

Abstract
PURPOSE
Despite advances in DNA sequencing technology and expanded medical guidelines, the vast majority of individuals carrying pathogenic variants of common cancer susceptibility genes have yet to be identified. An alternative to population-wide genetic screening of healthy individuals would exploit the trend for genetic testing at the time of cancer diagnosis to guide therapy and prevention, combined with augmented familial diffusion or “cascade” of genomic risk information.
METHODS
Using a multiple linear regression model, we derived the time interval to detect an estimated 3.9 million individuals in the United States with a pathogenic variant in 1 of 18 cancer susceptibility genes. We analyzed the impact of the proportion of incident patients sequenced, varying observed frequencies of pathogenic germline variants in patients with cancer, differential rates of diffusion of genetic information in families, and family size.
RESULTS
The time to detect inherited cancer predisposing variants in the population is affected by the extent of cascade to first-, second-, and third-degree relatives (FDR, SDR, TDR, respectively), family size, prevalence of mutations in patients with cancer, and the proportion of patients with cancer sequenced. In a representative scenario, assuming a 7% prevalence of pathogenic variants across cancer types, an average family size of 3 per generation, and 15% of incident patients with cancer in the United States undergoing germline testing, the time to detect all 3.9 million individuals with pathogenic variants in 18 cancer susceptibility genes would be 46.2, 22.3, 13.6, and 9.9 years if 10%, 25%, 50%, and 70%, respectively, of all FDR, SDR, and TDR were tested for familial mutations.
CONCLUSION
Peridiagnostic and cascade cancer genetic testing offers an alternative strategy to achieve population-wide identification of cancer susceptibility mutations.
INTRODUCTION
More than 2 decades after the discovery of genetic variants conferring substantial risks for breast, ovarian, prostate, colon, and other malignancies, and despite advances in DNA sequencing technologies, it has become increasingly clear that the promise of genomics as a tool for cancer prevention has yet to be realized.1 Fewer than 1 in 5 individuals with a family history of breast or ovarian cancer who meet established criteria for genetic testing have received it, and most have never discussed their hereditary cancer risk with a health care provider.2 For Lynch syndrome, the most common colon cancer predisposition syndrome, only 1 in 4 individuals who met criteria were screened in the Cancer Research Network,3 and 64%-85% of Community Hospital Cancer Programs do not screen for this syndrome.4
The scale of these shortcomings is measured in lives lost because for Lynch syndrome, as for hereditary breast and ovarian cancer, medical or surgical interventions have been shown to decrease mortality.1,5 To address these challenges more broadly, there have been renewed calls for population-wide testing for BRCA and other genes,6,7 and in the United States and abroad, efforts have begun to sequence the DNA of large cohorts to screen for a broad spectrum of inherited risks.8,9 At the same time, for-profit companies have sought to market genomic testing outside of the traditional medical context, despite public and professional diminished trust in the accuracy and privacy of direct-to-consumer genetic testing.10-14
Because population-based approaches to genomic screening remain costly and involve challenges in high through-put sequencing, obtaining informed consent, and interpretation of genomic variants,15,16 alternative strategies have been proposed, including testing isolated populations at higher risk for cancer,17,18 as well as testing family members of those found to harbor pathogenic variants (mutations) in disease predisposition genes (Data Supplement). An emerging strategy, emphasized in this article, takes its origin from the increasing use of genetic testing at the time of a diagnosis of cancer, affecting both preventive and therapeutic management.19,20 Rapidly expanding peridiagnostic analysis of patients with cancer has the potential to allow physician-facilitated family outreach to disseminate risk information and promote cancer prevention. We seek to model various parameters that would affect the time needed to identify all individuals heterozygous for pathogenic variants of clinically actionable cancer susceptibility genes in the United States through a strategy of peridiagnostic cancer genetic testing and family diffusion.
METHODS
To model the time course of familial transmission of testing for a pathogenic variant after peridiagnostic tumor-normal or germline panel testing, we assumed various rates of detection of pathogenic variants in probands and diffusion of this information in families, starting with 1.7 million patients with cancer diagnosed per year in the United States,21 assuming these to be among unrelated individuals, with a fraction of these patients offered tumor-normal or germline sequencing for a panel of cancer predisposition genes. For this analysis, a subset of 18 “clinically actionable” genes (Table 1) was chosen from a larger group of genes used in a prior analysis.20 Clinical actionability of pathogenic variants was defined by evidence for their utility in cancer prevention and/or potential utility as therapeutic targets.20 For this analysis, CHEK2, MUTYH, CDKN2A, and NBN were not included because of low penetrance of some common variants (CHEK2) or uncertain clinical actionability. However, all “high penetrance” genes proposed in a recent population-screening proposal7 were included.
TABLE 1.
Selected Cancer Susceptibility Genes Used in Model
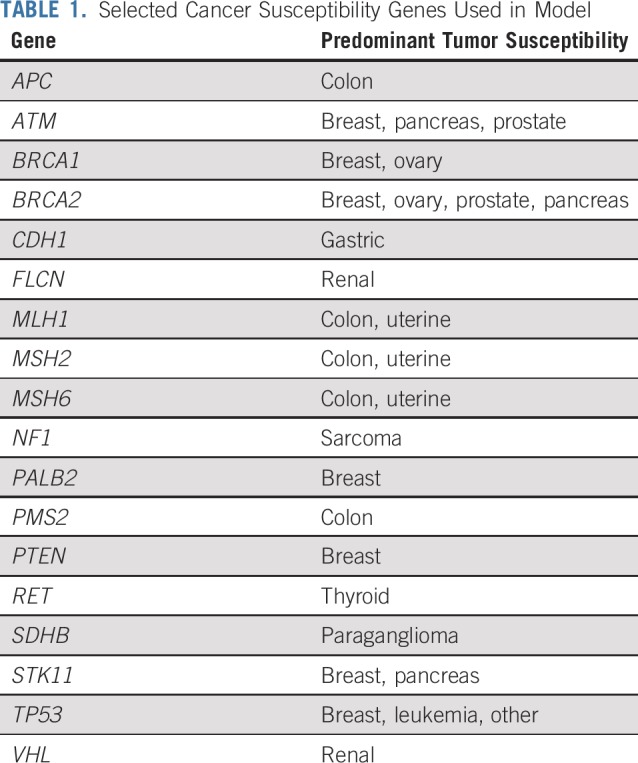
We assumed the prevalence of germline mutations in patients sequenced ranged from 5% to 15%.20-22 We next considered extended families across 3 generations with 2 to 4 offspring per generation23 (Data Supplement), with communication progressing from proband to first-, second-, and third-degree relatives (FDR, SDR, TDR, respectively) and then in successive years to FDR, SDR, and TDR of those contacted the prior year (Data Supplement). We projected that from 0% to 75% of relatives potentially heterozygous for an inherited mutation would be tested. We estimated the number of individuals carrying pathogenic variants already identified (Data Supplement). Driven by “cascade” testing of incident patients with cancer, we measured the time to pass a calculated threshold of 3.9 million individuals older than 25 years of age in the United States, representing the number heterozygous for pathogenic variants in this panel of 18 genes, as directly estimated by frequencies in the ExAC resource (Data Supplement). We assumed a steady state of mutation carriers with countervailing influences of compromised fitness, death due to hereditary cancer, and preimplantation genetic diagnosis, balanced by the birth and immigration of new carriers of pathogenic variants.
