

Abstract
Extracellular vesicles (EVs) are increasingly being recognized as important vehicles for intercellular communication and as promising sources for biomarker discovery. Because the state of protein post-translational modifications (PTMs) such as phosphorylation and glycosylation can be a key determinant of cellular physiology, comprehensive characterization of protein PTMs in EVs can be particularly valuable for early-stage diagnostics and monitoring of disease status. However, the analysis of PTMs in EVs has been complicated by limited amounts of purified EVs, low-abundance PTM proteins, and interference from proteins and metabolites in biofluids. Recently, we developed an approach to isolate phosphoproteins and glycoproteins in EVs from small volumes of human plasma that enabled us to identify nearly 10,000 unique phosphopeptides and 1,500 unique N-glycopeptides. The approach demonstrated the feasibility of using these data to identify potential markers to differentiate disease from healthy states. Here we present an updated workflow to sequentially isolate phosphopeptides and N-glycopeptides, enabling multiple PTM analyses of the same clinical samples. In this updated workflow, we have improved the reproducibility and efficiency of EV isolation, protein extraction, and phosphopeptide/N-glycopeptide enrichment to achieve sensitive analyses of low-abundance PTMs in EVs isolated from 1 mL of plasma. The modularity of the workflow also allows for the characterization of phospho- or glycopeptides only and enables additional analysis of total proteomes and other PTMs of interest. After blood collection, the protocol takes 2 d, including EV isolation, PTM/peptide enrichment, mass spectrometry analysis, and data quantification.
Introduction
Extracellular vesicles, generally including exosomes and microvesicles1–3, are membrane-enclosed vesicles containing proteins and nucleic acid cargos. EVs are released by almost all cell types and provide an effective and ubiquitous path for intercellular communication and the transmission of pathogenic and signaling molecules among cells3–5. Their potentially important cellular functions in disease onset and progression make them intriguing sources for biomarker discovery and disease diagnosis6–9. In particular, these EV-based disease markers can be identified well before the onset of symptoms or physiological detection of an ailment, making them promising candidates for early-stage disease detection9–14. Recently, proteins in EVs obtained from cell culture media15,16 and from biofluids such as plasma15–17 and urine18–21 have been reported to contain PTMs. In general, such modifications can alter protein conformation, stability, activity, cellular localization, and interaction with other cellular molecules. However, the effects of these PTMs on EVs and their potential use as biomarkers or in diagnostics has been largely unexplored until now.
Alterations in PTMs in proteins are thought to be major determinants in the early onset and progression of diseases such as cancer and neurodegeneration, and they therefore have become actively pursued targets as indicators of cellular states for disease diagnosis and treatment. However, these targets have been largely unexplored because of the limited availability of tools for studying low-abundance EV PTMs in highly complex clinically relevant samples such as plasma. Mass spectrometry (MS) is the major tool used to study PTMs, and multiple MS-based strategies and protocols have been introduced to efficiently analyze PTMs on a proteome-wide scale22–24. We have recently reported the identification and quantification of a large number of phosphoproteins and glycoproteins in EVs isolated from human plasma25,26. In this protocol, we describe a workflow for the sequential analysis of both types of modifications and total proteome analysis from a limited amount of starting material.
Development of the approach
Biofluids such as plasma are complex, and—owing to the presence of phosphatases in the blood-stream (e.g., alkaline phosphatase secreted by the liver)—previous attempts to identify phosphoproteins from plasma or serum samples identified only a small number of phosphorylation sites, of which the level of phosphorylation could not be connected to the biological status27,28. To overcome these challenges, we recently developed a sensitive strategy for the isolation of EVs from biobanked human plasma samples, enrichment of EV phosphopeptides or N-glycopetides, and corresponding EV phosphoproteome or N-glycoproteome analyses by nanoflow liquid chromatography (LC)-MS25,26. In these studies, we identified nearly 10,000 phosphorylation sites and 1,500 glycosylation sites from small (1-mL) amounts of plasma sample and demonstrated the possibility of using PTMs on EV proteins from biofluids as potential biomarkers. We reason that because EVs are membrane-encapsulated compartments, their contents are shielded from the activity of external phosphatases and other enzymes3,29,30, resulting in the successful identification of many PTMs from a limited volume of plasma sample. In addition, analyzing the glycoproteome from EVs instead of whole plasma or serum samples minimizes interference from highly abundant plasma components. This circumvents the challenge of analyzing samples with an extremely wide dynamic range of protein abundancies and enabled us to identify hundreds of glycoproteins that had not been previously discovered in blood25,26.
Since the publication of our separate approaches for EV phosphoproteomics22 and glycoproteomics25,26, we updated the workflow by developing a combined pipeline for the sequential isolation of both phosphopeptides and N-glycopeptides from the same plasma sample (Fig. 1). This allows us to efficiently utilize clinical plasma samples, such as plasma from patients, while maintaining comparable sensitivity for the identification of both phosphopeptides and N-glycopeptides (Anticipated results).
Fig. 1 |. Workflow for sequential EV phosphoproteomics and glycoproteomics.
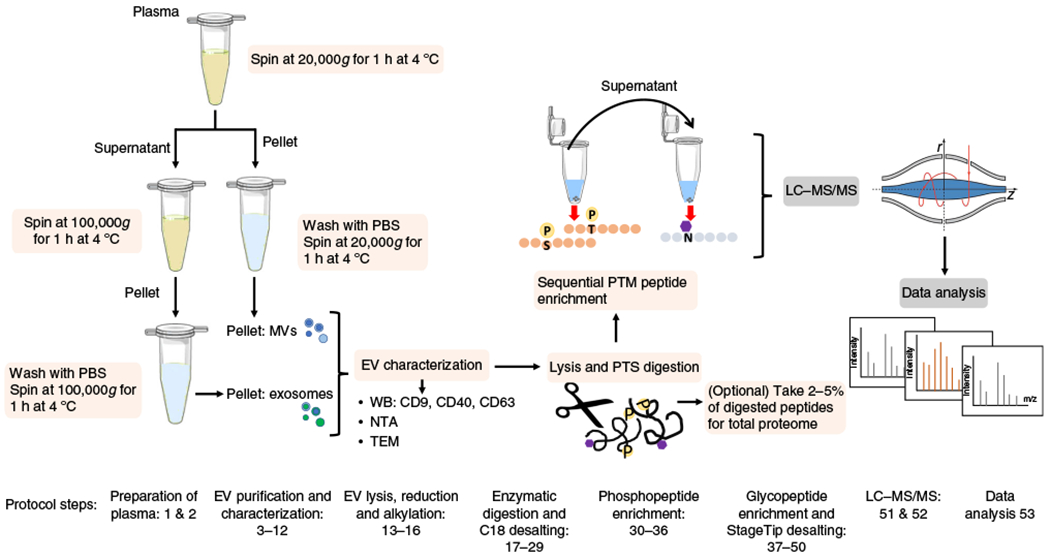
The workflow for the isolation of EVs, enrichment of phosphopeptides and glycopeptides, and nanoflow LC-MS analysis. Microvesicles and exosomes are isolated from human plasma through sequential high-speed and ultra-high-speed centrifugation. EVs are lysed and proteins are extracted and digested in SDC buffer. Phosphopeptides and N-glycopeptides are sequentially enriched, followed by LC-MS/MS analysis. Protocol steps are indicated. N, asparagine; P, phosphorylation; S, serine; T, threonine.
Overview of the procedure
Here, we describe a detailed workflow for the analysis of EV total proteome, phosphoproteome and glycoproteome from human plasma. We focus on several critical steps that improve the reproducibility and efficiency of EV isolation, protein extraction, and phosphopeptide/N-glycopeptide enrichment. We first describe how to prepare the plasma from biobanked or freshly collected human blood (Steps 1 and 2) and how to isolate exosomes and microvesicles (MVs) separately using differential centrifugation (Steps 3–11). The isolation efficiency and specificity can be characterized via multiple techniques such as western blot (WB) using antibodies against EV markers, nanoparticle tracking analysis (NTA), and transmission electron microscopy (TEM) (Step 12). EVs are then subjected to lysis, protein extraction, and digestion (Steps 13–21), followed by C18 desalting (Steps 22–29). Phosphopeptides are enriched through metal ion-affinity capture (Steps 30–36), and the unbound material is used to enrich glycopeptides through hydrazide chemistry capture followed by enzymatic release of formerly N-glycosylated peptides (Steps 37–44) and StageTip desalting (Steps 45–50). Samples are subsequently analyzed by nanoflow LC-MS analysis (Steps 51 and 52) and we provide detailed instructions for quantitative data analysis (Step 53).
Comparison with alternative approaches
Efficient isolation of EVs from cell culture media or biofluids is the first and most critical step of the downstream EV proteome analysis10,19. Several methods for EV isolation have been introduced19,31, including differential ultracentrifugation (UC)32, size-exclusion chromatography (SEC)33, polymer precipitation34, sucrose density gradient35, and affinity purification that can be either antibody36 or chemical based37. None of the existing isolation methods is perfect; each approach has its own advantages and problems related to, for example, efficiency, purity, isolation time, cost, and demands upon the instruments. For example, subpopulations of EVs can be isolated according to their size or specific marker proteins, but high selectivity also leads to lower EV yields, which can result in incomplete coverage of EV proteomes by MS analysis and potential loss of valuable biological information. EV proteome analyses, in particular PTM analyses, are highly sensitive to contamination from high-abundance plasma/serum proteins, lipids, and reagents in the case of polymer-based precipitation. As a result, some of the EV isolation methods may be suitable for downstream analyses such as nucleic acid sequencing or immunoassays but may not work for MS-based analysis. In this workflow, we chose to use UC for the isolation of EVs as an unbiased approach with low contamination of plasma proteins. Although there are EV proteins that can be used as markers (such as CD9, CD81 and CD63) to evaluate isolation efficiency and specificity, these markers were established using relatively homogeneous cell culture systems and may not be applicable to biofluids, in which many more different types of EVs exist38–40.
