

Abstract
Renal cell carcinoma (RCC) comprises a diverse group of malignancies arising from the nephron. The most prevalent type, clear cell renal cell carcinoma (ccRCC), is characterized by genetic mutations in factors governing the hypoxia signaling pathway, resulting in metabolic dysregulation, heightened angiogenesis, intratumoral heterogeneity, and deleterious tumor microenvironmental (TME) crosstalk. Identification of specific genetic variances has led to therapeutic innovation and improved survival for patients with ccRCC. Current barriers to effective long-term therapeutic success highlight the need for continued drug development using improved modeling systems. ccRCC preclinical models can be grouped into three broad categories: cell line, mouse, and 3D models. Yet, the breadth of important unanswered questions in ccRCC research far exceeds the accessibility of model systems capable of carrying them out. Accordingly, we review the strengths, weaknesses, and therapeutic implications of each model system that are relied upon today.
Keywords: Renal cell carcinoma, Clear cell RCC, Cell lines, Genetically engineered mouse models, 3D Organoids, Tumor microenvironment, Angiogenesis
Introduction
Biological models enable researchers to mimic discrete tumor attributes and have paved the way for therapeutic innovations in cancer. The hallmarks of cancer, outlined by Hanahan & Weinberg, describe a dynamic set of characteristics meant to simplify the complexities of tumor progression and set the stage for more in-depth understanding of narrow research questions1. Tumor-specific characteristics, including mutational burden, tissue of derivation, intratumoral heterogeneity, and microenvironmental interactions, are just a few variables that make a catch-all platform for basic cancer research and therapeutic exploration impractical2,3.
Renal Cell Carcinoma (RCC), cancer arising from nephric epithelial cells, constitutes a group of diseases characterized on the basis of anatomical origin, histological features, molecular hallmarks, and therapeutic outcomes4,5. The majority of RCC falls into three major histologically defined subsets including: clear cell RCC (ccRCC), papillary RCC (pRCC), and chromophobe RCC (chRCC); however, several other rare subtypes are also recognized6–8. Cell lines, 3D organoid systems, and both xenograft and genetically engineered mouse models (GEMMs), have all been exploited in RCC research (Figure 1). Here, we outline current and developing model systems, and describe the implications and opportunities for each.
Figure 1.
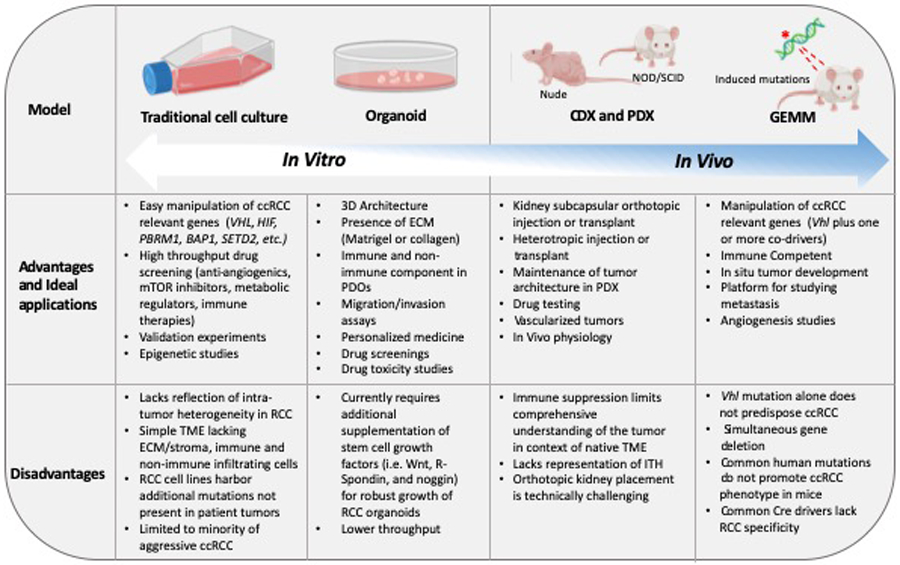
Advantages and disadvantages of model system platforms in RCC.
Advantages and ideal applications, and disadvantages of traditional cell line, organoid, xenograft, and GEM models in RCC. As physiological relevance increases, ease of manipulation decreases.
Defining the Genetic and Histological Landscape of ccRCC
ccRCC is the most common of the RCC subsets, accounting for ~80% of adult clinical cases9. Due to the high incidence of ccRCC, we focus our attention to model systems attempting to mimic the ccRCC phenotype, noting that some highly cited RCC cell lines are of pRCC origin10. ccRCC is characterized by loss or mutation of the von Hippel Lindau (VHL) gene, which is responsible for encoding the oxygen sensing mediator protein VHL (pVHL)11,12. pVHL is a crucial component of the E3 ubiquitin ligase complex, necessary for exposing the family of hypoxia-inducible transcription factors (HIFs) for proteasomal degradation in the presence of oxygen8,12,13. Without pVHL, HIF-1α and HIF-2α accumulation promotes gene expression programs that ultimately dysregulate glycolysis, angiogenesis, and lipolysis regardless of oxygen status11,14–16. Greater than 90% of human ccRCC types occur following VHL loss of heterozygosity (LOH) in a 2-hit phenomenon14,17. Thus, models of ccRCC are typically defined by the biallelic loss of VHL (Figure 2). Recently, multi-regional sampling and whole genome sequencing (WGS) enabled researchers to track landmark mutagenic events in ccRCC18. Results indicated that deletion of chromosome 3p typically occurs decades before diagnosis, usually during childhood or adolescence; this event is the projected ‘first-hit’ to VHL and the initial driver of sporadic ccRCC types18. A subsequent ‘second-hit’ occurs as the result of a somatic intragenic mutation or gene silencing event, in the remaining VHL allele19,20.
Figure 2.
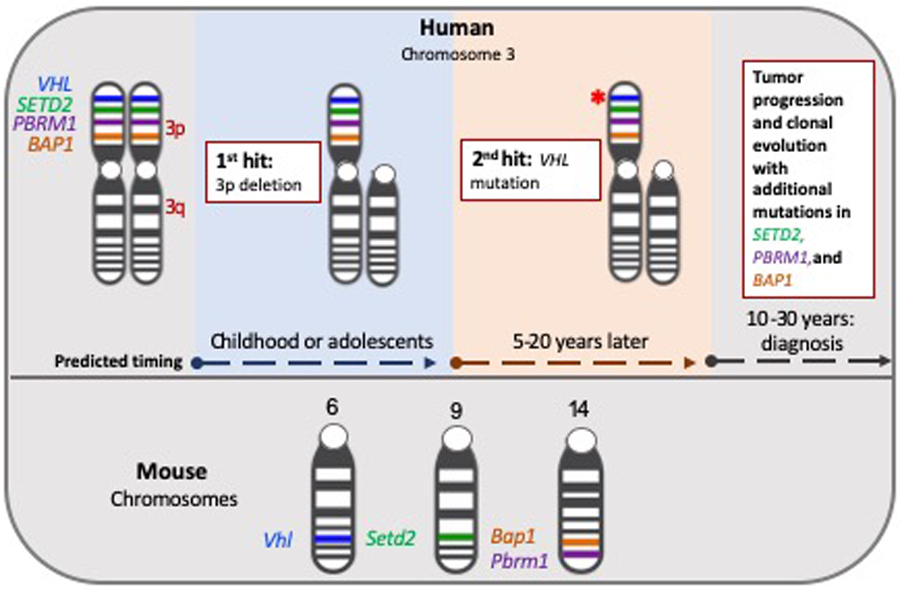
Location of ccRCC Associated Genes Human and Mouse Genome
VHL and additional driver genes (SETD2, PBRM1, and BAP1) are located on chromosome 3p in the human genome. WGS identified two key events in the evolutionary progression of ccRCC: loss of one 3p arm during childhood or adolescents, followed an additional mutation in the remaining VHL allele18. Corresponding genes in the mouse are located on chromosomes 6,9, and 14.
Intracellular HIF stabilization promotes over-expression of HIF-related pro-angiogenic and glycolytic target genes that lead to nutrient and oxygen imbalances in the tumor microenvironment (TME)21,22. A classic example is the accumulation of the angiogenic growth factor, VEGF-A, which promotes activation of its cognate receptor, VEGFR, on endothelial cells to induce a pro-angiogenic switch21. VEGF-A and VEGFR have been successfully exploited as therapeutic targets for angiogenesis in the treatment of ccRCC23. Hypoxia-driven dysregulation of glycolytic modulators, including glucose uptake transporter-1 (GLUT1) and pyruvate kinase-M2 (PKM2), have also been explored as therapeutic targets in ccRCC24,25.