We performed a sensitivity analysis on time (in years) to reach a threshold of detection of 3.9 million heterozygotes for these pathogenic variants by varying parameters, including the proportion of patients with cancer in the United States tested (range, 7.5%-75%), the proportion of patients with cancer with germline mutations (5%-15%), and the proportion of FDR, SDR, and TDR tested for familial pathogenic variants (0% to 75%). Multiple linear regression was performed in RStudio version 1.0.14324 on log-transformed response variables to define significant determinants/parameters and the magnitude of their effects (Data Supplement). A tornado plot was generated for the graphic representation of the degree to which the number of years (the result) was sensitive to the specific parameters, thus depicting the univariate sensitivity analysis (Fig 1).
FIG 1.

Tornado plot depicting univariate sensitivity analysis performed using baseline model: 7% prevalence of pathogenic variants across cancer types, an average family size of 3 per generation, 15% of incident patients with cancer undergoing germline testing, and 25% first-degree relatives, 25% second-degree relatives, and 25% third-degree relatives cascading, the time to detect 3.9 million individuals with a germline cancer susceptibility mutation was 22.2 years. The plot shows the effect on the output (number of years) of varying each input variable at a time, keeping all the other input variables at their baseline value. The cascading rate here is defined by the proportion of first-degree, second-degree, and third-degree relatives transmitting information, ranging from 10% to 75%; family size per generation refers to the number of siblings per generation, ranging from 2 to 4; proportion of patients with cancer screened refers to the proportion of incident patients with cancer subjected to germline sequencing of 18 selected genes, ranging from 7.5% to 75%; prevalence of pathogenic mutations refers to the prevalence of pathogenic variants of 18 cancer susceptibility genes in patients with cancer sequenced, ranging from 5% to 15%. The upper and lower bounds for each variable are labeled in the tornado plot.
RESULTS
Figure 2 and the Data Supplement show the time to detect all cancer predisposing pathogenic variants of 18 cancer susceptibility genes in the US population, assuming a 5%-15% prevalence of germline variants in cohorts with cancer, varying proportions of family sizes, varying proportions of incident patients with cancer germline sequenced each year, and 0% to 75% rates of cascade of mutation detection to FDR, SDR, and TDR. Sensitivity analysis using a multiple regression model demonstrates that the time to detect pathogenic variants in the population is significantly affected by rates of FDR, SDR, and TDR tested by cascade, family size, and prevalence of mutations in patients with cancer, followed by the proportion of patients with cancer tested; Table 2 and Figure 1 demonstrate model fit metrics for the simulated data. As a base case, assuming a 7% frequency of clinically actionable germline pathogenic variants in 18 cancer susceptibility genes across all cancer types, based on a SEER weighted adjustment of data derived from patients tested at our Center (Data Supplement), and assuming 15% of incident cancers (the proportion of patients treated at US Comprehensive Cancer Centers) receiving tumor-normal or germline panel testing, for an average family size of 3 offspring per generation (as measured empirically; Data Supplement), the time to detect 3.9 million individuals with germline cancer susceptibility mutations would be 46.2, 22.3, 13.6, and 9.9 years, if 10%, 25%, 50%, and 70%, respectively, of FDR, SDR and TDR were tested for the familial mutation. In such a scenario (Fig 3), the 9.9-year interval to detect all mutations in the population compares with 59.5 years if there is no cascade testing of relatives (Data Supplement). The time to detect all mutations in the population decreased from more than 30 years if only 70% of FDR relatives are tested, to 15 years if 70%, 70%, and 25% of FDR, SDR, and TDR, respectively, are tested and 9.9 years if the proportion of TDRs tested increases from 25% to 70% (Fig 4).
FIG 2.

Sensitivity analysis; Prevalence (%), patients with mutations of 18 cancer susceptibility genes; Proportion, of 1.7 million incident cancers sequenced; Years to detect 3.9 million mutation carriers. FDR, first-degree relatives; SDR, second-degree relatives; TDR, third-degree relatives.
TABLE 2.
Multiple Linear Regression Model to Predict Outcome as No. of Years to Detect All Pathogenic Variants of Selected Cancer Predisposition Genes in the At-Risk Population

FIG 3.

Diagram of peridiagnostic genetic testing and familial diffusion of information under the assumptions of a family size of 3 siblings per generation, 15% of patients with cancer receiving germline genetic testing at the time of diagnosis, a 7% frequency of pathogenic mutations found at the time of testing, and 70% diffusion of familial genetic information to first-degree relatives (F), second-degree relatives (S), and third-degree relatives (T). In this example, it is assumed that 220,000 carriers of pathogenic variants have been identified in the United States, with 44,000 additional individuals identified each year by virtue of expanded commercial clinical and direct-to-consumer testing. Yn represents the cumulative number of individuals with pathogenic variants identified each year. Under these assumptions, the time to detect all 3.9 million individuals with pathogenic variants in the United States is 9.9 years. (*) 419K heterozygous for pathogenic variants identified each year after Y3 (Data Supplement).
FIG 4.

Sensitivity analysis of the example of a family structure of 3 siblings per generation and a 7% prevalence of pathogenic variants in 1 of 18 clinically actionable cancer susceptibility genes; Proportion, of 1.7 million incident cancers sequenced; Years to detect 3.9 million mutation carriers. FDR, first-degree relatives; SDR, second-degree relatives; TDR, third-degree relatives.
