

Abstract
SsnB previously showed a promising role to lessen liver inflammation observed in a mouse model of NAFLD. Since NAFLD can progress to fibrosis, studies were designed to unravel its role in attenuating NAFLD associated fibrosis. Using both in vivo and in vitro approaches, the study probed the possible mechanisms that underlined the role of SsnB in mitigating fibrosis. Mechanistically, SsnB, a TLR4 antagonist, decreased TLR4-PI3k akt signaling by upregulating PTEN protein expression. It also decreased MDM2 protein activation and increased p53 and p21 gene and protein expression. SsnB also downregulated pro-fibrogenic hedgehog signaling pathway, inhibited hepatic stellate cell proliferation and induced apoptosis in hepatic stellate cells, a mechanism that was LPS dependent. Further, SsnB decreased fibrosis by antagonizing TLR4 induced TGFβ3 signaling pathway. Alternatively, SsnB augmented BAMBI (a TGFβ3 pseudo-receptor) expression in mice liver by inhibiting TLR4 signaling pathway and thus reduced TGFβ3 signaling, resulting in decreased hepatic stellate cell activation and extracellular matrix deposition. In vitro experiments on human hepatic stellate cell line showed that SsnB increased gene and protein expression of BAMBI. It also decreased nuclear co-localization of phospho SMAD2/3 and SMAD4 protein and thus attenuated TGFβ3 signaling in vitro. We also observed a significant decrease in phosphorylation of SMAD2/3 protein, decreased STAT3 activation, alteration of focal adhesion protein and stress fiber disassembly upon SsnB administration in hepatic stellate cells which further confirmed the antagonistic effect of SsnB on TLR4-induced fibrogenesis.
Keywords: NAFLD, NASH, p53, p21, Cyclin, E Cell cycle, Apoptosis, TLR4, Hedgehog signaling
1. Introduction
Nonalcoholic steatohepatitis (NASH) is manifestation of metabolic syndrome in liver which arises from hepatic fat accumulation (Bohinc and Diehl, 2012; Kim and Younossi, 2008). NASH is characterized by steatosis (fat deposition) in liver along with increased liver inflammation, fibrosis and progressive endothelial dysfunction, which can lead to cirrhosis and hepatocellular carcinoma (Larrain and Rinella, 2012; Pasarin et ah, 2012). NASH is a silent liver disease and patients generally feel well until they develop irreversible excessive liver damage. As there is almost no medication available to treat NASH related excessive liver damage, liver transplant can bring the only hope for survival-given that there is always a short supply of liver and liver transplant has been a challenge due to graft rejection, graft steatosis, infection during the transplantation etc. (Zezos and Renner, 2014). Toll like receptors (TLRs) have been shown to play a major role in the pathogenesis of NASH and clinical studies show that endotoxemia as a result of leaky gut in NASH condition, can lead to TLR4 activation (Das et al., 2015; Petrasek et al., 2013). Increased oxidative stress, perox-initrite formation, HMGB1 protein, Fibrinogen or other DAMPs (damage associated molecular patterns) associated with liver injury, can also act as ligands for TLR4 signaling (Petrasek et al., 2013). TLR4 activation can lead to increased IKK and JNK activation, increased NFkB translocation into nucleus etc., which leads to inflammation, fibrosis, insulin resistance and other metabolic dysregulation during NASH (Nace et al., 2013). Abrogation of TLR4 trafficking to lipid rafts helps to decrease inflammation in murine NASH models and had better histological outcomes (Petrasek et al., 2013). As TLR4 activation is a major event that leads to several inflammatory pathway activations and fibrogenesis, a potent TLR4 antagonist can be useful to treat such condition.
Different inflammatory signaling pathways are pivotal mediators of liver fibrogenesis and control the transition of quiescent hepatic stellate cells (HSC) to collagen-secreting myofibroblasts (Yi and Jeong, 2013). Systemic levels of endotoxins have been detected in patients with liver cirrhosis (lipopolysaccharide, LPS) (Fukui et al., 1995). Systemic endotoxins can be recognized by TLR4 present in hepatocytes, Kupffer cells, stellate cells, sinusoidal endothelial cells, biliary epithelial cells in the liver (Weber et al., 2016). Upon detecting the LPS or PAMPS, the TLR4 receptors give rise to a myriad of inflammatory signaling pathways and increase the production of inflammatory cytokines, induce macrophage infiltration and increase oxidative stress (Carrillo-Sepulveda et al., 2015; Shen et al., 2007; Weber et al., 2016). Recent studies have indicated that tissue injury and matrix degradation can release some endogenous ligands also referred to as DAMPS which can activate TLR4 signaling. Injured liver has augmented expression of TLR4 and its co-receptors which makes it more sensitive to the cascade of inflammatory cell signaling pathway mediated by TLR4 signaling in the damaged tissue (Guo and Friedman, 2010). Induced TLR4 signaling can give rise to different downstream factors like pro-inflammatory cytokines, chemotactic cytokines, reactive oxygen species (ROS), adhesion molecules, cell cycle regulating proteins, TGF-β1 pseudoreceptor BAMBI-which can give rise to pro-fibrogenic signals. Hepatic stellate cells along with kupffer cells, may be a target for TLR4 ligand induced liver injury and offer a direct connection between inflammatory and fibrotic liver damage (Guo and Friedman, 2010). We have previously shown that, activation of NADPH oxidase induces Toll like receptor 4 (TLR4) recruitment to the lipid rafts and thus potentiates inflammation (Das et al., 2015). We have also shown that fibrogenesis in NASH is regulated by a NADPH oxidase dependent pathway which increases microRNA21 activation, hepatic stellate cell activation and accelerates TGFβ signaling (Dattaroy et al., 2015).
Our previous studies have characterized a plant derived compound SsnB as a TLR4 antagonist. SsnB has also been shown to have antiinflammatory, anti-angiogenic, anti-proliferative and anti-oxidant properties (Bateman et al., 2013; Kumar et al., 2014; Liang et al., 2015; Liu et al., 2015). Further, we have already shown that SsnB decreases oxidative stress, kupffer cell activation, cytokine production and macrophage infiltration in an early model of NASH (Dattaroy et al., 2016).
In this research, we study the therapeutic anti-fibrotic role of a novel plant derived compound, Sparstolonin B (SsnB), on a murine model of nonalcoholic steatohepatitis. The purpose of the present study was to investigate the impact of SsnB on hepatic fibrosis in vivo and in vitro through assessment of the effects of SsnB on the proliferation of hepatic stellate cells, extracellular matrix deposition and pro-fibrogenic signaling pathways. We also wanted to find out the effect of SsnB on anti-proliferative proteins and hedgehog signaling pathway. NASH is a significant cause of mortality as it can cause liver fibrosis, cirrhosis, portal hypertension, and hepatocellular carcinoma (Amarapurka et al., 2006; Fierbinteanu-Braticevici et al., 2002; Yoshioka et al., 2004). During NASH associated chronic liver inflammation, activated hepatic stellate cells (HSCs) deposits excessive extracellular matrix proteins in the liver resulting in fibrotic scars (Higashi et al., 2017). However, there is no therapy available to treat liver fibrosis in the patients with NASH except some life style modifications (Blond et al., 2017). Thus, it is important to understand the molecular mechanisms of hepatic fibrosis to design therapeutic strategies to prevent and treat liver fibrosis. Also, since TGFβ signaling activation can induce JAK-STAT pathway and upregulate stress fiber assembly in myofibroblasts, we investigated the role of SsnB in alteration of those pathways (Edlund et al., 2002; Tang et al., 2017). We used rat primary hepatic stellate cells and human transformed hepatic stellate cells (LX2) as our in vitro models. Our data shows that SsnB can decrease myofibroblast proliferation through modulating multiple signaling pathways that include attenuation of TLR-induced cell proliferation, fibroblast differentiation and TGF-beta signaling followed by a subsequent decrease in focal adhesion and stress fiber assembly, a critical process in epithelial to mesenchymal transition (EMT) found in NASH (Alsamman et al., 2017).
2. Materials and methods
2.1. Cell culture
Human immortalized stellate cell line (LX2) (kindly gifted by Dr. Ana Mae Diehl, Duke University) was grown in complete Dulbecco’s Modified Eagle’s Medium (DMEM, Corning, VA) with 10% fetal bovine serum (FBS, Atlas Biologicals, CO) and 1x Penicillin-Streptomycin solution (Gibco, Life Technologies, NY) at 37 °C in a humidified incubator with 5% C02. Tissue culture plastic wares, HBSS buffer were purchased from Corning (Corning, VA). Sparstolonin B (SsnB) compound was kindly gifted by our collaborator Dr. Daping Fan (Cell Biology and Anatomy, University of South Carolina School of Medicine). Stock solution of SsnB was prepared in 100% dimethyl sulfoxide (DMSO). The final concentrations of SsnB on cells were 10 μM (SsnB10) and 100 μM (SsnB100). The equal amounts of DMSO were added to control cells and LPS treated cells to normalize the DMSO effect in SsnB treated cells. The cells were plated on 6 well/12 well plates using DMEM medium supplemented with 10% FBS. Before treating the cells with LPS (100 ng/ml, in PBS) and SsnB (10 μM, 100 μM), cells were cultured in DMEM with 2% FBS (overnight). All treatments are given for 24 h in 2% FBS containing DMEM medium. Cells treated with 0.1% DMSO were used as control. Primary rat hepatic stellate cells (ScienCell Research Lab, Carlsbad, CA) were cultured in specific medium as instructed by the manufacturer, on poly-l-lysine (ScienCell Research Lab)-coated 6well/12-well plates and incubated at 37 °C in a humidified incubator with 5% CO2 to initiate the culture. Treatments of LPS and SsnB were given as discussed above. All treated cells were subjected to mRNA and protein extraction. Maintaining aforementioned conditions, cells were plated on coverslips by putting coverslips on each well of 12 well plates and the cells adhered on coverslips were used for immune-fluorescence dual labeling staining after the treatment.
2.2. Mouse models
Pathogen-free, male mice with C57BL/6J background (Jackson Laboratories, Bar Harbor, ME) were housed one per cage at 23–24 °C with a 12-h/12-h light/dark cycle at libitum access to food and water. They were fed with 60% kcal high fat diet (Research Diets, New Brunswick, NJ) from 6 weeks until 16 weeks to generate a model of steatosis. We will refer to this group as Control. A similar group of high fat fed mice were administered Bromodichloromethane BDCM (1 mmol/kg, diluted in corn oil) though intraperitoneal injection, twice a week for 4 weeks to generate BDCM induced Non-alcoholic steatohepatitis model of mice (NASH) that induced fibrosis (Seth et al., 2013). BDCM and corn oil were purchased from Sigma-Aldrich (St. Louis, MO). A group of NASH mice were administered with SsnB (3 mg/kg) intraperitoneally twice for 4 weeks (NASH + SsnB). NIH guideline for Humane Care and Use of Laboratory Animals and local IACUC standards were followed during animal handling. Animal experiments were approved by the University of South Carolina at Columbia. Upon completion of the treatment, all mice were killed for liver tissues and serum samples for further experiments.
2.3. Laboratory analysis
2.3.1. Picrosirius red staining
Picrosirius red staining of formalin-fixed, paraffin embedded liver tissue Section (5-μm-thick) was done using a Nova ultra-sirius red stain kit following manufacturer’s instructions (IHC-World). Liver sections were observed under a 10X objective of a light microscope. Morphometric analysis of the stained regions of the tissue sections was performed using cellSens software (Olympus). The degree of fibrosis was evaluated by following the METAVIR scoring system (F0: no fibrosis, FI: portal fibrosis without septum formation, F2: portal fibrosis with few septum formation, F3: portal fibrosis with several septum formations but no cirrhosis and F4: cirrhosis) on the basis of histological observation of the slides.
2.3.2. Immunohistochemistry
Formalin-fixed, paraffin embedded liver tissue Section (5-μm-thick) were subjected to de-paraffinization and antigen retrieval using standard protocol. Immunohistochemistry was performed on neutral buffer formalin (NBF) fixed liver tissues according to the protocol described in our early publication (Dattaroy et al., 2016). Mouse monoclonal p53 (Cell Signaling Technology, MA) and p21 antibody (Santa cruz Biotechnology, TX) were used in recommended dilution. Rest of the experiment was processed as described in our previous publication (Dattaroy et al., 2016). Antibodies against fibronectin, BAMBI (Abeam, MA) were used in recommended dilution. Morphometric analysis of the immunoreactivity of tissue sections was performed using cellSens software (Olympus).
2.4. Immunofluorescence microscopy
De-paraffinization and antigen retrieval was performed on formalin-fixed; paraffin embedded liver tissue Section (5-μm-thick) were done by using standard protocol. The primary antibodies MDM2 (Santa cruz Biotechnology, TX), Glil, α-SMA (Abeam, MA) were used in recommended dilutions. Species-specific secondary antibodies conjugated with Alexa Fluor 633 (Invitrogen, CA) were used against the appropriate primary antibodies. Rest of the experiment was processed as described in our previous publication (Dattaroy et al., 2016). After completion of the treatments as previously stated, the LX2 and/or Primary Rat hepatic stellate Cells attached coverslips were fixed and processed according to our previous publication (Dattaroy et al., 2016). Cells were incubated with α-SMA (Abeam, MA), p53 (CST, Danvers MA), and p21 (Santa cruz Biotechnology, TX) primary antibodies followed by species-specific Alexa Fluor 633 (R) and 488 (G) (described above), for immunofluorescence dual-labeling staining. Alexa Fluor 633 was used against p53 and p21 antibodies. Alexa Fluor 488 was used against α-SMA antibody. LX2 and/or Primary Rat hepatic stellate Cells were incubated with pSMAD2/3 (Abeam, MA) and SMAD4 (Santa cruz Biotechnology, TX) primary antibodies followed by species-specific Alexa Fluor 633 (red, R) and Alexa Fluor 488 (Green, G) for immunofluorescence dual-labeling staining. Alexa Fluor 633 was used against anti-p-SMAD2/3 antibody. Alexa Fluor 488 was used against anti-SMAD4 antibody. Paxillin, α-SMA and vinculin primary antibodies (Santa Cruz biotechnology, Inc. Santa Cruz, CA) were used in 1:250 dilutions. Species specific alexa Fluor 488 (G) was used against vinculin antibody and species specific alexa Fluor 633 (R) was used against α-SMA antibody. ProLong Gold antifade reagent with DAPI (Life Technologies) was used to mount the stained cells attached on the coverslips and viewed under 20 × or 40 × objective with an Olympus BX51 fluorescent microscope.