In 2013, The Cancer Genome Atlas (TCGA) put forward a comprehensive analysis of over 400 RCC specimens, alongside matching normal kidney tissue, to reveal a recurrent pattern of metabolic reprogramming that correlated with disease severity26. Integrative unbiased pathway analysis disclosed that aggressive tumors resembled a metabolic shift similar to the Warburg phenotype, as determined by downregulation of TCA cycle intermediates and upregulation of glycolysis and the pentose phosphate pathway26. In addition, decreased PTEN and AMPK protein activity and increased Acetyl-CoA carboxylase correlated with aggressive tumor phenotypes26.
ccRCC biology can be subdivided based on recurrent mutations in the chromatin modifying co-driver genes (PBRM1, SETD2, BAP1, KDM5C, KDM6A, and MLL2) believed to contribute to intratumoral heterogeneity in ccRCC and serve as a potential etiology of drug resistance9,14,20,27. At the genomic level, intratumoral heterogeneity (ITH) is defined as the outgrowth of genetically different clonal subpopulations within the same tumor28. Phylogenetic analyses of multiple unique regions within the same ccRCC tumor determined that only VHL mutations were similar and abundant across all regions and additional mutations in co-driver genes varied across these tumor regions29. A comprehensive study using TRAcking cancer evolution Rx (TRACERx) and multi-regional WGS in ccRCC exposed clonal and sub-clonal drivers that contribute to the evolution of intratumoral heterogeneity and genomic instability30. Seven evolutionary subtypes were classified according to their metastatic phenotype30,31. Primary tumors characterized as BAP1 driven, VHL wild type, or having multiple clonal drivers were shown to exhibit low ITH and rapid progression to lungs, liver, adrenal glands, and lymph nodes30. Tumors classified as PBRM1 plus SETD2, PBRM1 plus PI3K, and PBRM1 plus somatic copy number alteration (SCNA) displayed high ITH and attenuated metastatic progression to the bones and lungs30. Primary tumors classified as VHL monodriver did not exhibit metastasis30. Together, TRACERx Renal and TCGA studies highlight factors that have the potential to predict a myriad of disease phenotypes and associated level of aggression20,31.
Biomarker Development and Treatment in Advanced ccRCC
To date, no genetic biomarker has been implemented as a reliable predictor of ccRCC prognostic or therapeutic outcome. Prediction of ccRCC tumor recurrence, using the multigene assays, ClearCode34 and the 16-gene recurrence score (RS) assay, have demonstrated success in aiding clinicians in judging risk of recurrence beyond existing pathological parameters32,33. ClearCode34 is a 34 gene expression signature originally designed to define molecular variation but has demonstrated the capacity to predict tumor recurrence in primary RCC tumors33,34. Based on expression of the 34 gene signature, ClearCode34 classifies tumors as either indolent (ccA) or aggressive (ccB)34. The predictive value of ClearCode34 has also been validated clinically in connecting primary tumor biology with distant, metastatic sites in ccRCC35. A 16-gene recurrence score (RS) assay has also been used in predicting tumor recurrence and projecting patient benefit to adjuvant therapies32,36. The 16-gene assay includes ccRCC related genes involved in vascular, immune, cell-cycle division, and growth pathways32,36. ClearCode34 and the 16-gene RS assay are both valuable tools for research purposes and could be considered for personalized therapeutic selection based on molecular tumor characteristics32,34.
ccRCC is typically non-responsive to traditional cytotoxic chemotherapeutic drugs37. Prior to the introduction of small molecule targeted therapies and immune checkpoint inhibitors, cytokine therapy (IFN-α and IL-2) was the standard of care for patients with metastatic RCC (mRCC); however, response rates remained low38. A mainstay of therapy, since their introduction in 2005, are the anti-angiogenic small molecule tyrosine kinase inhibitors (TKI) primarily targeting the VEGF signaling pathway39,40. The following TKI small molecule inhibitor drugs are approved for clinical use: axitinib, cabozantinib, lenvatinib, pazopanib, sorafenib, and sunitinib40. Additionally, mTOR signaling is tightly linked to HIF activation and increased metabolic demands, which makes mTOR inhibition a viable therapeutic target for some RCC patients41. Everolimus and temsirolimus are both approved for use in advanced RCC42, and more recently, everolimus has been applied in combination with VEGF inhibition43.
Checkpoint inhibitors targeting CTLA-4, PD-1 and PD-L1 work by unleashing the adaptive immune system’s potential to target and kill cancer cells44,45. Ipilimumab blocks CTLA-4 while nivolumab and pembrolizumab block the negative regulator PD-1 on T cells. Avelumab targets the negative regulation of T cells by blocking PD-L1 on cancer and other immune cells46,47. Checkpoint blockade therapies initially showed improvement in patient survival with use of nivolumab in the second line treatment setting48. Shortly after, checkpoint inhibition moved to the first-line standard of care in metastatic ccRCC with a combination of antibodies targeting CTLA-4 and PD-146, and more recently, finding that combination of checkpoint inhibition and VEGF TKI increased objective response rates and overall survival47,49,50.
Newer generation immunotherapies represent a promising approach to patient care in ccRCC, with the potential to provide a long duration of response49. However, many patients harbor non-responsive tumors and are faced with a poor prognosis. Additionally, for patients who initially respond, many will progress while on treatment as tumor cells develop resistance49,51. Ongoing efforts seek to identify and confirm clinically relevant predictive biomarkers in ccRCC to aid clinicians in immunotherapy selection52,53. As such, model systems capable of recapitulating the TME are crucial to enhance our understanding of drug interactions.
Cell Line Models of Renal Cell Carcinoma
Immortalized tumor cell lines are the most frequently utilized biological system in cancer research, contributing to our current molecular and genetic understanding of RCC biology54. Cost-effectiveness, ease of maintenance and manipulation, and infinite replicative capacity made accessible by immortalized cell lines allow for the three “R’s” in humane research: replacement, reduction, and refinement55. In the 1980s, the National Cancer Institute’s 60 cell line (NCI-60) comprehensive anticancer drug screening study included the following RCC cell lines: 786-O, A-498, ACHN, CAKI-1, RXF 393, SN12C, TK-10, and UO-3156. The NCI-60 study led to intense characterization and publicly available RCC specific genetic and molecular cell line data56. Consequently, the eight RCC cell lines included were established as first-line research models.
786-O is among the earliest ccRCC cell lines established and remains the most frequently cited today57. The COSMIC Cell Lines Project (CCLP) and Broad Novartis Cell Line Encyclopedia (CCLE) report a p.G104fs VHL frameshift deletion in 786-Os, and consistently, 786-O cells display increased HIF-2α and VEGF protein expression, making this cell line a workhorse for ccRCC research58. The negative regulatory role of the pVHL-HIF axis was first demonstrated in 786-O cells. When transduced with wild type (WT)-pVHL, in vivo tumor growth was inhibited59,60. 786-Os were also used to show that HIF overexpression alone promotes tumor growth, thus validating that HIF activation occurs downstream from the pVHL interaction61. More recently, 786-O cells were employed to decipher the role of hypoxia induced resistance to the VEGF inhibitor, sorafenib, via the HIF-2α-Cox2 pathway, thereby illustrating a potential drug resistance mechanism62. Histologically, 786-O cells do not exhibit clear cytoplasm in mouse xenograft models; rather, tumors are poorly differentiated with spindle shaped sarcomatoid features, a quality indicative of genetic and morphological regression63,64. Stem cell-like side populations (SPs) have been observed in 786-O cells, making this line a potential model for cancer stem cell (CSC) studies in aggressive forms of ccRCC57. SPs are understood to be a population of cancer stem cells (CSCs) that, when enriched, confer resistance to chemotherapy drugs and promote tumorigenicity65.
The second most highly cited RCC line, A-498, is classified as ccRCC based on VHL mutation status. The CCLP and CCLE report p.G144fs*14 and p.V142fs VHL mutations, respectively58,66,67. A498 cells exhibit clear cytoplasmic histological signatures when xenografted into nude mice11,57. 786-O and A-498 cell lines were utilized in a landmark study implicating mTOR activation as a driver of tumorigenesis in ccRCC68. This finding fueled further investigations into mTOR activity as a prognostic indicator and therapeutic target41,42,69. A-498 and 786-O cells were also used to illustrate the functional redundancy and dominance of HIF protein subunits in transcribing the angiogenic factor, VEGF70. When HIF-1α and HIF-2α are both present and functional, VEGF is preferentially expressed by HIF-1α; however, when HIF-1α is deleted or nonfunctional, HIF-2α is upregulated and VEGF levels remain unchanged70.