DISCUSSION
Although referral rates for genetic testing have increased in the United States since 2004,25-28 only a modest proportion of individuals carrying pathogenic variants of common cancer susceptibility genes have been identified.29 Of 1.4 million breast cancer survivors eligible for genetic testing and at risk for subsequent cancers, 1.2 million have yet to be tested. An additional 10.7 million women who are cancer free but at risk for a primary cancer have not yet received genetic testing.2 For the most common form of hereditary colorectal cancer,30 it has been estimated that 98% of those carrying genetic variants predictive of Lynch syndrome have yet to be diagnosed,31,32 and DNA testing for other cancer syndromes is also less than optimal.33,34 These shortfalls are because of varying clinical guidelines as well as disparities in access to cancer prevention services.35-41 Even if universally accessible, it is now evident that 26%-56% of individuals with inherited pathogenic variants in cancer susceptibility genes will not be detected by clinical guidelines based on family history; this has been shown for “panel” genetic testing,20 as well as family history–targeted testing for BRCA1/2, RET, FH, BAP1, VHL, MET, SDHA, and SDHB germline mutations.17,18,42-49
Despite calls for population screening for common cancer predisposition genes such as BRCA1/2,6 and other high penetrance genes,7 there remain concerns about accessibility to diverse populations, including needs for intensive counseling and evaluation after detection of variants of unknown significance (VUS)15,16 and adverse psychological sequelae.50 For these and other reasons, the US Preventive Services Task Force in February 2019 reinforced recommendations against population-based genetic screening for BRCA mutations.51,52 The cost effectiveness of population sequencing depends on assumptions of the models53,54 reflecting varying costs of systems for monitoring and reclassifying VUS, raising public/health professional awareness, education, cost of medical interventions, delivery logistics, quality control, call-recall mechanisms, and fail-safe checks/processes for quality assurance.54 To provide BRCA1/2 tests for all women older than 30 years of age in the United States would cost more than $14 billion,55 mostly as a nonreimbursable charge within the current US medical system.56 The model proposed here combines 2 elements as an alternative to population-based testing: peridiagnostic cancer genetic testing and familial cascade of results.
Genetic testing at the time of diagnosis of cancer (referred to by some as “mainstreaming”57) has several advantages.20 It identifies those who may benefit from poly (ADP-ribose) polymerase inhibitors for treatment of breast, ovarian, prostate, and other cancers58-61; checkpoint blockade immunotherapy for those with mismatch repair deficiency62; or targeted treatment of those with inherited mutations in MTOR,63,64 RET,65 Hedgehog,66 or other pathways. We have reported that the prevalence of pathogenic variants in cancer predisposition genes is higher in patients with metastatic prostate and other malignancies, compared with those with localized forms of these diseases.20,61 Germline testing of those with advanced malignancies regardless of family history also offers potential cost advantages, shown in the Data Supplement.
A limitation of the peridiagnostic testing model proposed is that it assumes sequencing of incident patients with cancer only. In 2016, there were 15.5 million individuals with a previous diagnosis of cancer (prevalent patients), including 429,000 survivors of childhood cancer in the United States.21 Although such individuals would eventually be tested by cascade, active ascertainment, and testing, even a fraction of these prevalent patients will decrease the time to achieve population-wide detection of cancer-predisposing mutations. Oversampling of early-onset or pediatric cancers will increase germline mutation detection and also lead to cascade testing of parents. Pediatric cancers demonstrate 8%-12% pathogenic germline variants,22,67-70 and there was a 2-fold increase in germline mutations in those older than 40 years of age compared with those younger than 60 years in 12,823 patients we have analyzed (MSK IMPACT20 and unpublished data). The model also assumes a low proportion of “de novo” mutations, observed for common syndromes71; however, when such de novo events occur, they will transmit to offspring, offering subsequent opportunity for cascade. Another limitation of the model is that it did not include polygenic effects of common, but lower-risk, single-nucleotide polymorphisms, which may offer the promise of passing thresholds of clinical actionability.72 The model was sensitive to the proportion of patients with cancer carrying germline pathogenic variants, which we have shown to be affected by founder mutations as well as case mix20; such correlations can be used to prioritize peridiagnostic genetic testing toward those in genetic isolates or with hereditary forms of cancer.
First described for cystic fibrosis and hypercholesterolemia73-75, cascade testing is a process of diffusion of genetic information from an affected family member (proband or index patient) to family members, involving iterative rounds of testing of both close and more distant relatives.75 Although 75%-82% of index patients may share familial cancer information with relatives,76-78 only 35%-64% of these at-risk adult relatives actually undergo genetic testing,79-88 with one study finding that 47.5% of FDR agreed to be tested after contact by a commercial laboratory offering them Internet access to self-pay sequencing for their familial mutation.89 Other investigators have increased familial testing to 46%-92% by contacting the proband’s relatives by mail or phone,90-93 achieving 57%-94% cascade to relatives in breast and colon cancer syndromes, respectively, with genetics professionals playing a role in the family outreach.94-96 Family dissemination of genomic risk information appears to be lower among minority populations,97-99 with patients of African American and Asian/Pacific ancestry less likely to share results with relatives.81 Because a focus on cascade testing could actually widen existing disparities in cancer genetic testing, a multifaceted approach will be required. Tailored and culturally sensitive interventions, including community-based outreach, partnering to decrease distrust and improve genetic literacy, navigators to assist genetic counselors and families, and an emphasis on telegenetics and phone counseling to decrease barriers, have been suggested.100 Our recent study in the United States by Frey et al101 (in this issue of J Clin Oncol.), a prior study in the United Kingdom,96 and a recent study in Trinidad and Tobago102 all achieved 60%-70% levels of cascade testing among at-risk relatives using a strategy of outreach by health care providers, indicating the cross-cultural feasibility of this approach. At the time of counseling, outreach to distant family members, such as first cousins, is vital; our analysis shows that expanding cascade from FDR to include SDR and TDR decreased by half the time to detect all carriers of pathogenic variants in the population. The model used was sensitive to family size; the estimates for family size were derived from empiric measures in a cancer clinic (Data Supplement). Population-based estimates of biologic family size are not included in household composition measures in census data; a recent population-based study found a mean of 19.7 FDR, SDR, and TDR per family sampled, with only modest variation by income strata.23 Finally, a substantial advantage of cascade-based approaches is decreased detection of VUS compared with population-wide sequencing. Cascade testing includes only established pathogenic variants that segregate in families, diminishing the complexities of counseling and interpretation of VUS.