2.5. Quantitative real-time polymerase chain reaction
Gene expression (mRNA) levels from mice liver tissue and hepatic stellate cell samples (LX2 and primary rat hepatic stellate cells) were measured by quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR) by following our routine lab protocol. The primers used for Real time PCR in 5’ to 3’ orientations are rat p53 (Forward: GCACGGCCTTTGTGGTAAAA, Reverse: TTTGCCAGGGCTGAGTAACC) and rat p21 (Forward: TGCCTTAGCCTTCATTCAGTGT, Reverse: TATCGAATTGCACGAGGGGAG)., Rat Glil (Forward: AACTCCACGAGCACACAGG and Reverse: GCTCAGGTTTCTCCTCTCTC), Rat Gli2 (Forward: CCATCCATAAGCGGAGCAGA and Reverse: CCAGATCTTCCTTGAGATCAG), Rat IHH (Forward: CCTCGTCTTGGTGTAGAG and Reverse: GAATCGCAGTCAGAGCTAGC), Rat Ptc (Forward: AAAGATGGATGTGGGCAAACG and Reverse: TCACACCCTGAGCCTTCCAT), Rat BAMBI (Forward: GCGGGGCGTCAATGGATCGC, Reverse: GAACTCAGAAGGCCTTCAAGG), Human Glil (Forward: GGCTCGCCATAGCTACTGAT and Reverse: CCAGCGCCCAGACAGAG), Human Gli2 (Forward: AGTTAATGAGAACCTGGGCAGT and Reverse: TTGGCAAAGGCGGGATAGTC), Human IHH (Forward: CAGCCTGCTCTCACTACGAG and Reverse: CCCAAAGGGGCCTAAGATGG), Human Ptc (Forward: GGGTGGCACAGTCAAGAACAG and Reverse: TACCCCTTGAAGTGCTCGTACA).
2.6. miR21 expression levels in liver tissues
Total miRNA was isolated from Control, NASH and NASH + SsnB liver tissue by using Qiazol reagent and miRNAeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Purified miRNA (1 μg) was converted to cDNA using miScript cDNA synthesis kit (Qiagen, Valencia, CA). Quantitative real-time PCR was performed with the miR 21 specific primer using miScript SYBR Green PCR master mix (Qiagen, Valencia, CA) and CFX96 thermal cycler (Bio-rad, Hercules, CA). Threshold Cycle (Ct) values for the selected genes were normalized against RNU6-2 (internal expression control) values in the same sample.
2.7. Western blot
Western blot from in vivo and in vitro samples were performed according to our previous publication (Liang et al., 2015). Primary antibodies PTEN, BAMBI, β-actin, α-SMA, CTGF (Abeam, MA), p53, phospho SMAD2/4, total SMAD2/3 (Cell Signaling Technology, MA), phospho STAT3, Cyclin E, cleaved caspase3, total caspase3 and cleaved PARP1 (Santa Cruz biotechnology, Inc. Santa Cruz, CA), were used at recommended dilutions, and compatible horseradish peroxidase-conjugated secondary antibodies were used.
2.8. TUNEL assay
TUNEL based ApopTag® technology (EMD Millipore, MO) was used to detect apoptotic cells according to the manufacturer’s instructions. ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA) with DAPI was used to mount the coverslips. The cells were imaged using immunofluorescence microscopy under 20× objective.
2.9. Cell cycle analysis
After treating LX2 cells with LPS and/ SsnB (as described previously), the cells harvested and centrifuged for 10 min at 120 g at 25 °C in the medium. After aspirating the medium, cells were washed with PBS and again centrifuged for 10 min at 120 g at 25 °C. This step was repeated, and the cells were fixed with 70% ice cold ethanol (dropwise) for 30 min on ice. After fixing the cells, they were centrifuged again at 120 g for 5 min. Ethanol was aspirated and 1 ml of propidium iodide solution (containing RNase A and 0.1%Triton X) was added to the pellets and incubated in ice for 30 min on ice. The samples were analyzed by Beckman Coulter FC500 Flow Cytometer at Institutional Resource Facility at the University of South Carolina School of Medicine.
Confocal microscopy: Confocal microscopy,colocalization analysis and fluorescence data interpretations for HDAC1 in the nucleus were carried out at the NIEHS core facility.
2.10. Statistics
Data were represented as mean ± S.E.M Statistical significance was calculated by Student’s t-test and the graphs were plotted using Origin (OriginLab Corporation, MA). p ≤ 0.05 was considered statistically significant.
3. Results
3.1. SsnB treatment ameliorates liver fibrosis in NASH mice
Picrosirius red stain is widely used to stain extracellular collagen matrix to detect fibrotic scar deposition in tissues. This staining is based on the firm binding of this stain’s sulfonic acid groups with the basic moieties of collagen fibers. Histological evaluation of mice liver tissues in NASH group showed increased deposition of collagen matrix and fibrosis compared to Control group. SsnB treated group (NASH + SsnB) showed significant reduction of fibrosis (Fig. 1A, B)(P < 0.05). In Control group fibrosis scoring was F0 (no fibrosis), NASH group the fibrosis grade was F2 to F3 (periportal and septal fibrosis). Interestingly, fibrosis scoring in NASH + SsnB group was F0 to F1 (no fibrosis to mild fibrosis without septa) (Fig. 1C).
Fig. 1. SsnB treatment ameliorates liver fibrosis in NASH mice.
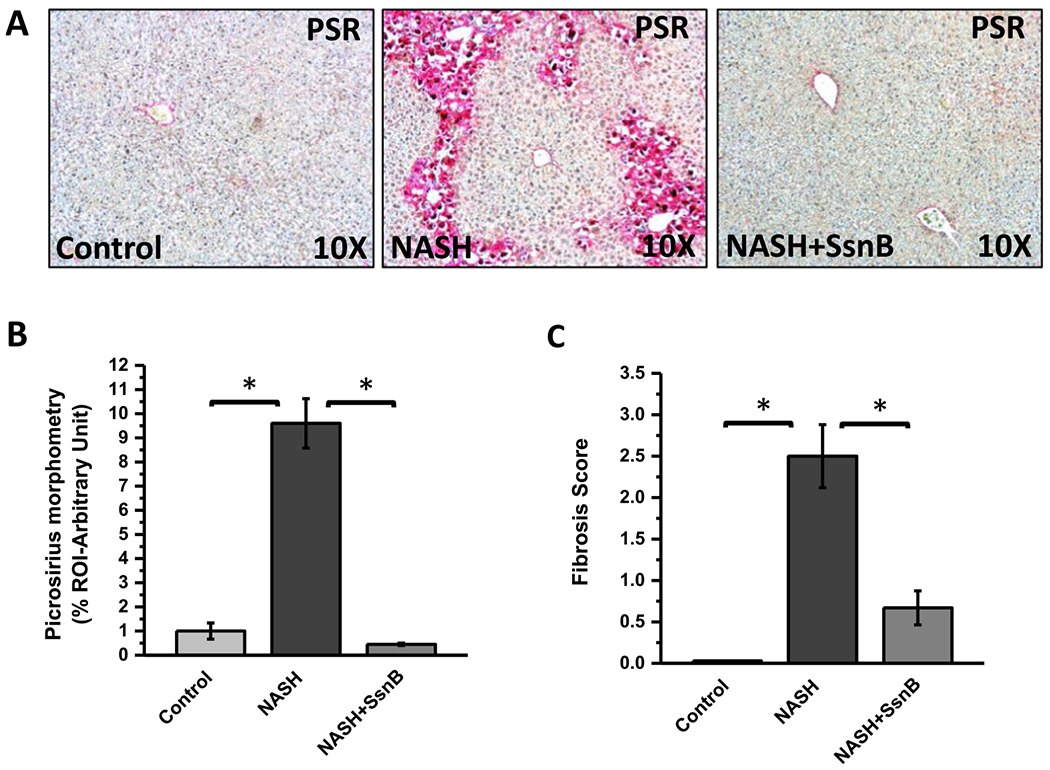
A: Representative images of picrosirius red stain of Control, NASH and NASH+SsnB mice. Images were taken at 20× magnification. (*P < 0.05). B: Morphometric analysis of picrosirius red immunohistochemistry in liver slices from Control, NASH and NASH+SsnB mice groups. (*P < 0.05). C: The degree of fibrosis by METAVIR scoring system. (*p < 0.05).
3.2. SsnB treatment decreased m1croRNA21 expression and upregulated PTEN protein expression in NASH liver
MicroRNA21 (miR21) is known to downregulate tumor suppressor protein PTEN (Zhang et al., 2010). We have previously shown that SsnB administration decreases miR21 expression and induces PTEN expression in an early NASH model where diet induced obese mice were subjected to BDCM treatment for 1 week (Dattaroy et al., 2016). Interestingly, we observed similar characteristics of SsnB when it was administered in our full-blown NASH model where diet induced obese mice were subjected to BDCM treatment for 4 weeks. We found that, SsnB treatment (NASH + SsnB group) significantly decreased miR21 expression compared to NASH group (Fig. 2A) (P < 0.05). PTEN protein expression was decreased in NASH group compared to Control. However, SsnB treatment (NASH + SsnB) augmented PTEN expression (Fig. 2B,C) which complemented the reduced expression of miR21 in that group (P < 0.05).
Fig. 2. SsnB treatment decreased microRNA21(miR21) expression and upregulated PTEN protein expression in NASH liver.
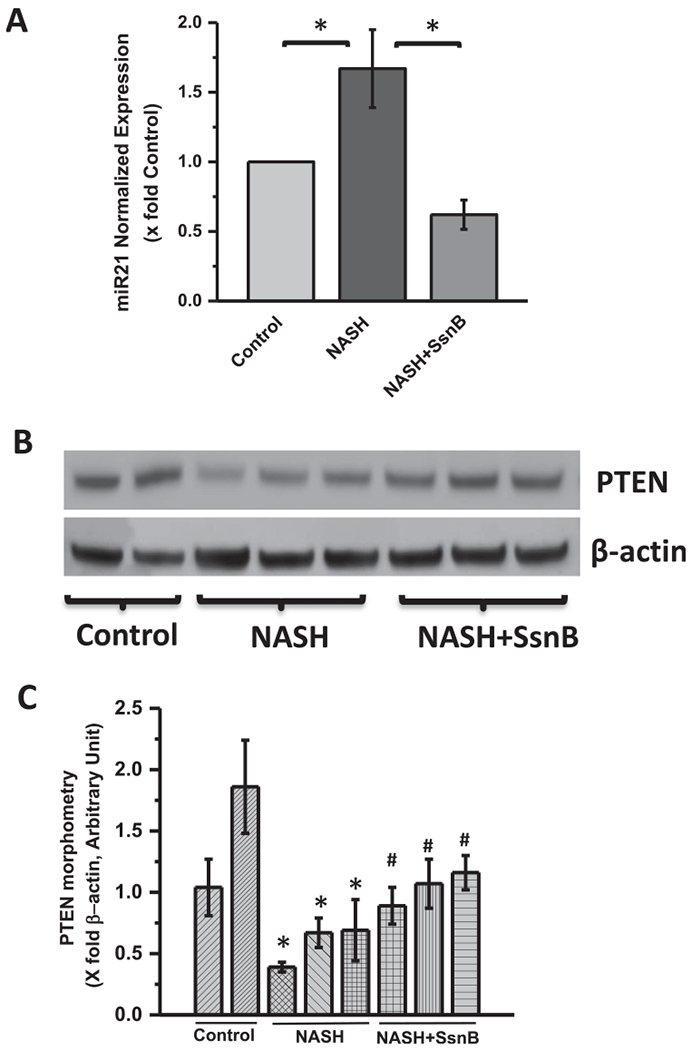
A: qRT-PCR analysis of miR21 expression of Control, NASH, and NASH+SsnB mouse liver samples normalized against Control (*P < 0.05). B: Western blot analysis of β-actin and PTEN protein levels of Control, NASH, and NASH+SsnB liver homogenates. C. Bar diagram representing levels of PTEN protein normalized against β-actin of respective samples. Y-axis represent arbitrary unit of PTEN band of mice groups from Control, NASH and NSH+SsnB liver homogenate. (*) P < 0.05 is considered statistically significant when compared with control. (#) P < 0.05 is considered statistically significant when compared with NASH.
3.3. SsnB treatment induced PTEN expression increased p53, p21 upregulation and decreased hedgehog signaling in liver
To detect the possible mechanism of PTEN induced expression of p53, we measured the immunoreactivity of MDM2 in paraffin embedded liver tissue sections though immunofluorescence microscopy. We observed significantly increased immunoreactivity of MDM2 in NASH liver section compared to Control liver (Figs. 3A, 4A). SsnB treated (NASH + SsnB) liver showed considerable decrease in MDM2 immunoreactivity compared to NASH liver (Figs. 3A, 4A). MDM2 is a known inhibitor of p53. Moreover, immunoreactivity against GUI, a pro-fibrogenic nuclear transcription factor, was significantly increased in NASH liver section compared to Control liver (Figs. 3B, 4B). SsnB treated (NASH + SsnB) liver showed remarkable decrease in Glil immunoreactivity compared to NASH liver as observed by immunofluorescence microscopy (Figs. 3B, 4B)) (P < 0.05). Immunohistochemistry against p21 also revealed significant decrease in p21 immunoreactivity in NASH liver compared to Control (Figs. 3C, 4C). SsnB treatment significantly increased p21 immunoreactivity in the sinusoidal area of liver in NASH + SsnB group compared to NASH group (Figs. 3C, 4C) (P < 0.05). Interestingly, Immunohistochemistry against p53 revealed decreased p53 protein expression in NASH mice liver in the sinusoidal area which was augmented with SsnB treatment (NASH + SsnB) (P < 0.05) (Figs. 3D, 4D). Morphometry of the immunoreactivities are shown in Fig. 4A–D.