ACHN, the third most highly cited line, was established from a metastatic pleural effusion and exhibits poorly differentiated sarcomatoid histological features57. ACHN cells harbor a pRCC specific mutation in c-MET and do not have classical VHL mutations57,71. Though not representative of the ccRCC genetic phenotype, ACHN cells were exploited to demonstrate VHL deficient sensitization to mTOR inhibition by shRNA knockdown72. Increased uptake of the glucose tracer fluorodeoxyglucose (FDG) was decreased in VHL knockdown tumors treated with the mTOR inhibitor, CCI-779, compared to the vehicle treated control tumors by positron emission tomography (PET)72. This study provided support for clinical use of mTOR dependent inhibition in the treatment of ccRCC. These features make ACHN cells an attractive model for studying aggressive tumor types but impractical for studies aimed at gaining a broad understanding of ccRCC diseases.
While the top three cited cells lines in RCC literature are reviewed above, many other useful cell line models exist and should be considered and applied depending on the biologic question. We have summarized the major classifications and characteristics of established cell lines used in RCC research today (Table 1)10,66,67,71,73–75. In summary, thoughtful consideration regarding experimental aims and well-established genomic and molecular signatures should be employed in the process of choosing an appropriate cell line for each RCC project.
Table 1.
Top cited RCC cell lines.
Histologic and genetic characteristics, including ccA and ccB sub-classification, and 3p deletion status as defined by Sinha et al., are outlined10. VHL and other known significantly mutated genes, determined by TCGA, are defined by CCLE and CCLP databases58,66,67,142.
| Cell line | Classification | Sub classification10 | Histological features | 3p status10 | VHL status66,67 | CCLE mutations66, 67 |
|---|---|---|---|---|---|---|
| (Sinha et al., 2017) | (Sinha et al., 2017) | (Barretina et al., 2012) | (Barretina et al., 2012) | |||
| 786-O | ccRCC | ccB | Poor differentiation with sarcomatoid features10 | Deleted | Null | PTEN, TP53, |
| (Sinha et al., 2017) | TSC2, MLL3 | |||||
| A-498 | ccRCC | ccB | Compact nests of tumor cells with clear cytoplasm10 | Deleted | Null | MLL3, SETD2 |
| (Sinha et al., 2017) | ||||||
| ACHN | pRCC | Unknown | Poor differentiation with sarcomatoid features10 | Deleted | Wildtype | PBRM1, NF2 |
| (Sinha et al., 2017) | ||||||
| CAKI-1 | ccRCC | ccB | Poor differentiation71 | Not Deleted | Wildtype | MET, MLL3, |
| (Korhonen et al., 1994) | SETD2 | |||||
| RCC-4 | ccRCC | Unknown | Unknown | Deleted | Null | NA |
| A-704 | ccRCC | ccA | Not tumorigenic in immunocompromised mice73 | Deleted | Null | MLL3, TP53, |
| (Giard et al., 1973) | PBRM1, SETD2 | |||||
| SN12C | Unknown | Unknown | Clear cell combined with granular morphology74 | Not Deleted | Wildtype | TP53, BAP1, |
| (Sanchez et al., 1994) | NF2 | |||||
| CAKI-2 | ccRCC | ccB | Well differentiated clear cell71 | Deleted | Wildtype | PBRM1 |
| (Korhonen et al., 1994) | ||||||
| 769-P | ccRCC | ccA | Unknown | Deleted | Null | BAP1, TSC2 |
| TK-10 | Unknown | Unknown | Unknown | Deleted | Wildtype | PIK3CA, TP53 |
| UMRC-2 | ccRCC | ccA | Unknown | Deleted | Null | NA |
| OS-RC-2 | ccRCC | ccB | Unknown | Deleted | Null | PTEN, TP53, |
| PBRM1 | ||||||
| CAL-54 | pRCC | Unknown | Well differentiated with papillary structures75 | Deleted | Wildtype | FH, MLL3 |
| (Gioanni et al., 1996) |
Overview of Preclinical Mouse Models
Mice are utilized extensively as a model system for the study of cancer biology. The mouse genome is experimentally accessible, allowing manipulation of relevant onco- and tumor suppressor genes that promote cancer hallmark characteristics in vivo76. Until recently, RCC murine models have been limited to cell-line-derived (CDX), or patient-derived xenografts (PDX). New developments in genetically engineered mouse models (GEMMs) have allowed for kidney-specific conditional knockout of Vhl with at least one other ccRCC associated co-driver gene. VHL bi-allelic inactivation is thought to occur during early stages of ccRCC development in humans with additional co-drivers leading to enhanced tumor progression at a later stage31,77. Most ccRCC GEMMs rely on complete gene deletion during early embryogenesis, thereby failing to model sequential mutagenic events through clonal selection30. Ultimately, the complexity involved with studying RCC cell mechanisms and therapeutic responses in mice is far too immense for one mouse model to encapsulate. Cell-line derived xenograft (CDX), Patient-derived xenograft (PDX), and Genetically engineered mouse models (GEMM) each provide unique opportunities to interrogate various questions in ccRCC.
Xenograft Models
CDX and PDX RCC models can be established by orthotopic placement in the subcapsular region of the kidney or by heterotopic transplantation: e.g. subcutaneous (s.c.), intraperitoneal (i.p.), or intramuscular (i.m.)78. To prevent rejection of the tumor cells from successful engraftment into the host animal, xenograft models using human cell lines or tissues rely on non-obese diabetic (NOD) severe combined immunodeficient (scid) mice, or equivalent severely immunocompromised models79,80. Xenograft models have been shown to maintain patient tumor histology, gene expression, DNA copy number alterations, mutagenic profiles, and general drug responsiveness81,82.
Successful xenograft transplantation of established RCC cell lines or primary patient-derived tissues has led to the development and current mechanistic understanding of the anti-angiogenic drugs heavily utilized to treat metastatic RCC (mRCC)83. Xenograft models have been shown to recapitulate important aspects of the TME, specifically the dense vascular bundling that is characteristic of ccRCC84. Rojas and colleagues were able to detect responses to sunitinib treatment and deduce tumor responsiveness one week prior to changes in tumor volume using ultrasound-derived microvascular density imaging84. In another study, CDX models using 786-O, A498 and CAKI-1 cell lines demonstrated changes in tumor blood flow rates correlating with anti-angiogenic resistance of sorafenib treatment85. In this model, arterial spin labeling (ASL) magnetic resonance (MR) imaging was used to measure changes in blood flow signal intensity and spatial patterning during blood vessel formation85. In 786-O xenografts, blood vessel growth in anti-angiogenic resistant tumors primarily occurred at the periphery of the tumor, whereas endothelial formation in A498 xenografts occurred closer to the tumor’s center85. ASL-MR imaging was also useful in identifying tumors with initial low blood flow intensity, as observed in CAKI-1 xenografts, a characteristic that could aid in treatment selection and prediction85. Overall, changes in blood flow intensity provided valuable diagnostic and mechanistic information at earlier timepoints than previously assessed, thereby highlighting the potential for measuring functional tumor activity, in addition to gross anatomical quantification86.
PDX models are created by direct transplantation and subsequent passaging of surgically resected RCC tissues into immunocompromised mice82,87,88. PDX models provide a closer to human model of tumor growth, but incur important selective pressures which likely skew the distribution of tumor phenotypes89. PDX modeling has demonstrated a propensity to select for the growth of aggressive forms of cancers, a phenomenon that allowed for the discovery of BAP1 as a tumor suppressor gene in ccRCC90. Patient tumors harboring BAP1 mutations have since been shown to associate with worse cancer specific survival and exhibit aggressive tumor features9,90,91.
Immune competent mice can be used to study ccRCC in syngeneic CDX models92. Murine renal adenocarcinoma (Renca) is a cell line that arose spontaneously from Balb/c lineage and exhibits progressive growth and metastasis to lymph nodes, liver, and lungs in transplantable subcapsular orthotopic models93. Though Renca cells do not fully mimic the genetic profile observed in human ccRCC, Vhl deletion in Renca cells leads to HIF-1α stabilization and epithelial-mesenchymal transition (EMT), both of which classically define human ccRCC94. The human kidney tumor-specific antigen, carbonic anhydrase-IX (CA-IX), has also been stably expressed on Renca cells, and has provided an ideal platform for metastatic targeting and tracking studies in ccRCC95. Similar to human RCC tumors, Renca xenograft models demonstrate high immune-infiltrating cell populations of both lymphoid and myeloid lineage96. In an orthotopic CDX model, Renca tumors were used to identify non-canonical expression of the T-regulatory-specific transcription factor, Foxp3, in tumor associated macrophages (TAMs) of the TME96,97. In addition, initial studies aimed at understanding functional mechanisms in which chemokine networks can promote tumor progression have identified chemokine receptor 4 (CCR4) as a potential target in RCC96.
Ultimately, xenograft models retain histology and genetic signatures of the original cell line or tissue type, thereby implementing an appropriate system for elucidating tumor mechanisms of response and resistance to candidate drugs98. However, the requirement of immune suppression in non-syngeneic models limits accurate representation of the TME and ITH in xenograft models.