Peridiagnostic and cascade testing has been shown to be cost effective for hypercholesterolemia103 and Lynch syndrome,30 where it is strongly influenced by the number of FDR tested.104 Importantly, the success of such strategies has been limited by the relative shortage of genetic counselors. As one means to address this need, digital media interventions to facilitate outreach of familial risk information have been piloted for colon cancer99 and pancreas cancer,103 and offer promise for implementing this proposed strategy. We have piloted digital learning tools in the context of a Web-based founder mutation testing program (www.bforstudy.com) that includes the option of follow-up with one’s primary health care provider.105 Integrating such tools in the peridiagnostic setting would offer at-home convenience and pretest education,106,107 and also avoid the logistic, privacy, and ethical barriers of “traceback” testing of DNA from paraffin archives of deceased patients.108-111 In contrast to population-screening approaches, patients with cancer can be offered germline genetic testing in a medical setting. The majority will want to know these results, emphasizing the potential advantages as well as the emotional and psychosocial components in a process that involves caregivers and family members.112
Testing for known inherited cancer susceptibility alleles in families is professionally endorsed,113 increasingly reimbursed by third-party carriers, and responsive to the legal and ethical “duty to warn” about familial disease risks.114 Although tumor genetic testing is now an Advanced Diagnostic Laboratory Test,115 Centers for Medicare & Medicaid Services is currently reevaluating including inherited germline testing in its coverage guidelines.116 These factors will help to mitigate disparities in access to genetic services, which constitute a barrier to peridiagnostic testing as well as cascade models.113,117 In the United States, peridiagnostic DNA sequencing has been initiated at many of the 71 National Cancer Institute (NCI)-designated cancer centers, which currently care for 15% of patients with cancer, and this effort could involve the more than 1,100 community cancer programs and oncology networks conducting clinical trials and approximately 250 academic and NCI-designated cancer research centers,118,119 with peridiagnostic genetic testing for prostate cancer to be offered through the US Veterans Administration.120 The Food and Drug Administration has approved targeting immunotherapy to a genetic defect in mismatch DNA repair,121 and a professional society recently provided a rationale for genetic testing of all patients with breast cancer.116
If adopted by clinicians, patients, and families, widespread peridiagnostic genetic testing and cascade to family members could identify all of the cancer predisposing mutations in the United States in a decade or less, diminishing the need for a costly effort to test and identify genetic variants in all healthy individuals in the population. As shown in recent studies (eg, Evans et al,96 Donenberg et al,102 and Frey et al101 [in this issue of J Clin Oncol.]), high rates of cascade of familial genetic information can be achieved by health care providers facilitating outreach and assisting patients and their families to communicate genetic risk information. Augmented by additional development, including Web-based applications, peridiagnostic cancer genetic testing, and familial transmission of genomic risk information can substantially decrease the burden of hereditary cancer.
PRIOR PRESENTATION
Presented at the American Society of Human Genetics Annual Meeting, Houston, TX, October 17, 2019. Presented at the Basser Center Symposium, Philadelphia, PA, May 6, 2019.
SUPPORT
Supported by the Robert and Kate Niehaus Center for Inherited Cancer Genomics at Memorial Sloan Kettering Cancer Center, the Andrew Sabin Family Foundation, the Breast Cancer Research Foundation, the Sharon Levine Corzine Cancer Research Fund, the American Cancer Society (MRSG-16-020-01-CPPB to J.G.H.), and the National Institutes of Health (5 P30 CA008748-49) and (K07CA216326, R01CA211723, and IHS-2017C3-9211 to R.N.S.).
See accompanying Editorial on page 1371
AUTHOR CONTRIBUTIONS
Conception and design: Kenneth Offit, Kaitlyn A. Tkachuk, Melissa K. Frey, Steven M. Lipkin, Mark E. Robson, Semanti Mukherjee
Financial support: Kenneth Offit, Semanti Mukherjee
Administrative support: Kenneth Offit, Jeffrey D. Levin
Provision of study materials or patients: Kenneth Offit
Collection and assembly of data: Kenneth Offit, Kaitlyn A. Tkachuk, Hector Diaz-Zabala, Vignesh Ravichandran, Semanti Mukherjee
Data analysis and interpretation: Kenneth Offit, Kaitlyn A. Tkachuk, Zsofia K. Stadler, Michael F. Walsh, Hector Diaz-Zabala, Jeffrey D. Levin, Zoe Steinsnyder, Vignesh Ravichandran, Ravi N. Sharaf, Melissa K. Frey, Steven M. Lipkin, Jada G. Hamilton, Joseph Vijai, Semanti Mukherjee
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Cascading After Peridiagnostic Cancer Genetic Testing: An Alternative to Population-Based Screening
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/journal/jco/site/ifc.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Kenneth Offit
Patents, Royalties, Other Intellectual Property: Diagnosis and treatment of ERCC3-mutant cancer; inventors: Joseph Vijai, Sabine Topka, Kenneth Offit; US National Stage Patent Application No.: 16/493,214; filing date: September 11, 2019 (Inst)
Zsofia K. Stadler
Consulting or Advisory Role: Allergan (I), Genentech (I), Regeneron (I), Optos (I), Adverum (I), Biomarin (I), Alimera Sciences (I), Novartis (I), Spark Therapeutics (I), Fortress Biotech (I), Regenxbio (I)
Jeffrey D. Levin
Employment: Indispensable Healthcare (I), Cardiology Consultants of Michigan (I)
Melissa K. Frey
Research Funding: Invitae (Inst)
Mark E. Robson
Honoraria: AstraZeneca
Consulting or Advisory Role: McKesson, AstraZeneca, AstraZeneca (Inst), Myriad Genetics (Inst), InVitae (Inst), Abbvie (Inst), Tesaro (Inst), Medivation (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Pfizer
Other Relationship: Research to Practice, Clinical Care Options, Physician Education Resource
Uncompensated Relationships: Merck, Pfizer, Daiichi Sankyo
Joseph Vijai
Patents, Royalties, Other Intellectual Property: Diagnosis and treatment of ERCC3-mutant cancer; inventors: Joseph Vijai, Sabine Topka, Kenneth Offit; US National Stage Patent Application No.: 16/493,214; filing date: September 11, 2019 (Inst)
Semanti Mukherjee
Stock and Other Ownership Interests: Regeneron
No other potential conflicts of interest were reported.