Fig. 3. SsnB treatment induced PTEN expression increases p53, p21 upregulation and decreases hedgehog signaling in liver.
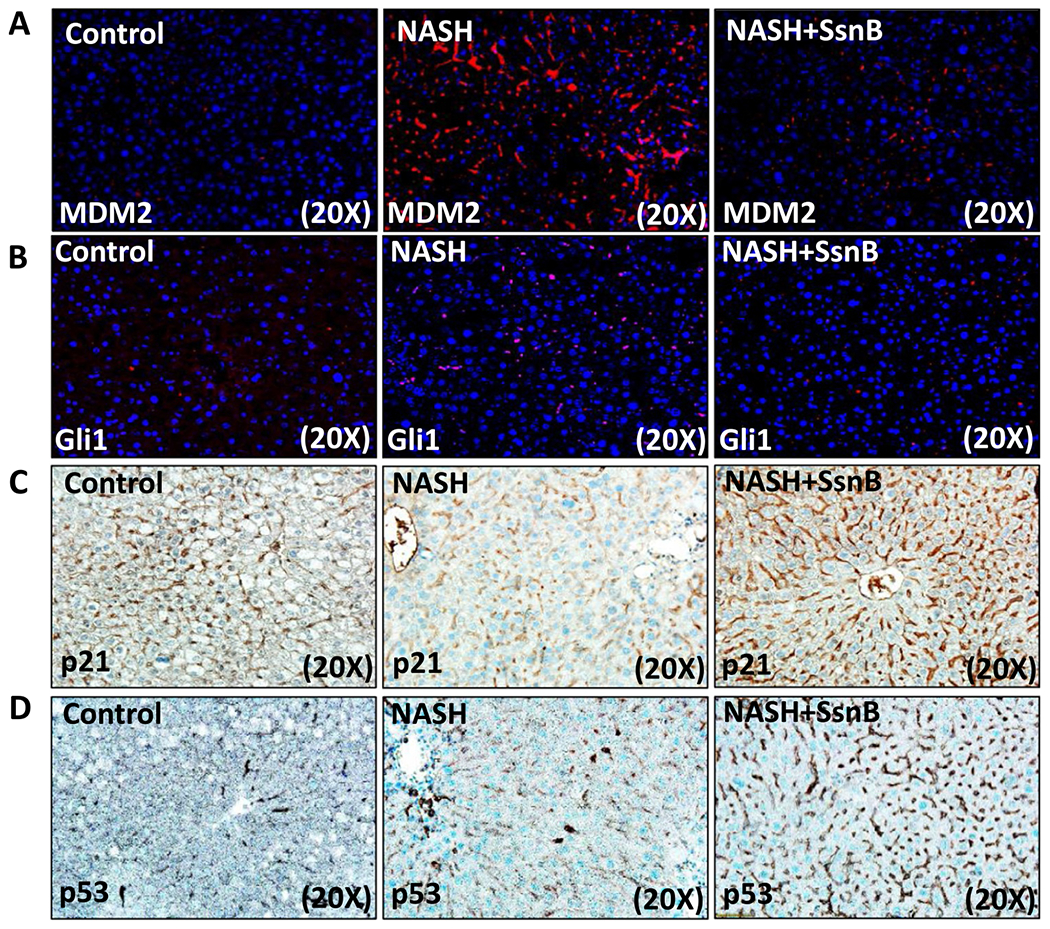
A and B: Representative images of MDM2 (A) and Gli1 (B) immunoreactivity as shown by immunofluorescence microscopy on liver slices of Control, NASH and NASH+SsnB mice, images taken at 20× magnification using immunofluorescence microscopy. C and D: Representative images of p21 (C) and p53 (D) immunoreactivity as shown by immunohistochemistry on liver slices of Control, NASH and NASH+SsnB mice, taken at 20× magnification.
Fig. 4. Morphometric analysis of Fig. 3.
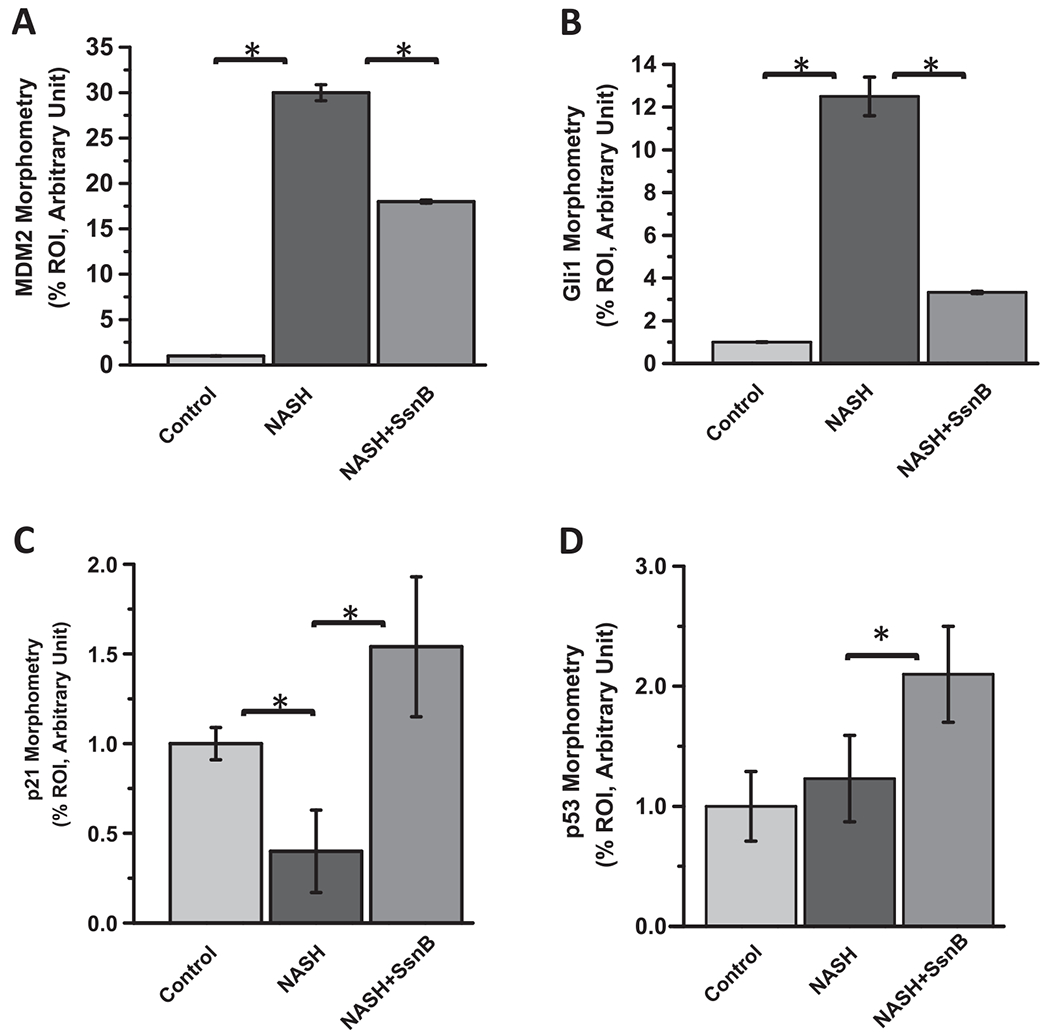
Morphometric analysis of immunoreactivities of (A)MDM2, (B)Gli1, (C)p21 and (D)p53. Between group comparisons were compared with a Students t-test and analyses with a *p < 0.05 were considered significant.
3.4. SsnB treatment increased p53, p21 expression in vitro
To see whether SsnB can upregulate expression of p53 and p21 in vitro as it does in vivo, we measured gene expression of p53 and p21 through qRT PCR experiments in primary rat hepatic stellate cells. LPS treatment significantly decreased the gene expression of p53 and p21 compared to untreated or control cells (Fig. 5A). However, SsnB treatment (100 μM) was able to improve LPS induced downregulation of those genes (Fig. 5A) (P < 0.05). Immunofluorescence microscopy also revealed increased immunoreactivity of p53 and p21 in SsnB (100 μM) treated primary rat hepatic stellate cells (LPS + SsnBlOO) compared to only LPS treatment (Fig. 5B, C respectively). The increased immunoreactivity of p53 and p21 in SsnB treated primary rat hepatic stellate cells (LPS + SsnB) was further confirmed by the immunoblots compared to β-actin (Fig. 5D–F). Due to unavailability of same batch of SsnB we were restricted to use 10 μM SsnB of the same batch for this experiment. We found that even 10μM SsnB was able to significantly increase the P53 and P21 protein expression as observed by immunoblot (p < 0.05).
Fig. 5. SsnB treatment increased p53, p21 expression in vitro.
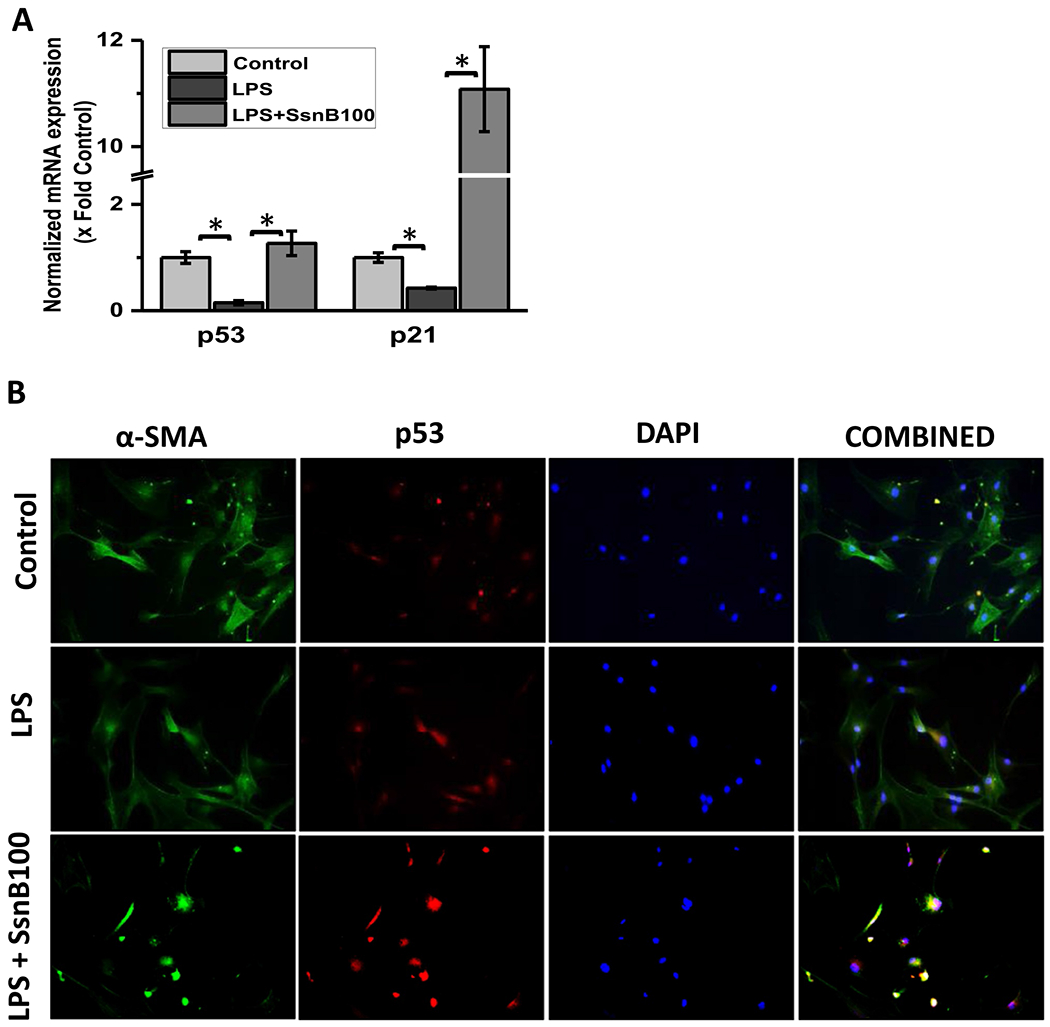
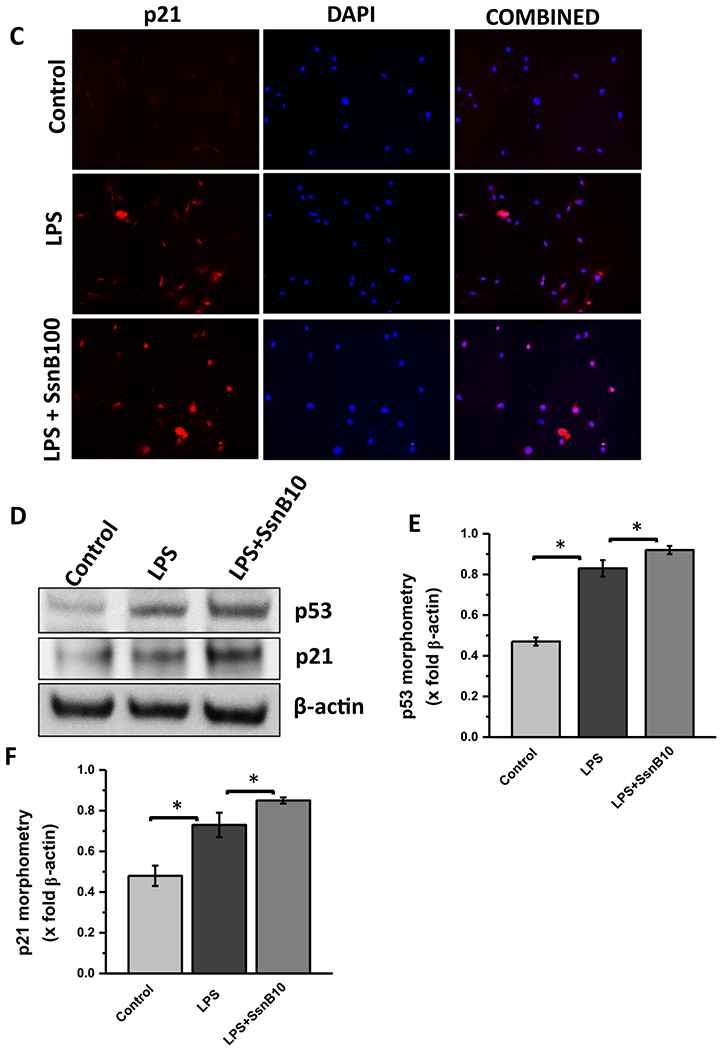
A: qRT-PCR analysis of mRNA expression of p53 and p21 from control (untreated), LPS-treated, and LPS+SsnB100 (100 μM) treated Rat primary hepatic stellate cells, normalized against control (*P < 0.05). B. Immunofluorescence dual labeling of Control (untreated), LPS-treated, and LPS+SsnB100 (100 μM) treated Rat primary hepatic stellate cells depicting α-SMA (green)-p53 (red) co-localization (yellow), taken at 20× magnification. C. Immunoreactivity of p21 (red) in control (untreated), LPS-treated, and LPS+SsnB100 (100 μM) treated Rat primary hepatic stellate as shown by immunofluorescence microscopy at 20× magnification. D. Western blot analysis of p53, p21 and β-actin protein levels in Control, LPS and LPS+SsnB10 (10 μM) primary Rat hepatic stellate cell homogenates. E and F. Bar diagram representing the levels of p53 protein (E) and p21 (F) normalized against β-actin of respective samples. (*) P < 0.05 is considered statistically significant.