The workflow also includes EV lysis, protein digestion, and PTM-peptide enrichment steps. Alternative strategies have been developed for each step; for example, EV lysis and the extraction of proteins can be achieved using methods developed for cultured cells, with or without any detergent41–45. Considering the high membrane content of EVs relative to cells, in this workflow we have adopted a lysis and extraction method for membrane protein analysis to improve EV lysis and protein extraction46. Once EV proteins have been extracted, different protocols can be applied to achieve efficient protein digestion47–49.
Multiple approaches have also been developed to enrich peptides with PTMs, such as phosphopeptides or glycopeptides, from a complex sample typically dominated by peptides without any modification. For instance, phosphopeptides can be efficiently enriched using immobilized-metal affinity chromatography (IMAC)50–52, metal oxides (e.g.,TiO2)51–53, or polymer-based affinity capture51. In this workflow, we used polymer-based metal-ion affinity capture (polyMAC)51, which improves the enrichment efficiency through homogeneous isolation of phosphopeptides from a complex mixture. For more complex samples such as a whole-cell extract, a fractionation step before or after the phosphopeptide enrichment has been shown to be particularly useful. However, we did not find that a fractionation step is needed for EV extracts and instead observed that a single-run EV phosphoproteomics workflow is sufficient. Using a single-run workflow can greatly improve sample throughput and is particularly suitable for label-free quantitation for EV-based biomarker discovery. Similarly, there are continuous efforts in the development of glycopeptide enrichment, which include approaches using classic hydrazide chemistry-based N-glycopeptide capture followed by enzymatic release for MS analysis54,55 and, more recently, the isolation of intact glycopeptides using affinity chromatography to identify peptide sequence, glycan structure, and glycosylation sites on the basis of multiple dissociation methods56–60. The latter approach can provide rich molecular information and determine the structure of intact glycopeptide, but the technique is still evolving, and the throughput is relatively low, which might be further developed for the identification of protein biomarkers. In this protocol, by primarily focusing on the protein portion, not on the glycan portion, we applied the hydrazide chemistry to enrich N-glycopeptides.
Limitations of the approach
The major limitation of the analysis of EVs obtained from plasma is the complexity of the samples. This complexity is a result of EV heterogeneity, possible contamination from high-abundance plasma components such as plasma proteins, lipids, apoptotic bodies, platelets and so on, and incomplete understanding of EV biogenesis. Compared to EVs isolated from cell culture media, plasma-derived EVs can be released by any type of cell, and quantitative analysis of the heterogeneity of the EVs will be critical to extracting important and relevant information from the high background of EVs released by all types of cells. If desired, different EV subpopulations can be isolated and their proteins and modifications can be analyzed separately, thereby allowing one to determine whether the presence of EV PTMs is restricted to specific subtypes of EVs. However, methods for isolating specific EV subtypes are often not efficient, and the amount of resulting EVs may be too low to be processed for measurement by MS.
The majority of EV isolation approaches (including in this protocol) are based on separation by UC. However, there are several drawbacks of this approach: the EV isolation time using UC is long; it requires the operation of delicate equipment; and the yield is typically low and is subject to the sample conditions, such as temperature and dilution factor. Together, these limitations make UC not the ideal method for EV isolation in clinical settings. Among different attempts to develop robust EV isolation methods, we have recently introduced a chemical affinity-based method to readily isolate EVs from urine samples on functionalized magnetic beads with the potential for future clinical applications37.
Finally, the relatively long procedure of the protocol, technical variability in plasma collection and EV purification, and the low-abundance, high-complexity, and highly dynamic nature of protein PTMs all greatly influence quantitative MS analysis and subsequent EV-based biomarker discovery. Because an increasing number of research groups are currently interested in measuring functional proteins that are specifically present in different EV subtypes, we expect that future studies may focus on targeted approaches such as multiple-reaction monitoring (MRM)61,62 for greater sensitivity and more accurate quantification.
Experimental design
Evaluating the specificity and reproducibility of EV isolation from plasma
The EV isolation needs to be specific, with minimal contamination from aggregated plasma/serum proteins, lipids, and nucleic acids that may affect the PTM enrichment, MS analysis and data interpretation. The isolation of EVs also requires a relatively high yield—because of the limited amount of clinical sample—for successful identification of low-abundance proteins and PTMs from EVs. Furthermore, EVs have to remain intact during the isolation process to prevent their internal cargo from being released. In this workflow, we use UC for EV isolation, which is the most commonly used approach and is considered a “gold standard” approach. The overall UC method is relatively straightforward and does not require additional chemicals or reagents. Characterization of isolated EVs on the basis of their shape, size, particle concentration and purity is typically required because of the imperfect nature of existing EV isolation methods63. Commonly used EV characterization approaches include antibody-based methods to demonstrate the presence of specific EV markers, NTA64,65 for EV size distribution and concentration, and electron microscopy (EM)65 for visual morphology and shape and size of EVs. Typical EV characterization requires at least two orthogonal techniques to obtain comprehensive information on the isolated EVs66. To verify whether the UC isolation method is suitable for downstream proteomics and phosphoproteomics, we analyzed two types of samples, one from breast cancer patients and another from healthy controls. Starting with 1 mL of plasma each, we applied the workflow in Fig. 1 to obtain EV phosphoproteomes. These phosphoproteomes were compared against an EV database downloaded from Vesiclepedia (http://microvesicles.org/). The results showed a 77% overlap between our identified phosphoproteins and the EV database (Supplementary Fig. 1), with relatively low contamination from plasma proteins. We also carried out Pearson correlation to examine the reproducibility of the isolation method. As shown in Supplementary Fig. 2, the reproducibility of the EV isolation method across different samples is relatively high, with Pearson correlations between 0.96 and 0.98.
Extracting EV proteins
The workflow also includes instructions for extracting and digesting EV proteins. With a membrane structure similar to that of cells, standard cell lysis and protein extraction methods have been applied to EVs. To improve the protein extraction efficiency for EVs (which contain a higher proportion of membrane contents as compared to cells), we adopted the phase-transfer surfactant (PTS) protocol67 for EV lysis and protein extraction. The protocol using sodium deoxycholate (SDC) and N-lauroylsarcosine sodium salt (SLS) as surfactants improves the number of identified peptides with PTMs46. In addition, the enzymatic activity is enhanced in the SDC-SLS buffer as compared to other digestion buffers, which results in a lower percentage of missed cleavages in peptides46,67.
Sequential enrichment of EV phosphoproteome and N-glycoproteome
In our original approaches25,26, EV phosphoproteomics and N-glycoproteomics were performed separately, using different samples of human plasma. Although the published methods work well for diverse biological and clinical samples, the quantities of valuable clinical samples are often limited. Instead of dividing clinical samples into several aliquots or increasing the number of samples by expanding cohorts of patients and corresponding controls, we therefore sought to simultaneously analyze several PTMs in EVs from a single clinical source through sequential enrichment of PTMs. The approach could also provide a unique opportunity to integrate studies of EV protein networks involving multiple PTMs.
In our previous studies25,26, we examined MVs and exosomes separately. Our results based on MS and WB analyses identified several exosome or MV protein markers in both MV and exosome fractions, suggesting that high- and ultra-high-speed centrifugation did not isolate plasma EV subpopulations specifically. In the updated protocol, we do not perform the isolation of MVs and exosome for separate MS analyses. This is consistent with the most recent recommendation from the International Society for Extracellular Vesicles63, which does not propose the use of molecular markers for characterizing a single EV population. We also updated the protocol by isolating phosphopeptides in spintips (StageTips)68, which can be made in-house by packing Ti(IV)-based solid phase into ordinary pipette tips or can be purchased from commercial sources (e.g., Sigma-Aldrich), enabling us to carry out single-run phosphoproteomics69 with low amounts of starting materials (30–50 μg) isolated from plasma EVs. Typically, an ‘enhancer’, a high concentration of glycolic acid or lactic acid, is used to improve the selectivity of phosphopeptide enrichment. The reagent, however, is not directly compatible with the downstream N-glycopeptide enrichment, because glycolic acid or lactic acid can also be oxidized to generate aldehyde when N-glycopeptides are oxidized by NaIO4 in the N-glycopeptide enrichment protocol55. It is possible to add one desalting step to remove the reagent, but the extra step will lead to potentially severe sample loss when minimal material is available. Instead, we introduce a low-pH condition using a high concentration of trifluoroacetic acid (TFA) to obtain similar selectivity, and the TFA can be later removed under vacuum or neutralized with base.
Analyzing total proteomes and additional PTMs
In addition to the analysis of phospho- and glycopeptides, we also provide guidance on how to perform total-proteome analysis (Fig. 1). A small portion of peptides (typically <1 μg) can be taken after the desalting step (Step 28) for the analysis of the total EV proteome, and the rest is subjected to sequential enrichment of PTM peptides. Furthermore, we expect it would be feasible to include the analysis of additional PTMs in the workflow described in this protocol. Such analysis could include immunoprecipitation with antibodies specific to acetylation or methylation to obtain multi-PTM-omics data using the same clinical samples. For example, EV peptide mixtures (obtained in Step 29) could be first incubated with beads immobilized with antibodies to enrich the peptides by acetylation or methylation, and the flow-through could be subjected to phosphopeptide and/or glycopeptide enrichment.
Materials
Biological materials
Patient and control plasma samples. The plasma samples used in this protocol were obtained from the Susan G. Komen Breast Cancer Foundation Tissue Bank under Standard Operating Procedure (SOP) 004V6.0 (https://komentissuebank.iu.edu/wp-content/uploads/2016/11/SOP-004-V6.0-Acquisition-of-Plasma-from-Whole-Blood.pdf?fbclid=IwAR0LAMm63gGjDrSG1GqrRCj-Jh7aOD-YE5SH6XBnN9BFTZPxYBDe5N6Uso) and also from Indiana Biobank. We anticipate that this protocol can also be adapted for cell lines, urine or other biofluids ! CAUTION All experiments involving animal or human samples should adhere to applicable institutional and governmental regulations and guidelines. All plasma samples used in this study were previously collected, de-identified, and stored at the Tissue Bank, not for the specific purpose of this project.