Genetically Engineered Mouse Models (GEMMs)
Reproducing common mutational events in genetically engineered mouse models (GEMMs) under kidney epithelial specific Cre drivers of deletion has proven challenging in ccRCC for several reasons (Figure 3). Contrary to human disease, Vhl heterozygosity has not been shown to predispose mice to ccRCC tumor development99–101. This dilemma is fueled by inter-species locational variances of commonly mutated ccRCC genes102. In humans, an initial ‘first-hit’ VHL mutagenic event is thought to occur when chromosome arm 3p is lost in near entirety; a subsequent ‘second-hit’ occurs when the remaining VHL allele is mutated, resulting in loss of function of VHL31. The ccRCC associated chromatin modifying genes PBRM1, BAP1, and SETD2 are all present on chromosome arm 3p in the human genome; thus, ‘first-hit’ and ‘second-hit’ mutagenic events described for VHL also apply to these secondary ccRCC related genes103. In the mouse genome, however, Vhl is located on chromosome 6, Setd2 on chromosome 9, and Pbrm1 and Bap1 reside on chromosome 14 (Figure 2)104. Additionally, models that alter genes atypically associated with classic human ccRCC have been shown to give rise to robust tumors resembling the disease in mice.105. Thus, differences observed in mouse versus human RCC tumor development highlight the need for consideration regarding basic species-specific kidney function.
Figure 3.
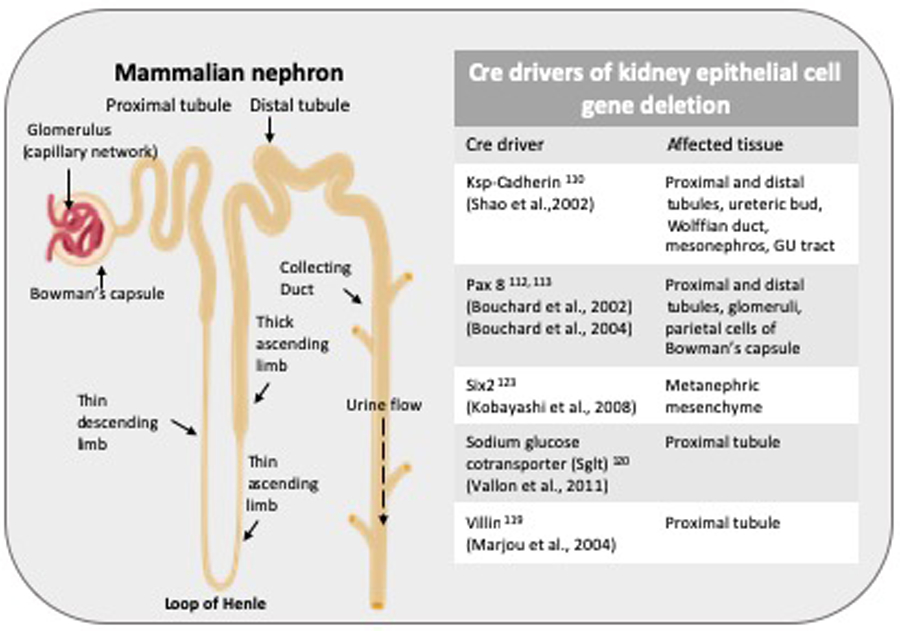
Cre drivers in kidney epithelial cell deletion.
Cre drivers in kidney epithelial cell gene specific deletion and tissues affected are outlined.
Despite present challenges, GEMMs are poised to impact our current therapeutic and basic biologic understanding of tumor development in a number of ways106,107. Considering the inevitability of drug resistance conferred by mono-therapeutic anti-cancer agents in ccRCC, immunocompetent GEMMs also offer an appropriate platform to address combination therapy. Importantly, GEM models mimicking ccRCC can develop de novo sporadic tumors, thereby providing an in vivo platform for thorough investigations of the complex interactions between tumor cells and the TME during tumorigenesis and progression. In this discussion, we highlight five of the most relevant GEM models currently available in ccRCC research (Figure 4).
Figure 4.
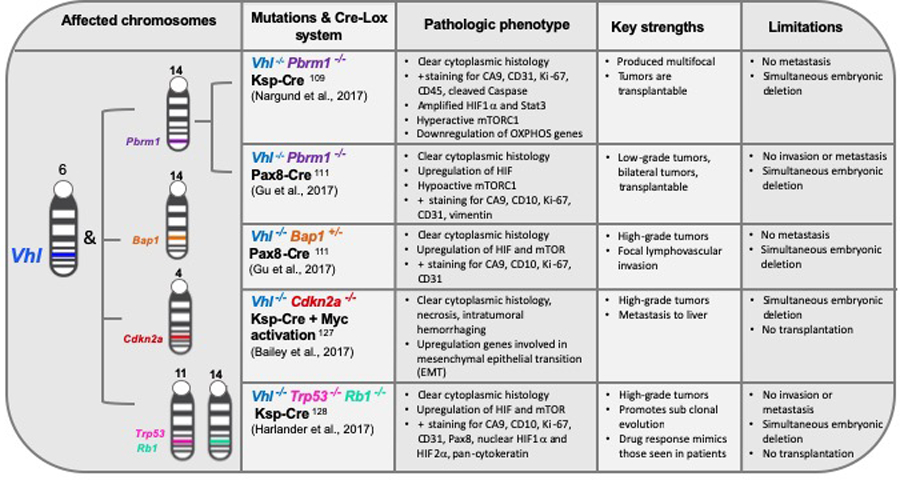
Genetically engineered RCC mouse models.
Strengths and limitations of relevant RCC GEM models, and the pathology recapitulated by each are outlined.
Vhl−/− Pbrm1−/−
Two independent groups have published their attempts to recapitulate human ccRCC in mice by deletion of both Vhl and Pbrm1, the 1st and 2nd most commonly altered genes observed in ccRCC, respectively. PBRM1, a tumor suppressor subunit of the SWI/SNF chromatin remodeling complex, located on chromosome 3p, is mutated in 45% of human ccRCC cases108. Nargund and colleagues utilized the Ksp-Cre system to conditionally delete Vhl and Pbrm1 in renal epithelial cells109,110. With this system, mice developed preneoplastic polycystic kidney disease (PKD) by 6 months of age and showed a 50% tumor incidence by 10 months, assessed by histological examination109. Tumors exhibited classic ccRCC characteristics, including clear cytoplasm, hyperactive mTORC1 signaling, downregulation of critical oxidative phosphorylation genes, and HIF-1α accumulation (Figure 2).
Gu and colleagues developed a combined Vhl and Pbrm1 conditional knockout model using paired box gene 8 (Pax8) as the Cre driver of gene deletion111–113. This approach gave rise to bilateral, homogenous tumors by 9 months of age in 85% of mice111. The author’s described the Vhl, Pbrm1 deleted tumors as low-grade, based on aggression and histologic analysis; a result consistent with loss of PBRM1 in human ccRCC111.
The nephron is made up of a complex capsular and tubular system rich in capillary beds and collecting ducts, specially designed to filter blood114. Based on immunohistochemical (IHC) staining and gene expression analysis of specialized nephric regions, the proximal convoluted tubule (PCT), consisting of at least 13 distinct epithelial cell types, has been recognized as the most likely site of origin of human ccRCC mutations115–119. However, Gu and colleagues found that Vhl and Pbrm1 deletion in a murine system using a Pax8 Cre promoted tumorigenesis, where the Cre driver expression was limited to the parietal cells of Bowman’s capsule111. In addition, the PCT-specific Cre drivers, Sglt2 and Villin, failed to promote ccRCC tumor growth111,120,121. IHC staining led the authors to propose that the parietal epithelial cells of Bowman’s capsule may be the location of tumor origin in their model. Therefore, future studies should seek to validate ccRCC cell specific nephric lineage and determine locational differences based on co-driver gene mutations. Importantly, both investigations targeting Vhl and Pbrm1 for deletion confirm the tumor suppressive role of Pbrm1 in vivo and will be valuable tools moving forward in ccRCC tumor therapeutic investigations.
Vhl−/− Bap1+/−
The histone deubiquitinase, BRCA1-associated protein-1 (BAP1), is a tumor suppressor that functions in DNA repair and chromatin remodeling in the nucleus103. BAP1 deletion or mutation occurs in 10–15% of ccRCC tumors90. BAP1 mutations were identified through next generation sequencing as strongly associated with high grade tumors and increased risk of tumor recurrence following surgical resection90,108. Combined targeting of Bap1 and Vhl, using the nephron progenitor transcription regulator, Six2, as a Cre driver, resulted in ccRCC development; however, pups only survived up to one month111,122,123. When the same group utilized the nephric specific transcription factor, Pax8, as the Cre driver of complete Vhl deletion and heterozygous manipulation of Bap1, features reminiscent of ccRCC were observed111. Vhl and Bap1 deficiency gave rise to high grade tumors with clear cell histology and positive carbonic anhydrase 9 (CAIX) staining111. Future studies should aim to validate tumor responsiveness to drug therapies in this model.