3.5. SsnB treatment decreased gene expression of hedgehog signaling markers and reduces Cyclin E protein expression in vitro
To correlate SsnB induced repression of Glil (a hedgehog transcription factor) in an in vitro model of human hepatic stellate cells (LX2), we measured gene expression of hedgehog signaling specific markers through qRT PCR experiments. LPS treatment increased the gene expression of Glil, Gli2, Indian hedgehog (IHH) and Patched (Ptc) compared to untreated or control cells (Fig. 6A). However, SsnB treatment was able to repress LPS induced upregulation of those genes (Fig. 6A) (P < 0.05). To see whether SsnB upregulation of PTEN, p53, p21 and repression of Glil alters proliferation of activated HSCs, we measured Cyclin E protein level from cell homogenates by western blot. LPS treatment had increased Cyclin E level compared to Control which decreased upon SsnB treatment (Fig. 6B–C). We also conducted the similar experiment in primary rat hepatic stellate cells. The results showed similar pattern of Glil where SsnB treatment ameliorate LPS induced Glil and IHH mRNA expression (Supplementary Fig. 1A). The LPS treated primary rat hepatic stellate cells showed a non-significant increase in Cyclin D1 expression and decreased in LPS + SsnB (10 μM) treated cells (Supplementary Fig. 1B, C).
Fig. 6. SsnB treatment decreased gene expression of hedgehog signaling markers and reduces Cyclin E protein expression in vitro.
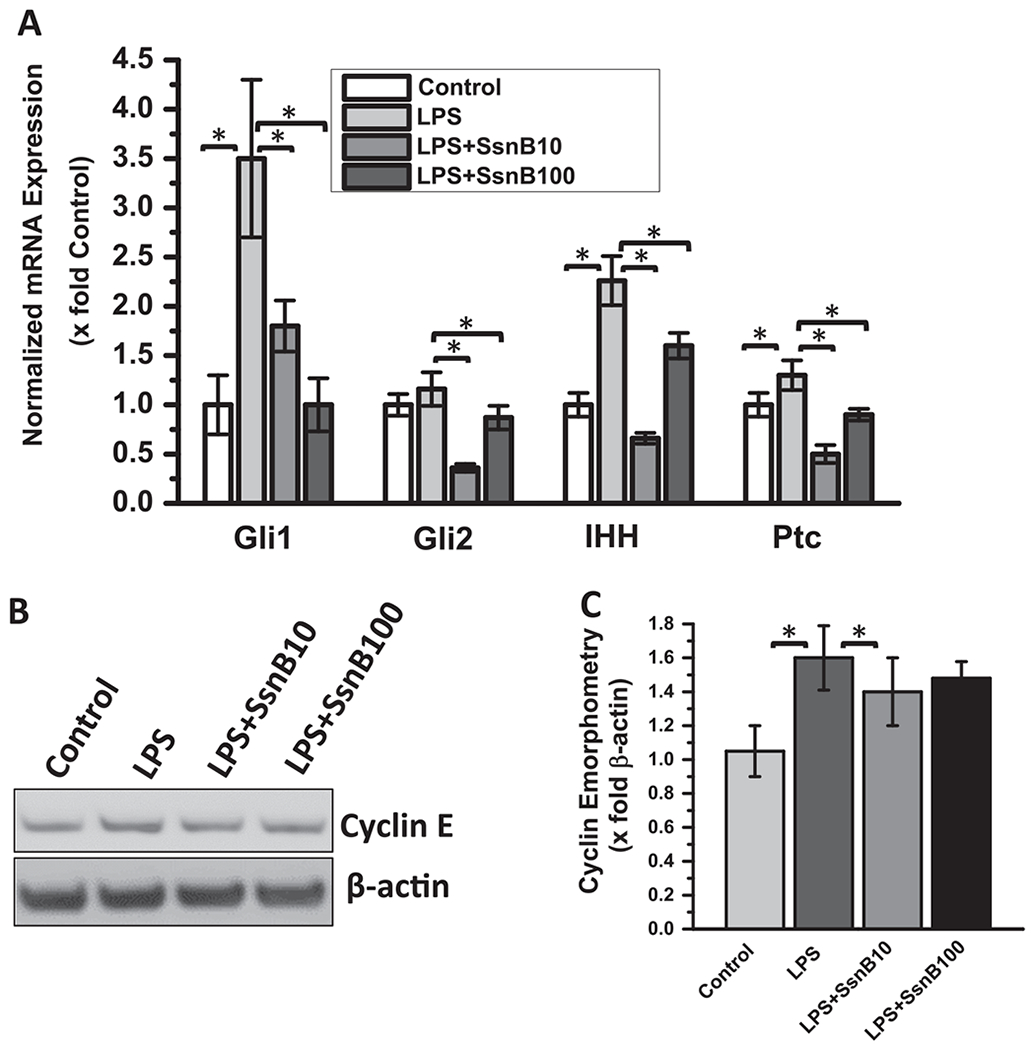
A: qRT-PCR analysis of mRNA expression of Gli1, Gli2, IHH, Ptc from Control (untreated), LPS-treated, LPS+SsnB10(10 μM) and LPS+SsnB100 (100 μM) treated human immortalized hepatic stellate cells (LX2), normalized against control (*P < 0.05). B: Western blot analysis of Cyclin E and β-actin protein levels of Control (untreated), LPS-treated, LPS+SsnB10(10 μM) and LPS+SsnB100(100 μM) treated human immortalized hepatic stellate cells (LX2). C. Morphometric analysis of western blot: Bar diagram represents the level of cyclin E normalized against β-actin of respective samples. (*) P < 0.05 is considered statistically significant.
3.6. SsnB treatment decreases proliferation and induces apoptosis in hepatic stellate cells
We performed flow cytometry to evaluate the effect of SsnB on hepatic stellate cell cycle progression. After treating the LX2 cells with vehicle, LPS and/ SsnB for 24 hs (as described previously), the cells were collected and stained with propidium iodide and then analyzed by Beckman Coulter FC500 Flow Cytometer. The resulting data clearly demonstrates that treatment of LX2 cells with SsnB (both 10 μM and 100 μM concentrations) induced accumulation of cells in G2/M phase (38.14% and 36.75% in SsnB 10 μM and 100 μM respectively) compared to LPS treated group (27.55%) accompanied by a decreased amount of cells in the G0-G1 phase (Fig. 7A). We also observed significantly higher number of apoptotic nuclei in SsnB treated cells (LPS + SsnB), compared to LPS treated and control cells (Fig. 7B) (P < 0.05). Western blot data clearly reveals increased p53 accumulation, increased cleaved caspase3 (Casp3) and cleaved PARP1 proteins in SsnB treated LX2 cells compared to LPS treated and control cells (Fig. 7C,D)), further establishing the pro-apoptotic role of SsnB. High apoptotic nuclei were also observed in the SsnB treated mouse liver (NASH + SsnB) as compared to NASH liver or control mouse liver (Fig. 7E,F). We also conducted the similar experiment in primary rat hepatic stellate cells. The western blot results showed similar pattern of increased p53, cleaved Caspase-3 (normalized with caspase 3) upon SsnB (10 μM) treatment in LPS primed cells (Supplementary Fig. 2A, B). Also high apoptotic nuclei were observed in SsnB (10 μM) treated LPS primed cells as compared to Control or LPS treated cells (Supplementary Fig. 2C).
Fig. 7. SsnB treatment decreases proliferation and induces apoptosis in hepatic stellate cells.
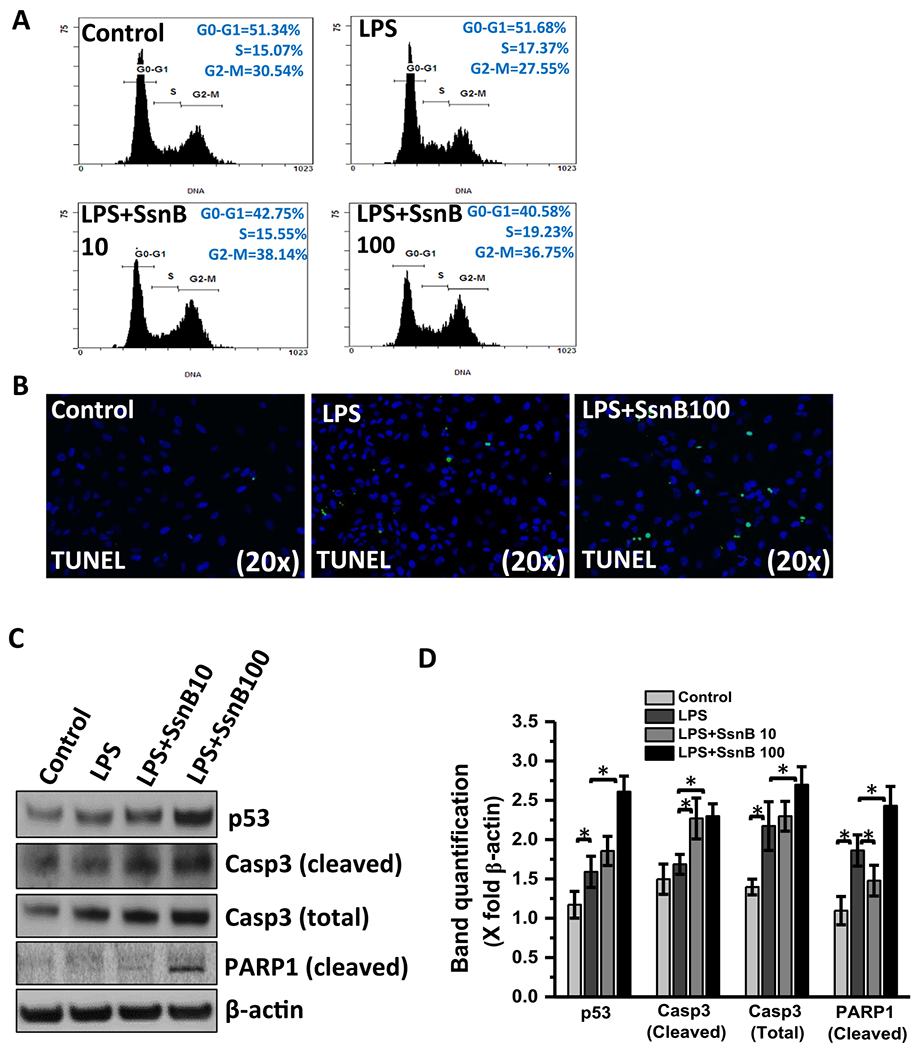
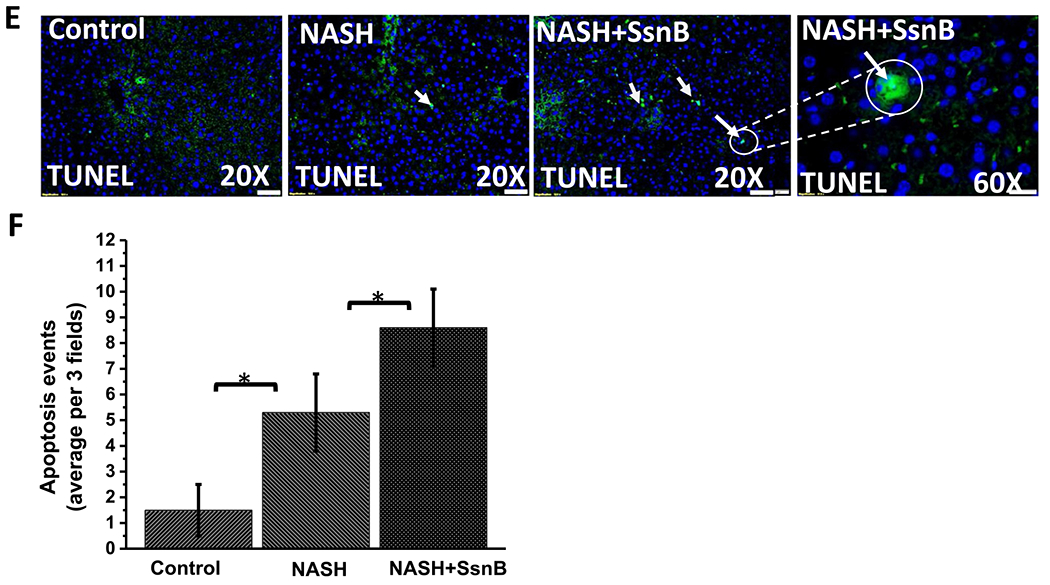
A. Cell cycle analysis of untreated cells (control), cells treated with LPS, LPS+SsnB10(10 μM), and LPS+SsnB100(100 μM). Quantitation of the PI staining data is presented as the cell cycle distribution percentages. B. Apoptosis is indicated by TUNEL based ApopTag® technology (EMD Millipore, MO) which labels 3’-OH ends of DNA fragments by fluorescent antibody as detected by immunofluorescence microscopy in Control (untreated), LPS-treated, and LPS+SsnB100 (100 μM) treated LX2 cells. C. Western blot analysis of p53, cleaved caspase3 (Casp3), total caspase3 (Casp3), cleaved PARP1 and β-actin protein levels of Control (untreated), LPS-treated, LPS+SsnB10 (10 μM), and LPS +SsnB100(100 μM) treated LX2 cells. D. Morphometric analysis of western blot where the bar diagram represents the level of p53, cleaved caspase3, total caspase3, cleaved PARP1 normalized against β-actin of respective samples. (*) P < 0.05 is considered statistically significant. E. TUNEL assay-based apoptosis analysis of Control, NASH, and NASH+SsnB treated mice liver samples. F. Morphometric analysis of apoptotic events/3 microscopic field. (*) P < 0.05 is considered statistically significant.
