, Makino H, Cetinyurek Yavuz A, Ben-Amor K, Roelofs M, Ishikawa E, … Knol J (2016). Early-life events, including mode of delivery and type of feeding, siblings and gender, shape the developing gut microbiota. PLOS ONE, 11(6), 1–30. doi: 10.1371/journal.pone.0158498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin VJ, Leonard MM, Fiechtner L, & Fasano A (2016). Transitioning from descriptive to mechanistic understanding of the microbiome: The need for a prospective longitudinal approach to predicting disease. The Journal of Pediatrics, 179, 240–248. doi: 10.1016/j.jpeds.2016.08.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez I, Maldonado-Gomez MX, Gomes-Neto JC, Kittana H, Ding H, Schmaltz R, … Walter J (2018). Experimental evaluation of the importance of colonization history in early-life gut microbiota assembly. eLife, 7, e36521. doi: 10.7554/eLife.36521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May RM (1975). Biological populations obeying difference equations: Stable points, stable cycles, and chaos. Journal of Theoretical Biology, 51(2), 511–524. doi: 10.1016/0022-5193(75)90078-8 [DOI] [PubMed] [Google Scholar]
- May RM (1977). Thresholds and breakpoints in ecosystems with a multiplicity of stable states. Nature, 269, 471 EP −. doi: 10.1038/269471a0 [DOI] [Google Scholar]
- McCord ALEIAI, CHAPMAN CA, WENY G, TUMUKUNDE A, HYEROBA D, KLOTZ K, …GOLDBERG TL (2014). Fecal microbiomes of non-human primates in western uganda reveal species-specific communities largely resistant to habitat perturbation. Am J Primatol, 76(4), 347–354. doi: 10.1002/ajp.22238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenney EA, Koelle K, Dunn RR, & Yoder AD (2018). The ecosystem services of animal microbiomes. Molecular Ecology, 27(8), 2164–2172. doi: 10.1111/mec.14532 [DOI] [PubMed] [Google Scholar]
- McKenney EA, Rodrigo A, & Yoder AD (2015). Patterns of gut bacterial colonization in three primate species. PLOS ONE, 10(5), 1–18. doi: 10.1371/journal.pone.0124618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metwally AA, Yang J, Ascoli C, Dai Y, Finn PW, & Perkins DL (2018). Metalonda: a flexible r package for identifying time intervals of differentially abundant features in metagenomic longitudinal studies. Microbiome, 6(1), 32–32. doi: 10.1186/s40168-018-0402-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell E, Davis A, Brass K, Dendinger M, Barner R, Gharaibeh R, … Kavanagh K (2017). Reduced intestinal motility, mucosal barrier function, and inflammation in aged monkeys. J Nutr Health Aging, 21(4), 354–361. doi: 10.1007/s12603-016-0725-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller AH, Caro-Quintero A, Mjungu D, Georgiev AV, Lonsdorf EV, Muller MN, … Ochman H (2016). Cospeciation of gut microbiota with hominids. Science, 353(6297), 380–382. doi: 10.1126/science.aaf3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller AH, Foerster S, Wilson ML, Pusey AE, Hahn BH, & Ochman H (2016). Social behavior shapes the chimpanzee pan-microbiome. Science Advances, 2(1). doi: 10.1126/sciadv.1500997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller AH, Li Y, Mpoudi Ngole E, Ahuka-Mundeke S, Lonsdorf EV, Pusey AE, … Ochman H (2014). Rapid changes in the gut microbiome during human evolution. Proceedings of the National Academy of Sciences, 111(46), 16431–16435. doi: 10.1073/pnas.1419136111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller AH, Peeters M, Ndjango J-B, Li Y, Hahn BH, & Ochman H (2013). Sympatric chimpanzees and gorillas harbor convergent gut microbial communities. Genome Res, 23(10), 1715–1720. doi: 10.1101/gr.154773.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller AH, Suzuki TA, Lin D, Lacey EA, Wasser SK, & Nachman MW (2017). Dispersal limitation promotes the diversification of the mammalian gut microbiota. Proceedings of the National Academy of Sciences, 114(52), 13768–13773. doi: 10.1073/pnas.1700122114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SE, Cole TJ, Poskitt EME, Sonko BJ, Whitehead RG, McGregor IA, & Prentice AM (1997). Season of birth predicts mortality in rural gambia. Nature, 388, 434 EP −. [DOI] [PubMed] [Google Scholar]
- Moore SE, Fulford AJ, Streatfield PK, Persson L. å., & Prentice AM (2004). Comparative analysis of patterns of survival by season of birth in rural bangladeshi and gambian populations. International Journal of Epidemiology, 33(1), 137–143. doi: 10.1093/ije/dyh007 [DOI] [PubMed] [Google Scholar]