Vhl−/− Cdkn2a−/− and c-Myc activation
MYC activation has been recognized as a common genetic anomaly in both pRCC and ccRCC. One study revealed a 3-fold increase in c-MYC expression when compared to matched healthy kidney tissue124. CDKN2A (INK4A/ARF), a tumor suppressor gene, encodes a protein that functions to promote cellular senescence. Consequently, CDKN2A loss-of-function promotes tumor growth and metastasis in many cancers125. Bailey and colleagues exploited a previously described doxycycline inducible c-Myc transgenic mouse126 to explore Myc activation alone, and with Ksp-Cre promotor driven Vhl and Cdkn2a deletion in renal tubular cells, thus generating three useful models127. Based on histological features and RNA-seq, c-Myc overexpression alone gave rise to renal tumors most representative of type 2 pRCC, assessed by pathologic staining. c-Myc overexpression plus Vhl deletion resulted in tumors demonstrating some ccRCC pathological features, such as cytoplasmic clearing, necrosis, and intratumor hemorrhaging127. Deletion of Cdkn2a, in addition to Vhl deletion and c-Myc overexpression, gave rise to an aggressive ccRCC phenotype, exhibiting liver metastasis in 30% of mice127. Though the authors demonstrated successful recapitulation of metastatic tumor sites resembling ccRCC, the genetic drivers in this model only occurs in 6% of human RCC cases127. This model is representative of a small aggressive ccRCC subset and is unique in its ability to metastasize.
Vhl−/−, Trp53−/−, and Rb1−/−
Single nucleotide alterations affecting the function of tumor suppressor genes, RB and TP53, during the G1-S cell cycle transition, are relatively infrequent in ccRCC128. However, copy number gains or losses in RB and TP53 are common, and TCGA patient data demonstrates independent evolution in these regulators from those occurring in VHL, PBRM1, BAP1, and SETD2128. Hence, the conditional triple-mutant GEMM model, inactivating Vhl, Trp53, and Rb1, was predicted to confer the evolution of genetically distinct tumors overtime128. This model utilized kidney specific cadherin (Ksp-cadherin), expressed on tubular epithelial cells of the kidney and genitourinary (GU) tract, as a Cre driver of deletion110. Sporadic tumors, formed in the mouse kidney, displayed classic ccRCC pathological features, including necrosis, tumor hemorrhaging, and nuclear accumulation of HIF-1α and HIF-2α128. Notably, a pathological phenotype reminiscent of ccRCC did not result from a Trp53, Rb1 double knockout model, thus confirming the role of Vhl loss as necessary for tumor progression128. Greater than 80% of the triple-mutant mice developed tumors within 25–61 weeks, with each mouse averaging five tumors128. Male mice developed more tumors than female mice in this model, mirroring the gender bias observed in human ccRCC128. Similar to human ccRCC responses to current therapies, Harlander et al. described heterogeneity in therapeutic responses to a panel of relevant drugs in the triple-mutant model128. Notably, VEGFR and mTOR treatment demonstrated a moderate decrease in tumor growth with development of drug resistance in some tumors128. These results implicate this model as a viable platform for investigating drug resistant mechanisms in an immunocompetent in vivo model128. Although the specific mutagenic profile driving tumor progression in this triple knockout is rarely seen in human ccRCC, this system does promote evolution of genetically distinct tumors with variable drug responses and classic pathological ccRCC features.
GEMM Limitations and Conclusions
GEMM models described here are unable to fully represent the genetic and pathological phenotypes identified in human disease. In addition, Setd2 GEM models have yet to be developed, despite the urgent need for qualitative understanding of SETD2 mutations in an in vivo setting. Most current RCC GEMMs rely on simultaneous deletion of target genes during early embryogenesis, rather than promoting sequential loss of function. Thus, future GEMMs in ccRCC should attempt to alter or delete genes of interest sequentially in an attempt to mimic clonal selection. This approach has already been utilized to dissect pancreatic ductal adenocarcinoma (PDAC) using CRISPR/Cas9 technology106.
For many genes residing on human 3p, notably SETD2, the ‘first-hit’ mutation results in a haploinsufficiency phenotype that is not yet technically feasible to recapitulate in a murine setting. Indeed, heterozygosity phenotypes for SETD2 have been implicated in tumorigenic processes as an early driver of genomic and microtubule instability129. Additionally, complete gene deletion, as opposed to mutation, limits investigations aimed at examining adverse interactions conferred by truncated or nonfunctional proteins, which may contribute to tumor progression in human ccRCC.
Comprehensive evaluation of the TME and ITH, in the above described GEMMs, have yet to be fully evaluated; however, researchers are actively exploiting the presence of a competent immune system to evaluate these features and associate the results with human disease. Specifically, future work should aim to determine if the tumor mutation status correlates with immune and non-immune cancer-associated cellular components in mice. Additionally, multi-regional sampling, whole exome sequencing, and single cell RNA sequencing, may uncover translatable tumor vulnerabilities that have not yet been identified in human tumors.
Ultimately, the complexity involved with studying ccRCC cell mechanisms and therapeutics in vivo suggests the need for current and continued development of xenograft and genetically modified mouse models, as each provide unique opportunities to interrogate various questions about the biology and treatment of RCC.
Patient-Derived Organoid (PDO) Model
Recently developed organoid technology promotes an in vitro structural organization that is comparable to an in vivo tumor architecture, while also maintaining the vascular-stromal-inflammatory milieu that makes up the TME130. Several studies have successfully created tumor organoid models using primary patient specimens from colorectal131, pancreatic132, bladder133, breast134, and lung cancers135. Grassi and colleagues became the first group to successfully establish and characterize organoid cultures from both normal and matched ccRCC adult-patient-derived-pluripotent stem cells of the kidney136. Differential organization was observed between ccRCC organoids, compared to their normal kidney counterparts, by immunofluorescence (IF)136. In addition, nephron region-specificity of the PCT and DCT, Bowman’s capsule, loop of Henle, thick ascending limb, and collecting duct tissues were all confirmed by cell type specific IF in both tumor and normal organoid cultures136. Successful transplant of RCC PDOs into immunocompromised mice, by orthotopic injection, demonstrated a 75% engraftment rate136. Furthermore, transplantation showed an increased capacity to invade kidney parenchyma and disorganized tissue structure, as compared to normal tissue PDO, thereby achieving tumor-propagating properties136. The authors contribution of organoids derived from normal adult kidney tissue is crucial, not only as a matched control for ccRCC, but also for the provision of a physiological relevant platform for basic kidney research136. Normal kidney tissue derived organoids also provide a foundation for future targeted gene editing in 3D culture utilizing CRISPR-Cas9 technology, an approach that has been established in organoids originating from intestinal and pancreatic tissue137,138. Initial selection of undifferentiated renal adult stem cells and rejection of hematopoietic derived cells limit this model’s ability to fully encompass the native TME in ccRCC.
Other groups, such as Neal and colleagues, developed an air liquid interface (ALI) method to cultivate patient derived organoids (PDOs) from surgically resected ccRCC biopsied human specimens. In a collagen-based matrix, the authors effectively retained primary tumor epithelium alongside immune and transformed non-immune TME components139. In addition, immune checkpoint blockade therapy with PD1 and PD-L1 propagated expansion of TILs and production of granzyme b cytotoxic CD8 T cells, thereby killing tumor cells and recapitulating immune checkpoint functionality in a 3D system139. Continued development of 3D organoid culture will likely provide opportunities to assess the efficacy of other immune- and metabolic-based therapies.
Challenges to Overcome in RCC PDOs
Technical challenges involved in RCC organoid development are not trivial, and standardization of optimized techniques to support experimental reproducibility have yet to be established. For unknown reasons, studies have observed a growth advantage of non-cancerous epithelial cells residing in the TME when exposed to supplemented cell culture conditions140. Modifications, including initial starvation of growth factors, followed by robust nutrient supplementation, have been shown to reduce contaminated normal epithelial cells in some tumor types139. This phenomenon is indicative of cell responses to in vitro culture conditions that may be different from those occurring in an in vivo tumor environment. In-depth analyses of the nutrients available in the native TME of human RCC tumors may be key to improving organoid development.
ITH is a challenging feature to capture in PDO models that rely on a small fraction of the resected tumor tissue. Given that ITH is a hallmark of ccRCC, methods aimed at sampling multiple regions of the tumor will likely provide a more representative view of any given tumor. Features common to RCC tumors, including necrosis, intratumoral hemorrhaging, inflammation, and hypoxia, make simultaneous retrieval and maintenance of viable tumor epithelial and TME cell populations challenging to obtain7,141. Despite current limitations, however, continued research development of PDOs will provide a reliable framework for investigations of RCC biology, therapeutic development, and evaluation of native TME/tumor crosstalk and organization.