3.7. SsnB treatment decreases hepatic stellate cell activation In murine NASH
Hepatic stellate cells reside in the sinusoidal endothelium space of the liver and play a key role to induce fibrosis in the liver. Upon activation in NASH, the quiescent hepatic stellate cells transform into fibrogenic myofibroblasts and produce alpha smooth muscle actin protein (α-SMA). α-SMA is a reliable marker to detect hepatic stellate cell activation in the liver. We have already shown in the previous chapter that SsnB administration ameliorates fibrotic scars in NASH liver. Our immunofluorescence data showed that SsnB administration significantly decreased α-SMA expression compared to NASH mouse liver (Fig. 8A,B) (P < 0.05).
Fig. 8. SsnB treatment decreases hepatic stellate cell activation in murine NASH.
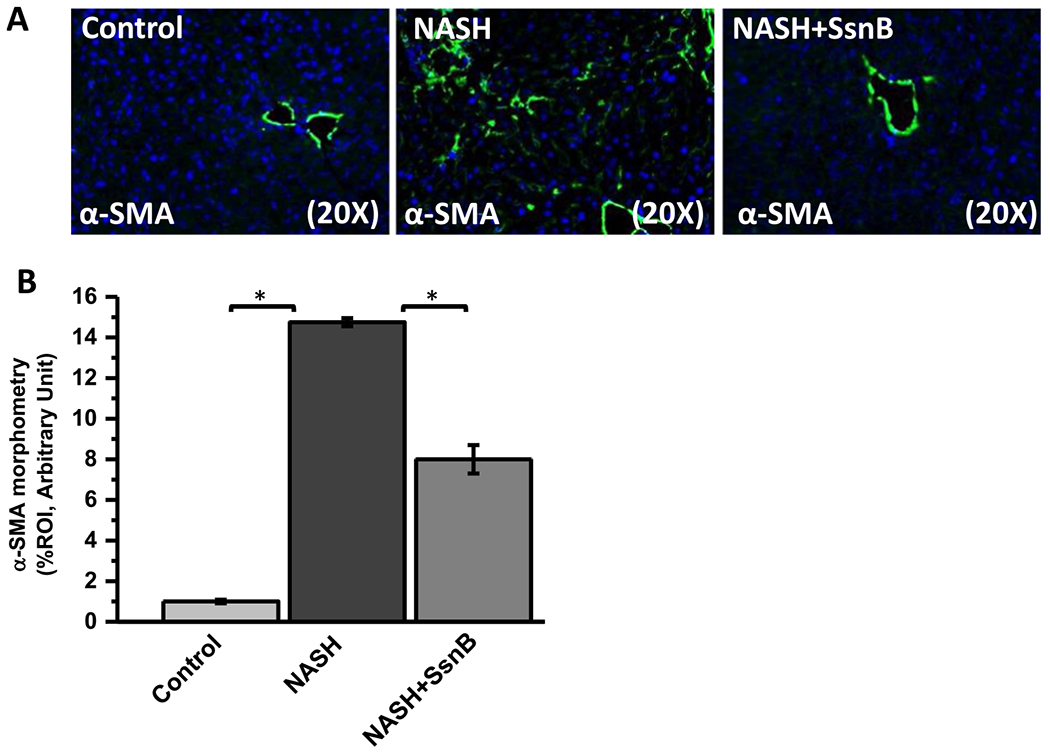
A. Representative images of α-SMA immunoreactivity as shown by immunofluorescence microscopy on liver slices of Control, NASH and NASH+SsnB mice, taken at 20× magnification using immunofluorescence microscopy. B. Morphometric analysis of α-SMA immunoreactivity in A. (*P < 0.05).
3.8. SsnB treatment In NASH mice upregulates BAMBI In liver
Studies have shown than TLR4 signaling activation can induce increased hepatic fibrosis by downregulating TGFβ pseudo receptor BAMBI. BAMBI is TGF-β type I receptor lacking an intracellular kinase domain. So it blocks signal transduction even after stimulation with TGF-beta superfamily ligands (Liu et al., 2014; Onichtchouk et al., 1999; Seki et al., 2007). Interestingly, immunohistochemistry of BAMBI on mouse liver sections showed that, BAMBI immunoreactivity was significantly decreased in NASH group compared to the Control group (P < 0.05). However, NASH mice treated with SsnB (NASH + SsnB group) showed considerably increased immunoreactivity of BAMBI compared to NASH liver tissues (Fig. 9A,B) (P < 0.05).
Fig. 9. SsnB treatment in NASH mice upregulates BAMBI in liver.
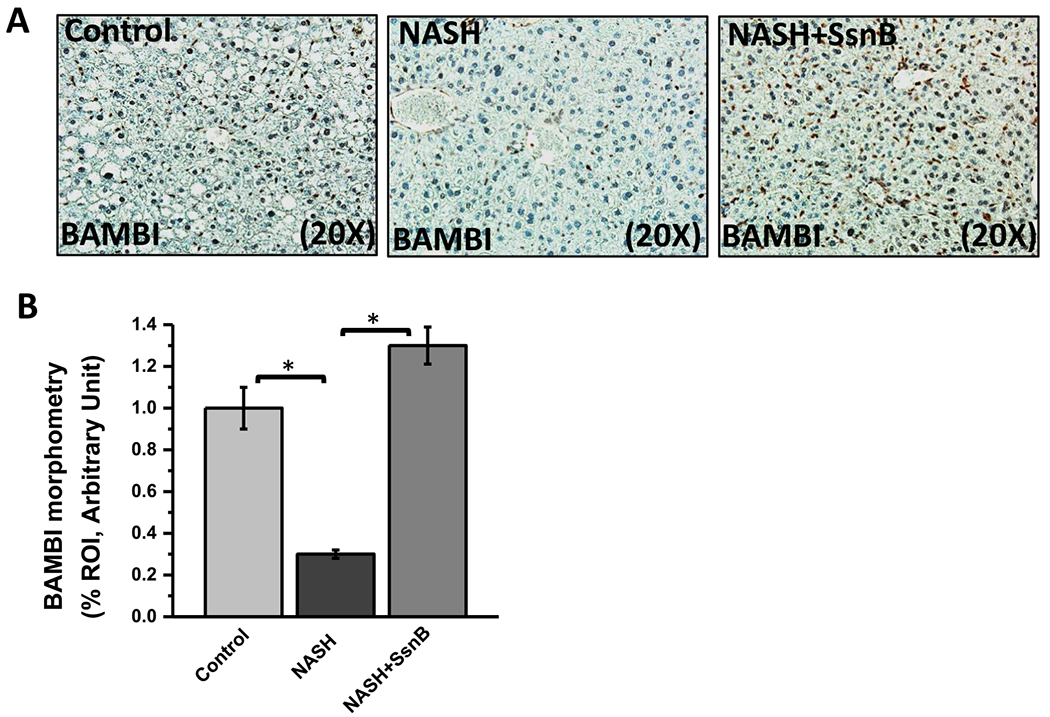
A. Representative images of BAMBI immunoreactivity as shown by immunohistochemistry on liver slices of Control, NASH and NASH+SsnB mice, taken at 20× magnification. B. Morphometric analysis of BAMBI immunoreactivity in A. (*P < 0.05).
3.9. SsnB treatment decreases flbronectln deposition in NASH liver
Fibronectin is an extracellular matrix (ECM) protein and its expression is stimulated by TGFβ signaling pathway (Dallas et al., 2005). It is also known to play an important role in liver fibrosis (Dattaroy et al., 2015). Interestingly, immunohistochemistry of fibronectin on mouse liver sections showed that, fibronectin immunoreactivity was significantly increased in NASH group in the sinusoidal area compared to the Control group. However, NASH mice treated with SsnB (NASH + SsnB group) showed considerably decreased immunoreactivity of fibronectin compared to NASH liver tissues (Fig. 10A, B) (P < 0.05).
Fig. 10. SsnB treatment decreases fibronectin deposition in NASH liver.
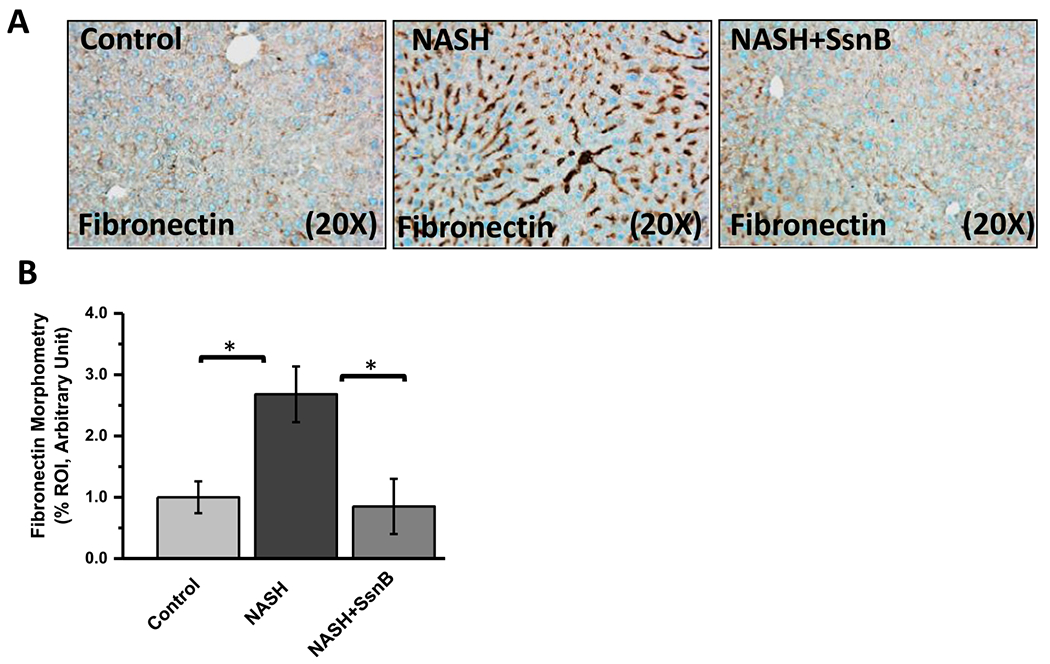
A. Representative images of fibronectin immunoreactivity as shown by immunohistochemistry on liver slices of Control, NASH and NASH+SsnB mice, taken at 20× magnification. B. Morphometric analysis of fibronectin immunoreactivity in A. (*P < 0.05).
3.10. SsnB upregulates BAMB1 and decreases TGFβ signaling in vitro
Upon activated by TGFβ ligands and PAMPS, quiescent hepatic stellate cells (HSC) become profibrogenic myofibroblasts and are primarily responsible to induce hepatic fibrogenesis in NASH by producing excessive extracellular matrix proteins (Friedman, 2008; Leask and Abraham, 2004). Interestingly, our in vitro data from rat primary hepatic stellate cells show that LPS treatment decreases BAMBI mRNA and protein expression compared to untreated or control cells (Fig. 11A, B) (P < 0.05). However, LPS + SsnB (100 μM) treated cells augmented BAMBI mRNA (Fig. 11A) and protein expression compared to only LPS treated cells (Fig. 11B, C) (P < 0.05). SsnB treatment also decreased SMAD2/3, SMAD4 co-localization compared to only LPS treated LX2 cells (transformed human hepatic stellate cells) (Fig. 11D). Phosphorylation of SMAD2/3 protein was also decreased upon SsnB treatment compared to the LPS treated cells (Fig. 11E,F).
Fig. 11. SsnB upregulates BAMBI and decreases TGFβ signaling in vitro.