REFERENCES
- 1.Offit K. The future of clinical cancer genomics. Semin Oncol. 2016;43:615–622. doi: 10.1053/j.seminoncol.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Childers CP, Childers KK, Maggard-Gibbons M, et al. National estimates of genetic testing in women with a history of breast or ovarian cancer. J Clin Oncol. 2017;35:3800–3806. doi: 10.1200/JCO.2017.73.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cross DS, Rahm AK, Kauffman TL, et al. Underutilization of Lynch syndrome screening in a multisite study of patients with colorectal cancer. Genet Med. 2013;15:933–940. doi: 10.1038/gim.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mange S, Bellcross C, Cragun D, et al. Creation of a network to promote universal screening for Lynch syndrome: The Lynch Syndrome Screening Network. J Genet Couns. 2015;24:421–427. doi: 10.1007/s10897-014-9770-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finch AP, Lubinski J, Møller P, et al. Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. J Clin Oncol. 2014;32:1547–1553. doi: 10.1200/JCO.2013.53.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.King MC, Levy-Lahad E, Lahad A. Population-based screening for BRCA1 and BRCA2: 2014 Lasker Award. JAMA. 2014;312:1091–1092. doi: 10.1001/jama.2014.12483. [DOI] [PubMed] [Google Scholar]
- 7. doi: 10.1038/s41588-018-0326-2. Turnbull C, Sud A, Houlston RS: Cancer genetics, precision prevention and a call to action. Nat Genet 50:1212-1218, 2018 [Erratum: Nat Genet 51:196, 2019] [DOI] [PubMed] [Google Scholar]
- 8.Sankar PL, Parker LS. The Precision Medicine Initiative’s All of Us Research Program: An agenda for research on its ethical, legal, and social issues. Genet Med. 2017;19:743–750. doi: 10.1038/gim.2016.183. [DOI] [PubMed] [Google Scholar]
- 9. doi: 10.1136/bmj.k1687. Turnbull C, Scott RH, Thomas E, et al: The 100 000 Genomes Project: Bringing whole genome sequencing to the NHS. BMJ 361:k1687, 2018 [Erratum: BMJ 361:k1952, 2018] [DOI] [PubMed] [Google Scholar]
- 10.Burke W, Trinidad SB. The deceptive appeal of direct-to-consumer genetics. Ann Intern Med. 2016;164:564–565. doi: 10.7326/M16-0257. [DOI] [PubMed] [Google Scholar]
- 11.van der Wouden CH, Carere DA, Maitland-van der Zee AH, et al. Consumer perceptions of interactions with primary care providers after direct-to-consumer personal genomic testing. Ann Intern Med. 2016;164:513–522. doi: 10.7326/M15-0995. [DOI] [PubMed] [Google Scholar]
- 12.Roberts JS, Gornick MC, Carere DA, et al. Direct-to-consumer genetic testing: User motivations, decision making, and perceived utility of results. Public Health Genomics. 2017;20:36–45. doi: 10.1159/000455006. [DOI] [PubMed] [Google Scholar]
- 13.Gray SW, Gollust SE, Carere DA, et al. Personal genomic testing for cancer risk: Results from the impact of personal genomics study. J Clin Oncol. 2017;35:636–644. doi: 10.1200/JCO.2016.67.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carere DA, VanderWeele T, Moreno TA, et al. The impact of direct-to-consumer personal genomic testing on perceived risk of breast, prostate, colorectal, and lung cancer: Findings from the PGen study. BMC Med Genomics. 2015;8:63. doi: 10.1186/s12920-015-0140-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yurgelun MB, Hiller E, Garber JE. Population-wide screening for germline BRCA1 and BRCA2 mutations: Too much of a good thing? J Clin Oncol. 2015;33:3092–3095. doi: 10.1200/JCO.2015.60.8596. [DOI] [PubMed] [Google Scholar]
- 16.Murray ML, Cerrato F, Bennett RL, et al. Follow-up of carriers of BRCA1 and BRCA2 variants of unknown significance: Variant reclassification and surgical decisions. Genet Med. 2011;13:998–1005. doi: 10.1097/GIM.0b013e318226fc15. [DOI] [PubMed] [Google Scholar]
- 17.Metcalfe KA, Poll A, Royer R, et al. Screening for founder mutations in BRCA1 and BRCA2 in unselected Jewish women. J Clin Oncol. 2010;28:387–391. doi: 10.1200/JCO.2009.25.0712. [DOI] [PubMed] [Google Scholar]
- 18.Manchanda R, Loggenberg K, Sanderson S, et al. Population testing for cancer predisposing BRCA1/BRCA2 mutations in the Ashkenazi-Jewish community: A randomized controlled trial. J Natl Cancer Inst. 2014;107:379. doi: 10.1093/jnci/dju379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zehir A, Benayed R, Shah RH, et al. Erratum: Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:1004. doi: 10.1038/nm0817-1004c. [DOI] [PubMed] [Google Scholar]
- 20.Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;318:825–835. doi: 10.1001/jama.2017.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. National Cancer Institute: Cancer statistics. https://www.cancer.gov/about-cancer/understanding/statistics.
- 22.Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373:2336–2346. doi: 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garceau A, Wideroff L, McNeel T, et al. Population estimates of extended family structure and size. Community Genet. 2008;11:331–342. doi: 10.1159/000133305. [DOI] [PubMed] [Google Scholar]
- 24. RStudio: Integrated Development for R. Boston, MA, RStudio, 2017. [Google Scholar]
- 25.Meyer LA, Anderson ME, Lacour RA, et al. Evaluating women with ovarian cancer for BRCA1 and BRCA2 mutations: Missed opportunities. Obstet Gynecol. 2010;115:945–952. doi: 10.1097/AOG.0b013e3181da08d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levy DE, Byfield SD, Comstock CB, et al. Underutilization of BRCA1/2 testing to guide breast cancer treatment: Black and Hispanic women particularly at risk. Genet Med. 2011;13:349–355. doi: 10.1097/GIM.0b013e3182091ba4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powell CB, Littell R, Hoodfar E, et al. Does the diagnosis of breast or ovarian cancer trigger referral to genetic counseling? Int J Gynecol Cancer. 2013;23:431–436. doi: 10.1097/IGC.0b013e318280f2b4. [DOI] [PubMed] [Google Scholar]
- 28.Febbraro T, Robison K, Wilbur JS, et al. Adherence patterns to National Comprehensive Cancer Network (NCCN) guidelines for referral to cancer genetic professionals. Gynecol Oncol. 2015;138:109–114. doi: 10.1016/j.ygyno.2015.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drohan B, Roche CA, Cusack JC, Jr, et al. Hereditary breast and ovarian cancer and other hereditary syndromes: Using technology to identify carriers. Ann Surg Oncol. 2012;19:1732–1737. doi: 10.1245/s10434-012-2257-y. [DOI] [PubMed] [Google Scholar]
- 30.Hampel H. Genetic counseling and cascade genetic testing in Lynch syndrome. Fam Cancer. 2016;15:423–427. doi: 10.1007/s10689-016-9893-5. [DOI] [PubMed] [Google Scholar]
- 31. Centers for Disease Control and Prevention: EGAPP: Informing the effective integration of genomics into health practice—Lynch syndrome. https://www.cdc.gov/genomics/gtesting/egapp/lynch_study.htm.