Reagents
Potassium dihydrogen phosphate (KH2PO4; Merck, cat. no. 5438410100)
Sodium chloride (NaCl; Fisher Chemical, cat. no. S271-1)
Potassium chloride (KCl; Fisher Chemical, cat. no. P217)
Sodium phosphate, dibasic, anhydrous (Na2HPO4; Sigma-Aldrich, cat. no. S7907)
Hydrochloric acid (HCl; Fisher Chemical, cat. no. A144SI-212)
Tris base (Fisher BioReagents, cat. no. BP152-1)
Sodium deoxycholate (SDC; Sigma-Aldrich, cat. no. D6750)
N-Lauroylsarcosine sodium salt (sarkosyl; Sigma-Aldrich, cat. no. L9150)
Tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl; Sigma-Aldrich, cat. no. C4706)
2-Chloroacetamide (CAA; Sigma-Aldrich, cat. no. C0267)
Phosphatase inhibitor cocktail (Sigma-Aldrich, cat. no. P2850)
Triethylammonium bicarbonate buffer (TEAB; Sigma-Aldrich, cat. no. T7408)
Bicinchoninic acid (BCA) Protein Assay Kit (Pierce, cat. no. 23225)
Endopeptidase Lys-C (Wako Chemicals, cat. no. 129-02541)
Trypsin (proteomics grade, modified; Sigma-Aldrich, cat. no. T6567)
Ethyl acetate (Fisher Chemical, cat. no. E145-4)
Ammonium hydroxide solution (28% (vol/vol) NH3 in H2O, NH4OH; Sigma-Aldrich, cat. no. 338818)
Glycolic acid (70% (wt/vol) solution in water, C2H4O3; Sigma-Aldrich, cat. no. 420581)
Sodium (meta)periodate (NaIO4; Sigma-Aldrich, cat. no. S1878)
Sodium sulfite (Na2SO3; Sigma-Aldrich, cat. no. S4672)
Trifluoroacetic acid (TFA; Merck, cat. no. 8082600100)
Acetonitrile (ACN; Fisher Scientific, cat. no. A955-4)
Methanol (MeOH; Fisher Scientific, cat. no. A456-1)
Formic acid (FA; Sigma-Aldrich, cat. no. F0507)
Water with 0.1% formic acid (vol/vol), LC/MS grade (Fisher Scientific, cat. no. LS118-500)
Deionized (DI) water
Equipment
Eppendorf ThermoMixer C (Eppendorf, cat. no. 5382000015). If an alternative mixer is used, make sure it can be heated to 95 °C, can accommodate 1.7- or 2-mL tubes, and can operate at mixing speeds of 1,500–2,000 r.p.m.
Microcentrifuge tubes (OmniSeal low-binding, clear, natural, RNase/DNase/Pyrogen/Human DNA/PCR inhibitor free, 1.7-mL; Life Science Products, cat. no. M-1700C-LB)
Precision water bath (Thelco, cat. no. 184). If an alternative water bath is used, ensure it can warm within the range of 30–37 °C
Nalgene syringe filter (0.2 μm, surfactant-free cellulose acetate (SFCA); Thermo Scientific, cat. no. 723-2520)
Refrigerated bench-top centrifuge (Eppendorf, cat. no. 5417R). If an alternative centrifuge is used, make sure it can accommodate 1.7- and 2-mL tubes and can operate at 4 °C at speeds of 20,000g (relative centrifugal force (RCF))
Ultracentrifuge (Beckman Coulter, Optima MAX-XP model). If an alternative centrifuge is used, make sure it can operate at 4 °C at speeds of 100,000g (RCF)
Acid-resistant benchtop concentrator (CentriVap; Labconco; cat. no. 7810016)
Sep-Pak C18 cartridges (1 cc, 50 mg; Waters, cat. no. WAT054955). Capacity will depend on the amount of protein digested
Styrene-divinylbenzene (SDB-XC 47-mm extraction disks (Empore Products, cat. no. 66884-U)
StageTips. We use in-house-made plugged StageTips because they are inexpensive and allow for the use of multiple layers according to the peptide amount being desalted68. Other alternatives (such as anion-exchange disks, ZipTips and C18 solid-phase extraction (SPE) cartridges) are available, however; buffer solutions may need to be changed as appropriate70,71
Spin-Tip PolyMAC-Ti Phosphopeptide Enrichment Kit (titanium-based; Tymora, cat. no. 707). Alternative phosphopeptide enrichment methods can be used (IMAC, TiO2, among others); however, buffers, washes and elutions may vary from the ones outlined in this protocol
PNGase F kit (New England BioLabs, cat. no. P0705L)
SiMAG-hydrazide (hydrazide magnetic beads; Chemicell, cat. no. 1404-5)
LC instrument (Thermo Fisher Scientific, model no. Easy-nLC 1000) with 45-cm packed column (360-μm o.d. × 75-μm i.d.) containing C18 resin (2.2 μm, 100Å; Michrom Bioresources), with a 30-cm column heater (Analytical Sales and Services) set to 50 °C
Mass spectrometer. Any mass spectrometer capable of MS1 and MS2 scans with high resolution and scanning speed. We use a Thermo LTQ Orbitrap Velos Pro model to profile PTMs in plasma-derived EVs and a QExactive hybrid quadrupole-Orbitrap mass spectrometer for parallel reaction monitoring (PRM) experiments
Syringe (16 gauge)
Pipette tips (200 μL)
Vacutainer K2EDTA tubes
Vacuum centrifuge
PCR strip tubes
Software
Proteome Discoverer or other software for analyzing raw proteomics data (https://www.thermofisher.com/store/products/OPTON-30945#/OPTON-30945)
Current MaxQuant release (http://maxquant.org) or other software for analyzing raw proteomics data
Current Perseus software platform (http://maxquant.net/perseus/)
Reagent setup
Filtered PBS
PBS should be filtered with a Nalgene syringe filter. Store the filtered PBS at 4 °C for up to 6–12 months.
Tris-HCl (100 mM, pH 8.5)
Dissolve 12.1 g of Tris base in 1 L of DI water and adjust the pH to 8.5 using 1 N HCl. This buffer can be stored at room temperature (RT; 20–25 °C) for up to 6–12 months.
Buffer A for Sep-Pak desalting
This buffer consists of 0.1% (vol/vol) TFA in DI water. This buffer is stable for >3 months at RT ! CAUTION TFA solutions are corrosive. Handle with gloves and prepare solutions in a fume hood.
Buffer A for SDB-XC desalting
This buffer consists of 5% (vol/vol) ACN–0.1% (vol/vol) TFA in DI water. This buffer is stable for >3 months at RT ! CAUTION TFA solutions are corrosive. Handle with gloves and prepare solutions in a fume hood.
Buffer B for both Sep-Pak and SDB-XC desalting
This buffer consists of 80% (vol/vol) ACN–0.1% (vol/vol) TFA in DI water. This buffer is stable for >3 months at RT ! CAUTION TFA solutions are corrosive. Handle with gloves and prepare solutions in a fume hood.
Phosphopeptide enrichment loading buffer for sequential enrichment
This buffer consists of 80% (vol/vol) ACN–1% (vol/vol) TFA in DI water. This buffer is stable for >3 months at RT ! CAUTION TFA solutions are corrosive. Handle with gloves and prepare solutions in a fume hood.
Phosphopeptide enrichment wash buffer 1
This buffer consists of 100 mM glycolic acid– 50% (vol/vol) ACN–1% (vol/vol) TFA in DI water. This buffer is stable for >3 months at RT ! CAUTION TFA and glycolic acid solutions are corrosive. Handle with gloves and prepare solutions in a fume hood.
Phosphopeptide enrichment wash buffer 2
This buffer consists of 25 mM glycolic acid–80% (vol/vol) ACN–0.2% (vol/vol) TFA in DI water. This buffer is stable for >3 months at RT ! CAUTION TFA and glycolic acid solutions are corrosive. Handle with gloves and prepare solutions in a fume hood.
Phosphopeptide enrichment wash buffer 3
This buffer consists of 80% (vol/vol) ACN in DI water. This buffer is stable for >3 months at RT.
Phosphopeptide enrichment elution buffer
This buffer consists of 400 mM ammonium hydroxide solution (NH4OH)–50% (vol/vol) ACN in DI water. This buffer is stable for >3 months at RT ! CAUTION Ammonia solutions are corrosive. Handle with gloves and prepare solutions in a fume hood.
Glycopeptide enrichment resuspension and wash buffer 1
This buffer consists of 50% (vol/vol) ACN–0.1% (vol/vol) TFA in DI water. This buffer is stable for >3 months at RT ! CAUTION TFA solutions are corrosive. Handle with gloves and prepare solutions in a fume hood.
Glycopeptide enrichment wash buffer 2
This buffer consists of 1.5 M NaCl in DI water. Add 2.19 g of NaCl to 25 mL of DI water. This buffer is stable for 6–12 months at RT.
Glycopeptide enrichment oxidation buffer
This buffer consists of 100 mM sodium (meta)periodate in DI water ▲CRITICAL Sodium periodate is light sensitive, so it must be prepared and kept in the dark. This buffer must be prepared fresh, within 1 h of use, and should be stored at 4 °C.
Glycopeptide enrichment quench buffer
This buffer consists of 1 M sodium sulfite in DI water. This buffer is stable at 4 °C for 1 week.
Hydrazide magnetic bead slurry
Combine 500 μL of hydrazide beads with 1 mL of DI water to make slurry. This slurry is stable for 12 months at 4 °C.
Trypsin buffer
This buffer consists of 50 mM TEAB in DI water. Add 100 μL of 1 M TEAB to 1,900 μL of DI water ▲CRITICAL this buffer must be prepared fresh (within 1 h of use) because the pH will fluctuate, therefore reducing its strength.
EV lysis, reduction, and alkylation buffer
This buffer contains 12 mM SDC, 12 mM sarkosyl, 10 mM TCEP-HCl, 40 mM CAA, and phosphatase inhibitor cocktail (1 μL per 100 μL of buffer) in 100 mM Tris-HCl, pH 8.5 ▲CRITICAL this buffer must be prepared fresh because SDC crystallizes and gelates when stored.