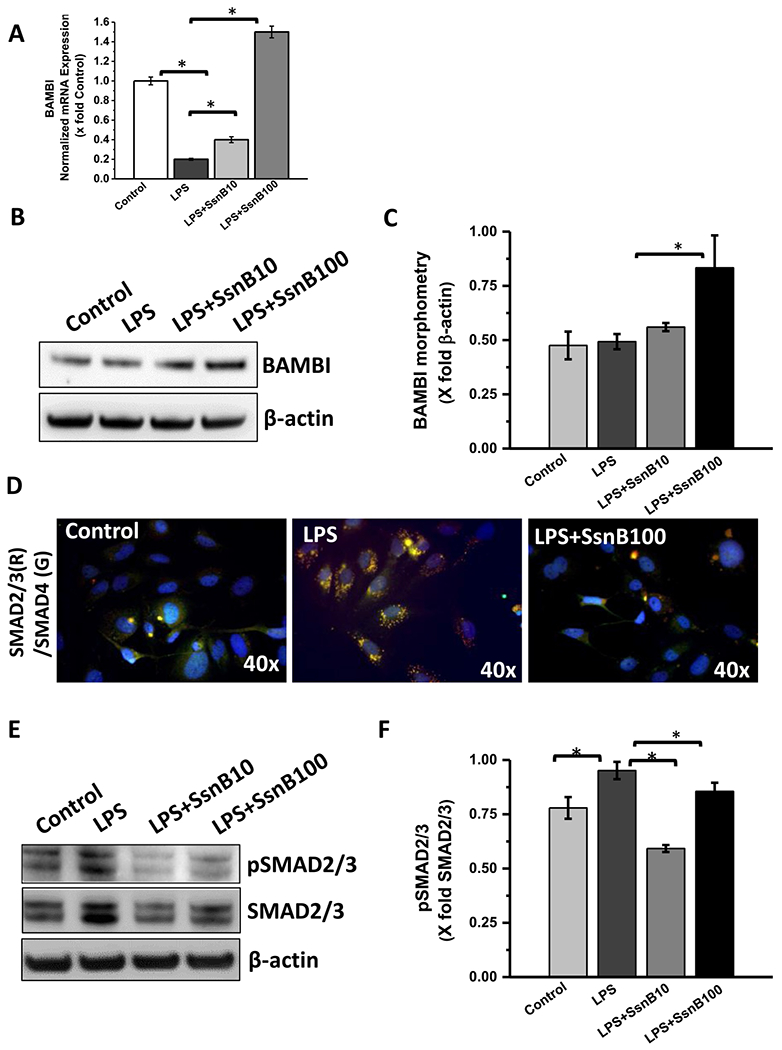
A. qRT-PCR analysis of mRNA expression of BAMBI from control (untreated), LPS-treated, LPS+SsnB10 (10 μM), and LPS+SsnB100 (100 μM) treated Rat primary hepatic stellate cells, normalized against control (*P < 0.05). B. Western blot analysis of β-actin and BAMBI protein levels of Control (untreated), LPS-treated, LPS+SsnB10 (10 μM), and LPS+SsnB100 (100 μM) treated rat primary hepatic stellate cells, C. Morphometric analysis of western blot where the bar diagram represents the level of BAMBI normalized against β-actin of respective samples. (*) P < 0.05 is considered statistically significant. D. Immunofluorescence dual labeling depicting SMAD2/3 (red)-SMAD4 (green) co-localization (yellow) on Control (untreated), LPS-treated, and LPS+SsnB100 (100 μM) treated human immortalized hepatic stellate cells (LX2) taken at 40× magnification. E. Western blot analysis of phosphor SMAD2/3, total SMAD2/3 and β-actin protein levels of Control (untreated), LPS-treated, LPS+SsnB10 (10 μM), and LPS+SsnB100 (100 μM) treated human immortalized hepatic stellate cells (LX2). F. Morphometric analysis of western blot where the bar diagram represents the level of pSMAD2/3 normalized against total SMAD2/3 of respective samples. (*) P < 0.05 is considered statistically significant.
3.11. SsnB downregulates STAT3 phosphorylation, decreases stellate cell activation and connective tissue growth factor in vitro
STAT3 inhibition is known to suppress hepatic stellate cell mediated fibrogenesis (Nunez Lopez et al., 2016). STAT3 also cooperates with TGFβ1 in activation and anti-apoptosis of hepatic stellate cells (Xu et al., 2014). TGFβ is also known to activate JAK1-STAT3 axis to augment liver fibrosis in coordination of SMAD mediated signaling pathway (Tang et al., 2017). We preferred to see whether SsnB modulates STAT3 activation in LX2 cells. Western blot analysis showed that SsnB treatment significantly decreased STAT3 phosphorylation, and CTGF protein expression compared to LPS treated LX2 cells (Fig. 12A,B). However, α-SMA protein expression also showed decreasing pattern upon SsnB treatment but the observation was not significant. (Fig. 12A,B). We also conducted the similar experiment in primary Rat hepatic stellate cells. The western blot results showed nonsignificant decrease of α-SMA and CTGF protein but no change in pSTAT3 upon SsnB (10 μM) treatment in LPS primed cells (Supplementary Fig. 3A, B).
Fig. 12. SsnB downregulates STAT3 phosphorylation, decreases stellate cell activation and connective tissue growth factor in vitro.
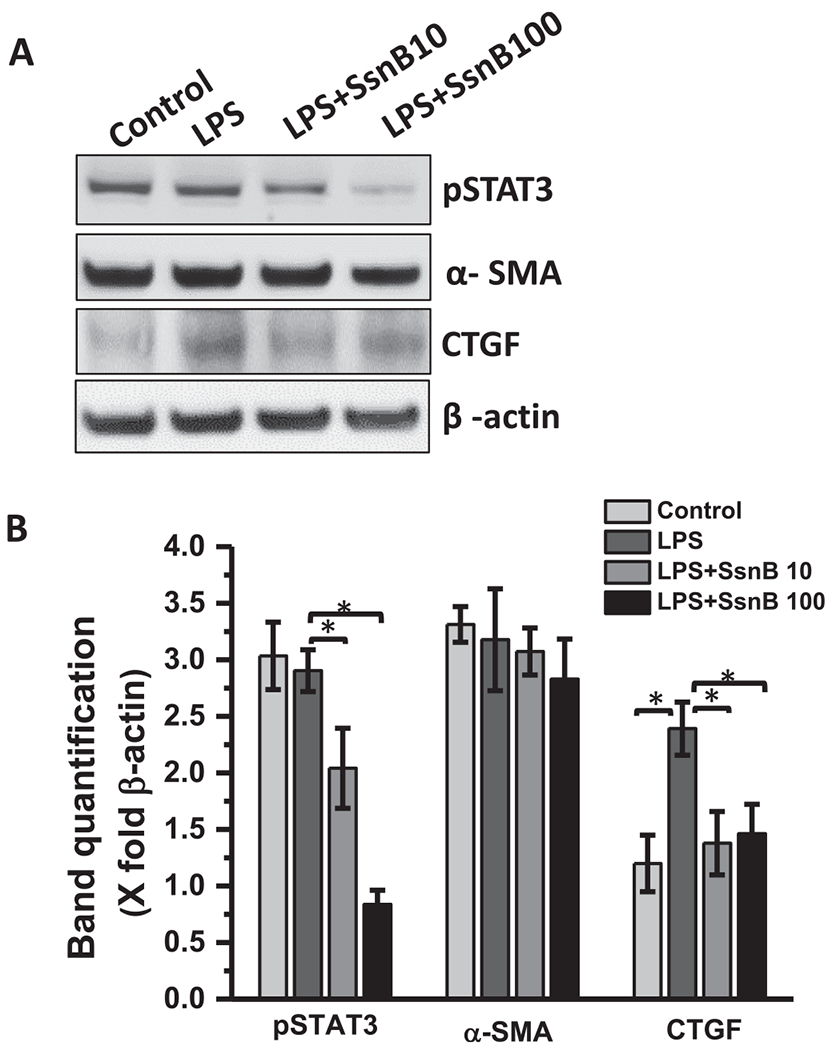
A. Western blot analysis of phospho STAT3, α-SMA, CTGF and β-actin protein levels of Control (untreated), LPS-treated, LPS+SsnB10 (10 μM), and LPS+SsnB100 (100 μM) treated human immortalized hepatic stellate cells (LX2). B. Morphometric analysis of western blot where the bar diagram represents the level of pSTAT3, α-SMA, and CTGF normalized against β-actin of respective samples. (*) P < 0.05 is considered statistically significant.
3.12. SsnB treatment decreases focal adhesion associated adaptor protein expression and inhibits stress fiber formation in vitro
Paxillin is a multi-domain focal-adhesion adaptor protein located at the edge between the plasma membrane and cytoskeleton. It helps in the process of cell adhesion and acts as a scaffold to bind many signaling proteins to the cell membrane (Turner, 2000). Immuno-fluorescent imaging of paxillin shows that Control and LPS stimulated LX2 cells (Fig. 13A,B) and Rat primary hepatic stellate cells (Fig. 13D) displayed heterogeneous adhesions which are large and elongated at the periphery of the cells. However, SsnB treatment on LX2 and primary cells significantly decreases paxillin immunoreactivity at the periphery. The focal adhesion regions in SsnB treated cells are smaller than Control or LPS treated cells. While lower concentration of SsnB (10 μM) decreased paxillin expression at the periphery of LX2 cells, higher concentration of SsnB further abrogated this focal adhesion adaptor protein expression and altered the cell morphology (Fig. 13A,B). Maturation of focal adhesions fuels the assemblage of adhesion-associated actin bundles known as radial stress fibers (Oakes et al., 2012). We have already seen in Fig. 12 that LPS treatment in LX2 cells increased a-SMA protein expression compared to untreated or control cells. SsnB treatment decreased α-SMA protein expression in LPS stimulated LX2 cells. In Fig. 13C (LX2 cells), Fig. 13E (primary cells), immunofluorescent staining of α-SMA specifically depicts the stress fiber morphology on both LX2 and primary cells. We observed that, SsnB treatment blocks the stress fiber formation compared to LPS treated cells as indicated by immunoreactivity of α-SMA (red) and vinculin (green). The cell morphology also changed upon SsnB treatment. They have smaller cell body, extended cell processes and they also lose stress fibers-similar to the quiescent stellate cells (Anthony et al., 2010).
Fig. 13. SsnB treatment decreases focal adhesion protein expression and inhibits stress fiber formation in vitro.
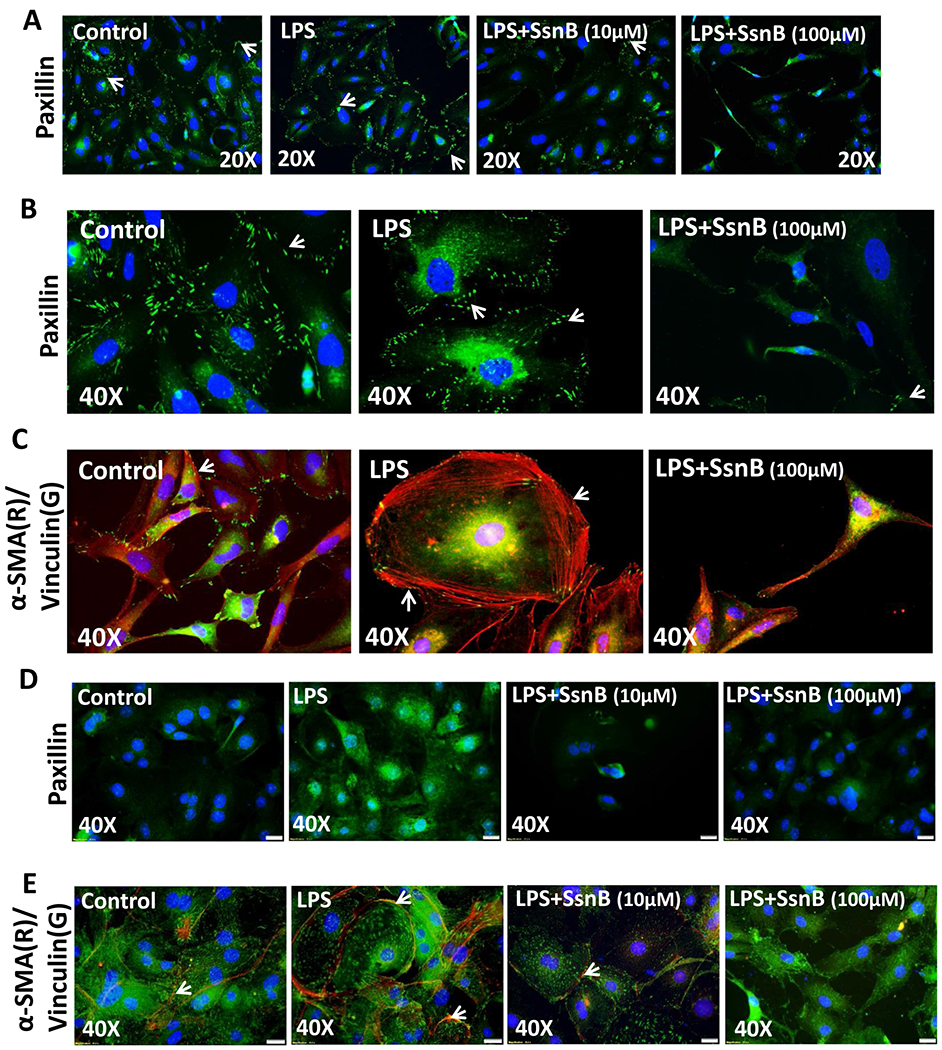
A. Immunofluorescence microscopy depicting paxillin (green) immunoreactivity in Control (untreated), LPS-treated, LPS+SsnB10(10 μM), and LPS+SsnB100(100 μM) treated human immortalized hepatic stellate cells (LX2) taken at 20× magnification B and D. Immunofluorescence microscopy depicting paxillin (green) immunoreactivity at 40× magnification in Control (untreated), LPS-treated, and LPS+SsnB100(100 μM) treated human immortalized hepatic stellate cells (LX2) (B) and Control (untreated), LPS-treated, LPS +SsnB10(10 μM), and LPS+SsnB100(100 μM) treated Rat primary hepatic stellate cells (D). Paxillin immunoreactivity (on the edges of the cells) is pointed by white arrows. C and E. Immunofluorescence dual labeling depicting α-SMA(red)-Vinculin(green) on Control (untreated), LPS-treated, and LPS+SsnB100(100 μM) treated human immortalized hepatic stellate cells (LX2) (C) and Control (untreated), LPS-treated, LPS+SsnB10(10 μM) and LPS+SsnB100(100 μM) treated in rat primary hepatic stellate cells (E) taken at 40× magnification. α-SMA immunoreactivity (as stress fibers) is pointed by white arrows. Nucleus stained with DAPI (Blue).