- 32.Hampel H, de la Chapelle A. The search for unaffected individuals with Lynch syndrome: Do the ends justify the means? Cancer Prev Res (Phila) 2011;4:1–5. doi: 10.1158/1940-6207.CAPR-10-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schultz KAP, Rednam SP, Kamihara J, et al. PTEN, DICER1, FH, and their associated tumor susceptibility syndromes: Clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res. 2017;23:e76–e82. doi: 10.1158/1078-0432.CCR-17-0629. [DOI] [PubMed] [Google Scholar]
- 34.Williams MD. Paragangliomas of the head and neck: An overview from diagnosis to genetics. Head Neck Pathol. 2017;11:278–287. doi: 10.1007/s12105-017-0803-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Canedo JR, Miller ST, Myers HF, et al. Racial and ethnic differences in knowledge and attitudes about genetic testing in the US: Systematic review. J Genet Couns. 2019;28:587–601. doi: 10.1002/jgc4.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hann KEJ, Freeman M, Fraser L, et al. Awareness, knowledge, perceptions, and attitudes towards genetic testing for cancer risk among ethnic minority groups: A systematic review. BMC Public Health. 2017;17:503. doi: 10.1186/s12889-017-4375-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters N, Rose A, Armstrong K. The association between race and attitudes about predictive genetic testing. Cancer Epidemiol Biomarkers Prev. 2004;13:361–365. [PubMed] [Google Scholar]
- 38.Suther S, Kiros GE. Barriers to the use of genetic testing: A study of racial and ethnic disparities. Genet Med. 2009;11:655–662. doi: 10.1097/GIM.0b013e3181ab22aa. [DOI] [PubMed] [Google Scholar]
- 39.Hall MJ, Olopade OI. Disparities in genetic testing: Thinking outside the BRCA box. J Clin Oncol. 2006;24:2197–2203. doi: 10.1200/JCO.2006.05.5889. [DOI] [PubMed] [Google Scholar]
- 40.Butrick M, Kelly S, Peshkin BN, et al. Disparities in uptake of BRCA1/2 genetic testing in a randomized trial of telephone counseling. Genet Med. 2015;17:467–475. doi: 10.1038/gim.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willis AM, Smith SK, Meiser B, et al. Sociodemographic, psychosocial and clinical factors associated with uptake of genetic counselling for hereditary cancer: A systematic review. Clin Genet. 2017;92:121–133. doi: 10.1111/cge.12868. [DOI] [PubMed] [Google Scholar]
- 42.Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997;336:1401–1408. doi: 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- 43.Moslehi R, Chu W, Karlan B, et al. BRCA1 and BRCA2 mutation analysis of 208 Ashkenazi Jewish women with ovarian cancer. Am J Hum Genet. 2000;66:1259–1272. doi: 10.1086/302853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King MC, Marks JH, Mandell JB: Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 45.Warner E, Foulkes W, Goodwin P, et al. Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer. J Natl Cancer Inst. 1999;91:1241–1247. doi: 10.1093/jnci/91.14.1241. [DOI] [PubMed] [Google Scholar]
- 46.Risch HA, McLaughlin JR, Cole DE, et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet. 2001;68:700–710. doi: 10.1086/318787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hartman AR, Kaldate RR, Sailer LM, et al. Prevalence of BRCA mutations in an unselected population of triple-negative breast cancer. Cancer. 2012;118:2787–2795. doi: 10.1002/cncr.26576. [DOI] [PubMed] [Google Scholar]
- 48.Parkhurst E, Calonico E, Abboy S. Utilization of genetic testing for RET mutations in patients with medullary thyroid carcinoma: A single-center experience. J Genet Couns. 2018;27:1411–1416. doi: 10.1007/s10897-018-0273-1. [DOI] [PubMed] [Google Scholar]
- 49.Carlo MI, Mukherjee S, Mandelker D, et al. Prevalence of germline mutations in cancer susceptibility genes in patients with advanced renal cell carcinoma. JAMA Oncol. 2018;4:1228–1235. doi: 10.1001/jamaoncol.2018.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Metcalfe KA, Poll A, Llacuachaqui M, et al. Patient satisfaction and cancer-related distress among unselected Jewish women undergoing genetic testing for BRCA1 and BRCA2. Clin Genet. 2010;78:411–417. doi: 10.1111/j.1399-0004.2010.01499.x. [DOI] [PubMed] [Google Scholar]
- 51.Moyer VA: Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2014;160:271–281. doi: 10.7326/M13-2747. [DOI] [PubMed] [Google Scholar]
- 52. US Preventive Services Task Force: Draft recommendation statement: BRCA-related cancer: Risk assessment, genetic counseling, and genetic testing. https://www.uspreventiveservicestaskforce.org/Page/Document/draft-recommendation-statement/brca-related-cancer-risk-assessment-genetic-counseling-and-genetic-testing1.
- 53.Long EF, Ganz PA. Cost-effectiveness of universal BRCA1/2 screening: Evidence-based decision making. JAMA Oncol. 2015;1:1217–1218. doi: 10.1001/jamaoncol.2015.2340. [DOI] [PubMed] [Google Scholar]
- 54.Manchanda R, Patel S, Gordeev VS, et al. Cost-effectiveness of population-based BRCA1, BRCA2, RAD51C, RAD51D, BRIP1, PALB2 mutation testing in unselected general population women. J Natl Cancer Inst. 2018;110:714–725. doi: 10.1093/jnci/djx265. [DOI] [PubMed] [Google Scholar]
- 55.Foulkes WD, Knoppers BM, Turnbull C. Population genetic testing for cancer susceptibility: Founder mutations to genomes. Nat Rev Clin Oncol. 2016;13:41–54. doi: 10.1038/nrclinonc.2015.173. [DOI] [PubMed] [Google Scholar]
- 56.Best AF, Tucker MA, Frone MN, et al. A pragmatic testing-eligibility framework for population mutation screening: The example of BRCA1/2. Cancer Epidemiol Biomarkers Prev. 2019;28:293–302. doi: 10.1158/1055-9965.EPI-18-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Slade I, Riddell D, Turnbull C, et al. Development of cancer genetic services in the UK: A national consultation. Genome Med. 2015;7:18. doi: 10.1186/s13073-015-0128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ashworth A, Lord CJ. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat Rev Clin Oncol. 2018;15:564–576. doi: 10.1038/s41571-018-0055-6. [DOI] [PubMed] [Google Scholar]
- 60.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110–120. doi: 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- 61.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sampson JR. Therapeutic targeting of mTOR in tuberous sclerosis. Biochem Soc Trans. 2009;37:259–264. doi: 10.1042/BST0370259. [DOI] [PubMed] [Google Scholar]
- 64.Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:817–824. doi: 10.1016/S0140-6736(12)61767-X. [DOI] [PubMed] [Google Scholar]