Concluding remarks
Each model system in RCC research offers a unique set of advantages and disadvantages that must be considered by investigators. Well established RCC cell lines have propelled our molecular and genetic understanding of renal cancers, both in cell line models and murine CDX models, thus providing a system for high-throughput drug screening. Results obtained from historical research utilizing cell lines, CDX, and PDX models identified angiogenic and mTOR signaling as candidate therapeutic targets that ultimately translated into significant improvements in patient care and prognosis. The relative ease of manipulation allowed by cell line and xenograft models are important for enhancing our understanding of established and newly identified RCC subtypes. Despite advancements permitted by cell line and xenograft models, unpredictable tumor recurrence and therapeutic resistance in mRCC remain challenging obstacles in the field. Therefore, sophisticated models are particularly critical as the TME emerges as a major factor in cancer control mechanisms. Complex crosstalk occurring within the TME, has prompted a shift toward immunotherapy-based treatment as the standard of care. Consequently, there is a need for improved immunocompetent in vivo models and PDO in vitro 3D systems to allow for accurate recapitulation of the TME.
GEMMs have demonstrated successful development of de novo sporadic ccRCC tumors, providing an in vivo platform for thorough investigations of tumorigenesis and progression. Continued development of GEMM models will result in an increased capacity to mimic the clonal evolution believed to occur in human ccRCC tumors and allow the underlying mechanisms that contribute to tumorigenesis and progression to be fully appreciated by researchers. ccRCC organoid systems are poised to uncover tumor specific vulnerabilities in the context of an intricate stromal-vascular-inflammatory interaction. As rapid scientific advancement continues to push the technical boundaries of cancer research toward the development of precision medicine and immune therapy-based treatment, so, too, should disease-specific model systems evolve.
Footnotes
The authors have no conflict of interest to report.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 2.Le Magnen C, Dutta A, Abate-Shen C. Optimizing mouse models for precision cancer prevention. Nat Rev Cancer 2016; 16: 187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer 2017; 16: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haake SM, Rathmell WK. Renal cancer subtypes: Should we be lumping or splitting for therapeutic decision making? Cancer 2017; 123: 200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs—Part A: Renal, Penile, and Testicular Tumours. Eur Urol 2016; 70: 93–105. [DOI] [PubMed] [Google Scholar]
- 6.Kovacs G, Akhtar M, Beckwith BJ, Bugert P, Cooper CS, Delahunt B et al. The Heidelberg classification of renal cell tumours. J Pathol 1997; 183: 131–133. [DOI] [PubMed] [Google Scholar]
- 7.Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M et al. Renal cell carcinoma. Nat Rev Dis Prim 2017; 3: 17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linehan WM, Walther MM, Zbar B. The Genetic Basis of Cancer of the Kidney. J Urol 2003; 170: 2163–2172. [DOI] [PubMed] [Google Scholar]
- 9.Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep 2018; 23: 313–326.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sinha R, Winer AG, Chevinsky M, Jakubowski C, Chen Y-B, Dong Y et al. Analysis of renal cancer cell lines from two major resources enables genomics-guided cell line selection. Nat Commun 2017; 8: 15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993; 260: 1317–20. [DOI] [PubMed] [Google Scholar]
- 12.Haake SM, Weyandt JD, Rathmell WK. Insights into the Genetic Basis of the Renal Cell Carcinomas from The Cancer Genome Atlas. Mol Cancer Res 2016; 14: 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore LE, Nickerson ML, Brennan P, Toro JR, Jaeger E, Rinsky J et al. Von Hippel-Lindau (VHL) Inactivation in Sporadic Clear Cell Renal Cancer: Associations with Germline VHL Polymorphisms and Etiologic Risk Factors. PLoS Genet 2011; 7: e1002312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M et al. Renal cell carcinoma. Nat Rev Dis Prim 2017; 3: 17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maxwell PH, Wiesener MS, Chang G-W, Clifford SC, Vaux EC, Cockman ME et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399: 271–275. [DOI] [PubMed] [Google Scholar]
- 16.Chappell JC, Payne LB, Rathmell WK. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J Clin Invest 2019; 129: 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 463: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, O’Brien T et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018; 173: 611–623.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jonasch E, Gao J, Rathmell WK. Renal cell carcinoma. BMJ 2014; 349: g4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013; 499: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis 2018; 9: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wettersten HI, Aboud OA, Lara PN, Weiss RH. Metabolic reprogramming in clear cell renal cell carcinoma. Nat Rev Nephrol 2017; 13: 410–419. [DOI] [PubMed] [Google Scholar]
- 23.Ebos JML, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 2011; 8: 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H et al. A Small-Molecule Inhibitor of Glucose Transporter 1 Downregulates Glycolysis, Induces Cell-Cycle Arrest, and Inhibits Cancer Cell Growth In Vitro and In Vivo. Mol Cancer Ther 2012; 11: 1672–1682. [DOI] [PubMed] [Google Scholar]
- 25.Shuch B, Amin A, Armstrong AJ, Eble JN, Ficarra V, Lopez-Beltran A et al. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. Eur Urol 2015; 67: 85–97. [DOI] [PubMed] [Google Scholar]
- 26.Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013; 499: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Cubas AA, Rathmell WK. Epigenetic modifiers: activities in renal cell carcinoma. Nat Rev Urol 2018; 15: 599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beksac AT, Paulucci DJ, Blum KA, Yadav SS, Sfakianos JP, Badani KK. Heterogeneity in renal cell carcinoma. Urol Oncol Semin Orig Investig 2017; 35: 507–515. [DOI] [PubMed] [Google Scholar]
- 29.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N Engl J Med 2012; 366: 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turajlic S, Xu H, Litchfield K, Larkin J. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 2018; 173: 595–607.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018; 173: 581–594.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rini BI, Escudier B, Martini J-F, Magheli A, Svedman C, Lopatin M et al. Validation of the 16-Gene Recurrence Score in Patients with Locoregional, High-Risk Renal Cell Carcinoma from a Phase III Trial of Adjuvant Sunitinib. Clin Cancer Res 2018; 24: 4407–4415. [DOI] [PubMed] [Google Scholar]
- 33.Ghatalia P, Rathmell WK. Systematic Review: ClearCode 34 - A Validated Prognostic Signature in Clear Cell Renal Cell Carcinoma (ccRCC). Kidney cancer 2018; 2: 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brooks SA, Brannon AR, Parker JS, Fisher JC, Sen O, Kattan MW et al. ClearCode34: A prognostic risk predictor for localized clear cell renal cell carcinoma. Eur Urol 2014; 66: 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Velasco G, Culhane AC, Fay AP, Hakimi AA, Voss MH, Tannir NM et al. Molecular Subtypes Improve Prognostic Value of International Metastatic Renal Cell Carcinoma Database Consortium Prognostic Model. Oncologist 2017; 22: 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rini B, Goddard A, Knezevic D, Maddala T, Zhou M, Aydin H et al. A 16-gene assay to predict recurrence after surgery in localised renal cell carcinoma: development and validation studies. Lancet Oncol 2015; 16: 676–685. [DOI] [PubMed] [Google Scholar]
- 37.Hartmann JT, Bokemeyer C. Chemotherapy for renal cell carcinoma. Anticancer Res; 19: 1541–3. [PubMed] [Google Scholar]
- 38.Motzer RJ, Mazumdar M, Bacik J, Berg W, Amsterdam A, Ferrara J. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999; 17: 2530–40. [DOI] [PubMed] [Google Scholar]
- 39.Sherwood LM, Parris EE, Folkman J. Tumor Angiogenesis: Therapeutic Implications. N Engl J Med 1971; 285: 1182–1186. [DOI] [PubMed] [Google Scholar]
- 40.Barata PC, Rini BI. Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J Clin 2017; 67: 507–524. [DOI] [PubMed] [Google Scholar]
- 41.Kucejova B, Peña-Llopis S, Yamasaki T, Sivanand S, Tran TAT, Alexander S et al. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Mol Cancer Res 2011; 9: 1255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rini BI. Temsirolimus, an Inhibitor of Mammalian Target of Rapamycin. Clin Cancer Res 2008; 14: 1286–1290. [DOI] [PubMed] [Google Scholar]