- Morimoto J, Simpson SJ, & Ponton F (2017). Direct and trans-generational effects of male and female gut microbiota in drosophila melanogaster. Biology Letters, 13(7). doi: 10.1098/rsbl.2016.0966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin PA, Wallis J, Moore JJ, Chakraborty R, & Woodruff DS (1993). Non-invasive sampling and dna amplification for paternity exclusion, community structure, and phylogeography in wild chimpanzees. Primates, 34(3), 347–356. doi: 10.1007/BF02382630 [DOI] [Google Scholar]
- Morowitz MJ, Denef VJ, Costello EK, Thomas BC, Poroyko V, Relman DA, & Banfield JF (2011). Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proceedings of the National Academy of Sciences, 108(3), 1128–1133. doi: 10.1073/pnas.1010992108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton ER, Lynch J, Froment A, Lafosse S, Heyer E, Przeworski M, … Ségurel L (2015). Variation in rural african gut microbiota is strongly correlated with colonization by entamoeba and subsistence. PLOS Genetics, 11(11), 1–28. doi: 10.1371/journal.pgen.1005658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Klein N, Heistermann M, Strube C, Morbach Z, Lilie N, Franz M, … Ostner J (2018). Physiological and social consequences of gastrointestinal nematode infection in a nonhuman primate. Behavioral Ecology. doi: 10.1093/beheco/ary168 [DOI] [Google Scholar]
- Murray CM, Stanton MA, Wellens KR, Santymire RM, Heintz MR, & Lonsdorf EV (2018). Maternal effects on offspring stress physiology in wild chimpanzees. American Journal of Primatology, 80(1), e22525. doi: 10.1002/ajp.22525 [DOI] [PubMed] [Google Scholar]
- Nayfach S, Rodriguez-Mueller B, Garud N, & Pollard KS (2016). An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res, 26(11), 1612–1625. doi: 10.1101/gr.201863.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussey DH, Froy H, Lemaitre J-F, Gaillard J-M, & Austad SN (2013). Senescence in natural populations of animals: Widespread evidence and its implications for bio-gerontology. Ageing Research Reviews, 12(1), 214–225. doi: 10.1016/j.arr.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Worobey M, Kuo C-H, Ndjango J-BN, Peeters M, Hahn BH, & Hugenholtz P (2010). Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLOS Biology, 8(11), 1–8. doi: 10.1371/journal.pbio.1000546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odamaki T, Kato K, Sugahara H, Hashikura N, Takahashi S, Xiao J. z., … Osawa R (2016). Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiology, 16(1), 90. doi: 10.1186/s12866-016-0708-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Byrd CCS, Allyson Land Deming, Program, N. C. S., Barnabas B, Blakesley R, Bouffard G, … Segre JA (2014). Biogeography and individuality shape function in the human skin metagenome. Nature, 514, 59 EP −. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyango PO, Gesquiere LR, Wango EO, Alberts SC, & Altmann J (2008). Persistence of maternal effects in baboons: Mother’s dominance rank at son’s conception predicts stress hormone levels in subadult males. Hormones and Behavior, 54(2), 319–324. doi: 10.1016/j.yhbeh.2008.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine RT (1969). A note on trophic complexity and community stability. The American Naturalist, 103(929), 91–93. doi: 10.1086/282586 [DOI] [Google Scholar]
- Perofsky AC, Lewis RJ, Abondano LA, Di Fiore A, & Meyers LA (2017). Hierarchical social networks shape gut microbial composition in wild verreaux’s sifaka. Proceedings of the Royal Society of London B: Biological Sciences, 284(1868). doi: 10.1098/rspb.2017.2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilosof S, Porter MA, Pascual M, & Kéfi S (2017). The multilayer nature of ecological networks. Nature Ecology &Amp; Evolution, 1, 0101 EP −. doi: 10.1038/s41559-017-0101 [DOI] [PubMed] [Google Scholar]
- Pimm SL (1984). The complexity and stability of ecosystems. Nature, 307, 321 EP −. doi: 10.1038/307321a0 [DOI] [Google Scholar]
- Pimm SL (1991). The balance of nature?: Ecological issues in the conservation of species and communities. University of Chicago Press. [Google Scholar]
- Preston FW (1948). The commonness, and rarity, of species. Ecology, 29(3), 254–283. doi: 10.2307/1930989 [DOI] [Google Scholar]