- 65.Wells SA, Jr, Gosnell JE, Gagel RF, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28:767–772. doi: 10.1200/JCO.2009.23.6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med. 2012;366:2180–2188. doi: 10.1056/NEJMoa1113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Walsh M, Wu G, Edmonson M, et al: Incidence of germline mutations in cancer-predisposition genes in children with hematologic malignancies: A report from the Pediatric Cancer Genome Project. Blood 124:127, 2014. [Google Scholar]
- 68.Parsons DW, Roy A, Yang Y, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016;2:616–624. doi: 10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mody RJ, Wu YM, Lonigro RJ, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 2015;314:913–925. doi: 10.1001/jama.2015.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walsh MF, Kennedy J, Harlan M, et al. Germline BRCA2 mutations detected in pediatric sequencing studies impact parents’ evaluation and care. Cold Spring Harb Mol Case Stud. 2017;3:a001925. doi: 10.1101/mcs.a001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Robson M, Scheuer L, Nafa K, et al. Unique de novo mutation of BRCA2 in a woman with early onset breast cancer. J Med Genet. 2002;39:126–128. doi: 10.1136/jmg.39.2.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chatterjee N, Shi J, García-Closas M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat Rev Genet. 2016;17:392–406. doi: 10.1038/nrg.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Super M, Schwarz MJ, Malone G. Screening for cystic fibrosis carriers. Lancet. 1992;340:490–491. doi: 10.1016/0140-6736(92)91816-q. [DOI] [PubMed] [Google Scholar]
- 74.Holloway S, Brock DJ. Cascade testing for the identification of carriers of cystic fibrosis. J Med Screen. 1994;1:159–164. doi: 10.1177/096914139400100305. [DOI] [PubMed] [Google Scholar]
- 75.Knowles JW, Rader DJ, Khoury MJ. Cascade screening for familial hypercholesterolemia and the use of genetic testing. JAMA. 2017;318:381–382. doi: 10.1001/jama.2017.8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taber JM, Chang CQ, Lam TK, et al. Prevalence and correlates of receiving and sharing high-penetrance cancer genetic test results: Findings from the Health Information National Trends Survey. Public Health Genomics. 2015;18:67–77. doi: 10.1159/000368745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Montgomery SV, Barsevick AM, Egleston BL, et al. Preparing individuals to communicate genetic test results to their relatives: Report of a randomized control trial. Fam Cancer. 2013;12:537–546. doi: 10.1007/s10689-013-9609-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stoffel EM, Ford B, Mercado RC, et al. Sharing genetic test results in Lynch syndrome: Communication with close and distant relatives. Clin Gastroenterol Hepatol. 2008;6:333–338. doi: 10.1016/j.cgh.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dilzell K, Kingham K, Ormond K, et al. Evaluating the utilization of educational materials in communicating about Lynch syndrome to at-risk relatives. Fam Cancer. 2014;13:381–389. doi: 10.1007/s10689-014-9720-9. [DOI] [PubMed] [Google Scholar]
- 80.Cheung EL, Olson AD, Yu TM, et al. Communication of BRCA results and family testing in 1,103 high-risk women. Cancer Epidemiol Biomarkers Prev. 2010;19:2211–2219. doi: 10.1158/1055-9965.EPI-10-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fehniger J, Lin F, Beattie MS, et al. Family communication of BRCA1/2 results and family uptake of BRCA1/2 testing in a diverse population of BRCA1/2 carriers. J Genet Couns. 2013;22:603–612. doi: 10.1007/s10897-013-9592-4. [DOI] [PubMed] [Google Scholar]
- 82.McGivern B, Everett J, Yager GG, et al. Family communication about positive BRCA1 and BRCA2 genetic test results. Genet Med. 2004;6:503–509. doi: 10.1097/01.gim.0000144014.91237.a1. [DOI] [PubMed] [Google Scholar]
- 83.Daly MB, Montgomery S, Bingler R, et al. Communicating genetic test results within the family: Is it lost in translation? A survey of relatives in the randomized six-step study. Fam Cancer. 2016;15:697–706. doi: 10.1007/s10689-016-9889-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Landsbergen K, Verhaak C, Kraaimaat F, et al. Genetic uptake in BRCA-mutation families is related to emotional and behavioral communication characteristics of index patients. Fam Cancer. 2005;4:115–119. doi: 10.1007/s10689-004-7991-2. [DOI] [PubMed] [Google Scholar]
- 85.Sharaf RN, Myer P, Stave CD, et al. Uptake of genetic testing by relatives of Lynch syndrome probands: A systematic review. Clin Gastroenterol Hepatol. 2013;11:1093–1100. doi: 10.1016/j.cgh.2013.04.044. [DOI] [PubMed] [Google Scholar]
- 86.Barrow P, Green K, Clancy T, et al. Improving the uptake of predictive testing and colorectal screening in Lynch syndrome: A regional primary care survey. Clin Genet. 2015;87:517–524. doi: 10.1111/cge.12559. [DOI] [PubMed] [Google Scholar]
- 87.Lerman C, Hughes C, Trock BJ, et al. Genetic testing in families with hereditary nonpolyposis colon cancer. JAMA. 1999;281:1618–1622. doi: 10.1001/jama.281.17.1618. [DOI] [PubMed] [Google Scholar]
- 88.Hadley DW, Jenkins J, Dimond E, et al. Genetic counseling and testing in families with hereditary nonpolyposis colorectal cancer. Arch Intern Med. 2003;163:573–582. doi: 10.1001/archinte.163.5.573. [DOI] [PubMed] [Google Scholar]
- 89.Caswell-Jin JL, Zimmer AD, Stedden W, et al. Cascade genetic testing of relatives for hereditary cancer risk: Results of an online initiative. J Natl Cancer Inst. 2019;111:95–98. doi: 10.1093/jnci/djy147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Suthers GK, Armstrong J, McCormack J, et al. Letting the family know: Balancing ethics and effectiveness when notifying relatives about genetic testing for a familial disorder. J Med Genet. 2006;43:665–670. doi: 10.1136/jmg.2005.039172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kerzin-Storrar L, Wright C, Williamson PR, et al. Comparison of genetic services with and without genetic registers: Access and attitudes to genetic counselling services among relatives of genetic clinic patients. J Med Genet. 2002;39:e85. doi: 10.1136/jmg.39.12.e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sermijn E, Delesie L, Deschepper E, et al. The impact of an interventional counselling procedure in families with a BRCA1/2 gene mutation: Efficacy and safety. Fam Cancer. 2016;15:155–162. doi: 10.1007/s10689-015-9854-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Forrest LE, Burke J, Bacic S, et al. Increased genetic counseling support improves communication of genetic information in families. Genet Med. 2008;10:167–172. doi: 10.1097/GIM.0b013e318164540b. [DOI] [PubMed] [Google Scholar]
- 94.Chivers Seymour K, Addington-Hall J, Lucassen AM, et al. What facilitates or impedes family communication following genetic testing for cancer risk? A systematic review and meta-synthesis of primary qualitative research. J Genet Couns. 2010;19:330–342. doi: 10.1007/s10897-010-9296-y. [DOI] [PubMed] [Google Scholar]