- 43.Harshman LC, Kroeger N, Rha SY, Donskov F, Wood L, Tantravahi SK et al. First-Line Mammalian Target of Rapamycin Inhibition in Metastatic Renal Cell Carcinoma: An Analysis of Practice Patterns From the International Metastatic Renal Cell Carcinoma Database Consortium. Clin Genitourin Cancer 2014; 12: 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leach DR, Krummel MF, Allison JP. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science (80-) 1996; 271: 1734–1736. [DOI] [PubMed] [Google Scholar]
- 45.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999; 11: 141–51. [DOI] [PubMed] [Google Scholar]
- 46.Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 2018; 378: 1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 2019; 380: 1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015; 373: 1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santoni M, Massari F, Di Nunno V, Conti A, Cimadamore A, Scarpelli M et al. Immunotherapy in renal cell carcinoma: latest evidence and clinical implications. Drugs Context 2018; 7: 212528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rini BI, Powles T. Immune Checkpoint Blockade plus Axitinib for Renal-Cell Carcinoma. N Engl J Med 2019; 380: 2581–2582. [DOI] [PubMed] [Google Scholar]
- 51.Beckermann KE, Johnson DB, Sosman JA. PD-1/PD-L1 blockade in renal cell cancer. Expert Rev Clin Immunol 2017; 13: 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lopez-Beltran A, Henriques V, Cimadamore A, Santoni M, Cheng L, Gevaert T et al. The Identification of Immunological Biomarkers in Kidney Cancers. Front Oncol 2018; 8: 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 2018; 24: 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kondo K, Yao M, Kobayashi K, Ota S, Yoshida M, Kaneko S et al. PTEN/MMAC1/TEP1 mutations in human primary renal-cell carcinomas and renal carcinoma cell lines. Int J cancer 2001; 91: 219–24. [DOI] [PubMed] [Google Scholar]
- 55.Baumans V Use of animals in experimental research: an ethical dilemma? Gene Ther 2004; 11: S64–S66. [DOI] [PubMed] [Google Scholar]
- 56.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006; 6: 813–823. [DOI] [PubMed] [Google Scholar]
- 57.Brodaczewska KK, Szczylik C, Fiedorowicz M, Porta C, Czarnecka AM. Choosing the right cell line for renal cell cancer research. Mol Cancer 2016; 15: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 2019; 47: D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iliopoulos O, Kibel A, Gray S, Kaelin WG. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995; 1: 822–6. [DOI] [PubMed] [Google Scholar]
- 60.Iliopoulos O, Levy AP, Jiang C, Kaelin WG, Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci 1996; 93: 10595–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 2002; 1: 237–246. [DOI] [PubMed] [Google Scholar]
- 62.Zhao C-X, Luo C-L, Wu X-H. Hypoxia promotes 786-O cells invasiveness and resistance to sorafenib via HIF-2α/COX-2. Med Oncol 2015; 32: 419. [DOI] [PubMed] [Google Scholar]
- 63.Huang B, Huang YJ, Yao ZJ, Chen X, Guo SJ, Mao XP et al. Cancer stem cell-like side population cells in clear cell renal cell carcinoma cell line 769P. PLoS One 2013; 8: e68293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pichler R, Compérat E, Klatte T, Pichler M, Loidl W, Lusuardi L et al. Renal Cell Carcinoma with Sarcomatoid Features: Finally New Therapeutic Hope? Cancers (Basel) 2019; 11. doi: 10.3390/cancers11030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou S, Schuetz JD, Bunting KD, Colapietro A-M, Sampath J, Morris JJ et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 2001; 7: 1028–1034. [DOI] [PubMed] [Google Scholar]
- 66.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012; 483: 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Broad Institute Cancer Cell Line Encyclopedia (CCLE). https://portals.broadinstitute.org/ccle (accessed 2 Feb 2020).
- 68.Robb VA, Karbowniczek M, Klein-Szanto AJ, Henske EP. Activation of the mTOR Signaling Pathway in Renal Clear Cell Carcinoma. J Urol 2007; 177: 346–352. [DOI] [PubMed] [Google Scholar]
- 69.Hudes GR. Targeting mTOR in renal cell carcinoma. Cancer 2009; 115: 2313–2320. [DOI] [PubMed] [Google Scholar]
- 70.Shinojima T, Oya M, Takayanagi A, Mizuno R, Shimizu N, Murai M. Renal cancer cells lacking hypoxia inducible factor (HIF)-1 expression maintain vascular endothelial growth factor expression through HIF-2. Carcinogenesis 2006; 28: 529–536. [DOI] [PubMed] [Google Scholar]
- 71.Korhonen M, Sariola H, Gould VE, Kangas L, Virtanen I. Integrins and laminins in human renal carcinoma cells and tumors grown in nude mice. Cancer Res 1994; 54: 4532–8. [PubMed] [Google Scholar]
- 72.Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med 2006; 12: 122–127. [DOI] [PubMed] [Google Scholar]
- 73.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H et al. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst 1973; 51: 1417–23. [DOI] [PubMed] [Google Scholar]
- 74.Sanchez Y, el-Naggar A, Pathak S, Killary AM. A tumor suppressor locus within 3p14-p12 mediates rapid cell death of renal cell carcinoma in vivo. Proc Natl Acad Sci U S A 1994; 91: 3383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gioanni J, Zanghellini E, Mazeau C, Amiel J, Poustis-Delpont C, Lagrange JL et al. [CAL 54, a new cell line derived from a human renal carcinoma: characterization and radiosensitivity]. Bull Cancer 1996; 83: 553–8. [PubMed] [Google Scholar]
- 76.Rangarajan A, Weinberg RA. Comparative biology of mouse versus human cells: modelling human cancer in mice. Nat Rev Cancer 2003; 3: 952–959. [DOI] [PubMed] [Google Scholar]
- 77.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014; 46: 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klinghammer K, Walther W, Hoffmann J. Choosing wisely – Preclinical test models in the era of precision medicine. Cancer Treat Rev 2017; 55: 36–45. [DOI] [PubMed] [Google Scholar]
- 79.Chen Q, Wang J, Liu WN, Zhao Y. Cancer Immunotherapies and Humanized Mouse Drug Testing Platforms. Transl Oncol 2019; 12: 987–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Prochazka M, Gaskins HR, Shultz LD, Leiter EH. The nonobese diabetic scid mouse: model for spontaneous thymomagenesis associated with immunodeficiency. Proc Natl Acad Sci U S A 1992; 89: 3290–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pavía-Jiménez A, Tcheuyap VT, Brugarolas J. Establishing a human renal cell carcinoma tumorgraft platform for preclinical drug testing. Nat Protoc 2014; 9: 1848–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grisanzio C, Seeley A, Chang M, Collins M, Di Napoli A, Cheng S-C et al. Orthotopic xenografts of RCC retain histological, immunophenotypic and genetic features of tumours in patients. J Pathol 2011; 225: 212–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee C-H, Motzer RJ. Kidney cancer in 2016: The evolution of anti-angiogenic therapy for kidney cancer. Nat Rev Nephrol 2017; 13: 69–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rojas JD, Papadopoulou V, Czernuszewicz TJ, Rajamahendiran RM, Chytil A, Chiang Y-C et al. Ultrasound Measurement of Vascular Density to Evaluate Response to Anti-Angiogenic Therapy in Renal Cell Carcinoma. IEEE Trans Biomed Eng 2019; 66: 873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schor-Bardach R, Alsop DC, Pedrosa I, Solazzo SA, Wang X, Marquis RP et al. Does Arterial Spin-labeling MR Imaging–measured Tumor Perfusion Correlate with Renal Cell Cancer Response to Antiangiogenic Therapy in a Mouse Model? Radiology 2009; 251: 731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cuenod CA, Fournier L, Balvay D, Guinebretière J-M. Tumor angiogenesis: pathophysiology and implications for contrast-enhanced MRI and CT assessment. Abdom Imaging 2006; 31: 188–193. [DOI] [PubMed] [Google Scholar]
- 87.Dong Y, Manley BJ, Becerra MF, Redzematovic A, Casuscelli J, Tennenbaum DM et al. Tumor Xenografts of Human Clear Cell Renal Cell Carcinoma But Not Corresponding Cell Lines Recapitulate Clinical Response to Sunitinib: Feasibility of Using Biopsy Samples. Eur Urol Focus 2017; 3: 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patel A, Cohen S, Moret R, Maresh G, Gobe GC, Li L. Patient-derived xenograft models to optimize kidney cancer therapies. Transl Androl Urol 2019; 8: S156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lai Y, Wei X, Lin S, Qin L, Cheng L, Li P. Current status and perspectives of patient-derived xenograft models in cancer research. J Hematol Oncol 2017; 10: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A, Leng N, Pavía-Jiménez A, Wang S et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 2012; 44: 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen Y-B, Gonen M et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res 2013; 19: 3259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lampreht Tratar U, Horvat S, Cemazar M. Transgenic Mouse Models in Cancer Research. Front Oncol 2018; 8: 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wiltrout RH, Hornung RL, Futami H, Back TT, Young HA, Sayers TJ. Murine Renal Cancer (Renca) Model: Background and Preclinical Studies In: Immunotherapy of Renal Cell Carcinoma. Springer Berlin Heidelberg: Berlin, Heidelberg, 1991, pp 13–19. [Google Scholar]