- Rapport DJ, Costanza R, & McMichael AJ (1998). Assessing ecosystem health. Trends in Ecology & Evolution, 13(10), 397–402. [DOI] [PubMed] [Google Scholar]
- Rawls JF, Mahowald MA, Goodman AL, Trent CM, & Gordon JI (2007). In vivo imaging and genetic analysis link bacterial motility and symbiosis in the zebrafish gut. Proceedings of the National Academy of Sciences, 104(18), 7622–7627. doi: 10.1073/pnas.0702386104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren T, Grieneisen LE, Alberts SC, Archie EA, & Wu M (2016). Development, diet, and dynamism: longitudinal and cross-sectional predictors of gut microbial communities in wild baboons. Environ Microbiol, 18(5), 1312–1325. doi: 10.1111/1462-2920.12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridenhour BJ, Brooker SL, Williams JE, Van Leuven JT, Miller AW, Dearing MD, & Remien CH (2017). Modeling time-series data from microbial communities. The Isme Journal, 11, 2526 EP −. doi: 10.1038/ismej.2017.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, … Segal E (2018). Environment dominates over host genetics in shaping human gut microbiota. Nature, 555, 210 EP −. [DOI] [PubMed] [Google Scholar]
- Saraswati S, & Sitaraman R (2014). Aging and the human gut microbiota–from correlation to causality. Front Microbiol, 5, 764. doi: 10.3389/fmicb.2014.00764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, Berry D, Rauch I, Rennisch I, Ramesmayer J, Hainzl E, … Urich T (2014). Longitudinal study of murine microbiota activity and interactions with the host during acute inflammation and recovery. ISME J, 8(5), 1101–1114. doi: 10.1038/ismej.2013.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silk JB, Beehner JC, Bergman TJ, Crockford C, Engh AL, Moscovice LR, … Cheney DL (2009). The benefits of social capital: close social bonds among female baboons enhance offspring survival. Proceedings of the Royal Society of London B: Biological Sciences, 276(1670), 3099–3104. doi: 10.1098/rspb.2009.0681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JD, Durand H, Bloom RJ, Mukherjee S, & David LA (2018). Dynamic linear models guide design and analysis of microbiota studies within artificial human guts. bioRxiv. doi: 10.1101/306597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JD, Roche K, Mukherjee S, & David LA (2018). Naught all zeros in sequence count data are the same. bioRxiv. doi: 10.1101/477794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JD, Shenhav L, Halperin EA, Mukherjee SA, & David LA (2018). Statistical considerations in the design and analysis of longitudinal microbiome studies. bioRxiv. doi: 10.1101/448332 [DOI] [Google Scholar]
- Smillie CS, Sauk J, Gevers D, Friedman J, Sung J, Youngster I, … Alm EJ (2018). Strain tracking reveals the determinants of bacterial engraftment in the human gut following fecal microbiota transplantation. Cell Host & Microbe, 23(2), 229–240.e5. doi: 10.1016/j.chom.2018.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, … Gordon JI (2013). Gut microbiomes of malawian twin pairs discordant for kwashiorkor. Science, 339(6119), 548. doi: 10.1126/science.1229000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P, Willemsen D, Popkes M, Metge F, Gandiwa E, Reichard M, & Valenzano DR (2017). Regulation of life span by the gut microbiota in the short-lived african turquoise killifish. eLife, 6, e27014. doi: 10.7554/eLife.27014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smuts B, Cheney D, Seyfarth R, & Wrangham R (1987). Primate societies. University of Chicago Press. [Google Scholar]
- Snyder-Mackler N, Majoros WH, Yuan ML, Shaver AO, Gordon JB, Kopp GH, … Tung J (2016). Efficient genome-wide sequencing and low-coverage pedigree analysis from noninvasively collected samples. Genetics, 203, 699–714. doi: 10.1534/genetics.116.187492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer F, Anderson JM, Bharti R, Raes J, & Rosenstiel P (2017). The resilience of the intestinal microbiota influences health and disease. Nature Reviews Microbiology, 15, 630 EP −. doi: 10.1038/nrmicro.2017.58 [DOI] [PubMed] [Google Scholar]
- Song SJ, Lauber C, Costello EK, Lozupone CA, Humphrey G, Berg-Lyons D, … Knight R (2013). Cohabiting family members share microbiota with one another and with their dogs. eLife, 2, e00458. doi: 10.7554/eLife.00458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spor A, Koren O, & Ley R (2011). Unravelling the effects of the environment and host genotype on the gut microbiome. Nature Reviews Microbiology, 9, 279 EP −. [DOI] [PubMed] [Google Scholar]