- 95.Menko FH, Ter Stege JA, van der Kolk LE, et al. The uptake of presymptomatic genetic testing in hereditary breast-ovarian cancer and Lynch syndrome: A systematic review of the literature and implications for clinical practice. Fam Cancer. 2019;18:127–135. doi: 10.1007/s10689-018-0089-z. [DOI] [PubMed] [Google Scholar]
- 96.Evans DG, Binchy A, Shenton A, et al. Comparison of proactive and usual approaches to offering predictive testing for BRCA1/2 mutations in unaffected relatives. Clin Genet. 2009;75:124–132. doi: 10.1111/j.1399-0004.2008.01146.x. [DOI] [PubMed] [Google Scholar]
- 97.Kinney AY, Gammon A, Coxworth J, et al. Exploring attitudes, beliefs, and communication preferences of Latino community members regarding BRCA1/2 mutation testing and preventive strategies. Genet Med. 2010;12:105–115. doi: 10.1097/GIM.0b013e3181c9af2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Etchegary H, Potter B, Perrier C, et al. Cultural differences in family communication about inherited cancer: Implications for cancer genetics research. J Cult Divers. 2013;20:195–201. [PubMed] [Google Scholar]
- 99.Trottier M, Lunn J, Butler R, et al. Prevalence of founder mutations in the BRCA1 and BRCA2 genes among unaffected women from the Bahamas. Clin Genet. 2016;89:328–331. doi: 10.1111/cge.12602. [DOI] [PubMed] [Google Scholar]
- 100.Hinchcliff EM, Bednar EM, Lu KH, et al. Disparities in gynecologic cancer genetics evaluation. Gynecol Oncol. 2019;153:184–191. doi: 10.1016/j.ygyno.2019.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Frey KM, Kahn R, Chapman-Davis E, et al. Prospective feasibility trial of a novel strategy of facilitated cascade genetic testing using telephone counseling. J Clin Oncol. 2020;38:1389–1397. doi: 10.1200/JCO.19.02005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Donenberg T, George S, Ali J, et al. A clinically structured and partnered approach to genetic testing in Trinidadian women with breast cancer and their families. Breast Cancer Res Treat. 2019;174:469–477. doi: 10.1007/s10549-018-5045-y. [DOI] [PubMed] [Google Scholar]
- 103.Hadfield SG, Humphries SE. Implementation of cascade testing for the detection of familial hypercholesterolaemia. Curr Opin Lipidol. 2005;16:428–433. doi: 10.1097/01.mol.0000174152.76554.d6. [DOI] [PubMed] [Google Scholar]
- 104.Grosse SD. When is genomic testing cost-effective? Testing for Lynch syndrome in patients with newly-diagnosed colorectal cancer and their relatives. Healthcare (Basel) 2015;3:860–878. doi: 10.3390/healthcare3040860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Morgan K, Gabriel C, Synmecko H, et al: Early results from the BRCA Founder Outreach (BFOR) study: Population genetic screening using a medical model. Presented at the ASCO Annual Meeting, Chicago, IL, May 31-June 4, 2019. [Google Scholar]
- 106.Singleton A, Erby LH, Foisie KV, et al. Informed choice in direct-to-consumer genetic testing (DTCGT) websites: A content analysis of benefits, risks, and limitations. J Genet Couns. 2012;21:433–439. doi: 10.1007/s10897-011-9474-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lachance CR, Erby LA, Ford BM, et al. Informational content, literacy demands, and usability of websites offering health-related genetic tests directly to consumers. Genet Med. 2010;12:304–312. doi: 10.1097/GIM.0b013e3181dbd8b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Samimi G, Bernardini MQ, Brody LC, et al. Traceback: A proposed framework to increase identification and genetic counseling of BRCA1 and BRCA2 mutation carriers through family-based outreach. J Clin Oncol. 2017;35:2329–2337. doi: 10.1200/JCO.2016.70.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. National Institutes of Health: Traceback testing: Increasing Identification and Genetic Counseling of Mutation Carriers Through Family-based Outreach (U01 Clinical Trial Optional). https://grants.nih.gov/grants/guide/pa-files/par-18-616.html.
- 110.Schwartz MD. Identification of BRCA1 and BRCA2 mutation carriers through a traceback framework: Consent, privacy, and autonomy. J Clin Oncol. 2017;35:2226–2228. doi: 10.1200/JCO.2017.72.8774. [DOI] [PubMed] [Google Scholar]
- 111.Moss HA, Samimi G, Havrilesky LJ, et al. Estimating the number of potential family members eligible for BRCA1 and BRCA2 mutation testing in a “traceback” approach. Genet Epidemiol. 2018;42:117–122. doi: 10.1002/gepi.22095. [DOI] [PubMed] [Google Scholar]
- 112.Hamilton JG, Shuk E, Genoff MC, et al. Interest and attitudes of patients with advanced cancer with regard to secondary germline findings from tumor genomic profiling. J Oncol Pract. 2017;13:e590–e601. doi: 10.1200/JOP.2016.020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Roberts MC, Dotson WD, DeVore CS, et al. Delivery of cascade screening for hereditary conditions: A scoping review of the literature. Health Aff (Millwood) 2018;37:801–808. doi: 10.1377/hlthaff.2017.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bombard Y, Offit K, Robson ME. Risks to relatives in genomic research: A duty to warn? Am J Bioeth. 2012;12:12–14. doi: 10.1080/15265161.2012.699157. [DOI] [PubMed] [Google Scholar]
- 115. Centers for Medicare & Medicaid Services: Decision memo for next generation sequencing (NGS) for Medicare beneficiaries with advanced cancer: (CAG-00450N). https://www.cms.gov/medicare-coverage-database/details/nca-decision-memo.aspx?NCAId=290.
- 116.Manahan ER, Kuerer HM, Sebastian M, et al. Consensus guidelines on genetic’ testing for hereditary breast cancer from The American Society of Breast Surgeons. Ann Surg Oncol. 2019;26:3025–3031. doi: 10.1245/s10434-019-07549-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nikolaidis C, Ming C, Pedrazzani C, et al: Challenges and opportunities for cancer predisposition cascade screening for hereditary breast and ovarian cancer and Lynch syndrome in Switzerland: Findings from an international workshop. Public Health Genomics 21:121-132, 2018. [DOI] [PubMed] [Google Scholar]
- 118. National Cancer Institute: Cancer Centers. https://cancercenters.cancer.gov/Center/CancerCenters.
- 119. Pavia L: Cancer clinical trials in the community: Hospitals & health networks: 2013: https://www.hhnmag.com/articles/5748-cancer-clinical-trials-in-the-community.
- 120. Prostate Cancer Foundation: PCF & the VA: Shepherding extraordinary care. https://www.pcf.org/va-partnership/
- 121.Boyiadzis MM, Kirkwood JM, Marshall JL, et al. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35. doi: 10.1186/s40425-018-0342-x. [DOI] [PMC free article] [PubMed] [Google Scholar]