Enzyme reconstitution
Dissolve the contents of a 20-μg tube of trypsin in 100 μL of trypsin buffer (depending on amount of trypsin to be added; trypsin buffer volume can vary). Separately, reconstitute a 10-AU (amidase units) vial of endopeptidase Lys-C in 3 mL of DI water. Vortex and centrifuge each vial at 1,000g for 1 min at RT ▲CRITICAL After reconstitution of enzymes, Lys-C should be divided into aliquots and stored for up to 3–6 months at −80 °C; trypsin can be stored for >3 months at −80 °C. Avoid multiple freeze–thaw cycles.
Easy-nLC buffer A
This buffer consists of 0.1% (vol/vol) FA in water, LC/MS grade. This buffer is stable for 6 months at RT.
Easy-nLC buffer B
This buffer consists of 0.1% (vol/vol) FA LC/MS grade in 80% (vol/vol) ACN. This buffer is stable for 6 months at RT.
MS loading buffer
MS loading buffer consists of 0.3% (vol/vol) FA and 3% (vol/vol) ACN in DI water. This buffer is stable for 6–12 months at 4 °C.
Equipment setup
StageTip setup
SDB-XC StageTips are prepared as described in the previous protocol68. We punched a disk using a 16-gauge syringe and placed it at the end of 200-μL pipette tips (~10-μg capacity). This desalting tip is used after glycopeptide enrichment. StageTips can be prepared and stored in a capped pipette box for months at RT.
LC-MS/MS setup
The flow rate is set to 300 nL/min and the parameters are set as follows:
| Time from start (min:s) | Duration (min:s) | Flow (nL/min) | % Buffer B |
|---|---|---|---|
| 00:00 | NA | 300 | 3 |
| 60:00 | 60:00 | 300 | 30 |
| 70:00 | 10:00 | 300 | 50 |
| 71:00 | 01:00 | 300 | 95 |
| 75:00 | 04:00 | 300 | 95 |
NA, not applicable.
The Easy-nLC 1000 is coupled online to a Velos Pro LTQ-Orbitrap mass spectrometer with a 45-cm in-house packed column (360-μm o.d. × 75-μm i.d.) containing C18 resin (2.2 μm, 100Å; Bischoff Chromatography) with a column heater set to 50 °C. The mass spectrometer should be operated in the data-dependent mode, in which the 10 most intense ions are subjected to collision-induced dissociation (CID) fragmentation (normalized collision energy (NCE), 30%; automatic gain control (AGC), 3 × 104; maximum injection time, 100 ms) for each full MS scan (from m/z 350–1,500 with a resolution of 30,000 at m/z 400). See table below for further parameters.
| Parameter | Value |
|---|---|
| Analyzer | FTMS |
| Mass range | Normal |
| Resolution | 30,000 |
| Scan type | Full |
| Polarity | Positive |
| Data type | Centroid |
| Acquire time (min) | 75.00 |
FTMS, Fourier transform mass spectrometry.
Proteome Discoverer parameters
Consensus workflow tree (default settings): MSF (Magellan storage file) files, PSM (peptide spectrum match) grouper, peptide validator, peptide and protein filter, protein scorer, protein grouping, peptide in protein annotation. The processing workflow is as follows:
| Spectrum Files RC | Dynamic modification: phosphorylation on serine, threonine or tyrosine. Deamidation of asparagine residues. Static modification: carbamidomethylation (C). |
| Spectrum Selector | Default |
| PMI-Byonic | Maximum number of missed cleavages: 2 Precursor mass tolerance: 10 p.p.m. Fragment mass tolerance: 0.6 Da Modifications: carbamidomethylation (C), oxidation (M), acetyl protein N-term, deamidation (N) or phosphorylation (S, T, Y). |
Make sure the modifications are changed accordingly. For glycosylation, the modification would be deamidation of asparagine residues, and for phosphorylation analysis, the variable modification would be phosphorylation on serine, threonine or tyrosine residues.
MaxQuant parameters
Initial precursor mass tolerance is set to 20 p.p.m., the final tolerance is set to 6 p.p.m., and ITMS (ion trap mass spectrometry) MS/MS tolerance is set to 0.6 Da. Search criteria include a static carbamidomethylation of cysteines (+57.0214 Da) and variable modifications of oxidation (+15.9949 Da) on methionine residues; acetylation (+42.011 Da) at the N-terminus of the protein; phosphorylation (+79.996 Da) on serine, threonine or tyrosine residues for phosphorylation; and deamidation (+0.984Da) on asparagine residues for glycosylation. Search is performed with trypsin/P digestion (‘trypsin/P’ means enzyme cleaves at carboxyl side of the amino acids lysine or arginine, also if a proline follows) and allows a maximum of two missed cleavages on the peptides analyzed from the sequence database. The false-discovery rates for proteins, peptides and PTM sites are set to 0.01. The minimum peptide length is six amino acids, and the minimum Andromeda score is set to 40 for modified peptides. The glycosylation sites are selected based on matching to the N-X-S/T (X not Pro) motif (N, asparagine; Pro, proline; S, serine; T, threonine; X, any amino acid). A site localization probability of 0.75 is used as the cutoff for localization of glycosylation sites. All the peptide spectral matches and MS/MS spectra can be viewed through MaxQuant viewer. Analyze all data using the Perseus software. For quantification of PTM-omic datasets, the intensities of proteins and PTM sites are derived from MaxQuant, and the missing values of intensities should be replaced by a normal distribution with a downshift of 1.8 s.d. and a width of 0.3 s.d.
Procedure
Preparation of plasma ● Timing 40 min
-
1
Centrifuge ~2 mL (for ~1 mL of plasma) of fresh blood in Vacutainer K2EDTA tubes at 1,300g for 10 min at RT to remove blood cells, debris and large apoptic bodies. Collect the supernatant.
▲CRITICAL STEP To obtain 1 mL of plasma from fresh blood, the optimal volume of starting material is ~2 mL. In our experience, this is the minimal amount of plasma (1 mL) that, upon processing, will produce a visible pellet (Step 7) and an optimal yield for PTM analysis and quantitation. On the basis of our experience, from 1 mL of plasma one can obtain 20–100 μg of protein in Step 16 (before digestion); this can vary depending on the source of plasma, disease, and other factors. For triplicates, we recommend tripling the initial amount of plasma (3 mL) to have enough sample for quantification.
-
2
Centrifuge the supernatant at 4,000g for 15 min at RT to deplete platelets. Collect the supernatant; this should be ~1 mL.
▲CRITICAL STEP Leave 30–50 μL of plasma behind above the buffy layer, taking care not to disturb it.
∎PAUSE POINT The plasma samples can be stored at −80 °C for up to 1–2 years until ready to process. Thaw the plasma samples in a 37 °C water bath immediately before use.
EV purification ● Timing 3–4 h
▲CRITICAL There are several methods for exosome and MV isolations34,37,72,73 (Introduction, ‘Comparison with alternative approaches’ for details). Here we focus on UC to isolate exosomes and MVs.
-
3
Centrifuge the plasma samples at 20,000g for 1 h at 4 °C.
-
4
Collect the supernatant for exosome purification and keep the sample on ice until Step 8. Follow Steps 5–7 in order to also isolate MVs; otherwise, proceed directly to Step 8.
-
5
Wash the pellet with filtered PBS (with the same volume as the initial plasma volume from Step 2).
? TROUBLESHOOTING
-
6
Centrifuge the resuspended pellet at 20,000g for 1 h at 4 °C.
-
7
Remove the supernatant; the pellet contains the MVs.
▲CRITICAL STEP Make sure that you have obtained a complete, solid, small pellet and do not disturb it. Pipette all supernatant carefully.
∎PAUSE POINT The MV pellet can be stored at −80 °C until ready for further analysis (preferably within a week).
-
8
Centrifuge the exosome-containing supernatant from Step 4 using an ultracentrifuge at 100,000g for 1 h at 4 °C.
-
9
Remove the supernatant and wash the pellet, using 1 mL of filtered PBS.
-
10
Centrifuge again at 100,000g for 1 h at 4 °C.
-
11
Remove the supernatant. The pellet contains the exosomes.
▲CRITICAL STEP Exosomes are not as visible as MVs; make sure to pipette carefully near where the pellet should be (it is imperceptible to the eye most of the time) and pipette all supernatant carefully.
∎PAUSE POINT The exosome pellet can be stored at −80 °C until ready for further analysis (preferably within a week).
Characterization of EVs ● Timing variable
-
12
Characterize the heterogeneity, composition and quantity of the EVs, using a method of choice. How extensive the characterization of the EVs should be depends on the specific scientific question asked and on the downstream applications used66. General characterization of EVs includes WB using antibodies against known EV markers such as CD9, CD63, CD81, α-actinin-4, CD40 and mitofilin64,74. The size distribution and concentration of EVs can be estimated using NTA64,65, tunable resistive pulse sensing (TRPS)64,75, or dynamic light scattering (DLS)64,65. In addition, the size and shape can be visualized using electron microscopy (TEM, cryo-EM)64,65,76, or atomic force microscopy (AFM)64,65.
EV lysis, reduction, and alkylation ● Timing 10–20 min
-
13
Solubilize the exosomes and MVs by resuspending them in 100 μL of EV lysis, reduction and alkylation buffer.
▲CRITICAL STEP Exosomes and MVs can be pooled or analyzed separately, depending on downstream applications. If you are pooling MVs and exosomes together, be careful because EV pellets are sticky, and lysis, reduction and alkylation buffer is prone to forming bubbles from the surfactants. Pipette slowly and carefully to avoid sample loss and excessive bubble formation.
▲CRITICAL STEP Protease inhibitors should be avoided because they will impact Lys-C and trypsin digestion (Steps 17–18), so we typically do not add any protease inhibitor to the lysis buffer.
? TROUBLESHOOTING
-
14
After solubilizing, heat the sample at 95 °C for 5 min.
-
15
Dilute the alkylated proteins fivefold by adding 400 μL of trypsin buffer.