- Springer A, Fichtel C, Al-Ghalith GA, Koch F, Amato KR, Clayton JB, … Kappeler PM (2017). Patterns of seasonality and group membership characterize the gut microbiota in a longitudinal study of wild verreaux’s sifakas (propithecus verreauxi). Ecology and Evolution, 7(15), 5732–5745. doi: 10.1002/ece3.3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprockett D, Fukami T, & Relman DA (2018). Role of priority effects in the early-life assembly of the gut microbiota. Nature Reviews Gastroenterology &Amp; Hepatology, 15, 197 EP −. doi: 10.1038/nrgastro.2017.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strier KB, Altmann J, Brockman DK, Bronikowski AM, Cords M, Fedigan LM, … Alberts SC (2010). The primate life history database: a unique shared ecological data resource. Methods in Ecology and Evolution, 1(2), 199–211. doi: 10.1111/j.2041-210X.2010.00023.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart MD, & Strier KB (1995). Primates and parasites: A case for a multidisciplinary approach. International Journal of Primatology, 16(4), 577–593. doi: 10.1007/BF02735282 [DOI] [Google Scholar]
- Su C, Zuo R, Liu W, Sun Y, Li Z, Jin X, … Zhang H (2016). Fecal bacterial composition of sichuan snub-nosed monkeys (rhinopithecus roxellana). International Journal of Primatology, 37(4), 518–533. doi: 10.1007/s10764-016-9918-9 [DOI] [Google Scholar]
- Suarez-Jimenez B, Hathaway A, Waters C, Vaughan K, Suomi SJ, Noble PL, … Nelson EE (2013). Effect of mother’s dominance rank on offspring temperament in infant rhesus monkeys (macaca mulatta). American Journal of Primatology, 75(1), 65–73. doi: 10.1002/ajp.22081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, … Gordon JI (2014). Persistent gut microbiota immaturity in malnourished bangladeshi children. Nature, 510, 417 EP −. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Gu Z, Wang X, Huffman MA, Garber PA, Sheeran LK, … Li J.-h. (2018). Season, age, and sex affect the fecal mycobiota of free-ranging tibetan macaques (macaca thibetana). American Journal of Primatology, 80(7), e22880. doi: 10.1002/ajp.22880 [DOI] [PubMed] [Google Scholar]
- Surbeck M, Mundry R, & Hohmann G (2010). Mothers matter! maternal support, dominance status and mating success in male bonobos (pan paniscus). Proceedings of the Royal Society of London B: Biological Sciences. doi: 10.1098/rspb.2010.1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Székely AJ, & Langenheder S (2014). The importance of species sorting differs between habitat generalists and specialists in bacterial communities. FEMS Microbiology Ecology, 87(1), 102–112. doi: 10.1111/1574-6941.12195 [DOI] [PubMed] [Google Scholar]
- Tilman D (1999). The ecological consequences of changes in biodiversity: A search for general principles. Ecology, 80(5), 1455–1474. doi: 10.1890/0012-9658(1999)080[1455:TECOCI]2.0.CO;2 [DOI] [Google Scholar]
- Trevelline BK, Fontaine SS, Hartup BK, & Kohl KD (2019). Conservation biology needs a microbial renaissance: a call for the consideration of host-associated microbiota in wildlife management practices. Proceedings of the Royal Society B, 286(1895), 20182448. doi: 10.1098/rspb.2018.2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trosvik P, & de Muinck EJ (2015). Ecology of bacteria in the human gastrointestinal tract–identification of keystone and foundation taxa. Microbiome, 3(1), 44. doi: 10.1186/s40168-015-0107-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trosvik P, de Muinck EJ, Rueness EK, Fashing PJ, Beierschmitt EC, Callingham KR, … Nguyen N (2018). Multilevel social structure and diet shape the gut microbiota of the gelada monkey, the only grazing primate. Microbiome, 6(1), 84. doi: 10.1186/s40168-018-0468-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsilimigras MC, & Fodor AA (2016). Compositional data analysis of the microbiome: fundamentals, tools, and challenges. Annals of Epidemiology, 26(5), 330–335. doi: 10.1016/j.annepidem.2016.03.002 [DOI] [PubMed] [Google Scholar]