- 94.Schokrpur S, Hu J, Moughon DL, Liu P, Lin LC, Hermann K et al. CRISPR-Mediated VHL Knockout Generates an Improved Model for Metastatic Renal Cell Carcinoma. Sci Rep 2016; 6: 29032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shvarts O, Janzen N, Lam JS, Leppert JT, Caliliw R, Figlin RA et al. RENCA/carbonic anhydrase-IX: A murine model of a carbonic anhydrase-IX-expressing renal cell carcinoma. Urology 2006; 68: 1132–1138. [DOI] [PubMed] [Google Scholar]
- 96.Berlato C, Khan MN, Schioppa T, Thompson R, Maniati E, Montfort A et al. A CCR4 antagonist reverses the tumor-promoting microenvironment of renal cancer. J Clin Invest 2017; 127: 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Devaud C, Yong CSM, John LB, Westwood JA, Duong CPM, House CM et al. Foxp3 expression in macrophages associated with RENCA tumors in mice. PLoS One 2014; 9: e108670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sivanand S, Peña-Llopis S, Zhao H, Kucejova B, Spence P, Pavia-Jimenez A et al. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Sci Transl Med 2012; 4: 137ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Espana-Agusti J, Warren A, Chew SK, Adams DJ, Matakidou A. Loss of PBRM1 rescues VHL dependent replication stress to promote renal carcinogenesis. Nat Commun 2017; 8: 2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kleymenova E, Everitt JI, Pluta L, Portis M, Gnarra JR, Walker CL. Susceptibility to vascular neoplasms but no increased susceptibility to renal carcinogenesis in Vhl knockout mice. Carcinogenesis 2004; 25: 309–15. [DOI] [PubMed] [Google Scholar]
- 101.Lee CM, Hickey M, Sanford CA, Mcguire CG, Cowey CL, Simon MC et al. VHL Type 2B gene mutation moderates HIF dosage in vitro and in vivo HHS Public Access. Oncogene 2009; 28: 1694–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cheval L, Pierrat F, Rajerison R, Piquemal D, Doucet A. Of Mice and Men: Divergence of Gene Expression Patterns in Kidney. PLoS One 2012; 7: e46876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liao L, Testa JR, Yang H. The roles of chromatin-remodelers and epigenetic modifiers in kidney cancer. Cancer Genet 2015; 208: 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bult CJ, Blake JA, Smith CL, Kadin JARJ. Mouse Genome Database (MGD). Nucleic Acids Res 2019; : D801–D806. [DOI] [PMC free article] [PubMed]
- 105.Sanchez DJ, Simon MC. Genetic and metabolic hallmarks of clear cell renal cell carcinoma. Biochim Biophys acta Rev cancer 2018; 1870: 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ideno N, Yamaguchi H, Okumura T, Huang J, Brun MJ, Ho ML et al. A pipeline for rapidly generating genetically engineered mouse models of pancreatic cancer using in vivo CRISPR-Cas9-mediated somatic recombination. Lab Investig 2019; 99: 1233. [DOI] [PubMed] [Google Scholar]
- 107.Kersten K, de Visser KE, van Miltenburg MH, Jonkers J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol Med 2017; 9: 137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011; 469: 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nargund AM, Pham CG, Dong Y, Wang PI, Osmangeyoglu HU, Xie Y et al. The SWI/SNF Protein PBRM1 Restrains VHL-Loss-Driven Clear Cell Renal Cell Carcinoma. Cell Rep 2017; 18: 2893–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shao X, Somlo S, Igarashi P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol 2002; 13: 1837–46. [DOI] [PubMed] [Google Scholar]
- 111.Gu Y-F, Cohn S, Christie A, McKenzie T, Wolff N, Do QN et al. Modeling Renal Cell Carcinoma in Mice: Bap1 and Pbrm1 Inactivation Drive Tumor Grade. Cancer Discov 2017; 7: 900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bouchard M, Souabni A, Mandler M, Neubüser A, Busslinger M. Nephric lineage specification by Pax2 and Pax8. Genes Dev 2002; 16: 2958–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bouchard M, Souabni A, Busslinger M. Tissue-specific expression of cre recombinase from thePax8 locus. genesis 2004; 38: 105–109. [DOI] [PubMed] [Google Scholar]
- 114.Capitanio U, Montorsi F. Renal cancer. Lancet 2016; 387: 894–906. [DOI] [PubMed] [Google Scholar]
- 115.Wallace AC, Nairn RC. Renal tubular antigens in kidney tumors. Cancer 1972; 29: 977–81. [DOI] [PubMed] [Google Scholar]
- 116.Schuetz AN, Yin-Goen Q, Amin MB, Moreno CS, Cohen C, Hornsby CD et al. Molecular Classification of Renal Tumors by Gene Expression Profiling. J Mol Diagnostics 2005; 7: 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen F, Zhang Y, Şenbabaoğlu Y, Ciriello G, Yang L, Reznik E et al. Multilevel Genomics-Based Taxonomy of Renal Cell Carcinoma. Cell Rep 2016; 14: 2476–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Frew IJ, Moch H. A Clearer View of the Molecular Complexity of Clear Cell Renal Cell Carcinoma. Annu Rev Pathol Mech Dis 2015; 10: 263–289. [DOI] [PubMed] [Google Scholar]
- 119.Young MD, Mitchell TJ, Vieira Braga FA, Tran MGB, Stewart BJ, Ferdinand JR et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 2018; 361: 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vallon V, Grahammer F, Richter K, Bleich M, Lang F, Barhanin J et al. Role of KCNE1-dependent K+ fluxes in mouse proximal tubule. J Am Soc Nephrol 2001; 12: 2003–11. [DOI] [PubMed] [Google Scholar]
- 121.El Marjou F, Janssen K-P, Hung-Junn Chang B, Li M, Hindie V, Chan L et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. genesis 2004; 39: 186–193. [DOI] [PubMed] [Google Scholar]
- 122.Wang S-S, Gu Y-F, Wolff N, Stefanius K, Christie A, Dey A et al. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proc Natl Acad Sci U S A 2014; 111: 16538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G et al. Six2 Defines and Regulates a Multipotent Self-Renewing Nephron Progenitor Population throughout Mammalian Kidney Development. Cell Stem Cell 2008; 3: 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yao M, Shuin T, Misaki H, Kubota Y. Enhanced expression of c-myc and epidermal growth factor receptor (C-erbB-1) genes in primary human renal cancer. Cancer Res 1988; 48: 6753–7. [PubMed] [Google Scholar]
- 125.Kim WY, Sharpless NE. The Regulation of INK4/ARF in Cancer and Aging. Cell 2006; 127: 265–275. [DOI] [PubMed] [Google Scholar]
- 126.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 1999; 4: 199–207. [DOI] [PubMed] [Google Scholar]
- 127.Bailey ST, Smith AM, Kardos J, Wobker SE, Wilson HL, Krishnan B et al. MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma. Nat Commun 2017; 8: 15770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Harlander S, Schönenberger D, Toussaint NC, Prummer M, Catalano A, Brandt L et al. Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice. Nat Med 2017; 23: 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chiang Y-C, Park I-Y, Terzo EA, Tripathi DN, Mason FM, Fahey CC et al. SETD2 Haploinsufficiency for Microtubule Methylation Is an Early Driver of Genomic Instability in Renal Cell Carcinoma. Cancer Res 2018; 78: 3135–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Clevers H Modeling Development and Disease with Organoids. Cell 2016; 165: 1586–1597. [DOI] [PubMed] [Google Scholar]
- 131.van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015; 161: 933–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Boj SF, Hwang C-I, Baker LA, Chio IIC, Engle DD, Corbo V et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015; 160: 324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018; 173: 515–528.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018; 172: 373–386.e10. [DOI] [PubMed] [Google Scholar]
- 135.Finnberg NK, Gokare P, Lev A, Grivennikov SI, MacFarlane AW, Campbell KS et al. Application of 3D tumoroid systems to define immune and cytotoxic therapeutic responses based on tumoroid and tissue slice culture molecular signatures. Oncotarget 2017; 8: 66747–66757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Grassi L, Alfonsi R, Francescangeli F, Signore M, De Angelis ML, Addario A et al. Organoids as a new model for improving regenerative medicine and cancer personalized therapy in renal diseases. Cell Death Dis 2019; 10: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y et al. Modeling colorectal cancer using CRISPR-Cas9–mediated engineering of human intestinal organoids. Nat Med 2015; 21: 256–262. [DOI] [PubMed] [Google Scholar]
- 138.Tiriac H, Bucobo JC, Tzimas D, Grewel S, Lacomb JF, Rowehl LM et al. Successful creation of pancreatic cancer organoids by means of EUS-guided fine-needle biopsy sampling for personalized cancer treatment. Gastrointest Endosc 2018; 87: 1474–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018; 175: 1972–1988.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J et al. Identification of Multipotent Luminal Progenitor Cells in Human Prostate Organoid Cultures. Cell 2014; 159: 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 2010; 40: 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013; 499: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]