-
16
Perform a BCA assay according to the manufacturer’s instructions to determine the protein concentration.
▲CRITICAL STEP Make sure to use the EV lysis, reduction and alkylation buffer diluted fivefold with trypsin buffer as a blank when performing BCA.
▲CRITICAL STEP We routinely obtain ~60 μg of EV proteins per 1 mL of plasma, and we assume ~<1% of the peptides are phosphorylated/glycosylated.
▲CRITICAL STEP When performing total proteome analysis, transfer ~2–5% (~1 μg of peptides) of the sample (Step 28) to a separate tube to be processed separately. The rest of the peptide sample is used for sequential phosphopeptide and glycopeptide enrichment.
Enzymatic digestion ● Timing 16–18 h
-
17
Digest 20–100 μg of the sample with Lys-C in a 1:100 (wt/wt) enzyme/protein ratio for 3 h at 37 °C. The total volume should be 500 μL.
-
18
Add trypsin to a final 1:50 (wt/wt) enzyme/protein ratio and incubate overnight at 37 °C.
-
19
Acidify the digested peptides by adding TFA to a final concentration of 0.5% (vol/vol) and add 500 μL of ethyl acetate to 500 μL of digested solution.
-
20
Vortex the solution for 2 min, and then centrifuge at 20,000g for 2 min at RT to obtain aqueous and organic phases. Discard the top organic layer.
▲CRITICAL STEP Remove the upper layer very carefully and do not disturb the lower layer or the interphase.
-
21
Repeat Steps 19 and 20 to remove as much detergent residue as possible (adding ethyl acetate, vortexing and discarding top organic layer; do not add TFA again). Collect the aqueous phase and dry it in a vacuum centrifuge.
∎PAUSE POINT If not processed immediately, digested peptides can be stored at −80 °C for up to 3 months.
C18 desalting68 ● Timing 1 h
▲CRITICAL The choice of Sep-Pak column/cartridge depends on the amount of recovered EV proteins and the experimental design. For one EV pellet from 1 mL, one typically obtains 20–100 μg of protein. We use a 50-mg column, which can desalt ~500 μg of peptides. Other columns, such as a 100-mg column, can desalt ~1,000 μg of peptides, and so on. Other desalting methods can be used as well, but buffers may need to be adjusted.
-
22
Reconstitute the dried sample from Step 21 in up to 1 mL of buffer A for Sep-Pak desalting.
-
23
Condition the cartridge by passing 1 mL of methanol through the packing bed.
▲CRITICAL STEP Do not allow air to enter the packing material at any step.
-
24
Equilibrate the cartridge by passing 1 mL of buffer B for Sep-Pak desalting through the packing bed.
-
25
Wash the cartridge by passing 1 mL of buffer A for Sep-Pak desalting through the packing bed.
-
26
Load the sample slowly onto the cartridge. Complete loading by slowly passing 1 mL of buffer A for Sep-Pak desalting onto the cartridge.
-
27
Wash the cartridge 3–5 times with 1 mL of buffer A for Sep-Pak desalting each time.
-
28
Elute the sample by slowly adding 1 mL of buffer B for Sep-Pak desalting. After elution, 2–5% of the collected sample can be separated for total proteomics analysis (Step 51 onward). The remaining sample is used for sequential phosphopeptide and glycopeptide enrichment.
-
29
Dry the total proteomics sample and the sample for sequential phosphopeptide/glycopeptide enrichment using a vacuum centrifuge.
∎PAUSE POINT If not processed immediately, both the proteomics sample and the sample for sequential enrichment can be stored at −80 °C for up to 3 months.
Phosphopeptide enrichment ● Timing 1–2 h
▲CRITICAL We typically enrich phosphopeptides using a Spin-Tip PolyMAC-Ti kit77. However, several other suitable phosphopeptide enrichment reagents are available78,79. For all steps involving tip and frit, centrifugation durations indicated are an estimate; spin times and speeds may need to be adjusted to pass buffers completely through.
-
30
Resuspend the dried peptides from Step 29 in 200 μL of phosphopeptide enrichment loading buffer for sequential enrichment and incubate with 50 μL of PolyMAC-Ti beads for 15 min.
-
31
Load the beads into the tip with a frit (included in the PolyMAC kit). Collect the flow-through for glycopeptide enrichment in Steps 37–44 and dry the sample using a vacuum centrifuge.
∎PAUSE POINT If not processed immediately, store the dried sample at −80 °C for up to a month.
-
32
Wash the beads in the tip with the frit by adding 200 μL of phosphopeptide enrichment wash buffer 1 to the tip and centrifuge at 20g for 2 min at RT, followed by 100g for 1 min at RT or until the beads are dry.
-
33
Wash the beads in the tip with the frit by adding 200 μL of phosphopeptide enrichment wash buffer 2 to the tip and centrifuge as described in Step 32 or until the beads are dry.
-
34
Wash the beads in the tip with the frit by adding 200 μL of phosphopeptide enrichment wash buffer 3 to the tip and centrifuge as described in Step 32 or until the beads are dry.
-
35
Elute the phosphopeptides from the beads by adding 50 μL of phosphopeptide enrichment elution buffer to the tip and centrifuging as described in Step 32. Collect the eluate.
-
36
Repeat Step 35, combine the two eluates and dry the sample in a vacuum centrifuge.
∎PAUSE POINT If not processed immediately, phosphopeptide dry samples can be stored at −80 °C for up to a week. On the basis of our experience, we recommend processing samples within 1–2 d of enrichment.
Glycopeptide enrichment ● Timing ~8 h
-
37
Resuspend the dried peptides from Step 31 in ~100 μL of glycopeptide enrichment resuspension and wash buffer 1. Make sure to vortex until the peptides are dissolved.
-
38
Oxidize the peptides by adding 11 μL of glycopeptide enrichment oxidation buffer (10 mM final concentration). Incubate for 1 h at RT with shaking in the dark at 1,500 r.p.m.
-
39
Quench excess sodium periodate by adding 7 μL of glycopeptide enrichment quench buffer (50 mM final concentration). Incubate for 15 min at RT with shaking in the dark at 1,500 r.p.m.
? TROUBLESHOOTING
-
40
Add 50 μL of hydrazide magnetic bead slurry to the sample and incubate for 3 h at RT with shaking at 1,500 r.p.m.
▲CRITICAL STEP The coupling reaction must be carried out for a minimum time of 3 h, but it can be left overnight without affecting the results.
-
41
Wash the magnetic beads three times with 400 μL of glycopeptide enrichment resuspension and wash buffer 1 for 1 min per wash, followed by three washes with 400 μL of glycopeptide enrichment wash buffer 2 to remove non-coupled peptides. For washes, incubate the sample for 1 min with glycopeptide enrichment resuspension and wash buffer 1 or glycopeptide enrichment wash buffer 2 with shaking at 1,500 r.p.m. (not in the dark), centrifuge at RT for 5 s at 20g (gently; this is only to bring the solution down), and pipette the buffer, using a magnetic rack. Remove the buffer after the final washing step.
-
42
Add 100 μL of 1× GlycoBuffer 2 (from the PNGase F kit) to the beads, vortex, and centrifuge as described in Step 41. Remove the supernatant and incubate the sample with 3 μL of PNGase F in 100 μL of 1× GlycoBuffer 2 (from the PNGase F kit) at 37 °C with shaking at 1500 r.p.m. for 2 h. After incubation, centrifuge the sample as described in Step 41.
-
43
Transfer the supernatant to a new tube and rinse the beads once with 100 μL of 1× GlycoBuffer 2 (from the PNGase F kit). Centrifuge as in Step 41 and transfer the supernatant to the new tube, using magnetic rack. Total volume in new tube should be ~200 μL.
-
44
Acidify the glycopeptides by adding TFA to a final concentration of 1% (vol/vol).
∎PAUSE POINT We recommend desalting after this step. If not possible, the sample can be stored at 4 °C for 2–3 h.
StageTip desalting of glycopeptides ● Timing 1 h
-
45
Activate the SDB-XC StageTip membrane by adding 20 μL of buffer B for both Sep-Pak and SDB-XC desalting to the prepared tip, and spin down for 1 min at 1,000g at RT.
-
46
Add 20 μL of buffer A for SDB-XC desalting, and spin down for 1 min at 1,000g at RT.
-
47
Load the acidified sample from Step 44, and spin it down for 5 min at 1,000g at RT.
-
48
Wash the membrane by adding 20 μL of buffer A for SDB-XC desalting, and spin down for 1 min at 1,000g at RT.
-
49
Elute by adding 20 μL of buffer B for both Sep-Pak and SDB-XC desalting, and spin down for 5 min at 500g at RT. Collect the eluates into clean PCR strip tubes.
? TROUBLESHOOTING
▲CRITICAL STEP Elution might take longer, depending on the amount of peptides or any remaining hydrazide beads.
-
50
Dry the eluates in a vacuum centrifuge
PAUSE POINT Dried peptides can be stored for several days at 4 °C, for a couple of weeks at −20 °C or for several months at −80 °C, provided that they are sealed airtight.
LC-MS/MS ● Timing ~1 h per measurement
-
51
Dissolve dried peptides from Step 29 (total proteome), Step 36 (phosphopeptides) and Step 50 (glycopeptides) each in ~4.5 μL of MS loading buffer and inject into the Easy-nLC 1000 LC instrument. Place the samples in an LC autosampler cooled to 4–8 °C. We usually inject samples directly from the PCR tubes in which they were dried.
-
52
Analyze each sample by mass spectrometry, using the parameters outlined in the ‘Equipment setup’ section. For each analysis, we usually load 4 μL of sample at a constant pressure of 650 bar.
Data analysis ● Timing >1 d
-
53
Search the raw files with Proteome Discoverer for ID numbers between technical replicates and search using MaxQuant software with the Andromeda search engine for quantitation and Pearson correlation scores between replicates (downstream bioinformatics analysis using the Perseus platform)80–83.
▲CRITICAL STEP For both MaxQuant and Proteome Discoverer, default settings are typically ideal, with most unchanged except for a few parameters, such as variable modifications, as described in the ‘Equipment setup’ section.