4. Discussion
This study unravels the anti-fibrotic and anti-proliferative mechanisms of a plant derived TLR4 antagonist, Sparstolonin B (SsnB). Mechanistically, SsnB reduces TLR signaling by inhibiting MyD88 recruitment to TLR4 (Liang et al., 2011). Our initial histochemical data from picrosirius red stained liver slices proved that SsnB prevents NASH induced fibrotic scars. We wanted to explore whether SsnB inhibits fibrosis through inhibiting TLR4 dependent pro-fibrogenic pathways. It is known that TLR4 signaling pathway activation can upregulate miR21 (Yang and Seki, 2012) which was reversed by SsnB, a TLR4 antagonist. PTEN (phosphatase and tension homolog deleted on chromosome ten) is known to be a tumor suppressor (Wei et al., 2013). It can also inhibit PI3K/Akt signaling by dephosphorylating phosphatidyllnositol (3,4,5)-trisphosphate (PIP3) at position 3 and also known to be involved in cell motility, proliferation, survival, metabolism and cellular architecture (Wei et al., 2013). It is known that PTEN can down regulate TLR4 induced pro-inflammatory pathways (Yin et al., 2016). Micro-RNA 21 is a known inhibitor of PTEN (Sheedy et al., 2010). Our study shows that PTEN expression is augmented in SsnB treated samples where miR21 was also downregulated (Fig. 2). This proves that, SsnB can upregulate PTEN expression by inhibiting TLR4 induced miR21 expression in our NASH model. It is known that activated TLR4-PI3K pathway can induce p53 degradation by upregulating MDM2 (Odkhuu et al., 2015). Recent research shows that senescence induction in fibrogenic cells by increasing p21, p53 can help to reduce excessive fibroblast proliferation and suppress hepatic tumor (Krizhanovsky et al., 2008). However, no research has focused on the possible route of repressing hepatic stellate cell proliferation by antagonizing TLR4-PI3K/Akt-MDM2 signaling pathway by SsnB. We found that SsnB, a TLR4 antagonist can possibly affect TLR4-PI3K/Akt signaling by upregulating PTEN protein expression. Our data suggested that SsnB treatment downregulated MDM2 which was otherwise activated in NASH liver. Interestingly, we found that, p53 was upregulated in SsnB treatment, which proves that SsnB induced p53 activation by suppressing MDM2 in a PTEN dependent mechanism (Brooks and Gu, 2006). We also found upregulated p21 protein expression in SsnB treated liver (NASH + SsnB) compared to NASH liver. It is known that increased p53 induces p21 expression (Benson et al., 2014) which further supports our findings. Induction of p53 and p21 in fibrotic liver can be instrumental to decrease uncontrolled proliferation of fibroblasts as our data might suggest. As uncontrolled HSC (a hepatic fibroblast) proliferation plays a major factor in hepatic fibrogenesis, we wanted to see whether SsnB induced activation of p53, p21 and decreased the proliferation of activated stellate cells. In vitro results showed that SsnB treatment increases mRNA and protein levels of p53 and p21 in HSC which was otherwise repressed by LPS-proving the anti-proliferative effect of SsnB. Hedgehog signaling plays a key role in liver fibrosis and is an important therapeutic target of anti-fibrotic drugs (Yang et al., 2014). Glioma-associated oncogene homologl (Glil) is a transcription factor which is a downstream target of hedgehog signaling pathway (Rimkus et al., 2016). Previous research has shown that increased p53 expression is known to inhibit Glil (Yoon et al., 2015). We found that SsnB treated mice liver tissue (NASH + SsnB) having upregulated p53 protein expression also had reduced expression of Glil compared to NASH mice liver. Activation of hepatic stellate cells (HSC) induces fibrosis in the liver and suppression of Hedgehog signaling in these cells is known to inhibit HSC activation (Li et al., 2015). We found that SsnB treatment in HSC culture downregulates LPS induced activation of Hedgehog signal specific gene expression (Fig. 6).
Hedgehog signaling pathway is known to induce proliferation by upregulating Cyclin D and Cyclin E. Shh proliferative signaling stimulates or maintain cyclin gene expression and activity of the Glcyclin-Rb axis in proliferating cells (Duman-Scheel et al., 2002; Kenney and Rowitch, 2000). Glil inhibition is also known to inhibit cell growth and cell cycle progression at G2/M phase and induced apoptosis (Sun et al., 2014). Several researchers have already shown that SsnB can inhibit angiogenesis and proliferation of cancer cells by inhibiting mitotic cyclins (Bateman et al., 2013; Benson et al., 2014). Similarly, our study found that SsnB treatment decreased Cyclin D activation in hepatic stellate cells (Fig. 6B). We also observed SsnB induced suppression of stellate cell proliferation at G2/M phase of cell cycle and apoptosis of hepatic stellate cells. Apoptosis induction in activated HSCs is one important therapeutic target to decrease HSC proliferation and hepatic fibrosis (Zhao et al., 2017). Anti-apoptotic role of SsnB has previously been shown in different cell types (Kumar et al., 2014). We observed significant number of apoptotic cells in SsnB treated group (LPS + SsnB) compared to untreated cells and LPS treated cells (Fig. 7). Inhibition of hepatic stellate cell proliferation can reduce liver fibrosis and is a major therapeutic target of anti-fibrotic drugs (Balta et al., 2015; Pan et al., 2004). Anti-proliferative and pro-apoptotic properties of SsnB could render it as a potential antifibrotic molecule. In future, it will be interesting to see therapeutic role of SsnB in other in vivo models of liver fibrosis as numerous studies have shown that no one murine model is a true representation of the human disease and could be viewed as a limitation in this study. Apart from HSCs, hepatic cholangiocytes and hepatocytes can also acquire phenotype of myofibroblasts through a process of epithelial to mesenchymal transition in the liver (Fausther et al., 2013; Forbes and Parola, 2011).
Intestinal microflora and a functional TLR4 are essential for hepatic fibrogenesis (Pradere et al., 2010). TLR4 activation can induce hepatic stellate cell proliferation and extracellular matrix deposition in the liver, resulting in liver scarring in chronic liver diseases. Increased TLR4 signaling in hepatic stellate cells induces chemokine secretion and chemotaxis of macrophages but downregulates TGFβ pseudoreceptor bone morphogenetic protein and activin membrane bound inhibitor (BAMBI) and thus sensitizes the HSCs to TGFβ induced activation and myofibroblastic differentiation (Seki et al., 2007). We observed that SsnB treatment decreased hepatic stellate cell activation in vivo as indicated by decreased αSMA immunoreactivity (Fig. 8). TLR4 activation induces NF-KBp50:HDACl interaction which represses transcription of BAMBI promoter (Liu et al., 2014). BAMBI is TGFβ type I receptor lacking an intracellular kinase domain. It blocks signal transduction even after timulation with TGFβ superfamily ligands and thus, decrease of BAMBI on hepatic stellate cells can increase TGFβ signaling and fibrogenesis (Liu et al., 2014; Seki et al., 2007). As abrogation of TLR4 signaling induces BAMBI mediated inhibition of pro-fibrogenic TGFβ signaling pathway, a TLR4 antagonist like SsnB might be assumed to have the potency to decrease TLR4 mediated hepatic fibrosis. Our results showed that SsnB treated NASH mice had upregulated BAMBI expression in liver which corresponds to decreased fibrogenesis (Fig. 9). Fibronectin, a multifunctional glycoprotein and extracellular matrix (ECM) component is produced by hepatic stellate cells and is required to support other extracellular matrix protein assembly (Kohan et al., 2010; Liu et al., 2016). We have previously shown that, BDCM, a chemical that initiated fibrogenesis induced TGFβ signaling and promoted fibronectin protein expression in the NASH liver (Dattaroy et al., 2015). Here we have observed that SsnB induced decreased stellate cell activation and TGFβ signaling through BAMBI upregulation which resulted in decreased fibronectin expression. From the histological data of picrosirius red stained liver slices in the previous dataset, we could correlate that SsnB treatment significantly decreased fibrotic scar deposition compared to NASH liver. Decreased fibronectin deposition upon SsnB treatment might be responsible in decreased collagen matrix accumulation in the liver. As hepatic stellate cells are the major fibrogenic cells in the liver, we wanted to see if SsnB mediated repression of TGFβ signaling by BAMBI upregulation is stellate cell dependent. Interestingly, we found that SsnB upregulates BAMBI in vitro (Fig. 11). TGFβ signals by binding to TGFβRI an TGFβRII transmembrane proteins, each having serine/threonine kinase Ligand binding to this receptor protein complex, phosphorylates and activates TGFβRI by the cytoplasmic kinase domains of TGFβRII Activated TGFβRI phosphorylates SMAD2 and SMAD3 which can form a complex with SMAD4 and assemble in the nucleus to initiate target gene expression in a SMAD dependent pathway (Hata and Chen, 2016). We found that SsnB decreased LPS induced TGFβ signaling by decreasing SMAD2/3 phosphorylation. SsnB induced reduction of TGFβ signaling was further confirmed by decreased co-localization of SMAD4 and phosphorylated SMAD2/3 in the nucleus in vitro. TGFβRI can also signal through non-SMAD pathways (Zhang, 2017). It has been recently discovered that activated TGFβ signaling can also phosphorylate STAT3 and thus activates JAK1-STAT3 axis in coordination with SMAD pathway to induce TGFβ mediated fibrotic response in hepatic stellate cells (Tang et al., 2017). Our in vitro data shows that SsnB decreased STAT3 phosphorylation in hepatic stellate cells and also downregulates αSMA and CTGF (connective tissue growth factor) protein levels (Fig. 12). Both of these proteins are key players in fibrotic proliferation of myofibroblasts and are downstream mediators of TGFβ signaling (Zhang et al., 2004). SsnB mediated downregulation of both αSMA and CTGF in hepatic stellate cells can be instrumental to decrease pro-fibrogenic cell proliferation. It is known that phosphorylated STAT3 localization to the focal adhesions increases the aggressive clinical behavior of proliferative cells. STAT3 is known to be required for cell motility (Silver et al., 2004). Focal adhesion plays an important role in liver fibrosis through activating hepatic stellate cells and induces TGFβ driven pro-fibrotic responses (Zhao et al., 2017). αSMA is vital for focal adhesion maturation in myofibroblasts Activated myofibroblasts incorporate αSMA in stress fibers. αSMA has been found to decrease the intracellular mechanical stress on focal adhesions and induces their supermaturation (Hinz et al., 2003). We found that, SsnB treatment changes the cell morphology of hepatic stellate cells. SsnB (100 μM) treated LX2 cells became smaller, possessed less stress fibers and acquired a dendritic morphology. SsnB also induced αSMA filament disassembly and abolished focal adhesions. Inhibition of focal adhesions and stress fibers of hepatic stellate cells by higher concentrations of SsnB can be crucial to inhibit stellate cell migration and it can also stop the stellate cells from acquiring a fibrogenic phenotype (Fig. 13).
In conclusion, SsnB inhibited liver fibrosis in murine NASH model and in HSC culture by modulating the expression of cell cycle related proteins and by downregulating the Hedgehog signaling pathway. Further SSnB targeted TGF-beta signaling-induced BAMBI repression, focal adhesion in stellate cells via a TLR4 dependent way. These results suggest that SsnB is a promising compound to attenuate liver fibrogenesis. To our knowledge, this is the first study reported to show the effects of SsnB on suppression of hepatic fibrogenesis through downregulation of a myriad of cell signaling pathways. In future, due to the strong anti-angiogenic and anti-proliferative properties of SsnB, it will be interesting test the therapeutic effect of SsnB to improve the pathophysiological conditions in liver cancer and other fibrotic disease.
Supplementary Material
Acknowledgement
The authors gratefully acknowledge the technical services of Benny Davidson at the IRF, University of South Carolina School of Medicine and AML Labs (Baltimore MD). We also thank the Instrumentation resource facility (IRF) at the University of South Carolina and the National Institute of Environmental Health Sciences, NIH for equipment usage and consulting services.
Grant support
This work has been supported NIH grants P01 AT-003961-Project 4, and NIH grant R00-ES19875-02 to Saurabh Chatterjee. R01DK053792 to Anna Mae Diehl, P01AT003961, P20GM103641, R01AT006888, R01ES019313, R01MH094755 to Mitzi Nagarkatti and Prakash S. Nagarkatti.