- Tung J, Barreiro LB, Burns MB, Grenier J-C, Lynch J, Grieneisen LE, … Archie EA (2015). Social networks predict gut microbiome composition in wild baboons. eLife, 4, e05224. doi: 10.7554/eLife.05224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, … Gordon JI (2008). A core gut microbiome in obese and lean twins. Nature, 457, 480 EP −. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tongeren SP, Slaets JPJ, Harmsen HJM, & Welling GW (2005). Fecal microbiota composition and frailty. Applied and Environmental Microbiology, 71(10), 6438. doi: 10.1128/AEM.71.10.6438-6442.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellend M (2016). The theory of ecological communities. Princeton University Press. [Google Scholar]
- Vellend M, & Agrawal A (2010). Conceptual synthesis in community ecology. The Quarterly Review of Biology, 85 ER(2), 183–206. doi: 10.1086/652373 [DOI] [PubMed] [Google Scholar]
- Voreades N, Kozil A, & Weir TL (2014). Diet and the development of the human intestinal microbiome. Frontiers in Microbiology, 5, 494. doi: 10.3389/fmicb.2014.00494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner MR, Lundberg DS, del Rio TG, Tringe SG, Dangl JL, & Mitchell-Olds T (2016). Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nature Communications, 7, 12151 EP −. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, & Ley R (2011). The human gut microbiome: Ecology and recent evolutionary changes. Annual Review of Microbiology, 65(1), 411–429. doi: 10.1146/annurev-micro-090110-102830 [DOI] [PubMed] [Google Scholar]
- Warton DI, Blanchet FG, O’Hara RB, Ovaskainen O, Taskinen S, Walker SC, & Hui FK (2015). So Many Variables: Joint Modeling in Community Ecology. Trends in Ecology and Evolution, 30, 1–14. doi: 10.1016/j.tree.2015.09.007 [DOI] [PubMed] [Google Scholar]
- Watts DJ, & Strogatz SH (1998). Collective dynamics of ‘small-world’ networks. Nature, 393, 440 EP −. doi: 10.1038/30918 [DOI] [PubMed] [Google Scholar]
- Weiss S, Van Treuren W, Lozupone C, Faust K, Friedman J, Deng Y, … Knight R (2016). Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J, 10(7), 1669–1681. doi: 10.1038/ismej.2015.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AG, Waite DW, Deines P, Bourne DG, Digby A, McKenzie VJ, & Taylor MW (2019). The microbiome in threatened species conservation. Biological Conservation, 229, 85–98. doi: 10.1016/j.biocon.2018.11.016 [DOI] [Google Scholar]
- West M, & Harrison J (1989). Bayesian forecasting and dynamic models. Springer. [Google Scholar]
- Whitten PL, Brockman DK, & Stavisky RC (1998). Recent advances in noninvasive techniques to monitor hormone-behavior interactions. American Journal of Physical Anthropology, 107(27), 1–23. doi: [DOI] [PubMed] [Google Scholar]
- Wilcox KR, Tredennick AT, Koerner SE, Grman E, Hallett LM, Avolio ML, … Zhang Y (2017). Asynchrony among local communities stabilises ecosystem function of metacommunities. Ecology Letters, 20(12), 1534–1545. doi: 10.1111/ele.12861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolak ME, Fairbairn DJ, & Paulsen YR (2011). Guidelines for estimating repeatability. Methods in Ecology and Evolution, 3(1), 129–137. doi: 10.1111/j.2041-210X.2011.00125.x [DOI] [Google Scholar]
- Wray N, & Visscher P (2008). Estimating trait heritability. Nature Education, 1(1), 29. doi: 10.1038/nrg2322 [DOI] [Google Scholar]
- Yassour M, Vatanen T, Siljander H, Hämäläinen A-M, Härkönen T, Ryhänen SJ, … Xavier RJ (2016). Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Science Translational Medicine, 8(343), 343ra81–343ra81. doi: 10.1126/scitranslmed.aad0917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, … Gordon JI (2012). Human gut microbiome viewed across age and geography. Nature, 486, 222 EP −. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim S, Yeoman CJ, Sipos M, Torralba M, Wilson BA, Goldberg TL, … Nelson KE (2010). Characterization of the fecal microbiome from non-human wild primates reveals species specific microbial communities. PLOS ONE, 5(11), 1–11. doi: 10.1371/journal.pone.0013963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wang G, Siegel P, He C, Wang H, Zhao W, … Meng H (2013). Quantitative genetic background of the host influences gut microbiomes in chickens. Scientific Reports, 3, 1163 EP −. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Shan G, Sodergren E, Weinstock G, Walker WA, & Gregory KE (2015). Longitudinal analysis of the premature infant intestinal microbiome prior to necrotizing enterocolitis: A case-control study. PLOS ONE, 10(3), 1–16. doi: 10.1371/journal.pone.0118632 [DOI] [PMC free article] [PubMed] [Google Scholar]