? TROUBLESHOOTING
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 5 | No pellet observed after MV isolation | A different collection or storage method for plasma (different collection tubes, centrifugation times, among others) was used | If plasma is obtained from a third party, make sure the isolation method for plasma is consistent with Steps 1 and 2 |
| An insufficient amount of plasma (<1 mL) was initially used | Make sure to use at least 1 mL of plasma | ||
| Isolation of plasma after blood draw took too much time, causing loss/degradation of the sample | Be sure to isolate the plasma promptly after blood draw | ||
| Plasma was frozen and then was not thawed according to the instructions above, causing loss/degradation of the sample | Follow the instructions given for thawing of plasma | ||
| 13 | Highly viscous and/or cloudy samples are obtained after lysis | Some patients’ plasma is more viscous than others. This can be associated with different diseases | A certain level of cloudiness is normal. Sonicate the samples to reduce viscosity before proceeding to the next step and/or increase the EV lysis volume |
| Samples from different patients were pooled | Sonicate the samples to reduce viscosity before proceeding to the next step and/or increase the EV lysis volume | ||
| The samples contain a high protein content | Sonicate the samples to reduce viscosity before proceeding to the next step and/or increase the EV lysis volume | ||
| 39 | A yellowish sample was obtained after quenching with sodium sulfite | The solution was not quenched completely or the sample contains a low amount of glycopeptides | Add an extra 2-3 μL of 1 M sodium sulfite and incubate for 10 min with shaking in the dark |
| 49 | The StageTips clog | If a StageTip flow is slow or stops during glycopeptide desalting, it usually is due to remaining hydrazide beads or any other material in the solution | Make sure to avoid taking any remaining material, especially beads, while transferring the eluates from the hydrazide beads. You can also increase the spin time by a couple of minutes and set the centrifugation speed 200g higher |
| StageTip clogging can also be due to excessive force used to press the material into the tip. | Make sure to change packed tip and that the material in the tip is not pressed too tightly. | ||
| 53 | Plasma proteins are highly abundant | This usually happens when not all supernatant is removed at the time of EV isolation (Steps 3-11). | The MV pellet is usually solid enough to pipette everything out without the pellet being disturbed. The same is true for the exosome pellet, even if this pellet is not visible (most of the time). Make sure to remove all of the supernatant |
| The yield of glycopeptides and phosphopeptides is low | Some common issues that lead to poor recovery are inefficient proteome digestion, use of old buffers, and sample contamination during the enrichment steps | If the yield is lower than expected, several parameters should be revised in the data search, including the digestion parameters, and phosphopeptide and glycopeptide enrichment specificity (selectivity). If large numbers of non-phosphorylated or non-glycosylated peptides are observed, it is most likely due to inefficient washing of the beads |
Timing
Steps 1 and 2, preparation of plasma: 40 min
Steps 3–11, EV purification: 3–4 h
Step 12, characterization of EVs: variable
Steps 13–16, EV lysis, reduction, and alkylation: 10–20 min
Steps 17–21, enzymatic digestion: 16–18 h
Steps 22–29, C18 desalting: 1 h
Steps 30–36, phosphopeptide enrichment: 1–2 h
Steps 37–44, glycopeptide enrichment: ~8 h
Steps 45–50, StageTip desalting of glycopeptides: 1 h
Steps 51 and 52, LC-MS/MS: ~1 h per measurement
Step 53, data analysis: >1 d
Anticipated results
We previously identified nearly 10,000 unique phosphopeptides22 and 1,500 unique glycopeptides23 in EVs using several milliliters of human plasma by analyzing the samples separately. Here, we have described an updated protocol that allows simultaneous analysis of the EV phosphoproteome and N-glycoproteome. This combined workflow is especially suitable for situations in which the amount of starting material is limited, as is often the case for clinical samples. To compare the performance of the separate phosphoproteomics and N-glycoproteomics approaches to the sequential workflow, we carried out label-free quantitation of the sequential and separate enrichment procedures using the same plasma samples. We started with a total of 9 mL of human plasma (Fig. 2): 3 mL for three technical replicates of sequential enrichment of phosphopeptides and N-glycopeptides, 3 mL for three technical replicates of the enrichment of phosphopeptides only, and 3 mL for three technical replicates of the enrichment of N-glycopeptides only. Using a 60-min LC gradient on a high-resolution Orbitrap mass spectrometer, we identified ~4,000 unique EV phosphopeptides for each 1 mL of human plasma, representing ~1,300 phosphoproteins in each sequential or separate replicate (Fig. 3). Similar to the previous study25, the selectivity (the percentage of PTM peptides over total peptides) for EV phosphopeptide identification from plasma samples (65–70%) was lower than phosphopeptide identification from a typical whole-cell extract (>90%), which is probably due to the small amount of starting materials and potential interference from EV components. The low-pH buffer condition for sequential enrichment led to only a 7% decrease in phosphopeptide identification number, and the correlation coefficients decreased from 0.94 (average) among technical replicates to 0.87 (average) between sequential and separate enrichments. Similarly, we identified virtually the same number of unique EV N-glycopeptides for each 1 mL of human plasma, representing ~650 glycoproteins in either sequential or separate replicates. We saw a moderate decrease in the selectivity (from 54 to 42%; Fig. 4) and correlation coefficients (from 0.97 on average to 0.85 on average between sequential and separate workflows; Fig. 5). Overall, our data indicate that the sequential enrichment of phosphopeptides and N-glycopeptides offers greater benefit by achieving similar numbers of identified PTMs with half the amount of starting material.
Fig. 2 |. Schematic of experimental setup for sequential and separate phosphopeptide and glycopeptide enrichments.
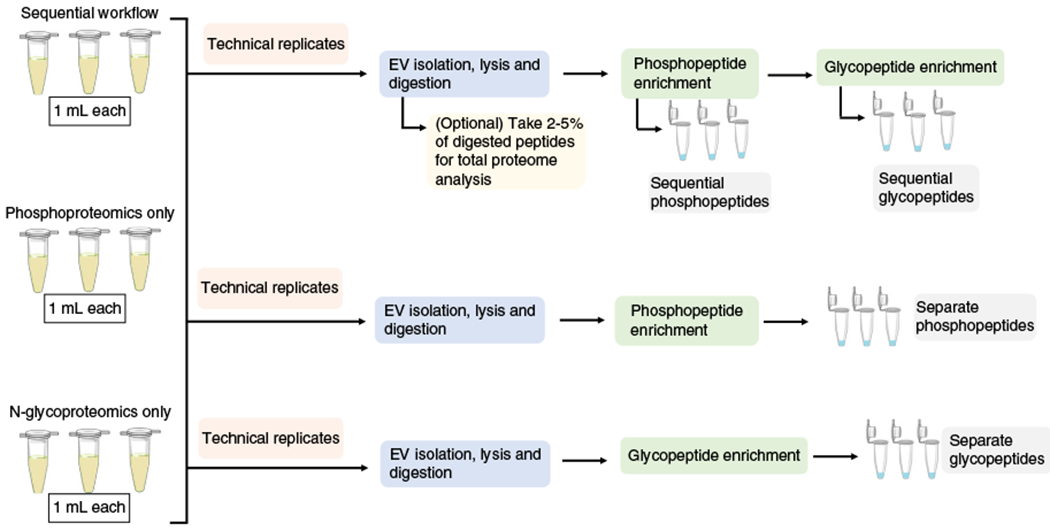
For the sequential analysis of both phosphopeptides and glycopeptides, the workflow as shown in Fig. 1 is used. For the separate enrichment procedures, only one modification is enriched, by either phosphorylation (by skipping Steps 37-44) or glycosylation (by skipping Steps 30-36).
Fig. 3 |. Venn diagrams showing the phosphopeptide and phosphoprotein overlap between replicates and the overlap between the separate and sequential workflows.
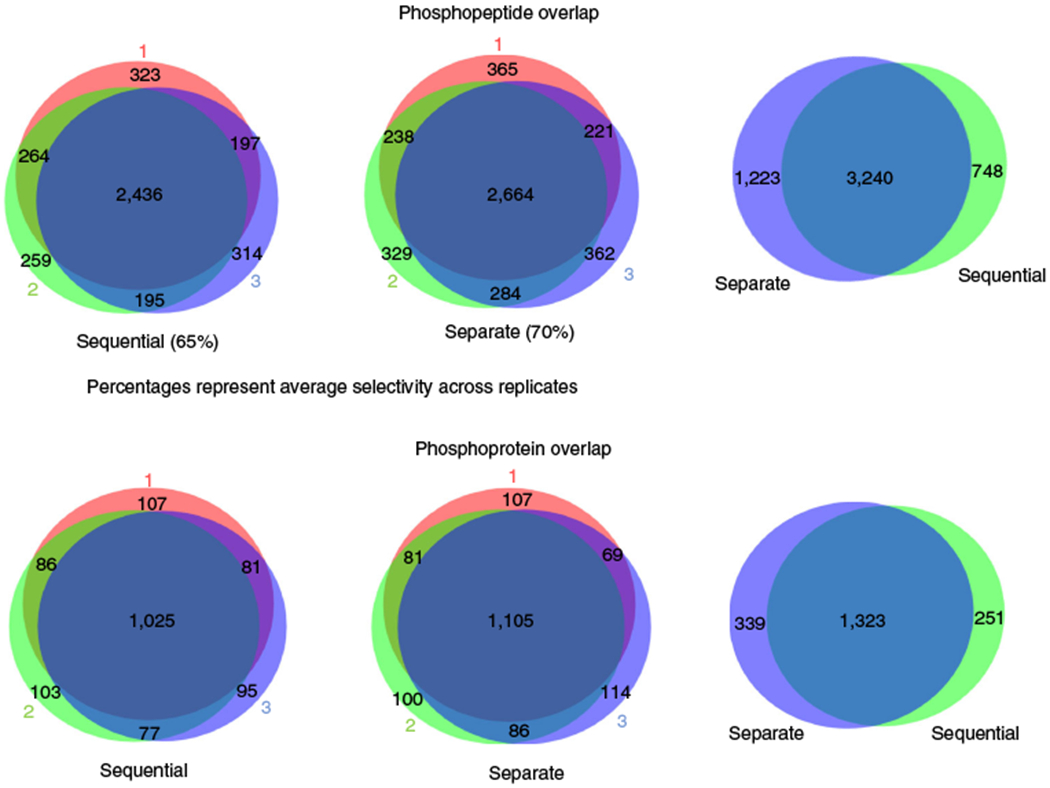
The overlaps between replicates are shown at the phosphoprotein and phosphopeptide level (left-hand and center diagrams). Selectivity across replicates and the overlap between the results obtained from the separate workflows and the sequential workflows (right-hand diagrams) are depicted.