Footnotes
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.ejphar.2018.08.040.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Alsamman M, Sterzer V, Meurer SK, Sahin H, Schaeper U, Kuscuoglu D, Strnad P, Weiskirchen R, Trautwein C, Scholten D, 2017. Endoglin in human liver disease and murine models of liver fibrosis-A protective factor against liver fibrosis. Liver Int.: Off. J. Int. Assoc. Study Liver [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarapurka DN, Amarapurkar AD, Patel ND, Agal S, Baigal R, Gupte P, Pramanik S, 2006. Nonalcoholic steatohepatitis (NASH) with diabetes: predictors of liver fibrosis. Ann. Hepatol 5, 30–33. [PubMed] [Google Scholar]
- Anthony B, Allen JT, Li YS, McManus DP, 2010. Hepatic stellate cells and parasiteinduced liver fibrosis. Parasites Vectors 3, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balta C, Herman H, Boldura OM, Gasca I, Rosu M, Ardelean A, Hermenean A, 2015. Chrysin attenuates liver fibrosis and hepatic stellate cell activation through TGF-beta/Smad signaling pathway. Chem.-Biol. Interact 240, 94–101. [DOI] [PubMed] [Google Scholar]
- Bateman HR, Liang Q, Fan D, Rodriguez V, Lessner SM, 2013. Sparstolonin B inhibits pro-angiogenic functions and blocks cell cycle progression in endothelial cells. PLoS One 8, e70500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson EK, Mungamuri SK, Attie O, Kracikova M, Sachidanandam R, Manfredi JJ, Aaronson SA, 2014. p53-dependent gene repression through p21 is mediated by recruitment of E2F4 repression complexes. Oncogene 33, 3959–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blond E, Disse E, Cuerq C, Drai J, Valette PJ, Laville M, Thivolet C, Simon C, Caussy C, 2017. EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease in severely obese people: do they lead to overreferral? Diabetologia 60, 1218–1222. [DOI] [PubMed] [Google Scholar]
- Bohinc BN, Diehl AM, 2012. Mechanisms of disease progression in NASH: new paradigms. Clin. Liver Dis 16, 549–565. [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W, 2006. p53 ubiquitination: Mdm2 and beyond. Mol. Cell 21, 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Sepulveda MA, Spitler K, Pandey D, Berkowitz DE, Matsumoto T, 2015. Inhibition of TLR4 attenuates vascular dysfunction and oxidative stress in diabetic rats. J. Mol. Med 93, 1341–1354. [DOI] [PubMed] [Google Scholar]
- Dallas SL, Sivakumar P, Jones CJ, Chen Q, Peters DM, Mosher DF, Humphries MJ, Kielty CM, 2005. Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. J. Biol. Chem 280, 18871–18880. [DOI] [PubMed] [Google Scholar]
- Das S, Alhasson F, Dattaroy D, Pourhoseini S, Seth RK, Nagarkatti M, Nagarkatti PS, Michelotti GA, Diehl AM, Kalyanaraman B, Chatterjee S, 2015. NADPH oxidase-derived peroxynitrite drives inflammation in mice and human nonalcoholic Steatohepatitis via TLR4-lipid raft recruitment. Am. J. Pathol 185, 1944–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dattaroy D, Pourhoseini S, Das S, Alhasson F, Seth RK, Nagarkatti M, Michelotti GA, Diehl AM, Chatterjee S, 2015. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol 308, G298–G312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dattaroy D, Seth RK, Das S, Alhasson F, Chandrashekaran V, Michelotti G, Fan D, Nagarkatti M, Nagarkatti P, Diehl AM, Chatterjee S, 2016. Sparstolonin B attenuates early liver inflammation in experimental NASH by modulating TLR4 trafficking in lipid rafts via NADPH oxidase activation. Am. J. Physiol. Gastrointest. Liver Physiol 310, G510–G525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman-Scheel M, Weng L, Xin S, Du W, 2002. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature 417, 299–304. [DOI] [PubMed] [Google Scholar]
- Edlund S, Landstrom M, Heldin CH, Aspenstrom P, 2002. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 13, 902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausther M, Lavoie EG, Dranoff JA, 2013. Contribution of myofibroblasts of different origins to liver fibrosis. Curr. Pathobiol. Rep 1, 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierbinteanu-Braticevici C, Bengus A, Neamtu M, Usvat R, 2002. The risk factors of fibrosis in nonalcoholic steatohepatitis. Rom. J. Intern. Med 40, 81–88. [PubMed] [Google Scholar]
- Forbes SJ, Parola M, 2011. Liver fibrogenic cells. Best. Pract. Res. Clin. Gastroenterol 25, 207–217. [DOI] [PubMed] [Google Scholar]
- Friedman SL, 2008. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev 88, 125–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui H, Tsujita S, Matsumoto M, Morimura M, Kitano H, Kinoshita K, Kikuchi E, Okamoto Y, Tsujii T, 1995. Endotoxin inactivating action of plasma in patients with liver cirrhosis. Liver 15, 104–109. [DOI] [PubMed] [Google Scholar]
- Guo J, Friedman SL, 2010. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenes. Tissue Repair 3, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A, Chen YG, 2016. TGF-beta signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi T, Friedman SL, Hoshida Y, 2017. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Dugina V, Ballestrem C, Wehrle-Haller B, Chaponnier C, 2003. Alphasmooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol. Biol. Cell 14, 2508–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney AM, Rowitch DH, 2000. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol. Cell. Biol 20, 9055–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Younossi ZM, 2008. Nonalcoholic fatty liver disease: a manifestation of the metabolic syndrome. Clevel. Clin. J. Med 75, 721–728. [DOI] [PubMed] [Google Scholar]
- Kohan M, Muro AF, White ES, Berkman N, 2010. EDA-containing cellular fibronectin induces fibroblast differentiation through binding to alpha4beta7 integrin receptor and MAPK/Erk 1/2-dependent signaling. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol 24, 4503–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW, 2008. Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Fan D, Dipette DJ, Singh US, 2014. Sparstolonin B, a novel plant derived compound, arrests cell cycle and induces apoptosis in N-myc amplified and N-myc nonamplified neuroblastoma cells. PLoS One 9, e96343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrain S, Rinella ME, 2012. A myriad of pathways to NASH. Clin. Liver Dis 16, 525–548. [DOI] [PubMed] [Google Scholar]
- Leask A, Abraham DJ, 2004. TGF-beta signaling and the fibrotic response. FASEB J 18, 816–827. [DOI] [PubMed] [Google Scholar]
- Li T, Leng XS, Zhu JY, Wang G, 2015. Suppression of hedgehog signaling regulates hepatic stellate cell activation and collagen secretion. Int. J. Clin. Exp. Pathol 8, 14574–14579. [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Dong S, Lei L, Liu J, Zhang J, Li J, Duan J, Fan D, 2015. Protective effects of Sparstolonin B, a selective TLR2 and TLR4 antagonist, on mouse endotoxin shock. Cytokine 75, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Wu Q, Jiang J, Duan J, Wang C, Smith MD, Lu H, Wang Q, Nagarkatti P, Fan D, 2011. Characterization of sparstolonin B, a Chinese herb-derived compound, as a selective Toll-like receptor antagonist with potent anti-inflammatory properties. J. Biol. Chem 286, 26470–26479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Chen X, Yang L, Kisseleva T, Brenner DA, Seki E, 2014. Transcriptional repression of the transforming growth factor beta (TGF-beta) Pseudoreceptor BMP and activin membrane-bound inhibitor (BAMBI) by nuclear Factor kappaB (NFkappaB) p50 enhances TGF-beta signaling in hepatic stellate cells. J. Biol. Chem 289, 7082–7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Li J, Liang Q, Wang D, Luo Y, Yu F, Janicki JS, Fan D, 2015. Sparstolonin B suppresses rat vascular smooth muscle cell proliferation, migration, inflammatory response and lipid accumulation. Vasc. Pharmacol 67–69, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XY, Liu RX, Hou F, Cui LJ, Li CY, Chi C, Yi E, Wen Y, Yin CH, 2016. Fibronectin expression is critical for liver fibrogenesis in vivo and in vitro. Mol. Med Rep 14, 3669–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nace GW, Huang H, Klune JR, Eid RE, Rosborough BR, Korff S, Li S, Shapiro RA, Stolz DB, Sodhi CP, Hackam DJ, Geller DA, Billiar TR, Tsung A, 2013Cellular-specific role of toll-like receptor 4 in hepatic ischemia-reperfusion injury in mice. Hepatology 58, 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez Lopez O, Bohanon FJ, Wang X, Ye N, Corsello T, Rojas-Khalil Y, Chen H, Chen H, Zhou J, Radhakrishnan RS, 2016. STAT3 inhibition suppresses hepatic stellate cell fibrogenesis: HJC0123, a potential therapeutic agent for liver fibrosis. RSC Adv. 6, 100652–100663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes PW, Beckham Y, Stricker J, Gardel ML, 2012. Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol 196, 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odkhuu E, Mendjargal A, Koide N, Naiki Y, Komatsu T, Yokochi T, 2015. Lipopolysaccharide downregulates the expression of p53 through activation of MDM2 and enhances activation of nuclear factor-kappa B. Immunobiology 220, 136–141. [DOI] [PubMed] [Google Scholar]
- Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massague J, Niehrs C, 1999. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 401, 480–485. [DOI] [PubMed] [Google Scholar]
- Pan Q, Li DG, Lu HM, Lu LY, Wang YQ, Xu QF, 2004. Antiproliferative and proapoptotic effects of somatostatin on activated hepatic stellate cells. World J. Gastroenterol 10, 1015–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasarin M, La Mura V, Gracia-Sancho J, Garcia-Caldero H, Rodriguez-Vilarrupla A, Garcia-Pagan JC, Bosch J, Abraldes JG, 2012. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS One 7, e32785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrasek J, Csak T, Szabo G, 2013. Toll-like receptors in liver disease. Adv. Clin. Chem 59, 155–201. [DOI] [PubMed] [Google Scholar]
- Pradere J-P, Troeger JS, Dapito DH, Mencin AA, Schwabe RF, 2010. Toll-like receptor 4 and hepatic fibrogenesis. Semin. Liver Dis 30, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW, 2016. Targeting the Sonic Hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF, 2007. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med 13, 1324–1332. [DOI] [PubMed] [Google Scholar]
- Seth RK, Kumar A, Das S, Kadiiska MB, Michelotti G, Diehl AM, Chatterjee S, 2013. Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicol. Sci.: Off. J. Soc. Toxicol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, Johnson DS, Chen Y, O’Neill LA, 2010. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol 11, 141–147. [DOI] [PubMed] [Google Scholar]
- Shen XD, Ke B, Zhai Y, Gao F, Tsuchihashi S, Lassman CR, Busuttil RW, Kupiec-Weglinski JW, 2007. Absence of toll-like receptor 4 (TLR4) signaling in the donor organ reduces ischemia and reperfusion injury in a murine liver transplantation model. Liver Transpl. 13, 1435–1443. [DOI] [PubMed] [Google Scholar]
- Silver DL, Naora H, Liu J, Cheng W, Montell DJ, 2004. Activated signal transducer and activator of transcription (STAT) 3: localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 64, 3550–3558. [DOI] [PubMed] [Google Scholar]
- Sun Y, Guo W, Ren T, Liang W, Zhou W, Lu Q, Jiao G, Yan T, 2014. Gli1 inhibition suppressed cell growth and cell cycle progression and induced apoptosis as well as autophagy depending on ERK1/2 activity in human chondrosarcoma cells. Cell Death Dis 5, e979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang LY, Heller M, Meng Z, Yu LR, Tang Y, Zhou M, Zhang YE, 2017. Transforming growth factor-beta (TGF-beta) directly activates the JAK1-STAT3 axis to induce hepatic fibrosis in coordination with the SMAD pathway. J. Biol. Chem 292, 4302–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CE, 2000. Paxillin and focal adhesion signalling. Nat. Cell Biol 2, E231–E236. [DOI] [PubMed] [Google Scholar]
- Weber SN, Bohner A, Dapito DH, Schwabe RF, Lammert F, 2016. TLR4 Deficiency protects against hepatic fibrosis and diethylnitrosamine-induced pre-carcinogenic liver injury in fibrotic liver. PLoS One 11, e0158819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Feng L, Li Z, Xu G, Fan X, 2013. MicroRNA-21 activates hepatic stellate cells via PTEN/Akt signaling. Biomed. Pharmacother. = Biomed. Pharmacother 67, 387–392. [DOI] [PubMed] [Google Scholar]
- Xu MY, Hu JJ, Shen J, Wang ML, Zhang QQ, Qu Y, Lu LG, 2014. Stat3 signaling activation crosslinking of TGF-beta1 in hepatic stellate cell exacerbates liver injury and fibrosis. Biochim. Biophys. Acta 1842, 2237–2245. [DOI] [PubMed] [Google Scholar]
- Yang JJ, Tao H, Li J, 2014. Hedgehog signaling pathway as key player in liver fibrosis: new insights and perspectives. Expert Opin. Ther. Targets 18, 1011–1021. [DOI] [PubMed] [Google Scholar]
- Yang L, Seki E, 2012. Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms. Front. Physiol 3, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi HS, Jeong WI, 2013. Interaction of hepatic stellate cells with diverse types of immune cells: foe or friend? J. Gastroenterol. Hepatol 28 (Suppl 1), S99–S104. [DOI] [PubMed] [Google Scholar]
- Yin H, Tan Y, Wu X, Yan H, Liu F, Yao Y, Jiang J, Wan Q, Li L, 2016. Association between TLR4 and PTEN Involved in LPS-TLR4 Signaling Response. Biomed. Res. Int 2016, 6083178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JW, Lamm M, Iannaccone S, Higashiyama N, Leong KF, Iannaccone P, Walterhouse D, 2015. p53 modulates the activity of the GLI1 oncogene through interactions with the shared coactivator TAF9. DNA Repair 34, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka Y, Hashimoto E, Yatsuji S, Kaneda H, Taniai M, Tokushige K, Shiratori K, 2004. Nonalcoholic steatohepatitis: cirrhosis, hepatocellular carcinoma, and burnt-out NASH. J. Gastroenterol 39, 1215–1218. [DOI] [PubMed] [Google Scholar]
- Zezos P, Renner EL, 2014. Liver transplantation and non-alcoholic fatty liver disease. World J. Gastroenterol 20, 15532–15538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Meng X, Zhu Z, Liu J, Deng A, 2004. Connective tissue growth factor regulates the key events in tubular epithelial to myofibroblast transition in vitro. Cell Biol. Int 28, 863–873. [DOI] [PubMed] [Google Scholar]
- Zhang JG, Wang JJ, Zhao F, Liu Q, Jiang K, Yang GH, 2010. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin. Chim. Acta 411, 846–852. [DOI] [PubMed] [Google Scholar]
- Zhang YE, 2017. Non-smad signaling pathways of the TGF-beta family. Cold Spring Harb. Perspect. Biol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XK, Yu L, Cheng ML, Che P, Lu YY, Zhang Q, Mu M, Li H, Zhu LL, Zhu JJ, Hu M, Li P, Liang YD, Luo XH, Cheng YJ, Xu ZX, Ding Q, 2017. Focal adhesion kinase regulates hepatic stellate cell activation and liver fibrosis. Sci. Rep 7, 4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.