Fig. 4 |. Venn diagrams showing the glycopeptide and glycoprotein overlap between replicates and the overlap between the separate and sequential workflows.
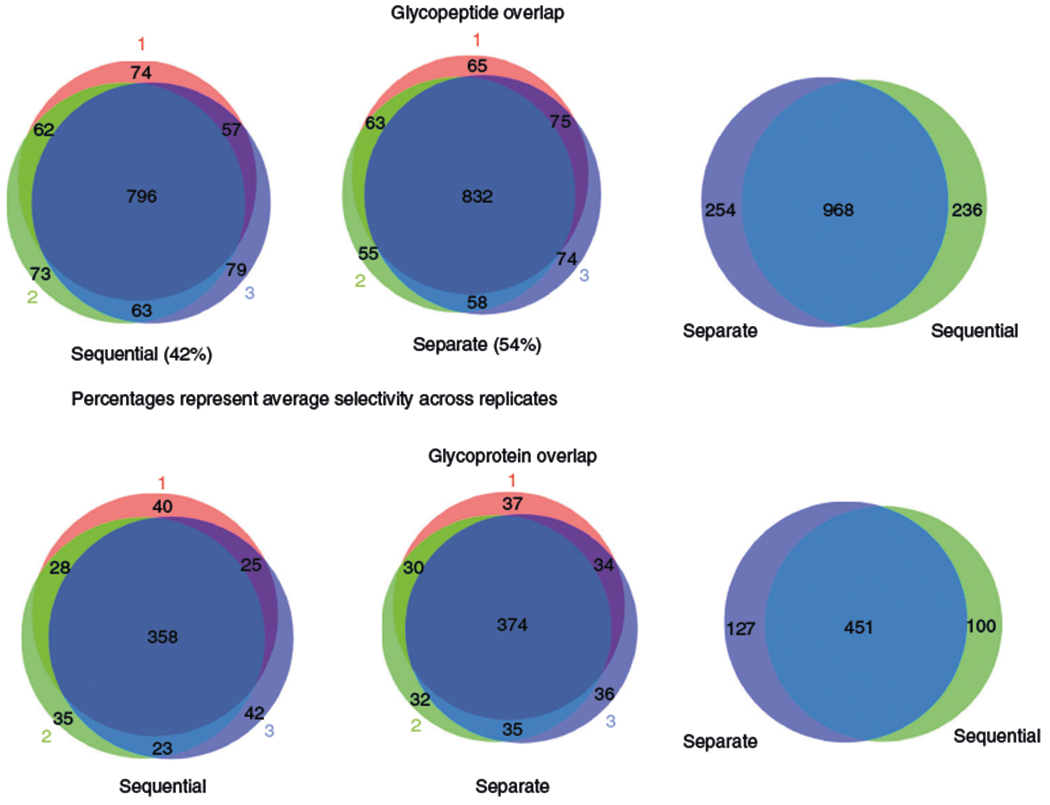
The overlaps between replicates are shown at the glycoprotein and glycopeptide level (left-hand and center diagrams). Selectivity across replicates and the overlap between the results obtained from the separate workflows and the sequential workflows (right-hand diagrams) are depicted.
Fig. 5 |. Quantitation results from MaxQuant and Perseus showing Pearson correlations across each condition and replicate.
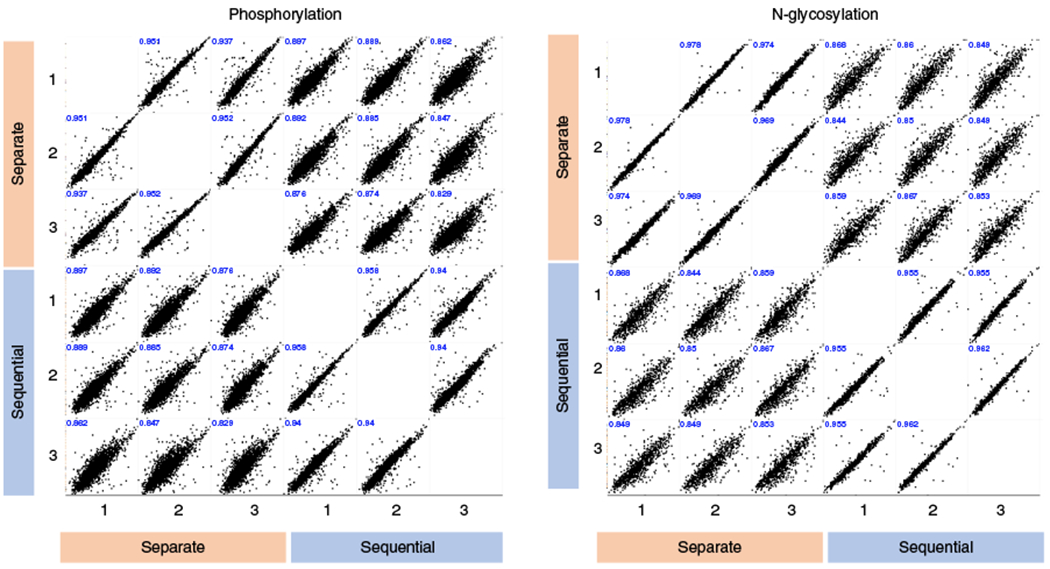
Scatterplots and Pearson correlation coefficients depicting the log2-tranformed intensities of phosphopeptides and glycopeptides across both workflows (sequential versus separate) and three technical replicates.
We further evaluated the level of variation in the composition of the EVs among individual samples from healthy donors. To do this, we carried out targeted analyses on five selected EV phosphopetides using PRM84–86 (Supplementary Table 2 and Supplementary Fig. 3). The measured coefficient of variation (CV) values between the samples from the five individuals are relatively high, indicating either a large variation in the concentration of these proteins in the EV and/or variation in the extent to which these peptides are modified between individuals.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The raw MS data from this study have been deposited into the ProteomeXchange Consortium via the PRIDE Archive with the identifier PXD013893 (https://www.ebi.ac.uk/pride/archive/projects/PXD013893).
Supplementary Material
Acknowledgements
This project was funded by NIH grants 5R01GM088317 and 1R01GM111788 and NSF grant 1506752.
Footnotes
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41596-019-0260-5.
Competing interests
A.B.I and W.A.T. are the co-founders of Tymora Analytical Operations, which commercialized a polyMAC kit used in parts of the described protocol.
References
- 1.Harel M, Oren-Giladi P, Kaidar-Person O, Shaked Y & Geiger T Proteomics of microparticles with SILAC Quantification (PROMIS-Quan): a novel proteomic method for plasma biomarker quantification. Mol. Cell Proteom. 14, 1127–1136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Milane L, Singh A, Mattheolabakis G, Suresh M & Amiji MM Exosome mediated communication within the tumor microenvironment. J. Control Release 219, 278–294 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Cocucci E & Meldolesi J Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. 25, 364–372 (2015). [DOI] [PubMed] [Google Scholar]
- 4.An T et al. Exosomes serve as tumour markers for personalized diagnostics owing to their important role in cancer metastasis. J. Extracell. Vesicles 4, 27522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobrowolski R & De Robertis EM Endocytic control of growth factor signalling: multivesicular bodies as signalling organelles. Nat. Rev. Mol. Cell Biol. 13, 53–60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melo SA et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 523, 177–182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzales PA et al. Large-scale proteomics and phosphoproteomics of urinary exosomes. J. Am. Soc. Nephrol. 20, 363–379 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boukouris S & Mathivanan S Exosomes in bodily fluids are a highly stable resource of disease biomarkers. Proteom. Clin. Appl. 9, 358–367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu AY, Ueda K & Lai CP Proteomic analysis of extracellular vesicles for cancer diagnostics. Proteomics 19, e1800162 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Xu R, Greening DW, Zhu HJ, Takahashi N & Simpson RJ Extracellular vesicle isolation and characterization: toward clinical application. J. Clin. Invest. 126, 1152–1162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bandu R, Oh JW & Kim KP Mass spectrometry-based proteome profiling of extracellular vesicles and their roles in cancer biology. Exp. Mol. Med. 51, 30 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emmanouilidi A, Paladin D, Greening DW & Falasca M Oncogenic and non-malignant pancreatic exosome cargo reveal distinct expression of oncogenic and prognostic factors involved in tumor invasion and metastasis. Proteomics 19, e1800158 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Hurwitz SN & Meckes DG Jr. Extracellular vesicle integrins distinguish unique cancers. Proteomes 7, 7020014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bae S, Brumbaugh J & Bonavida B Exosomes derived from cancerous and non-cancerous cells regulate the anti-tumor response in the tumor microenvironment. Genes Cancer 9, 87–100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh A et al. Rapid isolation of extracellular vesicles from cell culture and biological fluids using a synthetic peptide with specific affinity for heat shock proteins. PLoS One 9, e110443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lobb RJ et al. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 4, 27031 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallart-Palau X et al. Extracellular vesicles are rapidly purified from human plasma by potein organic solvent precipitation (PROSPR). Sci. Rep. 5, 14664 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moreno-Gonzalo O, Villarroya-Beltri C & Sanchez-Madrid F Post-translational modifications of exosomal proteins. Front. Immunol. 5, 383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Wu X & Tao WA Characterization and applications of extracellular vesicle proteome with post-translational modifications. Trends Analyt. Chem. 107, 21–30 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerlach JQ & Griffin MD Getting to know the extracellular vesicle glycome. Mol. Biosyst. 12, 1071–1081 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Oeyen E et al. Bladder cancer diagnosis and follow-up: the current status and possible role of extracellular vesicles. Int. J. Mol. Sci. 20, 821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mann M & Jensen ON Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Aebersold R & Goodlett DR Mass spectrometry in proteomics. Chem. Rev. 101, 269–295 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Kettenbach AN, Rush J & Gerber SA Absolute quantification of protein and post-translational modification abundance with stable isotope-labeled synthetic peptides. Nat. Protoc. 6, 175–186 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen IH et al. Phosphoproteins in extracellular vesicles as candidate markers for breast cancer. Pro. c. Natl Acad. Sci. USA 114, 3175–3180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen IH et al. Analytical pipeline for discovery and verification of glycoproteins from plasma-derived extracellular vesicles as breast cancer biomarkers. Anal. Chem. 90, 6307–6313 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Jaros JA et al. Clinical use of phosphorylated proteins in blood serum analysed by immobilised metal ion affinity chromatography and mass spectrometry. J. Proteom. 76(Spec No), 36–42 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Hu L et al. Profiling of endogenous serum phosphorylated peptides by titanium (IV) immobilized mesoporous silica particles enrichment and MALDI-TOFMS detection. Anal. Chem. 81, 94–104 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Sokolova V et al. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids Surf. B Biointerfaces 87, 146–150 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Palmisano G et al. Characterization of membrane-shed microvesicles from cytokine-stimulated beta-cells using proteomics strategies. Mol. Cell. Proteom. 11, 230–243 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Royo F et al. Different EV enrichment methods suitable for clinical settings yield different subpopulations of urinary extracellular vesicles from human samples. J. Extracell. Vesicles 5, 29497 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cvjetkovic A, Lotvall J & Lasser C The influence of rotor type and centrifugation time on the yield and purity of extracellular vesicles. J. Extracell. Vesicles 3, 1–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Enderle D et al. Characterization of RNA from exosomes and other extracellular vesicles isolated by a novel spin column-based method. PLoS One 10, e0136133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niu Z et al. Polymer-based precipitation preserves biological activities of extracellular vesicles from an endometrial cell line. PLoS One 12, e0186534 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu LL et al. A comparison of traditional and novel methods for the separation of exosomes from human samples. Biomed. Re. s. Int. 2018, 3634563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tauro BJ et al. Two distinct populations of exosomes are released from LIM1863 colon carcinoma cell-derived organoids. Mol. Cell. Proteom. 12, 587–598 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu X, Li L, Iliuk A & Tao WA Highly efficient phosphoproteome capture and analysis from urinary extracellular vesicles. J. Proteome Res. 17, 3308–3316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramirez MI et al. Technical challenges of working with extracellular vesicles. Nanoscale 10, 881–906 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Contreras-Naranjo JC, Wu HJ & Ugaz VM Microfluidics for exosome isolation and analysis: enabling liquid biopsy for personalized medicine. Lab Chip 17, 3558–3577 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosa-Fernandes L, Rocha VB, Carregari VC, Urbani A & Palmisano G A perspective on extracellular vesicles proteomics. Front Chem. 5, 102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wisniewski JR, Zougman A, Nagaraj N & Mann M Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Heath N et al. Rapid isolation and enrichment of extracellular vesicle preparations using anion exchange chromatography. Sci. Rep. 8, 5730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feist P & Hummon AB Proteomic challenges: sample preparation techniques for microgram-quantity protein analysis from biological samples. Int. J. Mol. Sci. 16, 3537–3563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gundry RL et al. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr. Protoc. Mol. Biol. Unit 10.25, supplement 88 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bereman MS, Egertson JD & MacCoss MJ Comparison between procedures using SDS for shotgun proteomic analyses of complex samples. Proteomics 11, 2931–2935 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsu CC et al. Universal plant phosphoproteomics workflow and its application to tomato signaling in response to cold stress. Mol. Cell. Proteom. 17, 2068–2080 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bayramoglu G, Celikbicak O, Arica MY & Salih B Trypsin Immobilized on Magnetic Beads via Click Chemistry: Fast Proteolysis of Proteins in a Microbioreactor for MALDI-ToF-MS Peptide Analysis. Ind. Eng. Chem. Res. 53, 4554–4564 (2014). [Google Scholar]
- 48.Sielaff M et al. Evaluation of FASP, SP3, and iST protocols for proteomic sample preparation in the low microgram range. J. Proteome Res. 16, 4060–4072 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Ludwig KR, Schroll MM & Hummon AB Comparison of in-solution, FASP, and S-trap based digestion methods for nottom-up proteomic studies. J. Proteome Res. 17, 2480–2490 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swaney DL & Villen J Enrichment of phosphopeptides via immobilized metal affinity chromatography. Cold Spring Harb. Protoc. 2016, 088005 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Fila J & Honys D Enrichment techniques employed in phosphoproteomics. Amino Acids 43, 1025–1047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yue X, Schunter A & Hummon AB Comparing multistep immobilized metal affinity chromatography and multistep TiO2 methods for phosphopeptide enrichment. Anal. Chem. 87, 8837–8844 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Montoya A, Beltran L, Casado P, Rodriguez-Prados JC & Cutillas PR Characterization of a TiO(2) enrichment method for label-free quantitative phosphoproteomics. Methods 54, 370–378 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang J et al. Highly efficient release of glycopeptides from hydrazide beads by hydroxylamine assisted PNGase F deglycosylation for N-glycoproteome analysis. Anal. Chem. 87, 10199–10204 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Zhang H, Li XJ, Martin DB & Aebersold R Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 21, 660–666 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Cao W, Huang J, Jiang B, Gao X & Yang P Highly selective enrichment of glycopeptides based on zwitterionically functionalized soluble nanopolymers. Sci. Rep. 6, 29776 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu R, Zacharias L, Wooding KM, Peng W & Mechref Y Glycoprotein enrichment analytical techniques: advantages and disadvantages. Methods Enzymol. 585, 397–429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang C et al. Evaluation of different N-glycopeptide enrichment methods for N-glycosylation sites mapping in mouse brain. J. Proteome Res. 15, 2960–2968 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Nilsson J et al. Enrichment of glycopeptides for glycan structure and attachment site identification. Nat. Methods 6, 809–811 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Yang W et al. Comparison of enrichment methods for intact N- and O-linked glycopeptides using strong anion exchange and hydrophilic interaction liquid chromatography. Anal. Chem. 89, 11193–11197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liebler DC & Zimmerman LJ Targeted quantitation of proteins by mass spectrometry. Biochemistry 52, 3797–3806 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Osinalde N, Aloria K, Omaetxebarria MJ & Kratchmarova I Targeted mass spectrometry: An emerging powerful approach to unblock the bottleneck in phosphoproteomics. J. Chromatogr. B 1055-1056, 29–38 (2017). [DOI] [PubMed] [Google Scholar]
- 63.Thery C et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7, 1535750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shao H et al. New technologies for analysis of extracellular vesicles. Chem. Rev. 118, 1917–1950 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Szatanek R et al. The methods of choice for extracellular vesicles (EVs) characterization. Int. J. Mol. Sci. 18, 1153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lotvall J et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 3, 26913 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Masuda T, Saito N, Tomita M & Ishihama Y Unbiased quantitation of Escherichia coli membrane proteome using phase transfer surfactants. Mol. Cell. Proteom. 8, 2770–2777 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rappsilber J, Mann M & Ishihama Y Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Humphrey SJ, Karayel O, James DE & Mann M High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nat. Protoc. 13, 1897–1916 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Mertins P et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 13, 1632–1661 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hernandez-Valladares M et al. Reliable FASP-based procedures for optimal quantitative proteomic and phosphoproteomic analysis on samples from acute myeloid leukemia patients. Biol. Proced. Online 18, 13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Witwer KW et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2, 20360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakai W et al. A novel affinity-based method for the isolation of highly purified extracellular vesicles. Sci. Rep. 6, 33935 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kowal J et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl Acad. Sci. USA 113, E968–977 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Coumans FA et al. Reproducible extracellular vesicle size and concentration determination with tunable resistive pulse sensing. J. Extracell. Vesicles 3, 25922 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yuana Y et al. Cryo-electron microscopy of extracellular vesicles in fresh plasma. J. Extracell. Vesicles 2, 21494 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iliuk AB, Martin VA, Alicie BM, Geahlen RL & Tao WA In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion-functionalized soluble nanopolymers. Mol. Cell. Proteom. 9, 2162–2172 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ye J et al. Optimized IMAC-IMAC protocol for phosphopeptide recovery from complex biological samples. J. Proteome Res. 9, 3561–3573 (2010). [DOI] [PubMed] [Google Scholar]
- 79.Larsen MR, Thingholm TE, Jensen ON, Roepstorff P & Jorgensen TJ Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteom. 4, 873–886 (2005). [DOI] [PubMed] [Google Scholar]
- 80.Tyanova S, Temu T & Cox J The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Cox J & Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 82.Tyanova S et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
- 83.Tyanova S & Cox J Perseus: a bioinformatics platform for integrative analysis of proteomics data in cancer tesearch. Methods Mol. Biol. 1711, 133–148 (2018). [DOI] [PubMed] [Google Scholar]
- 84.Rauniyar N Parallel reaction monitoring: a targeted experiment performed using high resolution and high mass accuracy mass spectrometry. Int. J. Mol. Sci. 16, 28566–28581 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cordwell SJ & White MY Targeted proteomics for determining phosphorylation site-specific associations in cardiovascular disease. Circulation 126, 1803–1807 (2012). [DOI] [PubMed] [Google Scholar]
- 86.Zauber H, Kirchner M & Selbach M Picky: a simple online PRM and SRM method designer for targeted proteomics. Nat. Methods 15, 156–157 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw MS data from this study have been deposited into the ProteomeXchange Consortium via the PRIDE Archive with the identifier PXD013893 (https://www.ebi.ac.uk/pride/archive/projects/PXD013893).